Role of Glia in Memory Deficits Following Traumatic Brain Injury: Biomarkers of Glia Dysfunction
 Venkata S. S. S. Sajja
Venkata S. S. S. Sajja Nora Hlavac
Nora Hlavac Pamela J. VandeVord
Pamela J. VandeVord- 1Cellular Imaging Section and Vascular Biology Program, Department of Radiology and Radiological Science, Institute for Cell Engineering, Johns Hopkins University School of Medicine, Baltimore, MA, USA
- 2Department of Biomedical Engineering and Mechanics, Virginia Tech University, Blacksburg, VA, USA
Historically, glial cells have been recognized as a structural component of the brain. However, it has become clear that glial cells are intimately involved in the complexities of neural networks and memory formations. Astrocytes, microglia, and oligodendrocytes have dynamic responsibilities which substantially impact neuronal function and activities. Moreover, the importance of glia following brain injury has come to the forefront in discussions to improve axonal regeneration and functional recovery. The numerous activities of glia following injury can either promote recovery or underlie the pathobiology of memory deficits. This review outlines the pathological states of glial cells which evolve from their positive supporting roles to those which disrupt synaptic function and neuroplasticity following injury. Evidence suggests that glial cells interact extensively with neurons both chemically and physically, reinforcing their role as pivotal for higher brain functions such as learning and memory. Collectively, this mini review surveys investigations of how glial dysfunction following brain injury can alter mechanisms of synaptic plasticity and how this may be related to an increased risk for persistent memory deficits. We also include recent findings, that demonstrate new molecular avenues for clinical biomarker discovery.
Introduction
It is generally accepted, that neurons make up less than 25% of the cells in the brain, yet are responsible for information processing and control of bodily functions. Astrocytes, which make up 30–65% of glia and are the most abundant cell type in the brain, are multifunctional cells whose roles include maintaining osmotic balance and optimal ionic conditions for neurons, information processing via neurotransmitter recycling, and metabolite homeostasis (Kimelberg, 2005; Buffo et al., 2010; Kimelberg and Nedergaard, 2010). Collectively, these functions, as well as others, make the astrocytes indirectly involved in all brain function including memory formation (Moraga-Amaro et al., 2014). Microglia compose approximately 10% of total glia in the brain and are mainly identified by their function as immune cells of the central nervous system (CNS), arriving first at the injury site to initiate the inflammatory cascade. However, evidence indicates that “resting” microglia play a critical role in regulating synaptic and structural plasticity during learning and memory (Kettenmann et al., 2011, 2013; Scheff et al., 2013). Lastly, oligodendrocytes provide support to axons with the production of the myelin sheath, which is vital for fast impulse conduction through the white matter (WM) tracts. These rapid interactions between brain regions are required for higher order brain functions like memory formation. Because of their high metabolic rates, oligodendrocytes are susceptible to the molecular consequences of tissue damage (McTigue and Tripathi, 2008). Oligodendrocyte death causes demyelination, impairment of axonal conduction, and ultimately axon death which contribute to memory impairment. Collectively, dysfunction of glia causes morphological and functional changes which effect the neural-glial and glial-glial interactions. Synaptic disconnections, imbalances of neurotransmitter homeostasis, and potential axonal degeneration and neuronal death can ultimately lead to memory impairment. Understanding the glia response, following injury at the molecular level may provide clues to decreasing chronic memory deficits.
Secondary Injury and Metabolic Dysfunction
Traumatic brain injury (TBI) is a complex, progressive condition that consists of primary and secondary injury mechanisms. Primary injury is due to direct mechanical insult and is the initiator of secondary molecular cascades. Secondary injury is characterized largely by metabolic imbalance and neuroinflammation (Figure 1). Following primary insult, brain cells experience energy depletion and a loss of calcium homeostasis, both of which are principal in mitochondrial function. Mitochondrial disruption is well documented in acute stages of TBI (Colicos and Dash, 1996; Xiong et al., 1997; Sullivan et al., 1998, 2005; Singh et al., 2006; Gilmer et al., 2009; Cheng et al., 2012). While these alterations are not glia-specific, they are intensified by activated glia. Because of the surge in extracellular ATP that results from damaged cells, glia are activated leading to downstream calcium release from endoplasmic reticulum (Locovei et al., 2006). Alterations in expression of various metabotropic receptors can occur as a result (Wang et al., 2012), contributing to surges of intracellular calcium. Increased cytosolic calcium is balanced by mitochondria at the expense of mitochondrial membrane potential. Eventually, mitochondria are driven to calcium overload and injury is exacerbated through generation of reactive oxygen species (ROS). Neurons are limited in their antioxidant capacity and thus rely on astrocytes to buffer ROS (Hamby and Sofroniew, 2010). Otherwise, they become susceptible to irreversible damage. Importantly, a pro-oxidative environment contributes to lipid, protein, and nucleic acid damage manifested largely in membrane disruption (Lewén and Hillered, 1998; Miller et al., 2015) and induction of neuroinflammation (Hsieh and Yang, 2013). Studies have concentrated on elucidating the roles of cellular sensors and enzymes that modulate intracellular calcium and ROS in metabolic dysfunction associated with death (Lu et al., 2014; Angeloni et al., 2015; Rao et al., 2015). Moreover, calcium signals in glial transmission are necessary for information processing and neuronal-glial coordination. Thus, impairment of glial-neuronal transmission contributes to memory loss (Walker and Tesco, 2013; Croft et al., 2015; Gundersen et al., 2015). In addition to calcium homeostasis, it is necessary to consider the consequence of potassium imbalances in secondary injury. Astrocytes normally uptake extracellular potassium via channels and Na+/K+/ATPase which in turn contributes to volume changes characteristic of TBI (Macaulay and Zeuthen, 2012; Larsen et al., 2014). Disruptions in potassium homeostasis, alongside neurotransmitter receptor activation, enhance neuronal impairment (D'Ambrosio et al., 1999; Pietrobon and Moskowitz, 2014).
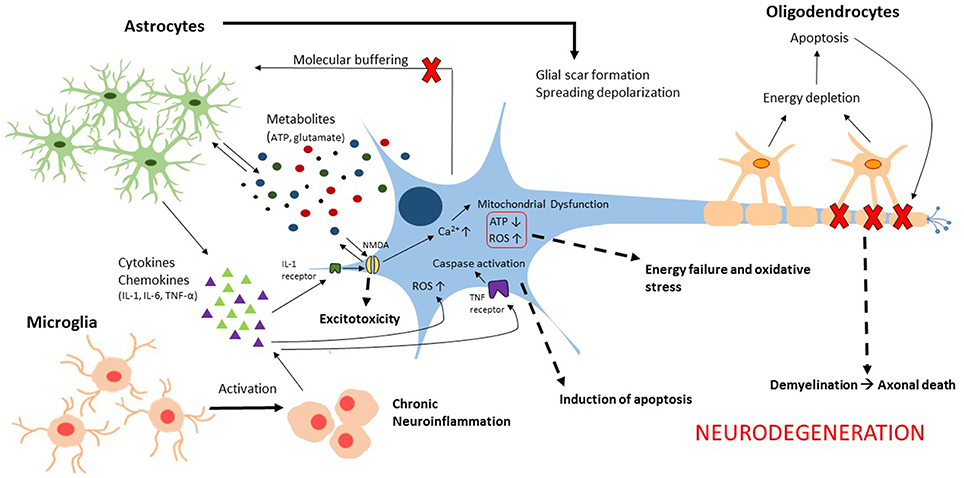
Figure 1. Illustration of the glial contributions to secondary injury mechanisms associated with neurodegeneration following traumatic brain injury.
Trauma-Associated Edema
Cerebral edema is induced by water imbalance in response to trauma. Cytotoxic edema occurs in acute stages of TBI as a result of dysregulated metabolism. Often, there is a biphasic edema response in which early cytotoxic edema is followed by vasogenic edema associated with compromised blood-brain-barrier (BBB). Glia play an integral role in regulation of water and other molecules that transverse the BBB. Astrocytic endfeet directly contact brain vessels and are localized with aquaporin (AQP) proteins, which are pore proteins for water passage (Nielsen et al., 1997). Moreover, astrocytes are highly susceptible to swelling due to expression of AQPs (Suzuki et al., 2006; Satoh et al., 2007; Rao et al., 2011). Expression of AQP following trauma suggests that sustained AQP expression is critical in alleviating edema however, it is dependent on location relative to the injury, time, and variations in TBI models (Kiening et al., 2002; Sun et al., 2003; Zhao et al., 2005). A recent study reported only a small reduction in brain volume in AQP4 knockout mice with no evidence of difference in BBB disruption between AQP4 knockout and wildtype (Yao et al., 2015). Varied results may be related to a differential role of AQPs in the biphasic edema response. New theories hypothesize that AQP4 facilitates bulk flow through the glymphatic system, which poses contradiction to edema formation localized in astrocytic endfeet (Thrane et al., 2014). Evidence also suggests, that crosstalk exists between microglia and astrocytes in the regulation of AQP4 via microglial pattern recognition receptor-mediated pathways (Laird et al., 2014). Other studies are aimed to understand the effect of modulation of AQPs to ameliorate neuronal injury and cognitive deficits associated with TBI-induced edema (Tran et al., 2010; Shenaq et al., 2012).
Extension of Cellular Death
Much of the intercellular molecular buffering required for homeostasis in the brain is mediated by gap junctions (GJs), which consist of connexin (Cx) hemichannels that transverse the plasma membrane directly connect adjacent cells. Cx30 and Cx43 are expressed by astrocytes while Cx32 is expressed only by oligodendrocytes (Rash et al., 2001). GJs are necessary for the formation of astrocytic networks that interconnect neurons synapses and vessels (Giaume, 2010; Giaume et al., 2010). The role of Cx43 in CNS injury has been debated as both protective and detrimental for GJ communications (Chew et al., 2010). GJs allow for the passage of ions, metabolites, and other small molecules. Thus, an injured cell can distribute its damaging components to adjacent healthy cells. While this is potentially protective for injured cells, it also exacerbates the spread of injury. Studies have investigated the role of Cx43 expression in the expansion of cellular death (Frantseva et al., 2002; Lin et al., 2008; Sun et al., 2014; Rovegno et al., 2015). Inflammatory cytokines secreted by microglia activate Cx43 in astrocytes and can enhance N-methyl-D-aspartate (NMDA) receptor-mediated excitotoxicity in surrounding neurons (Froger et al., 2010). Additionally, Cx hemichannels are a route for the release of ATP to extracellular space, which exacerbates metabolic dysfunction and inflammation (Cotrina et al., 1998; Frantseva et al., 2002; Davalos et al., 2005; Figiel et al., 2007). It is also known, that release of transmitters, including ATP and glutamate, can perturb intercellular calcium signaling within astrocytic networks, which in turn may contribute to neuroinflammation and cell death (Choo et al., 2013; De Bock et al., 2014). There is evidence that Cx expression influences functional and cognitive outcomes from injury (Huang et al., 2012; Sun et al., 2015) as well as progressive neurodegeneration (Orellana et al., 2009).
Reactive Gliosis
Subsequent to insult, glia are transformed into a reactive state. Reactive gliosis is characterized by specific molecular and morphologic changes in microglia and astrocytes. Upon activation, microglia in combination with macrophages and astrocytes secrete cytokines (interferon-γ, tissue necrosis factor-α, interleukins-1 and 6 as well as transforming growth factor-β (TGF-β)) (Morganti-Kossmann et al., 2001; Li et al., 2009; Kumar and Loane, 2012; Aungst et al., 2014; Sajja et al., 2014b). While activation is initiated immediately upon injury, it is often sustained chronically which is linked to damaging neuronal homeostasis and memory deficits (Hanisch and Kettenmann, 2007; Ramlackhansingh et al., 2011; Mannix and Whalen, 2012; Smith et al., 2012; Johnson et al., 2013). Neuroinflammation is associated with ROS and the exacerbation of astrocyte activation. Evidence of prolonged neurotrophic effects from activated microglia has been reported (Nagamoto-Combs et al., 2007). This chronic inflammation has detrimental effects and contributes to neurodegeneration and memory impairment (Faden and Loane, 2015). Approaches to molecular and genetic influence on decreased microglial activation have resulted in decreased neuropathology (Yi et al., 2008; Dohi et al., 2010) and improved cognitive and functional outcomes (Erlich et al., 2007; Li et al., 2009; Kabadi et al., 2012; Cho et al., 2013).
Astrocyte reactivity or astrogliosis, is characterized by three hallmarks: hypertrophy, increased expression of intermediate filaments (glial-fibrillary acidic protein (GFAP), nestin and vimentin), and increased proliferation (Baldwin and Scheff, 1996; Sahin Kaya et al., 1999; Vandevord et al., 2008). Astrogliosis is dependent on interplay with activated microglia (Di Giovanni et al., 2005; Myer et al., 2006). Reactive astrocytes secrete molecules for regulation of the existing neuroinflammatory response (Panenka et al., 2001; Gorina et al., 2011), are integral in creating physical barriers associated with the BBB, as well as contribute to scar formation around injured tissue. The astrocytic scar inhibits axonal regrowth as cells will secrete growth inhibitors, such as TGF-β, thus affecting long-term cognitive outcomes. Although, most research focuses on modulation of astrogliosis, both the protective and inhibitory effects have been evaluated in the context of improved neuronal survival and cognitive abilities over time (Smith et al., 1997; Hoane et al., 2003; Wu et al., 2010; Madathil et al., 2013).
Glial Contribution to Memory Deficits
Oligodendrocyte dysfunction due to inflammation or cellular death impairs neurotransmission via degeneration of WM tracts (Smith et al., 1997; Gorina et al., 2011). Pre-clinical and clinical studies have shown axonal disruption associated with functional impairment (Lu et al., 2012; MacDonald et al., 2013; Calabrese et al., 2014). A non-human primate study reported a loss of WM integrity and astrocytic hypertrophy with increased AQP-4 contributed to cell death associated with cognitive impairment (Lu et al., 2012). Specifically, learning and memory has been shown to be associated with abnormal levels of myo-inositol, which is an astrogliosis marker (Sajja et al., 2014b). Resultants of gliosis directed toward dementia, such as tau and DNA methylation markers are found to be upregulated following TBI (Bailey et al., 2015; Sajja et al., 2015; Shultz et al., 2015). Another indicator linked to memory deficits is the disrupted homeostasis of extra and intra-cellular K+ channels in glia (D'Ambrosio et al., 1999). Furthermore, it has shown, that by blocking glial activation, cognition was improved (Homsi et al., 2010; Bedi et al., 2013). New research has shown the role of ependymal cells in contributing to memory deficits. Ependymal cells are specialized glia, that line the ventricles of the CNS. Ependymal cell lose was found to decrease ventricular flow following TBI which could negatively affect the waste and nutrient exchange within the brain (Xiong et al., 2014). Additional research that helps decipher the molecular pathways between glia and memory deficits will be vital for development of better clinical tools for gauging memory loss.
Glia-Based Biomarkers
The response of glia to TBI is multifaceted, supporting the importance of these cells to recovery. However, the intricate chemical and physical reactions of glia are very difficult to detect in the clinical setting. It is technically challenging to diagnosis and study the involvement of the glia in the recovery stages following injury and their contribution to memory deficits. Most minor TBI cases have normal findings in conventional neuroimaging [computed tomography (CT) and magnetic resonance imaging (MRI)]. While both basic and clinical research have made significant improvements, advancements are vital to fill the translational gap. Innovative technologies have emerged, such as serum biomarkers and in vivo magnetic resonance spectroscopy (1H-MRS) which may provide the link needed to branch the basic and clinical arenas (Figure 2).
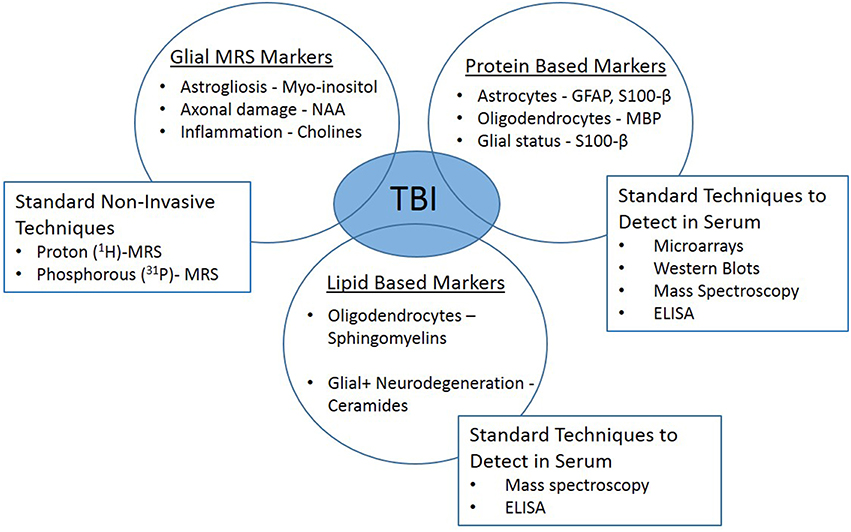
Figure 2. Clinically translatable biomarkers for traumatic brain injury. In vivo magnetic resonance spectroscopy (1H-MRS) and glial-specific serum biomarkers may provide the link needed to branch the basic and clinical research arenas.
Serum Biomarkers
Minimally invasive techniques, such as serum biomarkers, can be used to detect brain-specific pathologies. With technological advancements in proteomics and lipidomics, finding accurate biomarkers that reflect glial health status would be tremendously valuable. GFAP is a common astrocytic marker, that has been detected in serum following TBI in both pre-clinical and clinical studies (Fraser et al., 2011; Vajtr et al., 2012; Papa et al., 2014; Huang et al., 2015). Significant accumulation of GFAP persisted in blood up to 7 days post-injury (Svetlov et al., 2009; Boutte ́ et al., 2015). Some have suggested, that the use of GFAP as a TBI biomarker yields a net benefit above clinical screening alone and may help avoid costly imaging scans without sacrificing diagnostic sensitivity (McMahon et al., 2015). S100 calcium-binding protein B (S100-β) is another serum biomarker that is clinically used to help in diagnosis of neurological disorders (Bouvier et al., 2012; DeFazio et al., 2014; Thelin et al., 2014). S100-β is expressed primarily by mature astrocytes and is present in the extracellular space surrounding glia, assisting with regulation of the cell calcium influx/efflux, but also linked to apoptotic environments (Gyorgy et al., 2011; Vajtr et al., 2012). Studies have identified S100-β as biomarker that could potentially be used in TBI diagnosis, however, others suggest, that GFAP is a better evaluator of TBI without skull fractures (Papa et al., 2014). Myelin-basic protein (MBP) is a specific marker of oligodendrocytes and was detected in blood, indicating potential disruption in myelin, thus leading to axonal injury (Gyorgy et al., 2011; Yan et al., 2014; Papa et al., 2015).
Lipid-based biomarkers such as sphingolipids, specifically sphingomyelins and ceramides, have recently become an active area of biomarker research. Sphingomyelin is abundant in the myelin membrane and abnormal levels in blood can constitute changes in myelin health and associations with oligodendrocyte injury (Haughey, 2010; Abdullah et al., 2014; Novgorodov et al., 2014; Henriquez-Henriquez et al., 2015; Koal et al., 2015). In addition, ceramide is metabolized from sphingomyelins and vice versa by sphingomyelinase. Ceramide is known to serve as a secondary messenger for intracellular activation of caspase-3 levels in cellular apoptosis (Haughey et al., 2010). Therefore, combination of changes in ceramide and sphingomyelin levels can predict the overall lipid status of myelin in the brain. Lipids are highly sensitive to changes in brain health, so they offer new diagnostic possibilities due to the development of robust and sensitive analytical methods (Touboul and Gaudin, 2014).
1H-MRS
MRI is a non-invasive and widely accepted diagnostic modality to study brain abnormalities. While T1, T2, and T2* MRI can provide information related to gross anatomical changes, edema and cerebral hemorrhaging, 1H-MRS provides more detailed chemical insight into the functional status and pathological prognosis of the brain (Sajja et al., 2012; Kantarci, 2013). Pre-clinical 1H-MRS can resolve ~25 and clinical 1H-MRS about ~10 metabolites depending on peak-suppression parameters (Moore and Galloway, 2002; Moffett et al., 2007; Sofroniew and Vinters, 2010).
N-acetyl aspartate (NAA) is a neurometabolite synthesized from aspartate and acetyl co-enzyme A. NAA or NAA/creatine (Cre) is trans-regulated between oligodendrocytes and neurons and can provide insight to structural integrity of WM (Charlton et al., 2006; Ariyannur et al., 2008; Kantarci, 2013). Studies have shown NAA levels in brain correlate with altered WM integrity following TBI (Pendlebury et al., 2000; Brooks et al., 2001; Shiino et al., 2004). Disruption in neuron-oligodendrocyte homeostasis can affect axon potentials and eventually neurotransmission, leading to an altered cognitive status. Since, alterations in the levels of NAA in WM-rich regions could indicate health status of oligodendrocytes and it can be measured by both pre-clinical and clinical 1H-MRS, it has the potential to be an innovative translational avenue.
Reactive astrocytes rapidly accumulate in the injury region and alter their morphology, typically inducing swelling. This is related to osmolarity changes that result from edema or ischemia following TBI (Sofroniew and Vinters, 2010). Myo-inositol (Ins) is a primary metabolite that maintains brain osmolarity. Clinical studies have reported that an up-regulation of Ins correlates with astrogliosis in pathophysiological conditions such as TBI, dementia, and glioblastoma (Hattingen et al., 2008; Kantarci, 2013; Kierans et al., 2014). Pre-clinical studies have demonstrated that 1H-MRS-resolved Ins was associated with astrogliosis and impaired cognition following TBI (Kierans et al., 2014; Sajja et al., 2014a).
In conjunction with astrocytes, microglia actively participate in clearing debris resulting from neuroinflammation. Changes in metabolites such as phosphoryethanolamine (PEA), glycerophosphocholine (GPC), and cholines (Cho) have been linked to microglia. PEA and GPC are involved in cell membrane turnover indicating neuroinflammation and GPC/PEA levels change depending of cell activation status (Sajja et al., 2012). Thus, they indicate compromised cellular activities.
Resolving 1H-MRS peaks with lower signal-to-noise ratios depends on the field strength of the scanner, time of acquisition and number of repetitions of acquisition. Although, many metabolites can be resolved using pre-clinical MR scanners, only a small portion can be resolved with a clinical scanner which limits clinical translation. However, NAA, Ins, and Cho can be resolved with clinically available MR scanners. Thus, we highlighted the potential utility of clinical 1H-MRS in combination with other modalities for differential diagnosis.
Conclusion
We have reviewed several glial-based molecules, that give clues to glia health status following TBI. There is a general consensus that a panel of markers will provide the most clinically relevant diagnostic tool. Thus, understanding how glial dysfunction following injury can alter mechanisms of synaptic plasticity and its relationship to an increased risk for persistent memory deficits is necessary for advancement. Researchers are actively pursuing new targets for a minimally invasive tools which can accurately and objectively detect brain injury. Combining sophisticated tools, such as serum biomarkers and MRS, will provide for an accurate differential diagnosis following TBI. Moreover, a temporal pattern of these markers could offer prognostic clues as to neuronal plasticity leading to memory formations.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdullah, L., Evans, J. E., Ferguson, S., Mouzon, B., Montague, H., Reed, J., et al. (2014). Lipidomic analyses identify injury-specific phospholipid changes 3 mo after traumatic brain injury. FASEB J. 28, 5311–5321. doi: 10.1096/fj.14-258228
Angeloni, C., Prata, C., Vieceli Dalla Sega, F., Piperno, R., and Hrelia, S. (2015). Traumatic brain injury and NADPH Oxidase: a deep relationship. Oxid. Med. Cell. Longev. 2015, 10. doi: 10.1155/2015/370312
Ariyannur, P. S., Madhavarao, C. N., and Namboodiri, A. M. (2008). N-acetylaspartate synthesis in the brain: mitochondria vs. microsomes. Brain Res. 1227, 34–41. doi: 10.1016/j.brainres.2008.06.040
Aungst, S. L., Kabadi, S. V., Thompson, S. M., Stoica, B. A., and Faden, A. I. (2014). Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J. Cereb. Blood Flow Metab. 34, 1223–1232. doi: 10.1038/jcbfm.2014.75
Bailey, Z. S., Grinter, M. B., De La Torre Campos, D., and VandeVord, P. J. (2015). Blast induced neurotrauma causes overpressure dependent changes to the DNA methylation equilibrium. Neurosci. Lett. 604, 119–123. doi: 10.1016/j.neulet.2015.07.035
Baldwin, S. A., and Scheff, S. W. (1996). Intermediate filament change in astrocytes following mild cortical contusion. Glia 16, 266–275.
Bedi, S. S., Hetz, R., Thomas, C., Smith, P., Olsen, A. B., Williams, S., et al. (2013). Intravenous multipotent adult progenitor cell therapy attenuates activated microglial/macrophage response and improves spatial learning after traumatic brain injury. Stem Cells Transl. Med. 2, 953–960. doi: 10.5966/sctm.2013-0100
Boutté, A. M., Deng-Bryant, Y., Johnson, D., Tortella, F. C., Dave, J. R., Shear, D. A., et al. (2015). Serum glial fibrillary acidic protein predicts tissue glial fibrillary acidic protein break-down products and therapeutic efficacy after penetrating ballistic-like brain injury. J. Neurotrauma. 33, 147–156. doi: 10.1089/neu.2014.3672
Bouvier, D., Fournier, M., Dauphin, J. B., Amat, F., Ughetto, S., Labbé, A., et al. (2012). Serum S100B determination in the management of pediatric mild traumatic brain injury. Clin. Chem. 58, 1116–1122. doi: 10.1373/clinchem.2011.180828
Brooks, W. M., Friedman, S. D., and Gasparovic, C. (2001). Magnetic resonance spectroscopy in traumatic brain injury. J. Head Trauma Rehabil. 16, 149–164. doi: 10.1097/00001199-200104000-00005
Buffo, A., Rolando, C., and Ceruti, S. (2010). Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 79, 77–89. doi: 10.1016/j.bcp.2009.09.014
Calabrese, E., Du, F., Garman, R. H., Johnson, G. A., Riccio, C., Tong, L. C., et al. (2014). Diffusion tensor imaging reveals white matter injury in a rat model of repetitive blast-induced traumatic brain injury. J. Neurotrauma 31, 938–950. doi: 10.1089/neu.2013.3144
Charlton, R. A., Barrick, T. R., McIntyre, D. J., Shen, Y., O'sullivan, M., Howe, F. A., et al. (2006). White matter damage on diffusion tensor imaging correlates with age-related cognitive decline. Neurology 66, 217–222. doi: 10.1212/01.wnl.0000194256.15247.83
Cheng, G., Kong, R.-H., Zhang, L.-M., and Zhang, J.-N. (2012). Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 167, 699–719. doi: 10.1111/j.1476-5381.2012.02025.x
Chew, S. S. L., Johnson, C. S., Green, C. R., and Danesh-Meyer, H. V. (2010). Role of connexin43 in central nervous system injury. Exp. Neurol. 225, 250–261. doi: 10.1016/j.expneurol.2010.07.014
Cho, H. J., Sajja, V. S., Vandevord, P. J., and Lee, Y. W. (2013). Blast induces oxidative stress, inflammation, neuronal loss and subsequent short-term memory impairment in rats. Neuroscience 253, 9–20. doi: 10.1016/j.neuroscience.2013.08.037
Choo, A. M., Miller, W. J., Chen, Y. C., Nibley, P., Patel, T. P., Goletiani, C., et al. (2013). Antagonism of purinergic signalling improves recovery from traumatic brain injury. Brain 136, 65–80. doi: 10.1093/brain/aws286
Colicos, M. A., and Dash, P. K. (1996). Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: possible role in spatial memory deficits. Brain Res. 739, 120–131. doi: 10.1016/S0006-8993(96)00824-4
Cotrina, M. L., Lin, J. H. C., Alves-Rodrigues, A., Liu, S., Li, J., Azmi-Ghadimi, H., et al. (1998). Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. U.S.A. 95, 15735–15740. doi: 10.1073/pnas.95.26.15735
Croft, W., Dobson, K. L., and Bellamy, T. C. (2015). Plasticity of neuron-glial transmission: equipping glia for long-term integration of network activity. Neural Plast. 2015, 11. doi: 10.1155/2015/765792
D'Ambrosio, R., Maris, D. O., Grady, M. S., Winn, H. R., and Janigro, D. (1999). Impaired K(+) homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J. Neurosci. 19, 8152–8162.
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
De Bock, M., Decrock, E., Wang, N., Bol, M., Vinken, M., Bultynck, G., et al. (2014). The dual face of connexin-based astroglial Ca2+ communication: A key player in brain physiology and a prime target in pathology. Biochim. Biophys. Acta 1843, 2211–2232. doi: 10.1016/j.bbamcr.2014.04.016
DeFazio, M. V., Rammo, R. A., Robles, J. R., Bramlett, H. M., Dietrich, W. D., and Bullock, M. R. (2014). The potential utility of blood-derived biochemical markers as indicators of early clinical trends following severe traumatic brain injury. World Neurosurg. 81, 151–158. doi: 10.1016/j.wneu.2013.01.015
Di Giovanni, S., Movsesyan, V., Ahmed, F., Cernak, I., Schinelli, S., Stoica, B., et al. (2005). Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. U.S.A. 102, 8333–8338. doi: 10.1073/pnas.0500989102
Dohi, K., Ohtaki, H., Nakamachi, T., Yofu, S., Satoh, K., Miyamoto, K., et al. (2010). Gp91(phox)(NOX2) in classically activated microglia exacerbates traumatic brain injury. J. Neuroinflammation 7, 41–41. doi: 10.1186/1742-2094-7-41
Erlich, S., Alexandrovich, A., Shohami, E., and Pinkas-Kramarski, R. (2007). Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol. Dis. 26, 86–93. doi: 10.1016/j.nbd.2006.12.003
Faden, A. I., and Loane, D. J. (2015). Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 12, 143–150. doi: 10.1007/s13311-014-0319-5
Figiel, M., Allritz, C., Lehmann, C., and Engele, J. (2007). Gap junctional control of glial glutamate transporter expression. Mol. Cell. Neurosci. 35, 130–137. doi: 10.1016/j.mcn.2007.02.009
Frantseva, M. V., Kokarovtseva, L., Naus, C. G., Carlen, P. L., MacFabe, D., and Perez Velazquez, J. L. (2002). Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J. Neurosci. 22, 644–653.
Fraser, D. D., Close, T. E., Rose, K. L., Ward, R., Mehl, M., Farrell, C., et al. (2011). Severe traumatic brain injury in children elevates glial fibrillary acidic protein in cerebrospinal fluid and serum. Pediatr. Crit. Care Med. 12, 319–324. doi: 10.1097/PCC.0b013e3181e8b32d
Froger, N., Orellana, J. A., Calvo, C. F., Amigou, E., Kozoriz, M. G., Naus, C. C., et al. (2010). Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol. Cell. Neurosci. 45, 37–46. doi: 10.1016/j.mcn.2010.05.007
Giaume, C. (2010). Astroglial wiring is adding complexity to neuroglial networking. Front. Neuroenergetics 2:129. doi: 10.3389/fnene.2010.00129
Giaume, C., Koulakoff, A., Roux, L., Holcman, D., and Rouach, N. (2010). Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 11, 87–99. doi: 10.1038/nrn2757
Gilmer, L. K., Roberts, K. N., Joy, K., Sullivan, P. G., and Scheff, S. W. (2009). Early mitochondrial dysfunction after cortical contusion injury. J. Neurotrauma 26, 1271–1280. doi: 10.1089/neu.2008.0857
Gorina, R., Font-Nieves, M., Márquez-Kisinousky, L., Santalucia, T., and Planas, A. M. (2011). Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59, 242–255. doi: 10.1002/glia.21094
Gundersen, V., Storm-Mathisen, J., and Bergersen, L. H. (2015). Neuroglial transmission. Physiol. Rev. 95, 695–726. doi: 10.1152/physrev.00024.2014
Gyorgy, A., Ling, G., Wingo, D., Walker, J., Tong, L., Parks, S., et al. (2011). Time-dependent changes in serum biomarker levels after blast traumatic brain injury. J. Neurotrauma 28, 1121–1126. doi: 10.1089/neu.2010.1561
Hamby, M. E., and Sofroniew, M. V. (2010). Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7, 494–506. doi: 10.1016/j.nurt.2010.07.003
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hattingen, E., Raab, P., Franz, K., Zanella, F. E., Lanfermann, H., and Pilatus, U. (2008). Myo-inositol: a marker of reactive astrogliosis in glial tumors? NMR Biomed. 21, 233–241. doi: 10.1002/nbm.1186
Haughey, N. J. (2010). Sphingolipids in neurodegeneration. Neuromolecular Med. 12, 301–305. doi: 10.1007/s12017-010-8135-5
Haughey, N. J., Bandaru, V. V., Bae, M., and Mattson, M. P. (2010). Roles for dysfunctional sphingolipid metabolism in Alzheimer's disease neuropathogenesis. Biochim. Biophys. Acta 1801, 878–886. doi: 10.1016/j.bbalip.2010.05.003
Henríquez-Henríquez, M. P., Solari, S., Quiroga, T., Kim, B. I., Deckelbaum, R. J., and Worgall, T. S. (2015). Low serum sphingolipids in children with attention deficit-hyperactivity disorder. Front. Neurosci. 9:300. doi: 10.3389/fnins.2015.00300
Hoane, M. R., Akstulewicz, S. L., and Toppen, J. (2003). Treatment with Vitamin B3 improves functional recovery and reduces GFAP expression following traumatic brain injury in rats. J. Neurotrauma 20, 1189–1199. doi: 10.1089/089771503770802871
Homsi, S., Piaggio, T., Croci, N., Noble, F., Plotkine, M., Marchand-Leroux, C., et al. (2010). Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J. Neurotrauma. 27, 911–921. doi: 10.1089/neu.2009.1223
Hsieh, H.-L., and Yang, C.-M. (2013). Role of Redox Signaling in Neuroinflammation and Neurodegenerative Diseases. Biomed Res. Int. 2013, 18. doi: 10.1155/2013/484613
Huang, C., Han, X., Li, X., Lam, E., Peng, W., Lou, N., et al. (2012). Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. J. Neurosci. 32, 3333–3338. doi: 10.1523/JNEUROSCI.1216-11.2012
Huang, X. J., Glushakova, O., Mondello, S., Van, K., Hayes, R. L., and Lyeth, B. G. (2015). Acute temporal profiles of serum levels of UCH-L1 and GFAP and relationships to neuronal and astroglial pathology following traumatic brain injury in Rats. J. Neurotrauma 32, 1179–1189. doi: 10.1089/neu.2015.3873
Johnson, V. E., Stewart, J. E., Begbie, F. D., Trojanowski, J. Q., Smith, D. H., and Stewart, W. (2013). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42. doi: 10.1093/brain/aws322
Kabadi, S. V., Stoica, B. A., Byrnes, K. R., Hanscom, M., Loane, D. J., and Faden, A. I. (2012). Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. J. Cereb. Blood Flow Metab. 32, 137–149. doi: 10.1038/jcbfm.2011.117
Kantarci, K. (2013). Proton MRS in mild cognitive impairment. J. Magn. Reson. Imaging 37, 770–777. doi: 10.1002/jmri.23800
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kettenmann, H., Kirchhoff, F., and Verkhratsky, A. (2013). Microglia: new roles for the synaptic stripper. Neuron 77, 10–18. doi: 10.1016/j.neuron.2012.12.023
Kiening, K. L., van Landeghem, F. K. H., Schreiber, S., Thomale, U. W., von Deimling, A., Unterberg, A. W., et al. (2002). Decreased hemispheric Aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci. Lett. 324, 105–108. doi: 10.1016/S0304-3940(02)00180-5
Kierans, A. S., Kirov, I. I., Gonen, O., Haemer, G., Nisenbaum, E., Babb, J. S., et al. (2014). Myoinositol and glutamate complex neurometabolite abnormality after mild traumatic brain injury. Neurology 82, 521–528. doi: 10.1212/WNL.0000000000000105
Kimelberg, H. K. (2005). Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 50, 389–397. doi: 10.1002/glia.20174
Kimelberg, H. K., and Nedergaard, M. (2010). Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 7, 338–353. doi: 10.1016/j.nurt.2010.07.006
Koal, T., Klavins, K., Seppi, D., Kemmler, G., and Humpel, C. (2015). Sphingomyelin SM(d18:1/18:0) is significantly enhanced in cerebrospinal fluid samples dichotomized by pathological amyloid-beta42, tau, and phospho-tau-181 levels. J. Alzheimers. Dis. 44, 1193–1201. doi: 10.3233/JAD-142319
Kumar, A., and Loane, D. J. (2012). Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav. Immun. 26, 1191–1201. doi: 10.1016/j.bbi.2012.06.008
Laird, M. D., Shields, J. S., Sukumari-Ramesh, S., Kimbler, D. E., Fessler, R. D., Shakir, B., et al. (2014). High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 62, 26–38. doi: 10.1002/glia.22581
Larsen, B. R., Assentoft, M., Cotrina, M. L., Hua, S. Z., Nedergaard, M., Kaila, K., et al. (2014). Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4.1 to hippocampal K(+) clearance and volume responses. Glia 62, 608–622. doi: 10.1002/glia.22629
Lewén, A., and Hillered, L. (1998). Involvement of reactive oxygen species in membrane phospholipid breakdown and energy perturbation after traumatic brain injury in the Rat. J. Neurotrauma 15, 521–530. doi: 10.1089/neu.1998.15.521
Li, B., Mahmood, A., Lu, D., Wu, H., Xiong, Y., Qu, C., et al. (2009). Simvastatin attenuates microglia, astrocyte activation and decreases IL-1β Level following traumatic brain injury. Neurosurgery 65, 179–186. doi: 10.1227/01.NEU.0000346272.76537.DC
Lin, J. H., Lou, N., Kang, N., Takano, T., Hu, F., Han, X., et al. (2008). A central role of connexin 43 in hypoxic preconditioning. J. Neurosci. 28, 681–695. doi: 10.1523/JNEUROSCI.3827-07.2008
Locovei, S., Wang, J., and Dahl, G. (2006). Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett. 580, 239–244. doi: 10.1016/j.febslet.2005.12.004
Lu, J., Ng, K. C., Ling, G., Wu, J., Poon, D. J., Kan, E. M., et al. (2012). Effect of blast exposure on the brain structure and cognition in Macaca fascicularis. J. Neurotrauma. 229, 1434–1454. doi: 10.1089/neu.2010.1591
Lu, X.-Y., Wang, H.-D., Xu, J.-G., Ding, K., and Li, T. (2014). NADPH oxidase inhibition improves neurological outcome in experimental traumatic brain injury. Neurochem. Int. 69, 14–19. doi: 10.1016/j.neuint.2014.02.006
Macaulay, N., and Zeuthen, T. (2012). Glial K(+) clearance and cell swelling: key roles for cotransporters and pumps. Neurochem. Res. 37, 2299–2309. doi: 10.1007/s11064-012-0731-3
MacDonald, C., Johnson, A., Cooper, D., Malone, T., Sorrell, J., Shimony, J., et al. (2013). Cerebellar white matter abnormalities following primary blast injury in US military personnel. PLoS ONE 8:e55823. doi: 10.1371/journal.pone.0055823
Madathil, S. K., Carlson, S. W., Brelsfoard, J. M., Ye, P., D'Ercole, A. J., and Saatman, K. E. (2013). Astrocyte-specific overexpression of insulin-like growth factor-1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in Mice. PLoS ONE 8:e67204. doi: 10.1371/journal.pone.0067204
Mannix, R. C., and Whalen, M. J. (2012). Traumatic brain injury, microglia, and Beta amyloid. Int. J. Alzheimers. Dis. 2012:608732. doi: 10.1155/2012/608732
McMahon, P. J., Panczykowski, D. M., Yue, J. K., Puccio, A. M., Inoue, T., Sorani, M. D., et al. (2015). Measurement of the glial fibrillary acidic protein and its breakdown products GFAP-BDP biomarker for the detection of traumatic brain injury compared to computed tomography and magnetic resonance imaging. J. Neurotrauma 32, 527–533. doi: 10.1089/neu.2014.3635
McTigue, D. M., and Tripathi, R. B. (2008). The life, death, and replacement of oligodendrocytes in the adult CNS. J. Neurochem. 107, 1–19. doi: 10.1111/j.1471-4159.2008.05570.x
Miller, D. M., Singh, I. N., Wang, J. A., and Hall, E. D. (2015). Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp. Neurol. 264, 103–110. doi: 10.1016/j.expneurol.2014.11.008
Moffett, J. R., Ross, B., Arun, P., Madhavarao, C. N., and Namboodiri, M. A. A. (2007). N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog. Neurobiol. 81, 89–131. doi: 10.1016/j.pneurobio.2006.12.003
Moore, G. J., and Galloway, M. P. (2002). Magnetic resonance spectroscopy: neurochemistry and treatment effects in affective disorders. Psychopharmacol. Bull. 36, 5–23.
Moraga-Amaro, R., Jerez-Baraona, J. M., Simon, F., and Stehberg, J. (2014). Role of astrocytes in memory and psychiatric disorders. J. Physiol. Paris 108, 240–251. doi: 10.1016/j.jphysparis.2014.08.005
Morganti-Kossmann, M. C., Rancan, M., Otto, V. I., Stahel, P. F., and Kossmann, T. (2001). Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock 16, 165–177. doi: 10.1097/00024382-200116030-00001
Myer, D. J., Gurkoff, G. G., Lee, S. M., Hovda, D. A., and Sofroniew, M. V. (2006). Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129, 2761–2772. doi: 10.1093/brain/awl165
Nagamoto-Combs, K., McNeal, D. W., Morecraft, R. J., and Combs, C. K. (2007). Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J. Neurotrauma 24, 1719–1742. doi: 10.1089/neu.2007.0377
Nielsen, S., Nagelhus, E. A., Amiry-Moghaddam, M., Bourque, C., Agre, P., and Ottersen, O. P. (1997). Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 17, 171–180.
Novgorodov, S. A., Riley, C. L., Yu, J., Borg, K. T., Hannun, Y. A., Proia, R. L., et al. (2014). Essential roles of neutral ceramidase and sphingosine in mitochondrial dysfunction due to traumatic brain injury. J. Biol. Chem. 289, 13142–13154. doi: 10.1074/jbc.M113.530311
Orellana, J. A., Sáez, P. J., Shoji, K. F., Schalper, K. A., Palacios-Prado, N., Velarde, V., et al. (2009). Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid. Redox Signal. 11, 369–399. doi: 10.1089/ars.2008.2130
Panenka, W., Jijon, H., Herx, L. M., Armstrong, J. N., Feighan, D., Wei, T., et al. (2001). P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J. Neurosci. 21, 7135–7142.
Papa, L., Robertson, C. S., Wang, K. K., Brophy, G. M., Hannay, H. J., Heaton, S., et al. (2015). Biomarkers improve clinical outcome predictors of mortality following non-penetrating severe traumatic brain injury. Neurocrit. Care 22, 52–64. doi: 10.1007/s12028-014-0028-2
Papa, L., Silvestri, S., Brophy, G. M., Giordano, P., Falk, J. L., Braga, C. F., et al. (2014). GFAP out-performs S100beta in detecting traumatic intracranial lesions on computed tomography in trauma patients with mild traumatic brain injury and those with extracranial lesions. J. Neurotrauma 31, 1815–1822. doi: 10.1089/neu.2013.3245
Pendlebury, S. T., Lee, M. A., Blamire, A. M., Styles, P., and Matthews, P. M. (2000). Correlating magnetic resonance imaging markers of axonal injury and demyelination in motor impairment secondary to stroke and multiple sclerosis. Magn. Reson. Imaging 18, 369–378. doi: 10.1016/S0730-725X(00)00115-6
Pietrobon, D., and Moskowitz, M. A. (2014). Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 15, 379–393. doi: 10.1038/nrn3770
Ramlackhansingh, A. F., Brooks, D. J., Greenwood, R. J., Bose, S. K., Turkheimer, F. E., Kinnunen, K. M., et al. (2011). Inflammation after trauma: microglial activation and traumatic brain injury. Ann. Neurol. 70, 374–383. doi: 10.1002/ana.22455
Rao, K. V. R., Reddy, P. V. B., Curtis, K. M., and Norenberg, M. D. (2011). Aquaporin-4 expression in cultured astrocytes after fluid percussion injury. J. Neurotrauma 28, 371–381. doi: 10.1089/neu.2010.1705
Rao, W., Zhang, L., Peng, C., Hui, H., Wang, K., Su, N., et al. (2015). Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 1852, 2402–2413. doi: 10.1016/j.bbadis.2015.08.014
Rash, J. E., Yasumura, T., Davidson, K. G., Furman, C. S., Dudek, F. E., and Nagy, J. I. (2001). Identification of cells expressing Cx43, Cx30, Cx26, Cx32 and Cx36 in gap junctions of rat brain and spinal cord. Cell Commun. Adhes. 8, 315–320. doi: 10.3109/15419060109080745
Rovegno, M., Soto, P. A., Sáez, P. J., Naus, C. C., Saez, J. C., and von Bernhardi, R. (2015). Connexin43 hemichannels mediate secondary cellular damage spread from the trauma zone to distal zones in astrocyte monolayers. Glia 63, 1185–1199. doi: 10.1002/glia.22808
Sahin Kaya, S., Mahmood, A., Li, Y., Yavuz, E., and Chopp, M. (1999). Expression of nestin after traumatic brain injury in rat brain. Brain Res. 840, 153–157. doi: 10.1016/S0006-8993(99)01757-6
Sajja, V. S., Galloway, M. P., Ghoddoussi, F., Thiruthalinathan, D., Kepsel, A., Hay, K., et al. (2012). Blast-induced neurotrauma leads to neurochemical changes and neuronal degeneration in the rat hippocampus. NMR Biomed. 25, 1331–1339. doi: 10.1002/nbm.2805
Sajja, V. S., Hubbard, W. B., Hall, C. S., Ghoddoussi, F., Galloway, M. P., and VandeVord, P. J. (2015). Enduring deficits in memory and neuronal pathology after blast-induced traumatic brain injury. Sci. Rep. 5:15075. doi: 10.1038/srep15075
Sajja, V. S., Perrine, S. A., Ghoddoussi, F., Hall, C. S., Galloway, M. P., and VandeVord, P. J. (2014a). Blast neurotrauma impairs working memory and disrupts prefrontal myo-inositol levels in rats. Mol. Cell. Neurosci. 59, 119–126. doi: 10.1016/j.mcn.2014.02.004
Sajja, V. S. S. S., Ereifej, E. S., and VandeVord, P. J. (2014b). Hippocampal vulnerability and subacute response following varied blast magnitudes. Neurosci. Lett. 570, 33–37. doi: 10.1016/j.neulet.2014.03.072
Satoh, J.-I., Tabunoki, H., Yamamura, T., Arima, K., and Konno, H. (2007). Human astrocytes express aquaporin-1 and aquaporin-4 in vitro and in vivo. Neuropathology 27, 245–256. doi: 10.1111/j.1440-1789.2007.00774.x
Scheff, S. W., Price, D. A., Schmitt, F. A., Roberts, K. N., Ikonomovic, M. D., and Mufson, E. J. (2013). Synapse stability in the precuneus early in the progression of Alzheimers disease. J. Alzheimers. Dis. 35, 599–609. doi: 10.3233/JAD-122353
Shenaq, M., Kassem, H., Peng, C., Schafer, S., Ding, J. Y., Fredrickson, V., et al. (2012). Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin and HIF1alpha after traumatic brain injury (TBI). J. Neurol. Sci. 323, 134–140. doi: 10.1016/j.jns.2012.08.036
Shiino, A., Nishida, Y., Yasuda, H., Suzuki, M., Matsuda, M., and Inubushi, T. (2004). Magnetic resonance spectroscopic determination of a neuronal and axonal marker in white matter predicts reversibility of deficits in secondary normal pressure hydrocephalus. J. Neurol. Neurosurg. Psychiatr. 75, 1141–1148. doi: 10.1089/neu.2013.3110
Shultz, S. R., Wright, D. K., Zheng, P., Stuchbery, R., Liu, S. J., Sashindranath, M., et al. (2015). Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 138, 1297–1313. doi: 10.1093/brain/awv053
Singh, I. N., Sullivan, P. G., Deng, Y., Mbye, L. H., and Hall, E. D. (2006). Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J. Cereb. Blood Flow Metab. 26, 1407–1418. doi: 10.1038/sj.jcbfm.9600297
Smith, D. H., Chen, X.-H., Pierce, J. E. S., Wolf, J. A., Trojanowski, J. Q., Graham, D. I., et al. (1997). Progressive atrophy and neuron death for one year following brain trauma in the Rat. J. Neurotrauma 14, 715–727. doi: 10.1089/neu.1997.14.715
Smith, J. A., Das, A., Ray, S. K., and Banik, N. L. (2012). Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 87, 10–20. doi: 10.1016/j.brainresbull.2011.10.004
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Sullivan, P. G., Keller, J. N., Mattson, M. P., and Scheff, S. W. (1998). Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function. J. Neurotrauma 15, 789–798. doi: 10.1089/neu.1998.15.789
Sullivan, P., Rabchevsky, A., Waldmeier, P., and Springer, J. (2005). Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J. Neurosci. Res. 79, 231–239. doi: 10.1002/jnr.20292
Sun, L., Gao, J., Zhao, M., Cui, J., Li, Y., Yang, X., et al. (2015). A novel cognitive impairment mechanism that astrocytic p-connexin 43 promotes neuronic autophagy via activation of P2X7R and down-regulation of GLT-1 expression in the hippocampus following traumatic brain injury in rats. Behav. Brain Res. 291, 315–324. doi: 10.1016/j.bbr.2015.05.049
Sun, L. Q., Gao, J. L., Cui, C. M., Cui, Y., Jing, X. B., Zhao, M. M., et al. (2014). Astrocytic p-connexin 43 regulates neuronal autophagy in the hippocampus following traumatic brain injury in rats. Mol. Med. Rep. 9, 77–82. doi: 10.3892/mmr.2013.1787
Sun, M. C., Honey, C. R., Berk, C., Wong, N. L., and Tsui, J. K. (2003). Regulation of aquaporin-4 in a traumatic brain injury model in rats. J. Neurosurg. 98, 565–569. doi: 10.3171/jns.2003.98.3.0565
Suzuki, R., Okuda, M., Asai, J., Nagashima, G., Itokawa, H., Matsunaga, A., et al. (2006). “Astrocytes co-express aquaporin-1,-4, and vascular endothelial growth factor in brain edema tissue associated with brain contusion,” in Brain Edema XIII, eds J. T. Hoff, R. F. Keep, G. Xi, and Y. Hua (Vienna: Springer), 398–401.
Svetlov, S. I., Larner, S. F., Kirk, D. R., Atkinson, J., Hayes, R. L., and Wang, K. K. (2009). Biomarkers of blast-induced neurotrauma: profiling molecular and cellular mechanisms of blast brain injury. J. Neurotrauma 26, 913–921. doi: 10.1089/neu.2008.0609
Thelin, E. P., Nelson, D. W., and Bellander, B. M. (2014). Secondary peaks of S100B in serum relate to subsequent radiological pathology in traumatic brain injury. Neurocrit. Care 20, 217–229. doi: 10.1007/s12028-013-9916-0
Thrane, A. S., Rangroo Thrane, V., and Nedergaard, M. (2014). Drowning stars: reassessing the role of astrocytes in brain edema. Trends Neurosci. 37, 620–628. doi: 10.1016/j.tins.2014.08.010
Touboul, D., and Gaudin, M. (2014). Lipidomics of Alzheimers disease. Bioanalysis 6, 541–561. doi: 10.4155/bio.13.346
Tran, N. D., Kim, S., Vincent, H. K., Rodriguez, A., Hinton, D. R., Bullock, M. R., et al. (2010). Aquaporin-1-mediated cerebral edema following traumatic brain injury: effects of acidosis and corticosteroid administration. J. Neurosurg. 112, 1095–1104. doi: 10.3171/2009.8.JNS081704
Vajtr, D., Benada, O., Linzer, P., Sámal, F., Springer, D., Strejc, P., et al. (2012). Immunohistochemistry and serum values of S-100B, glial fibrillary acidic protein, and hyperphosphorylated neurofilaments in brain injuries. Soud. Lek. 57, 7–12.
Vandevord, P. J., Leung, L. Y., Hardy, W., Mason, M., Yang, K. H., and King, A. I. (2008). Up-regulation of reactivity and survival genes in astrocytes after exposure to short duration overpressure. Neurosci. Lett. 434, 247–252. doi: 10.1016/j.neulet.2008.01.056
Walker, K. R., and Tesco, G. (2013). Molecular Mechanisms of Cognitive Dysfunction following Traumatic Brain Injury. Front. Aging Neurosci. 5:29. doi: 10.3389/fnagi.2013.00029
Wang, J.-W., Wang, H.-D., Zhong, W.-Z., Li, N., and Cong, Z.-X. (2012). Expression and cell distribution of metabotropic glutamate receptor 5 in the rat cortex following traumatic brain injury. Brain Res. 1464, 73–81. doi: 10.1016/j.brainres.2012.05.014
Wu, H., Mahmood, A., Lu, D., Jiang, H., Xiong, Y., Zhou, D., et al. (2010). Attenuation of astrogliosis and modulation of endothelial growth factor receptor in lipid rafts by simvastatin after traumatic brain injury. J. Neurosurg. 113, 591–597. doi: 10.3171/2009.9.JNS09859
Xiong, G., Elkind, J. A., Kundu, S., Smith, C. J., Antunes, M. B., Tamashiro, E., et al. (2014). Traumatic brain injury-induced ependymal ciliary loss decreases cerebral spinal fluid flow. J. Neurotrauma. 31, 1396–1404. doi: 10.1089/neu.2013.3110
Xiong, Y., Gu, Q., Peterson, P. L., Muizelaar, J. P., and Lee, C. P. (1997). Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma 14, 23–34. doi: 10.1089/neu.1997.14.23
Yan, E. B., Satgunaseelan, L., Paul, E., Bye, N., Nguyen, P., Agyapomaa, D., et al. (2014). Post-traumatic hypoxia is associated with prolonged cerebral cytokine production, higher serum biomarker levels, and poor outcome in patients with severe traumatic brain injury. J. Neurotrauma 31, 618–629. doi: 10.1089/neu.2013.3087
Yao, X., Uchida, K., Papadopoulos, M. C., Zador, Z., Manley, G. T., and Verkman, A. S. (2015). Mildly reduced brain swelling and improved neurological outcome in aquaporin-4 knockout mice following controlled cortical impact brain injury. J. Neurotrauma 32, 1458–1464. doi: 10.1089/neu.2014.3675
Yi, J.-H., Park, S.-W., Brooks, N., Lang, B. T., and Vemuganti, R. (2008). PPARγ agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res. 1244, 164–172. doi: 10.1016/j.brainres.2008.09.074
Keywords: astrocytes, microglia, oligodendrocytes, traumtic brain injury (TBI), biomarkers, MRS spectroscopy, memory impairment, gliosis
Citation: Sajja VSSS, Hlavac N and VandeVord PJ (2016) Role of Glia in Memory Deficits Following Traumatic Brain Injury: Biomarkers of Glia Dysfunction. Front. Integr. Neurosci. 10:7. doi: 10.3389/fnint.2016.00007
Received: 31 October 2015; Accepted: 05 February 2016;
Published: 29 February 2016.
Edited by:
Ye Chen, Navy Medical Research Center, USAReviewed by:
Mikulas Chavko, Naval Medical Research Center, USAPeethambaran Arun, Walter Reed Army Institute of Research, USA
Esther Shohami, Hebrew Universtiy of Jerusalem, Israel
Copyright © 2016 Sajja, Hlavac and VandeVord. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela J. VandeVord, pvord@vt.edu