 Trevor R. Cardinal1,2* Kyle R. Struthers1 Thomas J. Kesler1 Matthew D. Yocum1 David T. Kurjiaka3† James B. Hoying4†
Trevor R. Cardinal1,2* Kyle R. Struthers1 Thomas J. Kesler1 Matthew D. Yocum1 David T. Kurjiaka3† James B. Hoying4†
- 1 Biomedical Engineering, California Polytechnic State University, San Luis Obispo, CA, USA
- 2 Physiological Sciences, University of Arizona, Tucson, AZ, USA
- 3 Department of Physiology, University of Arizona, Tucson, AZ, USA
- 4 Division of Microcirculation, University of Arizona, Tucson, AZ, USA
Vasodilation of lower leg arterioles is impaired in animal models of chronic peripheral ischemia. In addition to arterioles, feed arteries are a critical component of the vascular resistance network, accounting for as much as 50% of the pressure drop across the arterial circulation. Despite the critical importance of feed arteries in blood flow control, the impact of ischemia on feed artery vascular reactivity is unknown. At 14 days following unilateral resection of the femoral–saphenous artery–vein pair, functional vasodilation of the profunda femoris artery was severely impaired, 11 ± 9 versus 152 ± 22%. Although endothelial and smooth muscle-dependent vasodilation were both impaired in ischemic arteries compared to control arteries (Ach: 40 ± 14 versus 81 ± 11%, SNP: 43 ± 12 versus and 85 ± 11%), the responses to acetylcholine and sodium nitroprusside were similar, implicating impaired smooth muscle-dependent vasodilation. Conversely, vasoconstriction responses to norepinephrine were not different between ischemic and control arteries, −68 ± 3 versus −66 ± 3%, indicating that smooth muscle cells were functional following the ischemic insult. Finally, maximal dilation responses to acetylcholine, ex vivo, were significantly impaired in the ischemic artery compared to control, 71 ± 9 versus 97 ± 2%, despite a similar generation of myogenic tone to the same intravascular pressure (80 mmHg). These data indicate that ischemia impairs feed artery vasodilation by impairing the responsiveness of the vascular wall to vasodilating stimuli. Future studies to examine the mechanistic basis for the impact of ischemia on vascular reactivity or treatment strategies to improve vascular reactivity following ischemia could provide the foundation for an alternative therapeutic paradigm for peripheral arterial occlusive disease.
Introduction
The most common symptom in patients with peripheral arterial occlusive disease (PAOD) is intermittent claudication, an ischemic pain during locomotion or exercise (Aronow, 2005). Intermittent claudication is typically explained by poorly developed collaterals in PAOD patients (Ziegler et al., 2010), which is supported by the observation that exercise training in animal models of chronic peripheral ischemia improves exercise capacity (Yang et al., 1990) and induces arteriogenesis (Yang et al., 2008). However, the lack of correlation between the ankle-brachial index (a ratio of ankle and brachial arterial pressures) and physical performance in patients suggests that structural deficits are not sufficient to explain reduced limb function and the onset of intermittent claudication with PAOD (Coutinho et al., 2011). An alternative explanation is that intermittent claudication is caused, or at least exacerbated, by poor vascular reactivity. Patients that experience intermittent claudication have sufficient muscle perfusion at rest, but experience ischemic pain at a time when functional vasodilation would be expected to induce hyperemia and increase oxygen delivery to contracting muscle, a phenotype that can be recapitulated in rodent models (Brevetti et al., 2001). Additional support for the hypothesis that intermittent claudication is caused, or affected, by impaired vascular reactivity, can be found in the observation that exercise training increases walking distance and improves vascular reactivity in patients with PAOD (Brendle et al., 2001) and animal models of chronic peripheral ischemia (Hudlicka et al., 1994; Kelsall et al., 2004).
The possibility that impaired vascular reactivity plays a causal role in the pathophysiology of PAOD warrants investigation into the impact of arterial occlusion on resistance vessel reactivity. Understanding the mechanism of impaired vascular reactivity with chronic peripheral ischemia could provide the foundational knowledge for a new paradigm in the treatment of PAOD. Most effort in this area has been directed toward examining perfusion through the collateral circuit (Yang et al., 1990; Colleran et al., 2010), which is the primary site of resistance to blood flow in the distal ischemic zone (Taylor et al., 2008a). However, the collateral circuit is not the only significant site of resistance in the ischemic hindlimb. Perfusion in the distal hindlimb of Rac2−/− and Nox2−/− mice is reduced despite normal collateral enlargement (Distasi et al., 2009), suggesting that the resistance vasculature in the ischemic zone makes a significant contribution to the overall resistance to blood flow. This is further supported by computational studies indicating that vasodilation, angiogenesis, capillary arterialization, and collateral enlargement are necessary to explain perfusion recovery following unilateral ligation of the femoral artery (Benedict, Experimental Biology, 2011).
Although numerous reports have examined the impact of chronic ischemia on regional blood flow (Bauters et al., 1995; Walder et al., 1996; Yang et al., 1996; Song et al., 2004), it is difficult to discern the relative impacts of vascular architecture (i.e., enhanced arteriogenesis) and vascular reactivity (i.e., improved vasodilation) when measuring whole muscle or entire limb blood flow. Therefore, determining the impact of chronic ischemia on vascular reactivity requires measuring response in individual arterioles and feed arteries, each of which contributes approximately 50% of the overall resistance to tissue blood flow (Lash, 1994; Segal, 2000; Sweeney et al., 2007). In rat models of chronic peripheral ischemia, arterioles of the lower leg exhibit impaired functional (Dawson et al., 1990; Hudlicka et al., 1994) and endothelial-dependent vasodilation at early time points and impaired smooth muscle-dependent vasodilation at late time points (Kelsall et al., 2001). Reactivity of angiographically identifiable arterial vessels in the ischemic rat hindlimb demonstrate impaired endothelial- (Takeshita et al., 1998) and smooth muscle-dependent vasodilation (Takeshita et al., 1997), but the location of these vessels in the hindlimb vascular circuit cannot be readily determined. The impact of chronic ischemia on vascular reactivity of feed arteries has only been assessed ex vivo (Taylor et al., 2008b). In these studies, feed artery reactivity to endothelial- and smooth muscle-dependent dilators was unaffected by ischemia, even when examined at low and high intraluminal pressures (Taylor et al., 2008b). The divergence of observations regarding the impact of ischemia on arterioles and feed arteries, ex vivo and in vivo warrants further investigation into the impact of chronic ischemia on vascular reactivity. Even though the collateral circuit is the primary site of resistance in the ischemic hindlimb, the critical role of feed arteries in blood flow control suggests that improving vascular reactivity in patients with PAOD will require functional restoration of intramuscular arterioles and resistance arteries in the ischemic zone.
To test the hypothesis that ischemia impairs vasodilation in feed arteries, we assessed functional vasodilation and vascular reactivity (in vivo and ex vivo) in the profunda femoris artery (aka muscular branch) at day-14 following unilateral resection of the femoral artery and vein in the mouse hindlimb.
Materials and Methods
Animals
Male and female C57Bl/6 or C57Bl/6 hybrid mice (SW-129SV) were used for all experiments between 2 and 4 months of age, according to protocols approved by the University of Arizona and Cal Poly State University Institutional Animal Care and Use Committees. Male and female hybrid mice were used for the ex vivo vascular reactivity experiments. The control group included three animals one male and two females. The experimental group included six animals four males and two females. C57 male mice were used for all of the in vivo studies, with eight mice per group. C57Bl/6 hybrid mice were obtained from a breeding colony maintained at the University of Arizona while C57Bl/6 mice were obtained from Taconic Farms. Mice were maintained on a 12:12-h light:dark cycle and given water and rodent chow ad libitum.
Chronic Hindlimb Ischemia Surgery
Chronic hindlimb ischemia was induced by unilateral ligation and resection of the left femoral and saphenous artery–vein pair, from ∼2 mm upstream to the profunda femoris artery (aka muscular branch) to halfway between the knee and ankle, as described (Couffinhal et al., 1998; Sullivan et al., 2002) with some modifications. Animals were anesthetized in an induction chamber with 5% isoflurane administered through an isoflurane vaporizer and balanced with oxygen flowing at ∼3.5 l · min−1. Following induction, anesthesia was maintained throughout animal preparation and surgery with 1–2% isoflurane balanced with oxygen flowing at 0.5–1.0 l · min−1 though an anesthesia mask. Prior to surgery, left hindlimb hair was removed with trimming clippers and depilatory cream and rubbed with chlorhexidine diacetate to disinfect. Animals were maintained at ∼35°C with a rectal-temperature-controlled heat pad (TR-200, FST, Foster City, CA, USA). The epigastric neurovascular bundle and fat pad were dissected free with a heat cautery to create access for the upstream ligation site. Following resection of the artery–vein pair, the skin incision was closed with 7-0 polypropylene suture. Animals were given buprenorphine as an analgesic subcutaneously at a dose of 0.075 mg kg−1 and allowed to recover with warming.
Functional Vasodilation
At day-14 following the hindlimb ischemia surgery, animals were anesthetized and prepared as described above; a skin incision and gentle blunt dissection were used to expose the profunda femoris artery. A tungsten stimulating electrode (1–5 μm tip diameter and 250 μm shaft diameter, FHC, Bowdoinham, ME, USA) was rested across the mid-belly (approximate location of the motor end plate) of the gracilis muscles; the ground electrode was placed subcutaneously at the periphery of the incision and a brief set of contractions was induced to confirm proper electrode placement. The exposed tissue was covered with mineral oil to prevent desiccation and minimize oxygen exchange (Young et al., 1986). The intravital microscope (Microscan, Microvision Medical, Amsterdam, Netherlands) was then placed over the distal profunda femoris (the field of view was immediately anterior to the gracilis anterior muscle), electrode placement was evaluated a second time, and the preparation was given 30 min to stabilize before measurements were made. To elicit functional vasodilation, the gracilis muscles were stimulated with 1 mA square waves of 0.2 ms duration at 8 Hz for 90 s using a stimulus isolator and PowerLab 4-channel data acquisition system (ADInstruments, Colorado Springs, CO, USA). Profunda diameters were determined from merged still frames of videos of the microscope field using Automated Vascular Analysis (Microvision Medical, Amsterdam, Netherlands). To account for animal-to-animal variation in baseline artery diameter, resting diameter was set to 0%; increases above 0% represent vasodilation.
In vivo Vascular Reactivity
At day-14 following the hindlimb ischemia surgery, animals were anesthetized and prepared as described above. The intravital microscope was placed over the profunda femoris and the exposed tissue was continually irrigated with a physiological salt solution (PSS) that contained (in mM) NaCl 137, KCl 4.7, MgSO4 1.2, CaCaL2 2, NaHCO3 18, and was bubbled with 5% CO2-95% N2. The PSS was maintained at ∼35°C and flowed over the muscle at ∼2 ml min−1, as described previously (Moore et al., 2010). The preparation was given 30 min to stabilize before the application of vasoactive agents. For the first study, increasing doses of Ach were applied to generate a dose response curve, prior to application of SNP. In the second study, maximal doses of the vasoactive agents were applied in the following order (all at 10−4 M): norepinephrine, acetylcholine, and sodium nitroprusside. Profunda diameter was given 5 min to stabilize between the application of the each pharmacological agent. Diameter measurements were obtained as described above. Decreases below 0% represent vasoconstriction.
Ex vivo Vascular Reactivity
At day-14 following the hindlimb ischemia surgery, animals were anesthetized and prepared as described above. The distal aspect of the profunda femoris artery, running deep to the gracilis anterior muscle, was dissected from the ischemic hindlimb and non-ischemic contralateral hindlimb, or from a non-ischemic control, as described (Sun et al., 1994). Following dissection, the feed artery was cleaned of connective tissue in a PSS solution maintained below 10°C. PSS was composed of the following (in mM) 119 NaCl, 4.7 KCl, 1.2 MgSO4, 1.7 KH2PO4, 1.6 CaCl, 5 glucose, and 18 NaHCO3. Cleaned arteries were cannulated and prepared for microscopic viewing, as described (Kurjiaka et al., 2005). The cannulated vessel was then warmed to 35°C and pressurized to 80 mmHg to induce myogenic tone development prior to the application of acetylcholine (Ach) in PSS. Vessels were warmed to 35°C, as opposed to 37–38°C because the distal profunda femoris arteries used in these studies were isolated from the peripheral skeletal muscle, which at rest is less than core-body temperature, and tends to be closer to 34–35°. Following collection of dose response curves to Ach, Ca+2-free PBS with 3 mM EGTA was applied to obtain the maximal passive diameter. To account for animal-to-animal variation in baseline artery diameter, all diameter measurements were fit within a scale ranging from 0% (myogenic tone) to 100% (maximal passive diameter) for statistical analysis and graphic display.
Statistics
Data are presented as mean ± SEM. Statistical significance was set at p ≤ 0.05. Differences in maximal profunda functional vasodilation were assessed by paired t-test. Differences in in vivo profunda reactivity were assessed by one-way ANOVA, followed by Student’s t-tests. Differences in functional vasodilation recovery and ex vivo profunda reactivity were assessed by repeated measures ANOVA followed by Student’s t-tests. EC50 values were obtained using the non-linear regression procedure in SAS.
Results
Functional Vasodilation in the Ischemic Profunda
To determine if ischemia impaired endogenous vasodilation pathways, we measured the diameter of the profunda femoris feed artery before and after electrical stimulation of gracilis muscle contraction, Figures 1A,B. Ischemia significantly increased the resting diameter in the profunda femoris, 76 ± 4 versus 50 ± 4 μm. However, the increase in resting diameter did not reflect a maximal dilation, and ischemia profoundly reduced functional vasodilation in the profunda femoris, which exhibited an 11 ± 9% increase above resting diameter immediately following gracilis muscle contraction, compared to a 152 ± 22% increase in the profunda from the contralateral, sham operated limb, Figures 1C,D.
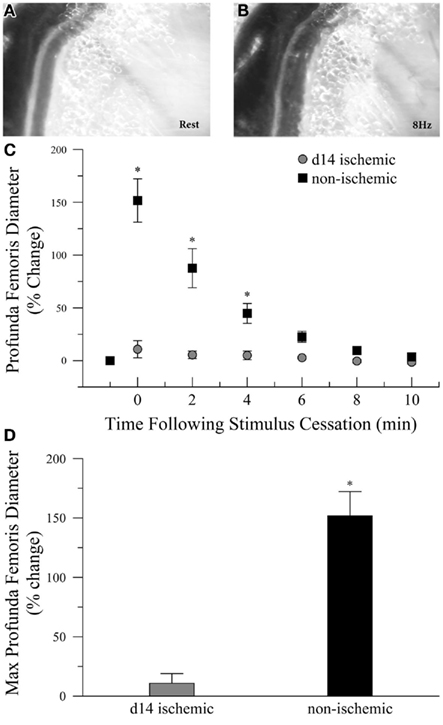
Figure 1. Intravital microscopy imaging field and functional vasodilation responses in the profunda femoris arteries, in vivo. (A) Representative photomicrographs of the distal profunda femoris artery–vein pair imaging site for functional vasodilation and vascular reactivity studies at rest and (B) immediately following the cessation of 8 Hz muscle contraction. (C) Resting diameter and 10 min following 90 s of 8 Hz muscle contraction in day-14 ischemic (gray circles) and contralateral control (black boxes) profunda femoris arteries. (D) Maximal functional vasodilation in day-14 ischemic (gray) and contralateral control (black) profunda femoris arteries following 90 s of 8 Hz muscle contraction. Data points are on a relative scale, with resting diameter represented by 0%. *p ≤ 0.05 versus day-14 ischemic.
In vivo Vascular Reactivity in the Ischemic Profunda
Next, we tested the hypothesis that impaired functional vasodilation following ischemia could be explained by endothelial dysfunction. To test this hypothesis, we measured the diameter of the profunda femoris feed artery before and after application of acetylcholine (Ach) and sodium nitroprusside (SNP), endothelial- and smooth muscle-dependent vasodilators, respectively. In the first study, we generated dose response curves to Ach (10−8–10−4) prior to the application of SNP (10−4). In this study, resting diameter was significantly larger in ischemic than non-ischemic vessels, 116 ± 11 versus 80 ± 6 μm, respectively. However, ischemia significantly reduced vascular reactivity, as percent increase in artery diameter was significantly lower at 10−6 (22 ± 6 versus 53 ± 12%), 10−5 (24 ± 7 versus 65 ± 11%), and 10−4 M Ach (25 ± 7 versus 69 ± 11%), Figure 2A. EC50 was not different between ischemic and control vessels, 2.55 × 10−7 versus 1.62 × 10−7 M Ach, respectively, but only seven non-ischemic and four ischemic replicates were able to achieve convergence in the non-linear regression model. Dilation responses to SNP were also significantly reduced in the ischemic artery as compared to the control artery, 31 ± 7 versus 72 ± 11%, respectively. To ensure that the reduced reactivity of the ischemic vessels was not due to the increased resting diameter of the profunda femoris, we completed a second study in which we delivered maximal doses of Ach (10−4) and SNP (10−4). In these animals, resting diameter was not different between ischemic and non-ischemic vessels, 82 ± 10 versus 76 ± 6 μm, respectively. Ischemia still significantly reduced vasodilation following the application of Ach, 40 ± 14 versus 81 ± 11%. The roughly 50% reduction in vasodilation to Ach was mirrored by the responsiveness to SNP, which was 43 ± 12% in the ischemic artery versus 85 ± 11% in the control, Figure 2B. Although there was a significant reduction in vasodilation responses in the ischemic arteries, there was no difference between the response to acetylcholine and sodium nitroprusside in either study.
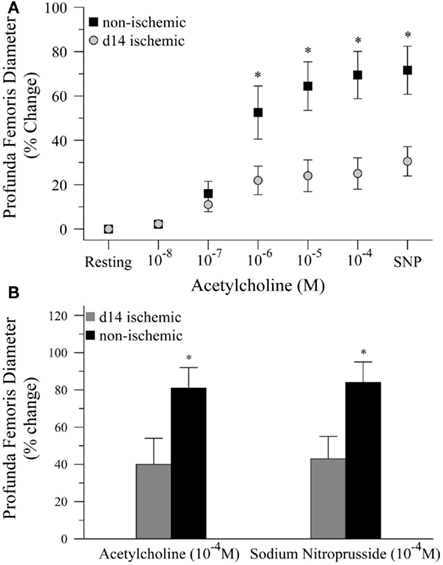
Figure 2. Vasodilation responses to endothelial- and smooth muscle-dependent dilating agents in ischemic and non-ischemic profunda femoris arteries, in vivo. (A) Vasodilation responses to increasing doses of acetylcholine (Ach), applied in 10−x M increments, and a maximal dose of sodium nitroprusside (SNP) in day-14 ischemic (gray) and non-ischemic (black) arteries. (B) Vasodilation responses to maximal doses of Ach (10−4 M) and SNP (10−4 M) in day-14 ischemic (gray) and contralateral control (black) profunda femoris arteries. Data points are on a relative scale, with resting diameter represented by 0%. *p ≤ 0.05 versus day-14 ischemic.
To complete our in vivo studies, we tested the hypothesis that the reduction in smooth muscle-dependent vasodilation could not be explained by a generalized loss of smooth muscle function. To test this hypothesis, we first performed a pilot study to examine the dose response to norepinephrine (NE), Figure 3A. Based on the pilot data, we measured the diameter of the profunda femoris feed artery before and after application of a maximal dose of NE (10−4 M). Ischemia had no effect on norepinephrine-induced vasoconstriction in the profunda femoris artery, as demonstrated by a similar decrease in diameter compared to the contralateral artery, 26 ± 4 versus 25 ± 1 μm, Figure 3B. These responses represent a 66 ± 3 and 68 ± 3% decrease from resting diameters in the ischemic and non-ischemic vessels, respectively, which were not different.
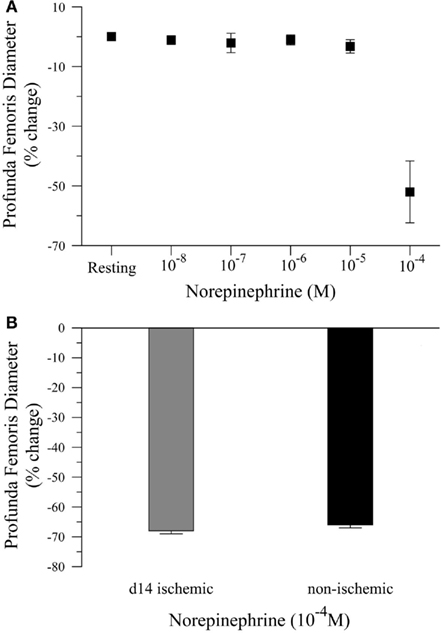
Figure 3. Vasoconstriction responses to norepinephrine in ischemic and non-ischemic profunda femoris arteries, in vivo. (A) Pilot study measuring the vasoconstriction responses to increasing doses of norepinephrine (NE), applied in 10−x M increments, and (B) vasoconstriction responses to maximal doses of NE (10−4 M) in day-14 ischemic (gray) and contralateral control (black) profunda femoris arteries. Data points are on a relative scale, with resting diameter represented by 0%.
Ex vivo Vasodilation in the Ischemic Profunda
Given that intravascular pressure in the profunda femoris is likely decreased by the femoral artery–vein resection, we tested the hypothesis that reduced arterial pressure could explain the reduced vascular reactivity in this vessel. To test this hypothesis, we measured the diameter response to acetylcholine (Ach) in isolated, distal profunda femoris arteries that were pressurized to the approximate pressure of the control profunda. Resting diameter was significantly larger in the ischemic profunda, 105 ± 6 versus 62 ± 3 μm. Given the individual variations in resting diameter, ex vivo artery responses were expressed as a percent change, with 0% representing myogenic tone and 100% representing maximal passive vasodilation in calcium-free conditions. Despite being exposed to an equal hydrostatic pressure (80 mmHg) and developing an equivalent level of myogenic tone (61 ± 5 versus 58 ± 1% of maximal passive diameter), vasodilation was still significantly impaired in the ischemic profunda femoris artery (71 ± 9 versus 97 ± 2% of maximal diameter at 3 × 10−5 M Ach), Figure 4. EC50 was significantly lower in the ischemic vessels, 2.82 × 10−7 versus 8.03 × 10−7 M Ach. The greater EC50 extrapolates that a higher concentration of Ach was necessary to induce vasodilation to 50% of the maximal response, indicating that the ischemic arteries were less sensitive to Ach.
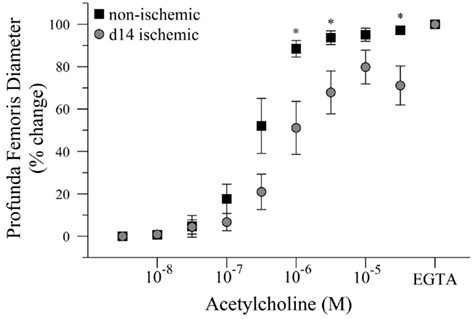
Figure 4. Dose response curve to acetylcholine in ischemic and non-ischemic distal profunda femoris arteries, ex vivo. Vessels were pressurized to 80 mmHg (baseline) and developed myogenic tone before exposure to the initial dose of Ach (10−8 M); increasing doses were applied at 3 × 10−x M increments. Data points are on a relative scale, with myogenic tone represented as 0% and maximal passive vasodilation represented as 100% (Ca+2-free PSS + EGTA). *p ≤ 0.05 versus day-14 ischemic.
Discussion
Patients with PAOD, who often present with intermittent claudication (Aronow, 2005), exhibit impaired vasodilation (Poredos et al., 2003), suggesting that improving vasodilation may be a reasonable strategy to alleviate ischemic pain associated with locomotion. Advancing this therapeutic paradigm requires understanding the mechanism of impaired vasodilation with chronic ischemia. Previous work demonstrated that chronic ischemia significantly impaired vasodilation in intramuscular arterioles (Hudlicka et al., 1994), but the effect of ischemia on feed arteries in vivo was unknown. Therefore, the objective of this work was to describe the effect of chronic ischemia on feed artery functional vasodilation and vascular reactivity.
As expected, chronic ischemia significantly impaired functional vasodilation in the profunda femoris feed artery, Figure 1. Given that ischemic arterioles demonstrate impaired endothelial-dependent vasodilation (Kelsall et al., 2001), we tested the hypothesis that impaired functional vasodilation in feed arteries could be explained by reduced endothelial-dependent responses. Surprisingly, both endothelial- and NO-mediated smooth muscle-dependent vasodilation were impaired in the profunda femoris feed artery following chronic ischemia, Figure 2.
Endothelial-dependent vasodilation was impaired in the profunda femoris following ischemia, but the degree of impairment was less than in intramuscular arterioles, which exhibit a slight vasoconstriction in response to acetylcholine (Kelsall et al., 2001). This reduced effect of ischemia on feed arteries, as compared to intramuscular arterioles, could be explained by the anatomical location of the feed arteries, which are outside of the parenchymal tissue. Intramuscular arterioles are likely affected to a greater extent by the hypoxia, elevated metabolites, and reduced pH resultant from skeletal muscle activity and the inflammation induced by myofiber repair (Scholz et al., 2002, 2003).
Even considering the explanation that feed arteries would be less affected by the ischemic tissue environment than arterioles, it is surprising that acetylcholine-dependent vasodilation was so much greater than functional vasodilation in the ischemic profunda, 40 versus 11% increase in diameter. In ischemic arterioles, functional vasodilation (Hudlicka et al., 1994) and acetylcholine (Kelsall et al., 2001) induced a slight vasoconstriction. The incongruence between functional- and acetylcholine-vasodilation could be explained by an impaired conducted response. As mentioned above, feed arteries lie outside of the metabolic field; therefore, the vasodilation response stimulated by an increase in tissue metabolism must be transmitted along the intramuscular arteriolar endothelium to the extra-parenchymal feed artery. If the conduction of vasodilation stimuli from the arterioles to the feed artery were impaired, then the feed artery would be capable of responding to exogenous vasodilators even if the same vessel were relatively unresponsive to increases in muscle metabolism. This hypothesis is supported by the observation that in the Cx40−/− mouse, arteriolar vasodilation responses to acetylcholine are normal, despite a reduction in both conducted responses (De Wit et al., 2000) and functional vasodilation (Milkau et al., 2010).
Although chronic ischemia had a more mild effect on endothelial-dependent vasodilation in feed arteries than on intramuscular arterioles (Kelsall et al., 2001), NO-mediated smooth muscle-dependent vasodilation was significantly impaired, and similar to endothelial-dependent vasodilation responses, Figure 2. Based on previous work, we expected vasodilation responses to sodium nitroprusside to remain intact at day-14 following the induction of chronic ischemia (Kelsall et al., 2001). This result cannot be explained by a generalized reduction in smooth muscle tone control, such as would be expected with the conversion to a synthetic phenotype during a structural remodeling event (Schaper, 2009), as the profunda femoris exhibited normal vasoconstriction responses to norepinephrine, Figure 3. Nor can the reduced vascular reactivity be explained solely by a reduced perfusion pressure, which would be expected following femoral artery resection, because ischemic profunda femoris arteries exhibited reduced vasodilation when they were isolated and pressurized to the same level as contralateral control arteries, Figure 4. Interestingly, these results are not without some precedent, as endothelial- and NO-mediated smooth muscle-dependent vasodilation is impaired in intramuscular arterioles at 5-weeks following induction of chronic ischemia (Kelsall et al., 2001) and in angiographically identifiable arteries of the rat thigh (Takeshita et al., 1997). The mechanism of impaired NO-mediated smooth muscle-dependent responses in these reports and in the data presented above is unknown, but may be related to oxidative stress, which increases with chronic ischemia (Pipinos et al., 2008) and impairs dilation responses to both acetylcholine and sodium nitroprusside in the obese Zucker rat (Frisbee and Stepp, 2001).
Our findings indicate that ischemia impairs the vascular smooth muscle cell signaling pathways involved in vasodilation. Much of the work in the area of smooth muscle-dependent tone regulation following arterial occlusion has addressed adrenergic signaling. Although adrenergic contractility appears to decrease in the collateral circulation (Taylor et al., 2008b) and in distal (ischemic) skeletal muscle arterioles (Kelsall et al., 2001) of rodent models, α-adrenergic contractility is exaggerated in ischemic skeletal muscle from patients (Jarajapu et al., 2001). The difference between rodent and patient responses is possibly due to the increased sympathetic tone observed with advanced age (Dinenno et al., 2005). Regardless, elevated sympathetic tone, which would be surprising in a rodent model of ischemia, does not appear to explain the impaired vasodilation observed in our studies, since norepinephrine-induced vasoconstriction is not altered, Figure 3. Although our finding appears contradictory to previous studies (Kelsall et al., 2001; Taylor et al., 2008b), it is not surprising that ischemia would have a limited effect on adrenergic sensitivity of the profunda femoris, because feed arteries are typically less sensitive to sympathetic tone than intramuscular arterioles (Vanteeffelen and Segal, 2003), which comprise most of the collateral circuit studied in previous reports.
Given the normal responses of ischemic arteries to norepinephrine (Figure 4), it would appear that ischemia specifically impairs the signaling cascades controlling smooth muscle cell relaxation in feed arteries. Few reports have assessed the impact of ischemia on smooth muscle cell signaling, as it pertains to vasodilation, but ischemia could be characterized by a reduction in NO-cGMP signaling, as supplementation with L-arginine (Murohara et al., 1998) or inhibition of PDE-5 increases vascular density and tissue perfusion following ischemia (Senthilkumar et al., 2007). Oxidative stress, induced by ischemia (Pipinos et al., 2008), could also contribute to impaired NO-dependent responses.
Having only assessed NO-mediated smooth muscle-dependent responses, we cannot definitively conclude that ischemia induces a generalized impairment in smooth muscle-dependent relaxation, as relaxation to other pathways may still be intact. Although acetylcholine is known to cause endothelial production of NO, prostanoid dilators, and hyperpolarizing agents (Fitzgerald et al., 2007), it is possible that chronic ischemia impairs endothelial production of dilating arachidonic acid derivatives without affecting their smooth muscle cell receptors. However, limited evidence suggests that chronic ischemia may also alter hyperpolarizing-channel function in feed arteries, as ischemia-reperfusion injury increases the sensitivity of calcium-activated potassium channels, or BK channels (Woodman and Wongsawatkul, 2004). Therefore, future studies are necessary to dissect the impact of chronic ischemia on the myriad of pathways capable of causing smooth muscle relaxation. This effort is challenged by the fact that a paracrine factor(s) has not been demonstrated to be both necessary and sufficient for functional vasodilation (Clifford and Hellsten, 2004; Segal, 2005), but the endothelium appears to be critical for the response (Duza and Sarelius, 2004), so future work would focus on the impact of ischemia on pathways involving endothelial-derived factors. Specifically, the functionality prostanoid-dependent dilation would be assessed by applying arachidonic acid to assess the functionality of the endothelial cylcooxygenase cascade (Frisbee et al., 2009), while a prostacyclin analog, such as iloprost, would be used to assess the functionality of smooth muscle receptors (Frisbee, 2004). Assessing the functional status of hyperpolarizing-dependent dilation is of greater difficulty, owing to the large number of factors apparently capable of inducing smooth muscle hyperpolarization (Feletou and Vanhoutte, 2006), and would therefore likely involve assessing the impact of potassium channel inhibitors on vasodilation responses to factors that are known to induce hyperpolarization, such as acetylcholine.
Our findings implicating smooth muscle dysfunction in impaired vascular reactivity with chronic ischemia may seem incongruent with preclinical (Bauters et al., 1995; Takeshita et al., 1998; Kelsall et al., 2004) and clinical reports (Poredos et al., 2003; Coutinho et al., 2011) that implicate endothelial dysfunction as the cause of impaired vascular reactivity. This incongruence could be explained in several ways. First, feed arteries may be differentially affected by ischemia than intramuscular arterioles, which exhibit impaired endothelial-dependent responses at day-14 following arterial occlusion (Kelsall et al., 2004). Support for this explanation comes from the observation that in patients, conduit vessels exhibit impaired endothelial- and smooth muscle-dependent responses, while resistance vessels exhibit only impaired endothelial-dependent responses (Liao et al., 1991). Second, the efficacy of endothelial-derived relaxing factors is dependent on the functionality of vascular smooth muscle cells. Therefore, defining endothelial dysfunction should require the observation of impaired endothelial-dependent dilation and intact smooth muscle-dependent dilation. However it is common in preclinical reports to conclude endothelial dysfunction after the application of only endothelial-dependent agents (Bauters et al., 1995; Takeshita et al., 1998) or in clinical reports based solely on flow-mediated dilation responses (Poredos et al., 2003). Finally, observations of endothelial dysfunction in patients could be explained by common co-morbidities to ischemic disease rather than the ischemic insult itself. For example, hypercholesterolemia and hyperglycemia are known to induce endothelial dysfunction (Granger et al., 2010; Ziegler et al., 2010).
One of the potential limitations of this study is the inclusion of C57Bl/6 hybrid mice of both sexes for the ex vivo vascular reactivity study. Although the variation in the study is small, and the findings are consistent with the in vivo studies that utilized only male C57Bl/6 mice, the data could not be stratified on the basis of sex and strain. Therefore, the absence of strain and sex-specific influences on the vascular response to ischemia cannot be confirmed, suggesting some caution when interpreting the findings.
In summary, the aim of these studies was to describe the impact of chronic ischemia on feed artery vascular reactivity. Feed artery functional vasodilation is severely impaired by chronic ischemia. The mechanism of this impairment cannot be explained by a simple reduction in intravascular pressure and appears to be due to impaired smooth muscle relaxation pathways. Interestingly, pathways controlling smooth muscle cell contraction remain intact. The dysfunctional signaling elements in smooth muscle relaxation will be identified in subsequent investigations. This overall area of research, determining the cellular basis for the impact of chronic ischemia on resistance vessel reactivity, has the potential to form the foundation for a new therapeutic paradigm in the treatment of PAOD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Office of Naval Research (N000140811209- Trevor R. Cardinal) and the National Institutes of Health (HL67067, HL63732, and HL07249- James B. Hoying). We would like to thank Drs. Steven Segal and Alex Moore for their advice and guidance on the vascular reactivity experimental preparation, Dr. Janis Burt for the use of her laboratory space for the ex vivo vascular reactivity studies, and Dr. Karen McGaughey for her assistance with the EC50 modeling.
References
Bauters, C., Asahara, T., Zheng, L. P., Takeshita, S., Bunting, S., Ferrara, N., Symes, J. F., and Isner, J. M. (1995). Recovery of disturbed endothelium-dependent flow in the collateral-perfused rabbit ischemic hindlimb after administration of vascular endothelial growth factor. Circulation 91, 2802–2809.
Brendle, D. C., Joseph, L. J., Corretti, M. C., Gardner, A. W., and Katzel, L. I. (2001). Effects of exercise rehabilitation on endothelial reactivity in older patients with peripheral arterial disease. Am. J. Cardiol. 87, 324–329.
Brevetti, L. S., Paek, R., Brady, S. E., Hoffman, J. I., Sarkar, R., and Messina, L. M. (2001). Exercise-induced hyperemia unmasks regional blood flow deficit in experimental hindlimb ischemia. J. Surg. Res. 98, 21–26.
Clifford, P. S., and Hellsten, Y. (2004). Vasodilatory mechanisms in contracting skeletal muscle. J. Appl. Physiol. 97, 393–403.
Colleran, P. N., Li, Z., Yang, H. T., Laughlin, M. H., and Terjung, R. L. (2010). Vasoresponsiveness of collateral vessels in the rat hindlimb: influence of training. J. Physiol. (Lond.) 588, 1293–1307.
Couffinhal, T., Silver, M., Zheng, L. P., Kearney, M., Witzenbichler, B., and Isner, J. M. (1998). Mouse model of angiogenesis. Am. J. Pathol. 152, 1667–1679.
Coutinho, T., Rooke, T. W., and Kullo, I. J. (2011). Arterial dysfunction and functional performance in patients with peripheral artery disease: a review. Vasc. Med. 16, 203–211.
Dawson, J. M., Okyayuz-Baklouti, I., and Hudlicka, O. (1990). Skeletal muscle microcirculation: the effects of limited blood supply and treatment with torbafylline. Int. J. Microcirc. Clin. Exp. 9, 385–400.
De Wit, C., Roos, F., Bolz, S. S., Kirchhoff, S., Kruger, O., Willecke, K., and Pohl, U. (2000). Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ. Res. 86, 649–655.
Dinenno, F. A., Masuki, S., and Joyner, M. J. (2005). Impaired modulation of sympathetic alpha-adrenergic vasoconstriction in contracting forearm muscle of ageing men. J. Physiol. 567, 311–321.
Distasi, M. R., Case, J., Ziegler, M. A., Dinauer, M. C., Yoder, M. C., Haneline, L. S., Dalsing, M. C., Miller, S. J., Labarrere, C. A., Murphy, M. P., Ingram, D. A., and Unthank, J. L. (2009). Suppressed hindlimb perfusion in Rac2−/− and Nox2−/− mice does not result from impaired collateral growth. Am. J. Physiol. Heart Circ. Physiol. 296, H877–H886.
Duza, T., and Sarelius, I. H. (2004). Increase in endothelial cell Ca(2+) in response to mouse cremaster muscle contraction. J. Physiol. 555, 459–469.
Feletou, M., and Vanhoutte, P. M. (2006). Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler. Thromb. Vasc. Biol. 26, 1215–1225.
Fitzgerald, S. M., Bashari, H., Cox, J. A., Parkington, H. C., and Evans, R. G. (2007). Contributions of endothelium-derived relaxing factors to control of hindlimb blood flow in the mouse in vivo. Am. J. Physiol. Heart Circ. Physiol. 293, H1072–H1082.
Frisbee, J. C. (2004). Enhanced arteriolar alpha-adrenergic constriction impairs dilator responses and skeletal muscle perfusion in obese Zucker rats. J. Appl. Physiol. 97, 764–772.
Frisbee, J. C., Hollander, J. M., Brock, R. W., Yu, H. G., and Boegehold, M. A. (2009). Integration of skeletal muscle resistance arteriolar reactivity for perfusion responses in the metabolic syndrome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R1771–R1782.
Frisbee, J. C., and Stepp, D. W. (2001). Impaired NO-dependent dilation of skeletal muscle arterioles in hypertensive diabetic obese Zucker rats. Am. J. Physiol. Heart Circ. Physiol. 281, H1304–H1311.
Granger, D. N., Rodrigues, S. F., Yildirim, A., and Senchenkova, E. Y. (2010). Microvascular responses to cardiovascular risk factors. Microcirculation 17, 192–205.
Hudlicka, O., Brown, M. D., Egginton, S., and Dawson, J. M. (1994). Effect of long-term electrical stimulation on vascular supply and fatigue in chronically ischemic muscles. J. Appl. Physiol. 77, 1317–1324.
Jarajapu, Y. P., Coats, P., Mcgrath, J. C., Macdonald, A., and Hillier, C. (2001). Increased alpha(1)- and alpha(2)-adrenoceptor-mediated contractile responses of human skeletal muscle resistance arteries in chronic limb ischemia. Cardiovasc. Res. 49, 218–225.
Kelsall, C. J., Brown, M. D., and Hudlicka, O. (2001). Alterations in reactivity of small arterioles in rat skeletal muscle as a result of chronic ischaemia. J. Vasc. Res. 38, 212–218.
Kelsall, C. J., Brown, M. D., Kent, J., Kloehn, M., and Hudlicka, O. (2004). Arteriolar endothelial dysfunction is restored in ischaemic muscles by chronic electrical stimulation. J. Vasc. Res. 41, 241–251.
Kurjiaka, D. T., Bender, S. B., Nye, D. D., Wiehler, W. B., and Welsh, D. G. (2005). Hypertension attenuates cell-to-cell communication in hamster retractor muscle feed arteries. Am. J. Physiol. Heart Circ. Physiol. 288, H861–H870.
Lash, J. M. (1994). Contribution of arterial feed vessels to skeletal muscle functional hyperemia. J. Appl. Physiol. 76, 1512–1519.
Liao, J. K., Bettmann, M. A., Sandor, T., Tucker, J. I., Coleman, S. M., and Creager, M. A. (1991). Differential impairment of vasodilator responsiveness of peripheral resistance and conduit vessels in humans with atherosclerosis. Circ. Res. 68, 1027–1034.
Milkau, M., Kohler, R., and De Wit, C. (2010). Crucial importance of the endothelial K+ channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J. 24, 3572–3579.
Moore, A. W., Bearden, S. E., and Segal, S. S. (2010). Regional activation of rapid onset vasodilatation in mouse skeletal muscle: regulation through adrenoreceptors. J. Physiol. (Lond.) 588, 3321–3331.
Murohara, T., Asahara, T., Silver, M., Bauters, C., Masuda, H., Kalka, C., Kearney, M., Chen, D., Symes, J. F., Fishman, M. C., Huang, P. L., and Isner, J. M. (1998). Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Invest. 101, 2567–2578.
Pipinos, I. I., Swanson, S. A., Zhu, Z., Nella, A. A., Weiss, D. J., Gutti, T. L., Mccomb, R. D., Baxter, B. T., Lynch, T. G., and Casale, G. P. (2008). Chronically ischemic mouse skeletal muscle exhibits myopathy in association with mitochondrial dysfunction and oxidative damage. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R290–R296.
Poredos, P., Golob, M., and Jensterle, M. (2003). Interrelationship between peripheral arterial occlusive disease, carotid atherosclerosis and flow mediated dilation of the brachial artery. Int. Angiol. 22, 83–87.
Scholz, D., Thomas, S., Sass, S., and Podzuweit, T. (2003). Angiogenesis and myogenesis as two facets of inflammatory post-ischemic tissue regeneration. Mol. Cell. Biochem. 246, 57–67.
Scholz, D., Ziegelhoeffer, T., Helisch, A., Wagner, S., Friedrich, C., Podzuweit, T., and Schaper, W. (2002). Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J. Mol. Cell. Cardiol. 34, 775–787.
Segal, S. S. (2000). Integration of blood flow control to skeletal muscle: key role of feed arteries. Acta Physiol. Scand. 168, 511–518.
Senthilkumar, A., Smith, R. D., Khitha, J., Arora, N., Veerareddy, S., Langston, W., Chidlow, J. H. Jr., Barlow, S. C., Teng, X., Patel, R. P., Lefer, D. J., and Kevil, C. G. (2007). Sildenafil promotes ischemia-induced angiogenesis through a PKG-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 27, 1947–1954.
Song, J., Cottler, P. S., Klibanov, A. L., Kaul, S., and Price, R. J. (2004). Microvascular remodeling and accelerated hyperemia blood flow restoration in arterially occluded skeletal muscle exposed to ultrasonic microbubble destruction. Am. J. Physiol. Heart Circ. Physiol. 287, H2754–H2761.
Sun, D., Kaley, G., and Koller, A. (1994). Characteristics and origin of myogenic response in isolated gracilis muscle arterioles. Am. J. Physiol. 266, H1177–H1183.
Sullivan, C. J., Doetschman, T., and Hoying, J. B. (2002). Targeted disruption of the Fgf2 gene does not affect vascular growth in the mouse ischemic hindlimb. J. Appl. Physiol. 93, 2009–2017.
Sweeney, T. E., Bagher, P., Bailey, J., Cherra, S., Grisafi, F. N., Pauli, E. M., Riley, K., and Soares, S. (2007). Intravascular pressure and diameter profile of the utero-ovarian resistance artery network: estrous cycle dependent modulation of resistance artery tone. Am. J. Physiol. Heart Circ. Physiol. 293, H2937–H2944.
Takeshita, S., Isshiki, T., Mori, H., Tanaka, E., Tanaka, A., Umetani, K., Eto, K., Miyazawa, Y., Ochiai, M., and Sato, T. (1997). Microangiographic assessment of collateral vessel formation following direct gene transfer of vascular endothelial growth factor in rats. Cardiovasc. Res. 35, 547–552.
Takeshita, S., Isshiki, T., Ochiai, M., Eto, K., Mori, H., Tanaka, E., Umetani, K., and Sato, T. (1998). Endothelium-dependent relaxation of collateral microvessels after intramuscular gene transfer of vascular endothelial growth factor in a rat model of hindlimb ischemia. Circulation 98, 1261–1263.
Taylor, J. C., Li, Z., Yang, H. T., Laughlin, M. H., and Terjung, R. L. (2008a). Alpha-adrenergic inhibition increases collateral circuit conductance in rats following acute occlusion of the femoral artery. J. Physiol. (Lond.) 586, 1649–1667.
Taylor, J. C., Yang, H. T., Laughlin, M. H., and Terjung, R. L. (2008b). Alpha-adrenergic and neuropeptide Y Y1 receptor control of collateral circuit conductance: influence of exercise training. J. Physiol. (Lond.) 586, 5983–5998.
Vanteeffelen, J. W., and Segal, S. S. (2003). Interaction between sympathetic nerve activation and muscle fibre contraction in resistance vessels of hamster retractor muscle. J. Physiol. (Lond.) 550, 563–574.
Walder, C. E., Errett, C. J., Bunting, S., Lindquist, P., Ogez, J. R., Heinsohn, H. G., Ferrara, N., and Thomas, G. R. (1996). Vascular endothelial growth factor augments muscle blood flow and function in a rabbit model of chronic hindlimb ischemia. J. Cardiovasc. Pharmacol. 27, 91–98.
Woodman, O. L., and Wongsawatkul, O. (2004). Ischaemia enhances the role of Ca2+-activated K+ channels in endothelium-dependent and nitric oxide-mediated dilatation of the rat hindquarters vasculature. Clin. Exp. Pharmacol. Physiol. 31, 254–260.
Yang, H. T., Deschenes, M. R., Ogilvie, R. W., and Terjung, R. L. (1996). Basic fibroblast growth factor increases collateral blood flow in rats with femoral arterial ligation. Circ. Res. 79, 62–69.
Yang, H. T., Dinn, R. F., and Terjung, R. L. (1990). Training increases muscle blood flow in rats with peripheral arterial insufficiency. J. Appl. Physiol. 69, 1353–1359.
Yang, H. T., Prior, B. M., Lloyd, P. G., Taylor, J. C., Li, Z., Laughlin, M. H., and Terjung, R. L. (2008). Training-induced vascular adaptations to ischemic muscle. J. Physiol. Pharmacol. 59(Suppl. 7), 57–70.
Young, D. A., Chi, M. M., and Lowry, O. H. (1986). Energy metabolism of skeletal muscle biopsies stimulated anaerobically without load in vitro. Am. J. Physiol. 250, C813–C820.
Keywords: vasodilation, reactivity, chronic ischemia, hindlimb, mouse
Citation: Cardinal TR, Struthers KR, Kesler TJ, Yocum MD, Kurjiaka DT and Hoying JB (2011) Chronic hindlimb ischemia impairs functional vasodilation and vascular reactivity in mouse feed arteries. Front. Physio. 2:91. doi: 10.3389/fphys.2011.00091
Received: 03 September 2011; Accepted: 14 November 2011;
Published online: 06 December 2011.
Edited by:
John D. Imig, Medical College of Wisconsin, USAReviewed by:
R. Dan Rudic, Medical College of Georgia, USAMichael J. Ryan, University of Mississippi Medical Center, USA
Mairead Carroll, New York Medical College, USA
Copyright: © 2011 Cardinal, Struthers, Kesler, Yocum, Kurjiaka and Hoying. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Trevor R. Cardinal, Biomedical Engineering, California Polytechnic State University, 1 Grand Avenue, San Luis Obispo, CA 93407-0365, USA. e-mail: tcardina@calpoly.edu
†Present address: David T. Kurjiaka, Biomedical Sciences Department, Grand Valley State University, Allendale, MI, USA; James B. Hoying, Cardiovascular Therapeutics, Cardiovascular Innovation Institute, Louisville, KY, USA