Latent tuberculosis: models, computational efforts and the pathogen’s regulatory mechanisms during dormancy
- 1National Institute for Mathematical and Biological Synthesis, University of Tennessee, Knoxville, TN, USA
- 2Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA
- 3Computational Biology Group, Department of Clinical Laboratory Sciences, Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Cape Town, South Africa
Latent tuberculosis is a clinical syndrome that occurs after an individual has been exposed to the Mycobacterium tuberculosis (Mtb) Bacillus, the infection has been established and an immune response has been generated to control the pathogen and force it into a quiescent state. Mtb can exit this quiescent state where it is unresponsive to treatment and elusive to the immune response, and enter a rapid replicating state, hence causing infection reactivation. It remains a gray area to understand how the pathogen causes a persistent infection and it is unclear whether the organism will be in a slow replicating state or a dormant non-replicating state. The ability of the pathogen to adapt to changing host immune response mechanisms, in which it is exposed to hypoxia, low pH, nitric oxide (NO), nutrient starvation, and several other anti-microbial effectors, is associated with a high metabolic plasticity that enables it to metabolize under these different conditions. Adaptive gene regulatory mechanisms are thought to coordinate how the pathogen changes their metabolic pathways through mechanisms that sense changes in oxygen tension and other stress factors, hence stimulating the pathogen to make necessary adjustments to ensure survival. Here, we review studies that give insights into latency/dormancy regulatory mechanisms that enable infection persistence and pathogen adaptation to different stress conditions. We highlight what mathematical and computational models can do and what they should do to enhance our current understanding of TB latency.
Introduction
The pathogenesis of Mycobacterium tuberculosis (Mtb) is complex and involves an elaborate interaction with the host. Key factors, including the ability to survive in macrophages, the predilection for the lung, the formation of granulomas and long-term persistence, are poorly understood. The Mtb bacterial pathogen generally infects its mammalian host through the aerosol route. Inhalation of Mtb leads to phagocytosis by alveolar macrophages. After the onset of cell-mediated immune response, surviving bacteria are believed to enter a period of non-replicating persistence (NRP) in the phagosome until waning of host immunity results in reactivation from the latent state and the onset of disease. The ability of Mtb to persist for a long time in the latent state has been associated with the ability of the pathogen to maintain resistance against anti-microbial molecules and adaptation to host-induced metabolic constraints such as low nutrients, nitrogen, and oxygen stress (Wayne and Hayes, 1996; Betts et al., 2002; Hampshire et al., 2004; Voskuil et al., 2004b; Deb et al., 2009). In latency, Mtb are less metabolically active, and they have replication rates that are drastically diminished compared to bacilli in an active infection (Lillebaek et al., 2002, 2003). Tubercle bacilli require oxygen for growth; oxygen deprivation is lethal to them unless they have time to adapt to its gradual depletion. In the oxygen deprived environment if the bacteria are to survive, they need to alter their program of gene expression (Park et al., 2003; Schnappinger et al., 2003; Voskuil et al., 2003, 2004b; Rustad et al., 2008, 2009; Larsson et al., 2012), change their metabolic pathways, and depend on anaerobic respiration or to develop alternative mechanisms for generating energy. All these factors are potential contributors to latent TB infection (LTBI) development in humans.
The interaction between a pathogen and its host occurs on different scales. These range from molecular interactions, including the recognition of specific molecular patterns on innate immune cells by toll-like receptors, to interactions between individual cells, which, in turn, can range from the phagocytosis of bacteria by macrophages to the spread of disease through a host population and the emergence of different strains of pathogens in response to different host conditions. Current approaches to detection, prevention, and treatment of LTBI are inadequate, and rational development of new tools has been limited by poor understanding of the fundamental biology of LTBI, the Bacillus-host interaction, the immunologic parameters that are involved in establishing persistent infection, and the mechanisms of pathogen and host that lead to reactivation of Mtb and development of active disease.
LTBI represents a state of equilibrium in which the host shows no apparent symptoms, but is infected and where the immune system is only robust enough to contain the infection but is unable to clear it. When the immune system gets compromised, this balance is lost, leading to infection reactivation. From the host perspective, LTBI is clinically considered to be an asymptomatic, non-infectious state from which progression to active, infectious disease may occur at any time. Such progression occurs more often soon after the initial infection, and during times of host immune-suppression (e.g., human immunodeficiency virus infection, aging, therapy with tumor necrosis-alpha blockers) (Stead and Lofgren, 1983; Corbett et al., 2003; Gardam et al., 2003). Although classically considered to be a state in which Mtb is either contained or metabolically dormant, it is increasingly understood that clinical LTBI may represent a wide spectrum of host responses (Barry et al., 2009). Our lack of accurate and specific diagnostic tools to differentiate these responses in vivo makes the development of corresponding in vitro models similarly challenging.
Global Prevalence of Latent TB
The global prevalence of LTBI is commonly cited as approximately 1 in 3, although no population-based evidence exists to verify this estimate. Such prevalence is roughly consistent with published population models of TB in globally representative settings (Abu-Raddad et al., 2009; Dowdy et al., 2013). The contribution of LTBI to population-level epidemiology differs widely by region; in high-incidence settings, recent infection contributes the majority of new cases of active TB, whereas in low-incidence settings, the majority of non-imported TB cases arise from reactivation of remote infection. Thus, while diagnosis and treatment of LTBI may substantially reduce the burden of active TB in settings of low or declining incidence (Comstock et al., 1979), it is unlikely to have similar population-level effects in high-burden areas until the ongoing annual risk of TB infection can be reduced. Nevertheless, as discussions regarding TB elimination garner increasing attention (Dye et al., 2012), it is becoming increasingly clear that this goal cannot be achieved without a strategy to diagnose and treat the massive reservoir of individuals with LTBI, in addition to those with active TB disease.
The lifetime risk of developing active TB after latent infection is often quoted as 10%, but modeling analyses suggest that, among individuals infected as adults, this number may be substantially higher (over 15%) (Vynnycky and Fine, 1997), and it is certainly much higher (up to 10% per year) among people living with untreated HIV (Corbett et al., 2003). The majority of this reactivation risk is experienced within the first 2 or 3 years following infection. Preventive therapy (traditionally with 6–9 months of daily isoniazid) can reduce this risk of reactivation by 60–70% (Smieja et al., 2000; Akolo et al., 2010) and is therefore a public health priority among individuals at high-risk of reactivation, including people living with HIV and people initiating biological therapy (e.g., TNF-alpha inhibitors) (Gardam et al., 2003) that affect the body’s ability to contain LTBI. However, in many parts of the world, such high-risk individuals comprise a relatively small proportion of the total population that will experience active TB from reactivation of latent infection. Plans for expanding access to preventive therapy among others with risk factors for reactivation (e.g., malnutrition, diabetes mellitus, adult household contacts) are therefore urgently needed and in most places lacking.
Macrophages, Granuloma, Mtb Pathogen and LTBI Development
Macrophages are phagocytes at the frontline of host immune defense against microbial pathogens. They are the primary habitat of Mtb, and the pathogen preferentially targets macrophage vacuoles. Thus, the adaptation of Mtb to the phagosomal compartment of the macrophages is an essential component of its pathogenesis, transmission, and continual survival (Smeulders et al., 1999; Hampshire et al., 2004). This apparent incongruity demands that Mtb either tolerate the macrophage’s anti-microbial effectors, that is (i) low pH, (ii) reactive oxygen, and (iii) nitrogen species, or actively subvert normal cellular mechanisms to avoid being killed. It implies that for the pathogen to have extended survival in the host in a state of latency, the pathogen must somehow evade or interfere with the immune surveillance and signaling pathways (Voskuil et al., 2003, 2004b; Bacon et al., 2004; Berney and Cook, 2010; Taneja et al., 2010). Alternatively, the Bacillus must tolerate host defense mechanisms, either through mobilization of repair or detoxification pathways or through phenotypic tolerance developed as a result of metabolic adaptation or quiescence. After Mtb phagocytosis by macrophages, phagocytosed micro-organisms are rapidly transferred from phagosomes to lysosomes, where there is fusion of the phagosome with the late endosome and lysosome, and the microbes should be destroyed (Clemens and Horwitz, 1996; Fratti et al., 2001; Dubnau and Smith, 2003). The phagosomal environment is nitrosative, oxidative, functionally hypoxic, carbohydrate-poor, and capable of perturbing the pathogen’s envelope (Schnappinger et al., 2003). However, pathogenic mycobacteria resist lysosomal delivery and survive within macrophages inside mycobacterial phagosomes, in an intraphagosomal environment that is friendly to the bacterium. Macrophages, instead of killing the pathogen, have been shown to be manipulated by the bacilli, creating an environment suitable for intracellular replication by progressively translocating from phagolysosomes into the cytosol. The bacilli have been shown to be both anti-apoptotic, keeping the host cell alive to avoid the anti-microbial effects of apoptosis, and pro-necrotic, killing the host macrophage to allow infection of neighboring cells. This Mtb induced macrophage death was shown to be dependent on mycobacterial expression of ESAT-6 (van der Wel et al., 2007; Abdallah et al., 2011; Yu and Xie, 2012). Inhibition of Mtb phagosomal maturation was proposed as another mechanism for the survival of the pathogen in macrophages (Clemens and Horwitz, 1996; Fratti et al., 2001). Mtb is among the micro-organisms most successful at adapting to long-term residence in macrophage phagosomes. Phagosome arrest is a complex process and is not fully understood Taneja et al. (2010) (Clemens and Horwitz, 1996; Fratti et al., 2001, 2003; Anes et al., 2003). It is also speculated that pathogenic mycobacteria (including Mtb) might interfere directly in host trafficking pathways through expression of their own signal transduction molecules. The Mtb genome encodes two eukaryotic-like serine/threonine kinases [reviewed in Av-Gay and Everett (2000)] and protein kinase G (PknG), which consists of a kinase domain flanked by a large N- and C-terminal domain, and was shown to inhibit phagosome-lysosome fusion. Rapid lysosomal transfer of mycobacteria lacking PknG results in intracellular killing by bactericidal activities present in the lysosomes. Therefore, the ability of PknG to prevent lysosomal transfer suggests that it may affect survival of bacteria that have been internalized in macrophages (Walburger et al., 2004).
In latency, lipids have been found to be a major source of nutrients to the Mtb pathogen (McKinney, 2000; Ehrt and Schnappinger, 2007). Fatty acids have been hypothesized to constitute a significant source of both carbon and energy for non-replicating bacilli and the glyoxylate by-pass may serve in the formation of carbohydrates from fatty acids (Muñoz-Elías and McKinney, 2005; Ehrt and Schnappinger, 2007, 2009). Mtb strain (H37Rv) has a gene that encodes an isocitrate lyse, icl1, expressing ICL activity in macrophage infection. Studies by Graham and Clark-Curtiss (1999) and McKinney (2000) showed that a strain of Mtb in which the icl gene had been insertionally inactivated, lost the ability to persist in tissues of mice or in activated macrophages even though it did not appear to have diminished ability to survive in vitro anaerobiosis (McKinney, 2000). Isocitrate lyases catalyze an essential reaction of the glyoxylate shunt, an anaplerotic pathway that by-passes the CO2-generating steps of the tricarboxylic acid cycle, and enables bacteria to synthesize carbohydrates and replenish tricarboxylic acid cycle intermediates from fatty acid-derived acetyl-coenzyme A (Bishai, 2000; Ehrt and Schnappinger, 2007). For many bacteria, this pathway is required for growth on fatty acids as a sole carbon source. The genomes of most Mtb strains encode not one but two functional isocitrate lyases: ICL1 and ICL2. Muñoz-Elías and McKinney (2005) showed that both icl1, icl2 Mtb mutants can grow on different carbon sources (e.g., glycerol, glucose, acetate, propionate).
Another important facet of LTBI development is the granuloma. A granuloma is a multicellular structure comprised of macrophages (resting, activated, and infected), immune effector T cells on the periphery, chemokines, cytokines, adhesion molecules, and a caseous necrotic center (Lin et al., 2006; Lin and Flynn, 2010). Macrophages within a granuloma have two main functions (i) primarily to contain infection and (ii) for bacterial multiplication (Mtb preferred environment for growth). In the event that they fail to control infection, they end up harboring large amounts of bacteria and the bacteria can persist for decades, which is the case in LTBI. An impairment of the immune system will result in the disruption of the granuloma structure and collapse of the granuloma center, hence infection dissemination and potential transmission of the bacteria to other individuals. LTBI in humans comprise of a heterogeneous mixture of granulomas in both lungs and lymph nodes that provide a range of physiological micro-environments associated with bacterial replication and persistence, and therefore the basis for TB clinical latency (Barry et al., 2009).
Mathematical Modeling and Latent Mtb Infection
Mathematical and computational efforts just like biological experiments have been key in unveiling many facets of infectious diseases. Their applications have immensely contributed to and shaped our current understanding of TB infection. In the same fashion as biological experiments, constructing, validating and analyzing mathematical, and computational models based on empirical observations can provide new insights into biological systems by making testable predictions and stir new directions for designing of biological experiments. Computational methods are our hope to handle and analyze vast amounts of high-throughput data. They are an essential tool in predictive studies of disease signatures, biomarkers, and vaccine and drug targets. Several mathematical schemes have been developed to provide a platform to study and understand host immune response mechanisms in active TB infection (Wigginton and Kirschner, 2001; Marino and Kirschner, 2004; Segovia-Juarez et al., 2004; Magombedze et al., 2006b, 2009, 2010; Sud et al., 2006; Ray et al., 2008, 2009). However, there is no substantial effort through mathematical modeling that has been put forth to enhance our understanding of LTBI (Magombedze and Mulder, 2011, 2012; Hegde et al., 2012; Magombedze et al., 2012).
In general, mathematical models have been useful in the evaluation, assessment, planning, and theoretical trial of vaccines and disease control intervention strategies of several infectious diseases and ailments (Scherer and McLean, 2002; Gandon and Day, 2007; Magombedze et al., 2008b; Smith and Schwartz, 2008; Heffernan and Keeling, 2009). The success or failure of a vaccine scheme can easily be predicted by use of such models when they are carefully developed and analyzed. This approach provides cost effective methods to give general insights and specific direction for implementing intervention programs (Long and Owens, 2011; Lugnér et al., 2012), hence providing direction for vaccinations. Application of optimal control and optimization techniques offers a platform to investigate and design optimal treatment, vaccine, and cost effective implementation protocols (Magombedze et al., 2011a,c). On the other hand, the effect of how these vaccines enhance the immune response in shrugging off the infection within the host has not been extensively studied compared to the extent of studies carried out at the population-level. Therapeutic and chemoprophylactic vaccines have been investigated in the studies (Koff et al., 2005; Smith and Schwartz, 2008; Johnson et al., 2011). Currently there is limited understanding for therapeutic vaccines and most of these vaccines offer partial immunity, which wanes over time. There has not been much new discovery for TB vaccines and BCG is still the vaccine that is recommended by WHO in infants. Several theories and debates exist as to what type of TB vaccine will be most effective, either an innate based (dendritic, macrophages, neutrophil) or a T cell based vaccine. A mathematical study (Magombedze et al., 2006a) was used to assess the efficacy of TB chemotherapy first line drug regimen following the directly observed therapy strategy (DOTs). This study was followed by models in which effects of treatment adherence and treatment when there is HIV coinfection were investigated (Magombedze et al., 2010, 2011b; Ramkissoon et al., 2012). This was designed to determine the possible treatment schemes when there are HIV and TB drug interactions, and several sequences of combinations of administering both HIV and TB drug cocktails, hence painting a better understanding of potential treatment schemes.
The current understanding of how the immune system interacts with the Mtb pathogen in TB infection owes credit to several mathematical studies (Wigginton and Kirschner, 2001; Marino and Kirschner, 2004; Magombedze et al., 2006b, 2008a, 2009; Sud et al., 2006; Ray et al., 2008, 2009; Day et al., 2009). However, the contribution of these studies toward the understanding of LTBI is still minimal. The first model of TB infection was in 1994 (Antia and Koella, 1994), which attempted to explain attributes of mycobacterial evolution and maintenance of micro-parasite virulence. Antia et al. (1996) developed another model on mycobacterial infections, where the mechanisms by which bacteria such as Mtb and Mycobacterium leprae persist at low densities for extended periods and attain high densities much later in the presence of an Ag specific immune response were investigated. Comprehensive and detailed modeling of Mtb was carried out in the studies (Wigginton and Kirschner, 2001; Marino and Kirschner, 2004; Segovia-Juarez et al., 2004; Magombedze et al., 2006b, 2008a, 2009, 2010; Sud et al., 2006), which described the interaction of the Mtb pathogen, macrophages, cytokines, and lymphocytes at the site of infection (lung compartment), and lung and lymph node compartments (Marino and Kirschner, 2004; Magombedze et al., 2009). These studies predicted attributes of the pathogen and immune response mechanisms that result in either LTBI or active TB, however without deciphering the mechanisms of LTBI development and persistence. Several other studies were carried, which showed the effects of TNF-alpha in granuloma formation and show how disruption of TNF-alpha mechanisms may enhance infection dissemination (Ray et al., 2008, 2009; Fallahi-Sichani et al., 2011). The mechanisms at the granuloma level that lead to different configurations of cells, however differential containment of the organisms are not well understood (Sharpe et al., 2009). There are current modeling efforts and methods for measuring and modulating TNF-alpha at the granuloma level to determine the effects on local control of Mtb infection using non-human primate data [reviewed in Kirschner et al. (2010)]. The studies (Ray et al., 2009; Fallahi-Sichani et al., 2011) present a way to study a single granuloma and the multi-scale molecular, cellular, and tissue events in the development of a granuloma, and show the critical role of TNF-alpha in control of bacterial growth in the granuloma.
There are studies that have started to incorporate details about signaling pathways into both immune cell and host-pathogen models, with the potential of identifying new drug targets (Goldstein et al., 2004; Franke et al., 2008; see review Kirschner and Linderman, 2009). Our study, Magombedze and Mulder (2011), is a first attempt to combine gene expression data, multiple stress factors, and bacterial cell densities using a mathematical frame work to investigate and explain several hypotheses on how persistence and reactivation of LTBI occurs. Results in this study echoes the results in the studies (Hegde et al., 2012; Magombedze and Mulder, 2012), were dynamics of several signaling and metabolic pathways mechanisms were explained using Boolean network modeling (Hegde et al., 2012), which used a bistable switch to help explain a potential mechanism on how the pathogen can switch between two different metabolic states. Our recent study (Magombedze and Mulder, 2012) predicted the regulation of the latency/dormancy program using a systems biology mathematical framework to be orchestrated by dynamic gene regulatory networks constituted of several DosR-regulon genes (Voskuil et al., 2003). This study predicted the role of DosR-regulon genes and other several genes as central in the regulation and maintenance of the latency program. In this study DosR-regulon genes were predicted to cluster with adaptation, detoxification, and virulence genes and several other genes of unknown or putative functions (Rv3131, Rv0569, Rv2032, Rv2530c, and Rv2694c), some of which were predicted both in the regulation of the stationary and non-replicating phase of the bacteria. This study furthermore predicted that other genes (Rv1133c, Rv2890c, Rv1177, Rv2710, Rv2532c, and Rv0982) to be also involved in the regulation of latency. These genes were shown in other studies (Sassetti and Rubin, 2003; Sassetti et al., 2003) to be essential for H37Rv growth and in vivo bacterial survival. Some of the genes predicted to be central in the regulation of latency are essential for bacterial growth (Rv3131), making them an attractive drug target. Other genes are known to have functions that are linked to human immune response mechanisms, such as induction of interferon-c (IFN-c) and interleukin-2 (IL-2) (Rv2032, Rv3133c, Rv3131, and Rv2031c), oxidative stress (Rv1909c), and nitrosative stress (Rv3131, Rv3133c, and Rv2031c). The latter are associated with nitrogen and oxygen reactive intermediate effector mechanisms of the immune response and were shown in other studies (Leyten et al., 2006; Roupie et al., 2007; Black et al., 2009) to have the potential to prime the human immune response. However, these predictions remain to be tested through biological experiments for potential LTBI drug design. With vast generation of data sets from different TB experimental infection models (mouse models, guinea pig, non-human primate models, and culture models) as well as data from real cases of infected people, mathematical and computational modeling (see Table 1 for current mathematical and computational methods) is our only hope to get sense of all data measured at different scales, through developing virtual models that can integrate data from different scales.
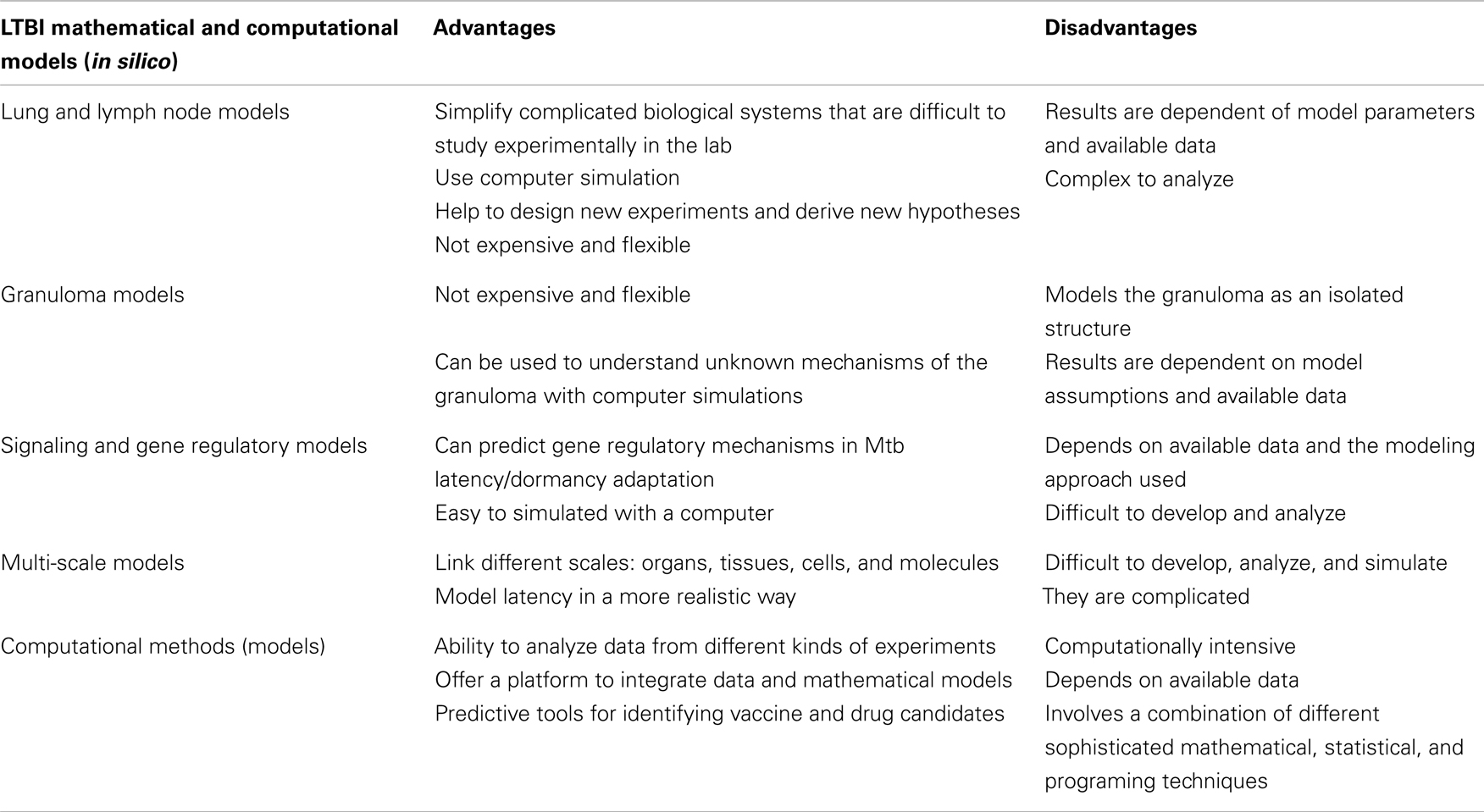
Table 1. LTBI mathematical and computational models.
Understanding Latent Mtb and Identifying New Drug Targets through Computational Efforts
Several computational models and bioinformatics techniques have revealed network interactions, metabolic, and signaling pathways associated with the development of the latent infection; and with prediction of gene functional groups, hence providing informative data sets for future experiments (Mazandu and Mulder, 2011a,b; Mazandu et al., 2011; Hegde et al., 2012; Magombedze and Mulder, 2012). Mazandu and Mulder (2011a) used a combination of sequence and functional genomics data to generate a functional interaction network for Mtb, which was used for drug target prediction and function prediction for the proteins of unknown function (Mazandu and Mulder, 2011b,c). The data integration method exploited sequence similarity and shared domains, gene synteny, phylogenetic profiles, co-occurrence in abstracts, co-expression and other protein–protein interaction prediction techniques to predict functional interactions between all possible protein pairs in the Mtb proteome. A combined score was attributed to each interaction based on the individual scores from each evidence type. Using only medium to high confidence interactions, a network of 4,136 proteins and 58,000 interactions was generated, and was found to have a small world property, which is common among biological networks. This network was used to predict functions for uncharacterized proteins in Mtb, which constitute a high percentage of the genome. Most of the uncharacterized proteins were found to have metabolic activities, but others were predicted to be involved in transcription regulation, signal transduction, and pathogenesis, suggesting that there are many currently uncharacterized proteins that could play a role in the survival and persistence of Mtb during infection and reacting to changes in the environment (Mazandu and Mulder, 2011b,c).
Functional interaction networks provide the advantage of enabling a systems level view of an organism. Proteins are not viewed in isolation, and for each protein we can establish its relative importance in the biological system by assessing its network properties. Network properties include degree (number of direct interacting partners), betweenness, closeness, and other measures of how well connected a protein is within the network and whether its removal will have a substantial effect on the rest of the network. Like mathematical modeling where we can model expression or metabolic changes and determine the effect of “knocking out” a gene or protein, in biological networks we can knock out proteins and determine the effect on the network. Mazandu and Mulder (2011a) used their Mtb functional interaction network to identify possible drug targets by ranking proteins by their betweenness and centrality measures. Using this technique, they identified 881 possible drug targets, whose elimination would affect a number of other proteins and interactions. These proteins included 114 previously identified drug targets in either UniProtKB or TDR targets (http://tdrtargets.org), as well as targets for some of the currently used anti-TB drugs (Mazandu and Mulder, 2011a). Zheng et al. (2012) used protein–protein interactions networks from STRING for identifying virulence proteins in several pathogens, including Mtb. Using the interaction network, they identified virulence factors based on number of neighbors and strength of interactions and compared this to a feature selection method and BLAST approaches. Their results were benchmarked against a database of validated virulence factors and the network-based method was found to out-perform the other two methods (Zheng et al., 2012).
These network studies, which are purely computational, allow us to predict effects of perturbations on the whole biological system and can help us to identify genes or proteins that known dormancy genes interact with. For example, the DosR protein has over 75 interactions with other proteins in the DosR-regulon, as well as with other regulators and proteins whose functions are not yet known. The host-pathogen interactions, which link two functional interaction networks, allow us to then determine how these proteins potentially interact with host proteins. Computational tools have also been used to identify new vaccine candidates for TB. For example, there are some new possible vaccine candidates on the horizon, such as the latency antigens α-crystallin (HspX) and Rv2660c of the DosR-regulon, which were identified from studies of gene expression changes during dormancy (Wang et al., 2013). In addition, Gideon et al. (2012), using a bioinformatics and empirical approach, identified several novel TB proteins that elicited an anti-tuberculosis T cell response.
Mtb Latency and Dormancy Models
The bacterial behavior during the growth phase (lag, log, stationary, and death phases) is well known, but little is known about how they behave in LTBI. The ability of the pathogen to enter and exit from different states has associated it with its ability to cause persistent infection. In this state it is not clearly known whether it will be in a persistent slow replicating state or a dormant non-replicating state, thus ultimately causing a latent infection with the potential to reactivate to active disease. The terminology for latent TB state and dormant bacterial state is somehow confusing and is used differently in different studies. TB Latency refers to an in vivo situation where the bacteria and the host have established a balanced state without causing apparent symptoms. Whereas the term dormant refers to the physiological and reversible metabolic shutdown state of the bacteria, which is normally simulated in in vitro culture models.
In vitro models have been used to model latency or investigate the adaptive processes of the Mtb to non-proliferating conditions which can be achieved by limiting at least one of the essential conditions required for Mtb growth (for a summary of the latency/dormancy models see Table 2). These include: (i) starvation of essential nutrients, such as carbon, nitrogen, or phosphorus and (ii) depletion of oxygen, which prevents aerobic respiration by the obligate aerobe. The adaptation to oxygen is the most widely studied of these conditions. Low-oxygen levels have long been recognized to limit Mtb replication while promoting long-term survival, which is associated with a distinct physiologic adaptation and marked by bacteriostasis in addition to metabolic, chromosomal, and structural changes of the bacilli (Wayne and Hayes, 1996; Wayne and Sohaskey, 2001). These states can be distinguished using a resuscitation reculture failure test (Sun and Zhang, 1999). Resuscitation of non-colony forming cultures only confirms the difference between dead cells and dormant cells. Truly dormant cells were shown to resume replication after stimulation with a resuscitation promoting factor (Rpf), a 16-kDa protein derived from a Gram-positive coccus Micrococcus luteus (M. luteus) [reviewed in Zhang (2004)]. The studies (Mukamolova et al., 2002; Tufariello et al., 2004; Downing et al., 2005; Shleeva et al., 2010) showed over-expression of five of the Mtb Rpf genes that could stimulate the growth of tubercle bacilli from an old culture with a small inoculum. It may be concluded that the Rpf proteins are directly involved in reactivation of persisting bacteria. However, the mechanism of the activating effect of Rpf on dormant cells is still not completely understood.
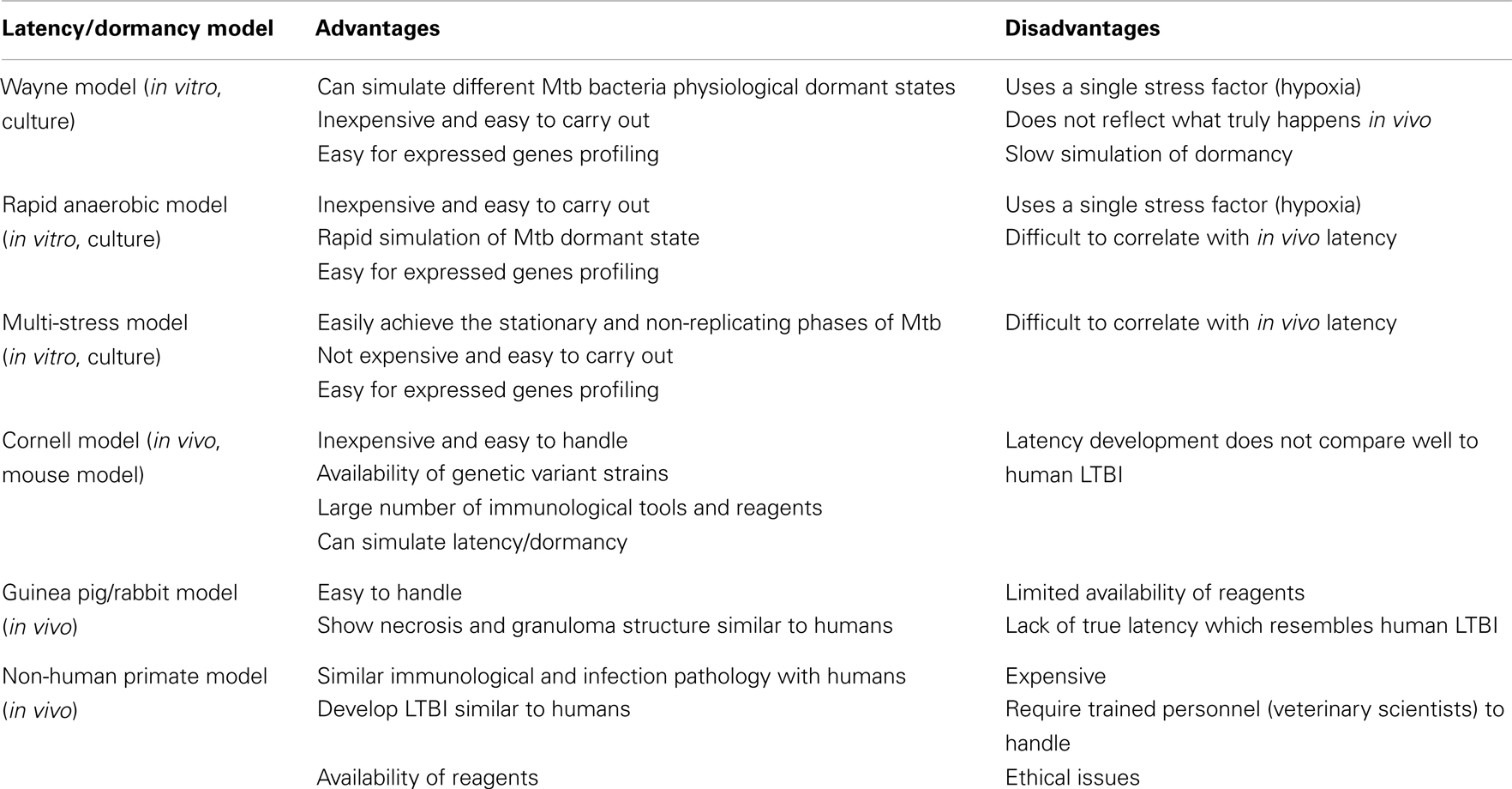
Table 2. Latency/dormancy in vitro and in vivo models.
The Wayne Model
Wayne and Hayes (1996), established an “in vivo model for dormancy” where in vitro growth TB cultures were subjected to gradual oxygen depletion to mimic tubercle bacilli in vivo. A sealed, standing culture is allowed to incubate over a period of days while the bacteria deplete the available oxygen. The culture becomes progressively more hypoxic with a concomitant shift in Mtb physiology (Wayne, 1977; Wayne and Lin, 1982). Gentle stirring and a defined culture-to-headspin ratio improve reproducibility (Wayne and Hayes, 1996). Two distinct states of NRP that reflect discrete metabolic and drug susceptibility states compared with log phase growth are illustrated. The first stage, designated NRP 1, occurred when the declining oxygen level reached 1% saturation. This stage is characterized by increased production of glycine dehydrogenase and steady ATP generation. The second stage, NRP 2, occurred when the oxygen level reached 0.06% saturation and this stage is characterized by a marked decline of glycine dehydrogenase and susceptibility to metronidazole. The dormant cells in the Wayne model remained responsive to heat shock and were culturable when transferred to fresh aerated medium (Shleeva et al., 2004, 2010). Therefore, it is thought that the Wayne model probably represents the initial stage of formation of a dormant state and essentially reflects bacterial adaptation to low-oxygen level rather than to the dormant state. A study by Shleeva et al. (2002) showed that longer incubation time of several months in the Wayne model can produce some dormant bacilli.
The Rapid Anaerobic Model
There are several other studies (Honaker et al., 2009; Leistikow et al., 2010) that have improved and modified the Wayne model to yield consistent and more precise results. In these studies, a rapid anaerobic dormancy model is implemented. This model differs from the Wayne model in the larger size of stir bars and faster rate of stirring resulting in a more homogeneous population of bacilli, and even oxygen distribution throughout the culture. The model exhausts available oxygen rapidly ensuring achievement of anaerobiosis in a relatively short time interval compared to the Wayne model.
Cornell Model
The most convincing evidence of dormant Mtb was demonstrated by McCune Jr. et al. (1956, 1966) in a mouse model (Zhang, 2004; Shleeva et al., 2010). Mice were infected with virulent Mtb and the infection was allowed to establish for 2 weeks, followed by treatment with a combination of INH and PZA for 3 months. Initially no bacilli could be detected in the mouse spleen; however, one third of the mice relapsed with culture-positive tubercle bacilli when the treatment was discontinued for 3 months (McCune Jr. et al., 1956; Hu et al., 2000) and more relapses were noticed after administration of immunosuppressive steroids (McCune Jr. et al., 1966). This was an indication that dormant bacilli insensitive to antibiotic treatment persisted. There are variations of the Cornell model that show the same disappearance and reappearance phenomenon of tubercle bacilli in mice with different infectious dose, route of infection (aerosol or intravenous), and different drug combinations (Scanga et al., 1999).
Multi-Stress Model
The limitation with single stress models, like hypoxia, nutrient starvation, and exposure to NO is that they cannot closely imitate the in vivo latency/dormancy stimulating conditions. Unfortunately there is no substantive repertoire of alternative models that can model latency with multiple stresses. To date, the study of Deb et al. (2009) is the only one that has achieved this. This multiple stress model used a combination of low-oxygen (5%), high CO2 (10%), low nutrients (10% Dubos medium), and an acidic pH of 5.0. This study illustrates how the Mtb pathogen adapts and prepares for the dormancy stage. Under these conditions the pathogen was shown to lose its acid-fastness characteristics, while at the same time it accumulated wax esters and lipids, a process thought to be an energy storing strategy as it shifts from aerobic respiration to anaerobic respiration. Several studies (Hampshire et al., 2004; Deb et al., 2009; Berney and Cook, 2010) have suggested that, continual survival of Mtb in the anaerobic respiration state is sustained through energy generation from metabolism of fatty acids. This model revealed high expression of genes involved in the glyoxylate cycle and energy metabolism, which is a strong indication that the pathogen had switched to anaerobic respiration (Deb et al., 2009; Patel et al., 2011).
Non-Human Primate Model
There are several in vivo models that are currently used to experimentally explain the development of LTBI. These include, (i) mouse models (for example the Cornell model), (ii) the Guinea pig/rabbit model, with the potential to simulate granulomas similar to those observed in humans. Unfortunately it is difficult to observe LTBI manifestation in Guinea pigs, which are highly susceptible to rapid disease progression proceeding low-dose aerosol infection. Although the pathology in lungs of Guinea pigs and rabbits is reminiscent of human pathology, there is relative lack of immunologic reagents for these animals (Flynn et al., 2003). The most attractive model to date is the non-human primate model. Non-human primates (Cynomolgus macaque) have physiological, infection pathology, immunological similarities with humans, and they make it possible to carry out experiments that are difficult or impossible to perform in the human population. Its ability to generate clinical correlates of infection has made the non-human primate model important in the study of LTBI. The downside of this model includes cost, difficulty with handling animals, containment of biohazards, and ethical considerations.
Latency and Dormancy Regulation
Survival of Mtb bacilli in vivo is attributed to the pathogen’s ability to adapt to the changing environment. This is orchestrated by a program of genes that sense changes of stress factors (hypoxia, NO-exposure, heat shock, nutrient starvation, pH change, and low iron) in its environment. The phagosome environment is associated with production of oxidative bursts, the synthesis of inducible nitric oxide (NO) synthase resulting in the production of NO and other reactive nitrogen intermediates, which are responsible for the killing of intracellular bacteria. This changing environment within the macrophages should elicit new programs of gene expression in the pathogen if it is to survive. The Mtb pathogens possess mechanisms to detect the ambient oxygen tension based on the DosT/DosS/DosR system and WhiB3 sensory systems, which enable them to adapt to changes in oxygen availability by adjusting their metabolism accordingly (Park et al., 2003; Kumar et al., 2007; Rustad et al., 2008; Honaker et al., 2009).
The DosS/DosT and DosR response regulator is a two component system, which consists of a sensor kinase (component 1) which autophosphorylates histidine, then transphosphorylates a conserved aspartate of its cognate transcriptional regulator (component 2). Mtb has eleven complete systems and seven orphans belonging to sensor kinase and response regulator families (Cole et al., 1998). DosR has been shown to act as a transcriptional regulator, which is responsible for the induction of nearly all hypoxia-induced genes of Mtb (Park et al., 2003; Voskuil et al., 2003, 2004b; Kumar et al., 2007; Rustad et al., 2008). It has been shown that, recombinant sensor kinases DosS and DosT autophosphorylate to stable phosphoproteins (Malhotra et al., 2006; Murphy and Brown, 2007), but rapidly ransphosphorylate DosR under conditions of low-oxygen tension or in the presence of NO or CO (Voskuil et al., 2003; Kumar et al., 2007; Sousa et al., 2007; Shiloh et al., 2008), confirming these as members of the two component family. DosS is a redox sensor and DosT is a hypoxia sensor. Aerobically, DosS is in its inactive Fe3+ form, as a result of its rapid oxidation by O2. When DosS is reduced to Fe2+, its autokinase activity is increased and uses this oxidation state to signal DosR activation (Honaker et al., 2009). DosT exists aerobically in the O2-bound form, which is inactive and its resistance to oxidation removes the possibility of its functioning as a redox sensor. The study of Honaker et al. (2009) shows that the DosR-regulon is induced in stepwise manner with DosT only important in early stages and DosS important in the late stages. This is because DosT is not able to signal DosR expression efficiently under low-oxygen. The early activity by DosT is thought to kick start induction of DosR-regulon genes, among them DosS. As the DosR-regulon is expressed, levels of DosS become elevated, enabling further induction of the regulon.
The ability of the Mtb bacteria to adapt to changing oxygen concentration has been linked with the role of the WhiB3 protein implicated in sensing oxygen tension and redox state by mycobacteria. The WhiB3 gene was found to be strongly induced in Mtb during acute infection of mouse lungs and also during growth in resting bone marrow-derived macrophages, but repressed after IFN-gamma activation of the macrophages (Banaiee et al., 2006). The study of Singh et al. (2007) was able to show that WhiB3 contains a 4Fe–4S cluster, which can bind NO. In the presence of oxygen, the WhiB3 [4Fe–4S]2+ cluster is degraded first to a [3Fe–4S]+ cluster, then to [2Fe–2S]2+, and subsequently lost altogether (Crack et al., 2004; Singh et al., 2007; Alam and Agrawal, 2008). WhiB3-mediated response to the presence of oxygen, therefore, is predicted to occur through direct control of the activity of metabolic proteins or through modification of transcriptional regulators (Alam and Agrawal, 2008). A role for WhiB3 in regulation of the transcriptional machinery in mycobacteria may be supported by the finding that WhiB3 interacts with the major sigma factor, SigA (RpoV) (Steyn et al., 2002), but the effect of this interaction on SigA activity is not known, and further studies are required to understand the precise role of WhiB3 in mediating any adaptation of mycobacteria to changes in oxygen tension.
The DosR-Regulon
The DosR-regulon is a significant regulator of genes in response to hypoxia and NO [see Table 3 for the list of DosR-regulon genes (Park et al., 2003; Kumar et al., 2008)]. This, combined with the production of NO by host immune cells and the likely hypoxic nature of the granuloma, makes a strong prediction that this regulon is a primary trigger of metabolic shift down to achieve the non-replicative state. Latency and dormancy models (Park et al., 2003; Voskuil et al., 2003, 2004b; Honaker et al., 2009) have predicted that LTBI is regulated through the DosR-regulon/system. Roberts et al. (2004) showed that deletion of DosS or DosT in the H37Rv strain causes a reduced expression of hypoxia reporter gene and complete failure of hspX stimulation in the double mutant. Studies (Voskuil et al., 2003; Kumar et al., 2007; Sousa et al., 2007; Shiloh et al., 2008) predicted that without the NO signal, the DosR-regulon will not be up-regulated, and cell division will continue. The DosR-regulon was shown to be weakly induced in the stationary phase indicating partial stimulation of the dormancy genes. With increased oxygen tension there is increased expression of the transcripts fdxA, pfkB, narX, and narK2, which indicate a switch to anaerobic metabolism from aerobic respiration (Bacon et al., 2004; Voskuil et al., 2004b). These genes were also observed to be induced in NO exposure and nutrient starvation (Betts et al., 2002; Bacon et al., 2004; Hampshire et al., 2004; Deb et al., 2009). Dormancy genes expressed in the stationary phase and the NRP were shown (Voskuil et al., 2003, 2004b) to be located in “operon like clusters.” One such cluster of genes (rsbW, Rv3288c, Rv3289c, and lat) is seen in the study Hampshire et al. (2004) and has previously been identified as up-regulated in the stationary phase survival study by Betts et al. (2002). Other gene clusters include Rv0982-Rv0984 (moaB2), Rv1460-Rv1463, Rv3138-Rv3141 (pflA, fadE24, fadE23, and fadB4, respectively), and Rv3458c-Rv3460c. The largest cluster of genes includes Rv3864, Rv3867-Rv3871, Rv3876, and Rv3878, the majority of which fall within the RD-1 region deleted from strain BCG Pasteur. A complete list of genes up-regulated and down regulated in hypoxia, NO exposure, nutrient starvation, and other stress conditions are found in the studies (Betts et al., 2002; Park et al., 2003; Voskuil et al., 2003, 2004b; Hampshire et al., 2004; Deb et al., 2009).
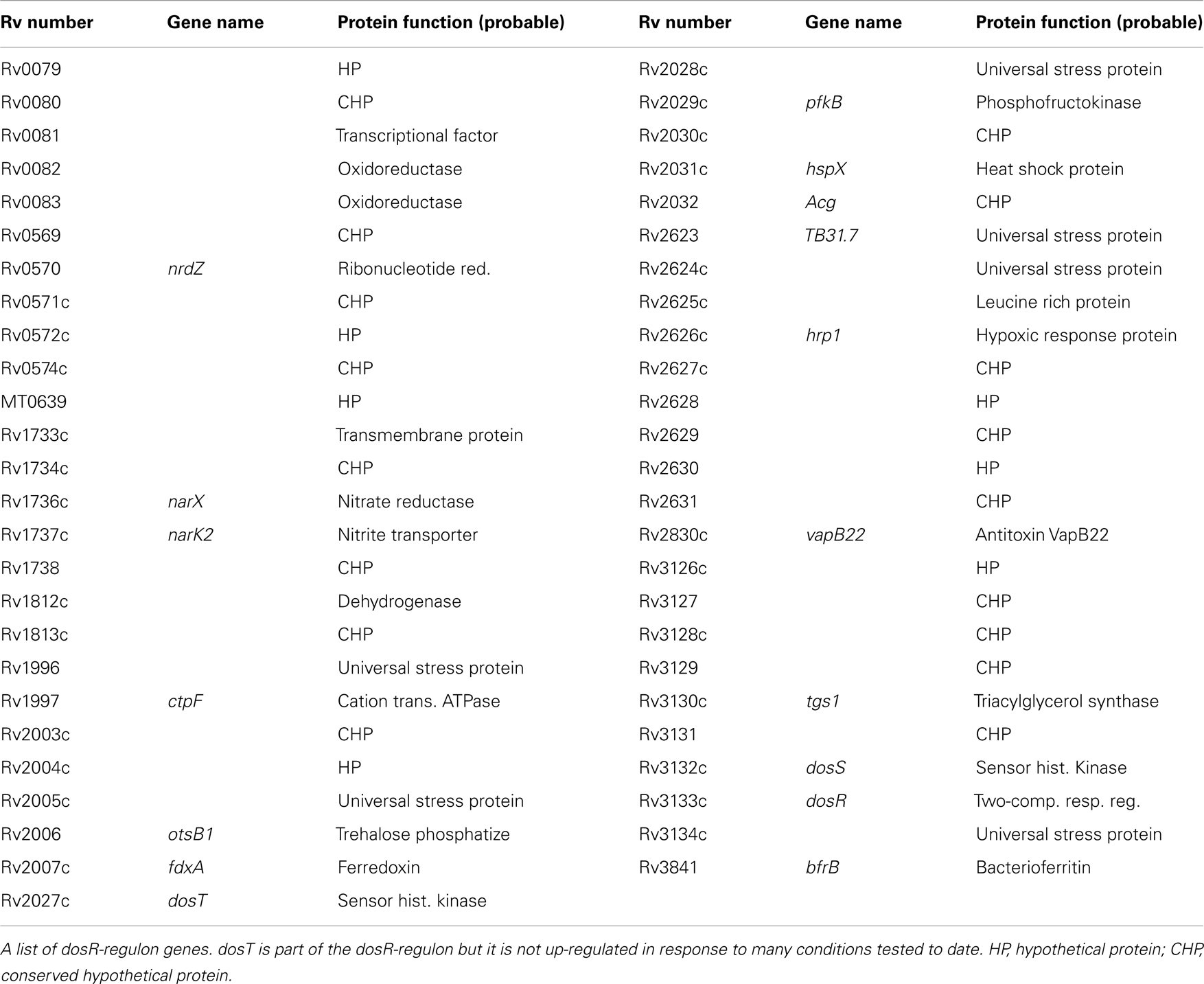
Table 3. Mtb DosR-regulon genes.
Latency/Dormancy Stationary Phase Operons
Using hierarchical clustering Voskuil et al. (2004b) showed that most stationary phase-specific induced genes are transcriptionally clustered and thus share a common regulatory profile. These genes include Rv0106, PPE3, Rv1535, PE20, PPE31, rpsR2, rpsN2, rpmG1, and rpmB2. The four ribosome encoding genes are separated from the majority of ribosome encoding genes on the Mtb genome, perhaps indicating a difference in function. By contrast, most ribosomal protein genes are repressed during stationary phase as well as in NRP. It is thought that, the ribosomal proteins encoded by rpsR2, rpsN2, rpmG1, and rpmB2 are involved in increasing ribosomal fidelity during non-proliferating conditions. The desA3 gene (encoding a probable fatty acid desaturase) is highly induced in the transition to stationary phase, but not during NRP. Interestingly, the other two fatty acid desaturase genes found in the Mtb genome are induced early during NRP (Bacon et al., 2004; Voskuil et al., 2004b). Also, the desA genes are induced, with desA3 being strongly repressed throughout NRP, while desA1 and desA2 are only induced during stationary phase. The fact that all three genes are induced during the transition to non-proliferating conditions while the bacilli are still replicating indicates the bacilli may be changing the degree of saturation of the cell wall mycolic acids in preparation for a non-proliferating state. Genes of the PE and PPE families (which constitute 8% of the genes in the Mtb genome) are also differentially regulated between stationary phase and NRP. PE3, PE20, and PPE31 are induced during stationary phase, while PE11, PE34, PE-PGRS11, PE-PGRS14, PPE17, PPE47, and PPE48 are induced during NRP, and many PE/PPE genes are repressed under both conditions (Voskuil et al., 2004a). There is speculation on the functions of these proteins that involve their ability to provide antigenic variation and interfere with immune responses, in addition to performing a purely structural role.
In addition to the dormancy regulon, several other genes are strongly up-regulated in the NRP model. Most of these genes are repressed late in the NRP model, with the exception of hsp, Rv0678, Rv0841c, Rv1734c, Rv1874, mez, and Rv2660c (Voskuil et al., 2003, 2004b). Interestingly, Rv1734c is a dormancy regulon gene that is weakly induced early in NRP but is the only dormancy gene that was noticed to remain expressed in the late stages of NRP. The continued induction of Rv1734c, and the induction of a few other dormancy genes, when most dormancy genes are no longer induced, indicates there may be another level of control for some of the dormancy genes other than through activation by DosR. Two strongly induced genes early in NRP (ahpC and ahpD) encode the alkyl hydroperoxide reductase and are strongly repressed throughout the adaptation to stationary phase. AhpC is a peroxidase and peroxynitrate reductase is involved in antioxidant defense mechanisms in Mtb. Genes encoding cytochrome bd oxidase (cydA, cydB, cydC, and cydD) are also expressed early in the NRP model. The cytochrome bd-1 oxidase is an alternative terminal oxidase for the aerobic respiratory chain that is believed to function at low-oxygen levels due to its higher affinity for oxygen than the primary cytochrome c oxidase. Therefore, the induction of these genes is consistent with the depletion of oxygen early in the NRP model (Voskuil et al., 2003, 2004b; Hampshire et al., 2004). Induction of the bfrB gene that encodes the iron storage protein bacterioferritin, bfrB is repressed when the mycobactin synthesis genes are induced under low iron conditions. The iron-dependent regulation of bfrB and the mycobactin genes require the presence of IdeR. The fact that the mycobactin and bfrB genes are co-induced indicates that bfrB can be regulated by a mechanism independent of the IdeR-dependent activation that is observed during conditions of low iron. During NRP, the bacilli may attempt to increase iron stores for use during long periods of dormancy. Therefore, Mtb appears to induce both uptake and storage functions simultaneously.
Transcriptome analysis of stationary phase Mtb cells incubated under standard conditions (Betts et al., 2002; Voskuil et al., 2004b; Shleeva et al., 2010) revealed activation of some of the DosR-regulon genes, including those encoding the two component regulatory system of stress response, as well as nitrate reductase, nitrite binding protein, and alpha crystalline. It may therefore be suggested that expression of the DosR-regulon not only acts as a marker of a hypoxic state, but is also responsible for development of the universal cellular stress response. However, transcriptome analysis of Mtb under oxygen limitation revealed that enhanced expression of the DosR operon genes under hypoxia was brief and occurred only at the initial stage. A bigger group of genes was then activated, reflecting the “true” cellular response to hypoxia (Rustad et al., 2008; Shleeva et al., 2010).
Latency and Mtb Adaptation to Changes in PH
The macrophage intracellular environment is associated with pH changes that are detrimental to bacterial survival, as such, a change in expression of a large number of Mtb genes over a range of pH values is noticed (Rao et al., 2001; Fisher et al., 2002; Saviola et al., 2003). This implies that mycobacteria are able to perceive the pH of their environment. Two adenylyl cyclases have been suggested to fulfill that role in Mtb. Rv1264 was observed to have an increased fold change of adenylyl cyclase activity at pH 6 compared to pH 8, mediated through a pH-responsive catalytic domain (Tews et al., 2005). In response to pH, the protein undergoes conformational rearrangements and these may involve an unsaturated fatty acid as a structural element (Tews et al., 2005; Findeisen et al., 2007). In the closely related adenylyl cyclase Rv2212, pH-sensing activity is strongly induced by binding of unsaturated fatty acids, but the mechanism of this effect is not fully understood (Motaal et al., 2006). While both Rv1264 and Rv2212 have increased activity at acidic pH, the mechanism for activation was reported to be different between the two proteins (Tews et al., 2005; Motaal et al., 2006).
Adaptation to Nutrient Depletion
A rapid reduction in the ability of bacteria to synthesize proteins leads to the accumulation of the stringent response signaling molecule (p)ppGpp which in turn reduces transcription of stable RNA genes (Dahl et al., 2003; Jain et al., 2006). The enzyme relAb (the only ppGpp synthase homolog in Mtb) is required for survival of mycobacteria under long-term in vitro starvation conditions. Microarray studies characterizing the response of Mtb to both a fall in available nutrients and hypoxia, have demonstrated a rise in relA (Betts et al., 2002; Hampshire et al., 2004). The study of Hampshire et al., 2004, showed that relA is involved in initial nutrient depletion adaptation in facilitating long-term survival (Primm et al., 2000; Chatterji and Kumar Ojha, 2001). The type of regulatory pattern seen for relA was also observed for aceAb. It has previously been suggested that increasing levels of (p)ppGpp in E. coli may be the sole inducer of qor, a gene shown to be up-regulated in response to conditions of stress, which seems to function by reducing the quinone pool (Chang et al., 2002; Dürrschmid et al., 2008). However, in the study of Hampshire et al. (2004), following the drop in expression of relA early on, qor stayed up-regulated throughout the time course and even showed a late substantial increase, suggesting that other levels of regulation are present.
Different types of stress tend to up-regulate different sigma factors, however sigB, sigE, and sigH are a subset of sigma factors associated with the entire period of stationary phase adaptation (Betts et al., 2002; Hampshire et al., 2004). Alternative sigma factors are a major category of transcriptional regulators responsible for orchestrating the gene expression responses to non-proliferating conditions in B. subtilis and E. coli. The sigma factor genes sigF, sigB, and sigJ are the ones that are induced significantly during stationary phase [reviewed in Wang et al. (2011)]. Voskuil et al. (2004b) showed that only sigB is induced during stationary phase, while sigF and sigJ are expressed relative to their levels in exponentially growing bacilli and there is a weak induction of sigE. SigC is induced early, while sigH is induced late in the NRP model. The study (Voskuil et al., 2004b) spells out the need to determine if the state of Mtb during clinical latency more closely resembles the E. coli Sigma S model or the Bacillus sporulation cascade paradigm.
The genes involved in Mtb persistence in vivo are believed to fall into two groups: those responsible for transition to the persisting state and those responsible for its maintenance (Betts et al., 2002; Shleeva et al., 2010). For example, the Mtb mutant lacking the relA gene (its product in ppGpp), a “strong response” mediator, regulating cellular activity (Dahl et al., 2003) was capable of normal growth in the macrophages and in vitro with citrate and phospholipids as carbon sources, but did not survive transition into the stationary phase or incubation under anaerobic conditions (Primm et al., 2000; Shleeva et al., 2010). The expression of a number of genes in the mutant strain and in the wild type was different, including the genes encoding the reactivating protein: rpfA (enhanced expression) and rpfC (inhibited expression). This demonstrates that the relA product is required for persistence under starvation, oxygen limitation, and in an extensive stationary phase, as well as in the chronic model and in the artificial granuloma in mice in vivo; it is one of the candidates for participation in the persistent state in humans.
Concluding Remarks
Understanding the development and persistence of LTBI infection is complex. Despite many years of research and several strides that have been made in identifying key Mtb persistence factors, gene signatures for vaccine and drug design, eradication of TB still seems to be out of reach. The main short coming of current studies is the inadequacy of the current in vivo and in vitro latency models [reviewed in Patel et al. (2011)] to closely imitate latency development in humans. Nevertheless, these in vitro single stress models have shaped our current understanding on bacilli adaptation to the stationary and non-replicating stages. This has revealed key functional groups of genes that regulate latency and dormancy development. Most of these models are not close to modeling the real human latency development, which in humans can persistent for the entire life span until an opportune moment for reactivation, since they are designed to simulate latency/dormancy over a few hours or days. Development of models that follow the framework proposed in the study Deb et al. (2009), in which several stress factors are considered, has the potential to illuminate and enhance our current understanding of Mtb persistence. The use of the non-human primate model is more encouraging since it simulates latency/dormancy close to that of humans (Flynn et al., 2003), and has become the most attractive model to study LTBI compared to other models. However, it is marred with genotype variability and small sample sizes which affects model results reproducibility. The complexity associated with handling this model makes it expensive and unmanageable for several scientists.
Several studies reviewed here suggest that under each different stress condition the bacteria has a unique set of genes that sense and report changes in its environment, hence facilitating necessary changes for adaptation. These mechanisms seem to be dynamic with the length of time spent in each condition as well as the type of the stress. However, overlaps of genes expressed in hypoxia, NO-exposure, and nutrient starvation are noticed (Betts et al., 2002; Voskuil et al., 2003, 2004b; Hampshire et al., 2004). These kinds of experiments generate lots of data and this is where we think mathematical and computational modeling should play a crucial role. Studies (Magombedze and Mulder, 2011, 2012; Mazandu and Mulder, 2011a,b,c; Hegde et al., 2012), show how computational and mathematical methods can be used to analyze, learn more, identify traits and properties of protein–protein interactions, gene regulatory mechanisms, and metabolic pathways that are active in latency development and maintenance, which cannot be identified by carrying out basic statistical analysis of the experimental data. A few strides have been made so far to achieve this goal. The study (Fallahi-Sichani et al., 2011) applied a multi-scale approach to study the granuloma using a granuloma model with multiple single cell entities modeled separately to understand TNF trafficking within the granuloma and how this correlates to control of infection. The studies (Magombedze and Mulder, 2011, 2012) are the only recent studies to attempt to model gene cross talk and regulation that occurs in latency adaptation and interface change in gene expression with Mtb cell populations to show how these gene regulations influence the final physiological and metabolic stages of the pathogen. The future should see development of more computational schemes that integrate data from different scales in an endeavor to understand the dynamics of LTBI. A multi-scale model that captures measurements of cells (T cells), cytokines (influence of TNF-alpha and IFN-gamma), signaling mechanisms of immune cells when there is an infection, pathogen mechanisms of interfering with the immune signaling pathways, pathogen gene regulatory mechanisms and how these mechanisms influence the pathogen phenotypic characteristics and metabolic pathways into one framework will be a good basis to generate and test several hypotheses and has potential to usher the study of LTBI on to a new level.
Existing models of the immune system do not cover much detail at the molecular level such as protein sequences and structures and the effect of polymorphisms. The use of bioinformatics is resourceful in this respect, it allows for analysis of pathogen genome sequences and prediction of protein–protein interactions within the pathogen, and between the pathogen and host (Leung and Cavalieri, 2003; Kendall et al., 2004). Availability of the genome sequence helps in compilation of all the potential gene products encoded by a particular organism, identification of functions that are missing or unique in a particular organism and identifying genes that are common between prokaryotes and eukaryotes. This enhances host-pathogen interaction studies which could provide important clues for development of more rational and specific methods to search for new drug targets, vaccine candidates and diagnostic tests.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Gesham Magombedze acknowledges the support of the National Institute for Mathematical and Biological Synthesis (NIMBioS), an Institute sponsored by the National Science Foundation, the U.S. Department of Homeland Security, and the U.S. Department of Agriculture through NSF Award #EF-0832858, with additional support from The University of Tennessee, Knoxville, TN, USA.
References
Abdallah, A. M., Bestebroer, J., Savage, N. D., de Punder, K., van Zon, M., Wilson, L., et al. (2011). Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J. Immunol. 187, 4744–4753. doi:10.4049/jimmunol.1101457
Abu-Raddad, L. J., Sabatelli, L., Achterberg, J. T., Sugimoto, J. D., Longini, I. M. Jr., Dye, C., et al. (2009). Epidemiological benefits of more-effective tuberculosis vaccines, drugs, and diagnostics. Proc. Natl. Acad. Sci. U.S.A. 106, 13980–13985. doi:10.1073/pnas.0901720106
Akolo, C., Adetifa, I., Shepperd, S., and Volmink, J. (2010). Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst. Rev. 1, CD000171.
Alam, S., and Agrawal, P. (2008). Matrix-assisted refolding and redox properties of WhiB3/Rv3416 of Mycobacterium tuberculosis H37Rv. Protein Expr. Purif. 61, 83–91. doi:10.1016/j.pep.2008.04.010
Anes, E., Kühnel, M. P., Bos, E., Moniz-Pereira, J., Habermann, A., and Griffiths, G. (2003). Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nat. Cell Biol. 5, 793–802. doi:10.1038/ncb1036
Antia, R., and Koella, J. C. (1994). A model of non-specific immunity. J. Theor. Biol. 168, 141–150. doi:10.1006/jtbi.1994.1094
Antia, R., Koella, J. C., and Perrot, V. (1996). Models of the within-host dynamics of persistent mycobacterial infections. Proc. R. Soc. Lond. B Biol. Sci. 263, 257–263. doi:10.1098/rspb.1996.0040
Av-Gay, Y., and Everett, M. (2000). The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 8, 238–244. doi:10.1016/S0966-842X(00)01734-0
Bacon, J., James, B. W., Wernisch, L., Williams, A., Morley, K. A., Hatch, G. J., et al. (2004). The influence of reduced oxygen availability on pathogenicity and gene expression in Mycobacterium tuberculosis. Tuberculosis 84, 205–217. doi:10.1016/j.tube.2003.12.011
Banaiee, N., Jacobs, W. Jr., and Ernst, J. (2006). Regulation of Mycobacterium tuberculosis whiB3 in the mouse lung and macrophages. Infect. Immun. 74, 6449–6457. doi:10.1128/IAI.00190-06
Barry, C. E., Boshoff, H. I., Dartois, V., Dick, T., Ehrt, S., Flynn, J., et al. (2009). The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7, 845–855. doi:10.1038/nrmicro2236
Berney, M., and Cook, G. M. (2010). Unique flexibility in energy metabolism allows mycobacteria to combat starvation and hypoxia. PLoS ONE 5:e8614. doi:10.1371/journal.pone.0008614
Betts, J. C., Lukey, P. T., Robb, L. C., McAdam, R. A., and Duncan, K. (2002). Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731. doi:10.1046/j.1365-2958.2002.02779.x
Bishai, W. (2000). Microbiology: lipid lunch for persistent pathogen. Nature 406, 683–685. doi:10.1038/35021159
Black, G. F., Thiel, B. A., Ota, M. O., Parida, S. K., Adegbola, R., Boom, W. H., et al. (2009). Immunogenicity of novel DosR regulon-encoded candidate antigens of Mycobacterium tuberculosis in three high-burden populations in Africa. Clin. Vaccine Immunol. 16, 1203–1212. doi:10.1128/CVI.00111-09
Chang, D.-E., Smalley, D. J., and Conway, T. (2002). Gene expression profiling of Escherichia coli growth transitions: an expanded stringent response model. Mol. Microbiol. 45, 289–306. doi:10.1046/j.1365-2958.2002.03001.x
Chatterji, D., and Kumar Ojha, A. (2001). Revisiting the stringent response, ppGpp and starvation signaling. Curr. Opin. Microbiol. 4, 160–165. doi:10.1016/S1369-5274(00)00182-X
Clemens, D. L., and Horwitz, M. A. (1996). The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J. Exp. Med. 184, 1349–1355. doi:10.1084/jem.184.4.1349
Cole, S., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi:10.1038/31159
Comstock, G. W., Baum, C., and Snider, D. E. Jr. (1979). Isoniazid prophylaxis among Alaskan Eskimos: a final report of the bethel isoniazid studies. Am. Rev. Respir. Dis. 119, 827–830.
Corbett, E. L., Watt, C. J., Walker, N., Maher, D., Williams, B. G., Raviglione, M. C., et al. (2003). The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch. Intern. Med. 163, 1009–1021. doi:10.1001/archinte.163.9.1009
Crack, J., Green, J., and Thomson, A. J. (2004). Mechanism of oxygen sensing by the bacterial transcription factor fumarate-nitrate reduction (FNR). J. Biol. Chem. 279, 9278–9286. doi:10.1074/jbc.M309878200
Dahl, J. L., Kraus, C. N., Boshoff, H. I., Doan, B., Foley, K., Avarbock, D., et al. (2003). The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc. Natl. Acad. Sci. U.S.A. 100, 10026–10031. doi:10.1073/pnas.1631248100
Day, J., Friedman, A., and Schlesinger, L. S. (2009). Modeling the immune rheostat of macrophages in the lung in response to infection. Proc. Natl. Acad. Sci. U.S.A. 106, 11246–11251. doi:10.1073/pnas.0904846106
Deb, C., Lee, C.-M., Dubey, V. S., Daniel, J., Abomoelak, B., Sirakova, T. D., et al. (2009). A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS ONE 4:e6077. doi:10.1371/journal.pone.0006077
Dowdy, D. W., Basu, S., and Andrews, J. R. (2013). Is passive diagnosis enough?: the impact of subclinical disease on diagnostic strategies for tuberculosis. Am. J. Respir. Crit. Care Med. 187, 543–551. doi:10.1164/rccm.201207-1217OC
Downing, K. J., Mischenko, V. V., Shleeva, M. O., Young, D. I., Young, M., Kaprelyants, A. S., et al. (2005). Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect. Immun. 73, 3038–3043. doi:10.1128/IAI.73.5.3038-3043.2005
Dubnau, E., and Smith, I. (2003). Mycobacterium tuberculosis gene expression in macrophages. Microbes Infect. 5, 629–637. doi:10.1016/S1286-4579(03)00090-X
Dürrschmid, K., Reischer, H., Schmidt-Heck, W., Hrebicek, T., Guthke, R., Rizzi, A., et al. (2008). Monitoring of transcriptome and proteome profiles to investigate the cellular response of E. coli towards recombinant protein expression under defined chemostat conditions. J. Biotechnol. 135, 34–44. doi:10.1016/j.jbiotec.2008.02.013
Dye, C., Glaziou, P., Floyd, K., and Raviglione, M. (2012). Prospects for tuberculosis elimination. Annu. Rev. Public Health 34, 271–286. doi:10.1146/annurev-publhealth-031912-114431
Ehrt, S., and Schnappinger, D. (2007). Mycobacterium tuberculosis virulence: lipids inside and out. Nat. Med. 13, 284–285. doi:10.1038/nm0307-284
Ehrt, S., and Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 11, 1170–1178. doi:10.1111/j.1462-5822.2009.01335.x
Fallahi-Sichani, M., El-Kebir, M., Marino, S., Kirschner, D. E., and Linderman, J. J. (2011). Multiscale computational modeling reveals a critical role for TNF-α receptor 1 dynamics in tuberculosis granuloma formation. J. Immunol. 186, 3472–3483. doi:10.4049/jimmunol.1003299
Findeisen, F., Linder, J. U., Schultz, A., Schultz, J. E., Brügger, B., Wieland, F., et al. (2007). The structure of the regulatory domain of the adenylyl cyclase Rv1264 from Mycobacterium tuberculosis with bound oleic acid. J. Mol. Biol. 369, 1282–1295. doi:10.1016/j.jmb.2007.04.013
Fisher, M. A., Plikaytis, B. B., and Shinnick, T. M. (2002). Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J. Bacteriol. 184, 4025–4032. doi:10.1128/JB.184.14.4025-4032.2002
Flynn, J. L., Capuano, S. V., Croix, D., Pawar, S., Myers, A., Zinovik, A., et al. (2003). Non-human primates: a model for tuberculosis research. Tuberculosis (Edinb) 83, 116–118. doi:10.1016/S1472-9792(02)00059-8
Franke, R., Müller, M., Wundrack, N., Gilles, E.-D., Klamt, S., Kähne, T., et al. (2008). Host-pathogen systems biology: logical modelling of hepatocyte growth factor and Helicobacter pylori induced c-Met signal transduction. BMC Syst. Biol. 2:4. doi:10.1186/1752-0509-2-4
Fratti, R. A., Backer, J. M., Gruenberg, J., Corvera, S., and Deretic, V. (2001). Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J. Cell Biol. 154, 631–644. doi:10.1083/jcb.200106049
Fratti, R. A., Chua, J., Vergne, I., and Deretic, V. (2003). Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc. Natl. Acad. Sci. U.S.A. 100, 5437–5442. doi:10.1073/pnas.0737613100
Gandon, S., and Day, T. (2007). The evolutionary epidemiology of vaccination. J. R. Soc. Interface 4, 803–817. doi:10.1098/rsif.2006.0207
Gardam, M. A., Keystone, E. C., Menzies, R., Manners, S., Skamene, E., Long, R., et al. (2003). Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. Lancet Infect. Dis. 3, 148–155. doi:10.1016/S1473-3099(03)00545-0
Gideon, H. P., Wilkinson, K. A., Rustad, T. R., Oni, T., Guio, H., Sherman, D. R., et al. (2012). Bioinformatic and empirical analysis of novel hypoxia-inducible targets of the human antituberculosis T cell response. J. Immunol. 189, 5867–5876. doi:10.4049/jimmunol.1202281
Goldstein, B., Faeder, J. R., and Hlavacek, W. S. (2004). Mathematical and computational models of immune-receptor signalling. Nat. Rev. Immunol. 4, 445–456. doi:10.1038/nri1374
Graham, J. E., and Clark-Curtiss, J. E. (1999). Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc. Natl. Acad. Sci. U.S.A. 96, 11554–11559. doi:10.1073/pnas.96.20.11554
Hampshire, T., Soneji, S., Bacon, J., James, B. W., Hinds, J., Laing, K., et al. (2004). Stationary phase gene expression of Mycobacterium tuberculosis following a progressive nutrient depletion: a model for persistent organisms? Tuberculosis (Edinb) 84, 228. doi:10.1016/j.tube.2003.12.010
Heffernan, J., and Keeling, M. J. (2009). Implications of vaccination and waning immunity. Proc. R. Soc. Lond. B Biol. Sci. 276, 2071–2080. doi:10.1098/rspb.2009.0057
Hegde, S. R., Rajasingh, H., Das, C., Mande, S. S., and Mande, S. C. (2012). Understanding communication signals during mycobacterial latency through predicted genome-wide protein interactions and Boolean modeling. PLoS ONE 7:e33893. doi:10.1371/journal.pone.0033893
Honaker, R. W., Leistikow, R. L. I., Bartek, L., and Voskuil, M. I. (2009). Unique roles of DosT and DosS in DosR regulon induction and Mycobacterium tuberculosis dormancy. Infect. Immun. 77, 3258–3263. doi:10.1128/IAI.01449-08
Hu, Y., Mangan, J. A., Dhillon, J., Sole, K. M., Mitchison, D. A., Butcher, P. D., et al. (2000). Detection of mRNA transcripts and active transcription in persistent Mycobacterium tuberculosis induced by exposure to rifampin or pyrazinamide. J. Bacteriol. 182, 6358–6365. doi:10.1128/JB.182.22.6358-6365.2000
Jain, V., Kumar, M., and Chatterji, D. (2006). ppGpp: stringent response and survival. J. Microbiol. 44, 1.
Johnson, P. L., Kochin, B. F., McAfee, M. S., Stromnes, M., Regoes, R. R., Ahmed, R., et al. (2011). Vaccination alters the balance between protective immunity, exhaustion, escape, and death in chronic infections. J. Virol. 85, 5565–5570. doi:10.1128/JVI.00166-11
Kendall, S. L., Rison, S. C., Movahedzadeh, F., Frita, R., and Stoker, N. G. (2004). What do microarrays really tell us about M. tuberculosis? Trends Microbiol. 12, 537–544. doi:10.1016/j.tim.2004.10.005
Kirschner, D. E., and Linderman, J. J. (2009). Mathematical and computational approaches can complement experimental studies of host–pathogen interactions. Cell. Microbiol. 11, 531–539. doi:10.1111/j.1462-5822.2008.01281.x
Kirschner, D. E., Young, D., and Flynn, J. L. (2010). Tuberculosis: global approaches to a global disease. Curr. Opin. Biotechnol. 21, 524–531. doi:10.1016/j.copbio.2010.06.002
Koff, W. C., Johnson, P. R., Watkins, D. I., Burton, D. R., Lifson, J. D., Hasenkrug, K. J., et al. (2005). HIV vaccine design: insights from live attenuated SIV vaccines. Nat. Immunol. 7, 19–23. doi:10.1038/ni1296
Kumar, A., Deshane, J. S., Crossman, D. K., Bolisetty, S., Yan, B.-S., Kramnik, I., et al. (2008). Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J. Biol. Chem. 283, 18032–18039. doi:10.1074/jbc.M802274200
Kumar, A., Toledo, J. C., Patel, R. P., Lancaster, J. R., and Steyn, A. J. C. (2007). Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc. Natl. Acad. Sci. U.S.A. 104, 11568–11573. doi:10.1073/pnas.0705054104
Larsson, C., Luna, B., Ammerman, N. C., Maiga, M., Agarwal, N., and Bishai, W. R. (2012). Gene expression of Mycobacterium tuberculosis putative transcription factors whiB1-7 in redox environments. PLoS ONE 7:e37516. doi:10.1371/journal.pone.0037516
Leistikow, R. L., Morton, R. A., Bartek, I. L., Frimpong, I., Wagner, K., and Voskuil, M. I. (2010). The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J. Bacteriol. 192, 1662–1670. doi:10.1128/JB.00926-09
Leung, Y. F., and Cavalieri, D. (2003). Fundamentals of cDNA microarray data analysis. Trends Genet. 19, 649–659. doi:10.1016/j.tig.2003.09.015
Leyten, E., Lin, M. Y., Franken, K., Friggen, A. H., Prins, C., Van Meijgaarden, K. E., et al. (2006). Human T-cell responses to 25 novel antigens encoded by genes of the dormancy regulon of Mycobacterium tuberculosis. Microbes Infect. 8, 2052. doi:10.1016/j.micinf.2006.03.018
Lillebaek, T., Dirksen, A., Baess, I., Strunge, B., Thomsen, V. Ø, and Andersen, ÅB. (2002). Molecular evidence of endogenous reactivation of Mycobacterium tuberculosis after 33 years of latent infection. J. Infect. Dis. 185, 401–404. doi:10.1086/338342
Lillebaek, T., Dirksen, A., Vynnycky, E., Baess, I., Thomsen, V. Ø, and Andersen, ÅB. (2003). Stability of DNA patterns and evidence of Mycobacterium tuberculosis reactivation occurring decades after the initial infection. J. Infect. Dis. 188, 1032–1039. doi:10.1086/378240
Lin, P. L., and Flynn, J. L. (2010). Understanding latent tuberculosis: a moving target. J. Immunol. 185, 15–22. doi:10.4049/jimmunol.0903856
Lin, P. L., Pawar, S., Myers, A., Pegu, A., Fuhrman, C., Reinhart, T. A., et al. (2006). Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect. Immun. 74, 3790–3803. doi:10.1128/IAI.74.4.2052-2062.2006
Long, E. F., and Owens, D. K. (2011). The cost-effectiveness of a modestly effective HIV vaccine in the United States. Vaccine 29, 6113–6124. doi:10.1016/j.vaccine.2011.04.013
Lugnér, A. K., van Boven, M., de Vries, R., Postma, M. J., and Wallinga, J. (2012). Cost effectiveness of vaccination against pandemic influenza in European countries: mathematical modelling analysis. BMJ 345:e4445. doi:10.1136/bmj.e4445
Magombedze, G., Chiyaka, C., and Mukandavire, Z. (2011a). Optimal control of malaria chemotherapy. Nonlinear Anal. Modell. Contr. 16, 415–434.
Magombedze, G., Garira, W., Mwenje, E., and Bhunu, C. (2011b). “Mycobacterium tuberculosis treatment and the emergence of a multi-drug resistant strain in the lungs,” in Infectious Disease Modelling Research Progress Series: Public Health in the 21st Century, eds J. M. Tchuenche and C. Chayaka (New York: Nova Publishers), 197–227.
Magombedze, G., Garira, W., Mwenje, E., and Bhunu, C. P. (2011c). Optimal control for HIV-1 multi-drug therapy. Int. J. Comput. Math. 88, 314–340.
Magombedze, G., Dube, N., and Mulder, N. (2012). “Mathematical modeling of infection and treatment of Mycobacterium tuberculosis latency,” in A Treatise of Biological Models. Series: Mathematics Research Developments, eds F. Nyabadza, M. Kgosimore, and E. Lungu (New York: Nova Publishers), Chap. 2, 9–34.
Magombedze, G., Garira, W., and Mwenje, E. (2006a). Mathematical modeling of chemotherapy of human TB infection. J. Biol. Syst. 14, 509–553. doi:10.1142/S0218339006001945
Magombedze, G., Garira, W., and Mwenje, E. (2006b). Modelling the human immune response mechanisms to Mycobacterium tuberculosis infection in the lungs. Math. Biosci. Eng. 3, 661. doi:10.3934/mbe.2006.3.661
Magombedze, G., Garira, W., and Mwenje, E. (2008a). In-vivo mathematical study of co-infection dynamics of HIV-1 and Mycobacterium tuberculosis. J. Biol. Syst. 16, 357–394. doi:10.1142/S0218339008002551
Magombedze, G., Garira, W., and Mwenje, E. (2008b). Modelling the immunopathogenesis of HIV-1 infection and the effect of multi-drug therapy: the role of fusion inhibitors in HAART. Math. Biosci. Eng. 5, 485–504. doi:10.3934/mbe.2008.5.485
Magombedze, G., Garira, W., and Mwenje, E. (2009). The role of dendritic cells and other immune mechanisms during human infection with Mycobacterium tuberculosis. Int. J. Biomath. 2, 69–105. doi:10.1142/S1793524509000534
Magombedze, G., Garira, W., and Mwenje, E. (2010). Modeling the TB/HIV-1 co-infection and the effects of its treatment. Math. Popul. Stud. 17, 12–64. doi:10.1080/08898480903467241
Magombedze, G., and Mulder, N. (2011). A mathematical representation of the development of Mycobacterium tuberculosis active, latent and dormant stages. J. Theor. Biol. 292, 44–59. doi:10.1016/j.jtbi.2011.09.025
Magombedze, G., and Mulder, N. (2012). Understanding TB latency using computational and dynamic modelling procedures. Infect. Genet. Evol. 13, 267–283. doi:10.1016/j.meegid.2012.09.017
Malhotra, V., Sharma, D., Ramanathan, V., Shakila, H., Saini, D. K., Chakravorty, S., et al. (2006). Disruption of response regulator gene, devR, leads to attenuation in virulence of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 231, 237–245. doi:10.1016/S0378-1097(04)00002-3
Marino, S., and Kirschner, D. E. (2004). The human immune response to Mycobacterium tuberculosis in lung and lymph node. J. Theor. Biol. 227, 463–486. doi:10.1016/j.jtbi.2003.11.023
Mazandu, G. K., and Mulder, N. J. (2011a). Generation and analysis of large-scale data-driven Mycobacterium tuberculosis functional networks for drug target identification. Adv. Bioinformatics 2011. doi:10.1155/2011/801478
Mazandu, G. K., and Mulder, N. J. (2011b). Scoring protein relationships in functional interaction networks predicted from sequence data. PLoS ONE 6:e18607. doi:10.1371/journal.pone.0018607
Mazandu, G. K., and Mulder, N. J. (2011c). Using the underlying biological organization of the Mycobacterium tuberculosis functional network for protein function prediction. Infect. Genet. Evol. 12, 922–932. doi:10.1016/j.meegid.2011.10.027
Mazandu, G. K., Opap, K., and Mulder, N. J. (2011). Contribution of microarray data to the advancement of knowledge on the Mycobacterium tuberculosis interactome: use of the random partial least squares approach. Infect. Genet. Evol. 11, 725–733. doi:10.1016/j.meegid.2010.09.003
McCune, R. M. Jr., Tompsett, R., and McDermott, W. (1956). The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J. Exp. Med. 104, 763–802. doi:10.1084/jem.104.5.737
McCune, R. M., Feldmann, F. M., Lambert, H. P., and McDermott, W. (1966). Microbial persistence I. The capacity of tubercle bacilli to survive sterilization in mouse tissues. J. Exp. Med. 123, 445–468. doi:10.1084/jem.123.3.445
McKinney, J. D. (2000). In vivo veritas: the search for TB drug targets goes live. Nat. Med. 6, 1330–1333. doi:10.1038/82142
Motaal, A. A., Tews, I., Schultz, J. E., and Linder, J. U. (2006). Fatty acid regulation of adenylyl cyclase Rv2212 from Mycobacterium tuberculosis H37Rv. FEBS J. 273, 4219–4228. doi:10.1111/j.1742-4658.2006.05420.x
Mukamolova, G. V., Turapov, O. A., Kazarian, K., Telkov, M., Kaprelyants, A. S., Kell, D. B., et al. (2002). The rpf gene of Micrococcus luteus encodes an essential secreted growth factor. Mol. Microbiol. 46, 611–621. doi:10.1046/j.1365-2958.2002.03184.x
Muñoz-Elías, E. J., and McKinney, J. D. (2005). Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 11, 638–644. doi:10.1038/nm1252
Murphy, D., and Brown, J. (2007). Identification of gene targets against dormant phase Mycobacterium tuberculosis infections. BMC Infect. Dis. 7:84. doi:10.1186/1471-2334-7-84
Park, H.-D., Guinn, K. M., Harrell, M. I., Liao, R., Voskuil, M. I., Tompa, M., et al. (2003). Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 48, 833–843. doi:10.1046/j.1365-2958.2003.03474.x
Patel, K., Jhamb, S. S., and Singh, P. P. (2011). Models of latent tuberculosis: their salient features, limitations, and development. J. Lab. Physicians 3, 75. doi:10.4103/0974-2727.86837
Primm, T. P., Andersen, S. J., Mizrahi, V., Avarbock, D., Rubin, H., and Barry, C. E. (2000). The stringent response of Mycobacterium tuberculosis is required for long-term survival. J. Bacteriol. 182, 4889–4898. doi:10.1128/JB.182.17.4889-4898.2000
Ramkissoon, S., Mwambi, H. G., and Matthews, A. P. (2012). Modelling HIV and MTB co-infection including combined treatment strategies. PLoS ONE 7:e49492. doi:10.1371/journal.pone.0049492
Rao, M., Streur, T. L., Aldwell, F. E., and Cook, G. M. (2001). Intracellular pH regulation by Mycobacterium smegmatis and Mycobacterium bovis BCG. Microbiology 147, 1017–1024.
Ray, J. C., Flynn, J. L., and Kirschner, D. E. (2009). Synergy between individual TNF-dependent functions determines granuloma performance for controlling Mycobacterium tuberculosis infection. J. Immunol. 182, 3706–3717. doi:10.4049/jimmunol.0802297
Ray, J. C., Wang, J., Chan, J., and Kirschner, D. E. (2008). The timing of TNF and IFN-gamma signaling affects macrophage activation strategies during Mycobacterium tuberculosis infection. J. Theor. Biol. 252, 24–38. doi:10.1016/j.jtbi.2008.01.010
Roberts, D. M., Liao, R. P., Wisedchaisri, G., Hol, W. G., and Sherman, D. R. (2004). Two sensor kinases contribute to the hypoxic response of Mycobacterium tuberculosis. J. Biol. Chem. 279, 23082–23087. doi:10.1074/jbc.M401230200
Roupie, V., Romano, M., Zhang, L., Korf, H., Lin, M. Y., Franken, K. L., et al. (2007). Immunogenicity of eight dormancy regulon-encoded proteins of Mycobacterium tuberculosis in DNA-vaccinated and tuberculosis-infected mice. Infect. Immun. 75, 941–949. doi:10.1128/IAI.01137-06
Rustad, T. R., Harrell, M. I., Liao, R., and Sherman, D. R. (2008). The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 3:e1502. doi:10.1371/journal.pone.0001502
Rustad, T. R., Sherrid, A. M., Minch, K. J., and Sherman, D. R. (2009). Hypoxia: a window into Mycobacterium tuberculosis latency. Cell. Microbiol. 11, 1151–1159. doi:10.1111/j.1462-5822.2009.01325.x
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi:10.1046/j.1365-2958.2003.03425.x
Sassetti, C. M., and Rubin, E. J. (2003). Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994. doi:10.1073/pnas.2134250100
Saviola, B., Woolwine, S. C., and Bishai, W. R. (2003). Isolation of acid-inducible genes of Mycobacterium tuberculosis with the use of recombinase-based in vivo expression technology. Infect. Immun. 71, 1379–1388. doi:10.1128/IAI.71.3.1379-1388.2003
Scanga, C. A., Mohan, V., Joseph, H., Yu, K., Chan, J., and Flynn, J. L. (1999). Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect. Immun. 67, 4531–4538.
Scherer, A., and McLean, A. (2002). Mathematical models of vaccination. Br. Med. Bull. 62, 187–199. doi:10.1093/bmb/62.1.187
Schnappinger, D., Ehrt, S., Voskuil, M. I., Liu, Y., Mangan, J. A., Monahan, M., et al. (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages insights into the phagosomal environment. J. Exp. Med. 198, 693–704. doi:10.1084/jem.20030846
Segovia-Juarez, J. L., Ganguli, S., and Kirschner, D. (2004). Identifying control mechanisms of granuloma formation during M. tuberculosis infection using an agent-based model. J. Theor. Biol. 231, 357–376. doi:10.1016/j.jtbi.2004.06.031
Sharpe, S., Eschelbach, E., Basaraba, R., Gleeson, F., Hall, G., McIntyre, A., et al. (2009). Determination of lesion volume by MRI and stereology in a macaque model of tuberculosis. Tuberculosis 89, 405–416. doi:10.1016/j.tube.2009.09.002
Shiloh, M. U., Manzanillo, P., and Cox, J. S. (2008). Mycobacterium tuberculosis senses host-derived carbon monoxide during macrophage infection. Cell Host Microbe 3, 323–330.
Shleeva, M., Bagramyan, K., Telkov, M., Mukamolova, G. V., Young, M., Kell, D. B., et al. (2002). Formation and resuscitation of ‘non-culturable’cells of Rhodococcus rhodochrous and Mycobacterium tuberculosis in prolonged stationary phase. Microbiology 148, 1581–1591.
Shleeva, M., Mukamolova, G. V., Young, M., Williams, H. D., and Kaprelyants, A. S. (2004). Formation of ‘non-culturable’cells of Mycobacterium smegmatis in stationary phase in response to growth under suboptimal conditions and their Rpf-mediated resuscitation. Microbiology 150, 1687–1697. doi:10.1099/mic.0.26893-0
Shleeva, M., Salina, E., and Kaprelyants, A. (2010). Dormant forms of mycobacteria. Microbiology 79, 1–12. doi:10.1134/S0026261710010017
Singh, A., Guidry, L., Narasimhulu, K., Mai, D., Trombley, J., Redding, K. E., et al. (2007). Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc. Natl. Acad. Sci. U.S.A. 104, 11562–11567. doi:10.1073/pnas.0700490104
Smeulders, M. J., Keer, J., Speight, R. A., and Williams, H. D. (1999). Adaptation of Mycobacterium smegmatis to stationary phase. J. Bacteriol. 181, 270–283.
Smieja, M. J., Marchetti, C. A., Cook, D. J., and Smaill, F. M. (2000). Isoniazid for preventing tuberculosis in non-HIV infected persons. Cochrane Database Syst. Rev. 2, CD001363.
Smith, R. J., and Schwartz, E. J. (2008). Predicting the potential impact of a cytotoxic T-lymphocyte HIV vaccine: how often should you vaccinate and how strong should the vaccine be? Math. Biosci. 212, 180–187. doi:10.1016/j.mbs.2008.02.001
Sousa, E. H. S., Tuckerman, J. R., Gonzalez, G., and Gilles-Gonzalez, M. A. (2007). DosT and DevS are oxygen switched kinases in Mycobacterium tuberculosis. Protein Sci. 16, 1708–1719. doi:10.1110/ps.072897707
Stead, W. W., and Lofgren, J. (1983). Does the risk of tuberculosis increase in old age? J. Infect. Dis. 147, 951–955. doi:10.1093/infdis/147.5.951
Steyn, A. J., Collins, D. M., Hondalus, M. K., Jacobs, W. R., Kawakami, R. P., and Bloom, B. R. (2002). Mycobacterium tuberculosis WhiB3 interacts with RpoV to affect host survival but is dispensable for in vivo growth. Proc. Natl. Acad. Sci. U.S.A. 99, 3147–3152. doi:10.1073/pnas.052705399
Sud, D., Bigbee, C., Flynn, J. L., and Kirschner, D. E. (2006). Contribution of CD8+ T cells to control of Mycobacterium tuberculosis infection. J. Immunol. 176, 4296–4314.
Sun, Z., and Zhang, Y. (1999). Spent culture supernatant of Mycobacterium tuberculosis H37Ra improves viability of aged cultures of this strain and allows small inocula to initiate growth. J. Bacteriol. 181, 7626–7628.
Taneja, N. K., Dhingra, S., Mittal, A., Naresh, M., and Tyagi, J. S. (2010). Mycobacterium tuberculosis transcriptional adaptation, growth arrest and dormancy phenotype development is triggered by vitamin C. PLoS ONE 5:e10860. doi:10.1371/journal.pone.0010860
Tews, I., Findeisen, F., Sinning, I., Schultz, A., Schultz, J. E., and Linder, J. U. (2005). The structure of a pH-sensing mycobacterial adenylyl cyclase holoenzyme. Science 308, 1020.
Tufariello, J. M., Jacobs, W. R. Jr., and Chan, J. (2004). Individual Mycobacterium tuberculosis resuscitation-promoting factor homologues are dispensable for growth in vitro and in vivo. Infect. Immun. 72, 515–526. doi:10.1128/IAI.72.1.515-526.2004
van der Wel, N., Hava, D., Houben, D., Fluitsma, D., van Zon, M., Pierson, J., et al. (2007). M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129, 1287–1298. doi:10.1016/j.cell.2007.05.059
Voskuil, M., Schnappinger, D., Rutherford, R., Liu, Y., and Schoolnik, G. (2004a). Regulation of the Mycobacterium tuberculosis PE/PPE genes. Tuberculosis 84, 256–262. doi:10.1016/j.tube.2003.12.014
Voskuil, M. I., Visconti, K. C., and Schoolnik, G. K. (2004b). Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis 84, 218–227. doi:10.1016/j.tube.2003.12.014
Voskuil, M. I., Schnappinger, D., Visconti, K. C., Harrell, M. I., Dolganov, G. M., Sherman, D. R., et al. (2003). Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713. doi:10.1084/jem.20030205
Vynnycky, E., and Fine, P. E. (1997). The natural history of tuberculosis: the implications of age-dependent risks of disease and the role of reinfection. Epidemiol. Infect. 119, 183–201. doi:10.1017/S0950268897007917
Walburger, A., Koul, A., Ferrari, G., Nguyen, L., Prescianotto-Baschong, C., Huygen, K., et al. (2004). Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304, 1800.
Wang, C. C., Zhu, B., Fan, X., Gicquel, B., and Zhang, Y. (2013). Systems approach to tuberculosis vaccine development. Respirology 18, 412–420. doi:10.1111/resp.12052
Wang, X., Wang, H., and Xie, J. (2011). Genes and regulatory networks involved in persistence of Mycobacterium tuberculosis. Sci. China Life Sci. 54, 300–310. doi:10.1007/s11427-011-4134-5
Wayne, L., and Lin, K. (1982). Glyoxylate metabolism and adaptation of Mycobacterium tuberculosis to survival under anaerobic conditions. Infect. Immun. 37, 1042–1049.
Wayne, L. G. (1977). Synchronized replication of Mycobacterium tuberculosis. Infect. Immun. 17, 528–530.
Wayne, L. G., and Hayes, L. G. (1996). An in vitro model for sequential study of shift down of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64, 2062–2069.
Wayne, L. G., and Sohaskey, C. D. (2001). Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55, 139–163. doi:10.1146/annurev.micro.55.1.139
Wigginton, J. E., and Kirschner, D. (2001). A model to predict cell-mediated immune regulatory mechanisms during human infection with Mycobacterium tuberculosis. J. Immunol. 166, 1951–1967.
Yu, X., and Xie, J. (2012). Roles and underlying mechanisms of ESAT-6 in the context of Mycobacterium tuberculosis–host interaction from a systems biology perspective. Cell. Signal. 24, 1841–1846. doi:10.1016/j.cellsig.2012.05.014
Zhang, Y. (2004). Persistent and dormant tubercle bacilli and latent tuberculosis. Front. Biosci. 9:1136–1156. doi:10.2741/1492
Keywords: Mycobacterium tuberculosis, latency and dormancy regulation, latency models, mathematical and computational modeling
Citation: Magombedze G, Dowdy D and Mulder N (2013) Latent tuberculosis: models, computational efforts and the pathogen’s regulatory mechanisms during dormancy. Front. Bioeng. Biotechnol. 1:4. doi: 10.3389/fbioe.2013.00004
Received: 24 May 2013; Accepted: 12 August 2013;
Published online: 27 August 2013.
Edited by:
Kathleen Dave, Harvard University, USAReviewed by:
Martin I. Voskuil, University of Colorado Denver, USADenise Krischner, University of Michigan Medical School, USA
Copyright: © 2013 Magombedze, Dowdy and Mulder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gesham Magombedze, National Institute for Mathematical and Biological Synthesis, University of Tennessee, 1122 Volunteer Blvd, Knoxville, TN 37996-3410, USA e-mail: gmagombedze@nimbios.org; gmagombedze@gmail.com