Catalytic conversion of carbon dioxide into dimethyl carbonate using reduced copper-cerium oxide catalysts as low as 353 K and 1.3 MPa and the reaction mechanism
- Department of Chemistry, Graduate School of Science, Chiba University, Chiba, Japan
Synthesis of dimethyl carbonate (DMC) from CO2 and methanol under milder reaction conditions was performed using reduced cerium oxide catalysts and reduced copper-promoted Ce oxide catalysts. Although the conversion of methanol was low (0.005–0.11%) for 2 h of reaction, DMC was synthesized as low as 353 K and at total pressure of as low as 1.3 MPa using reduced Cu–CeO2 catalyst (0.5 wt% of Cu). The apparent activation energy was 120 kJ mol−1 and the DMC synthesis rates were proportional to the partial pressure of CO2. An optimum amount of Cu addition to CeO2 was 0.1 wt% for DMC synthesis under the conditions at 393 K and total pressure of 1.3 MPa for 2 h (conversion of methanol: 0.15%) due to the compromise of two effects of Cu: the activation of H2 during reduction prior to the kinetic tests and the block (cover) of the surface active site. The reduction effects in H2 were monitored through the reduction of Ce4+ sites to Ce3+ based on the shoulder peak intensity at 5727 eV in the Ce L3-edge X-ray absorption near-edge structure (XANES). The Ce3+ content was 10% for reduced CeO2 catalyst whereas it increased to 15% for reduced Cu–CeO2 catalyst (0.5 wt% of Cu). Moreover, the content of reduced Ce3+ sites (10%) associated with the surface O vacancy (defect sites) decreased to 5% under CO2 at 290 K for reduced Cu–CeO2 catalyst (0.1 wt% of Cu). The adsorption step of CO2 on the defect sites might be the key step in DMC synthesis and thus the DMC synthesis rate dependence on the partial pressure of CO2 was proportional. Subsequent H atom subtraction steps from methanol at the neighboring surface Lewis base sites should combine two methoxy species to the adsorbed CO2 to form DMC, water, and restore the surface O vacancy.
Introduction
Carbon dioxide is one of major green house gases. The conversion of CO2 has been widely investigated to reduce the atmospheric concentration of CO2 (Izumi, 2013). In the viewpoint of global warming, fixation methods of CO2 and/or converted compounds from CO2 are also critical. Transferring captured CO2 to the bottom of the sea in a supercritical state is partially in practical use, but it incurs huge investment costs. The conversion of CO2 to dimethyl carbonate (DMC) is attractive because DMC can be used as an electrolytic solution of lithium ion battery, methylating reagent, and feedstock for engineering plastics (Ono, 1997).
DMC has been conventionally synthesized starting from phosgene, carbon monoxide, or oxirane, but these materials are toxic and/or explosive. From CO2 and methanol/acetals, DMC was synthesized using homogeneous Sn catalysts at 10–30 MPa and 353–453 K (Sakakura et al., 1998, 1999, 2000; Kalhor et al., 2011) and using homogeneous Ni catalysts at 353 K and 1.0 MPa (Shi et al., 2013). Catalyst separation was improved for DMC synthesis using heterogeneous CeO2 (Yoshida et al., 2006), ZrO2 (Tomishige et al., 1999), solid solution of ZrO2 and CeO2 (Tomishige et al., 2001; Zhang et al., 2011b), Ga2O3/Ce0.6Zr0.4O2 (Lee et al., 2011), CexZr0.9–xY0.1O2 (Zhang et al., 2011a), SnO2–ZrO2/SiO2 (Ballivet-Tkatchenko et al., 2011), Co1.5PW12O40 (Aouissi et al., 2010), H3PW12O40/CexTi1–xO2 (La et al., 2007), Cu–KF/MgSiO (Li and Zhong, 2003), Cu–Ni–diatomite (Chen et al., 2012), Cu–Ni–graphite (Bian et al., 2009a), Cu–Ni–V2O5–active carbon (Bian et al., 2009b), and Cu–Ni–V2O5–SiO2 (Wu et al., 2006; Wang et al., 2007) at 353–453 K and 0.1–60 MPa. The conversion of methanol to DMC was as much as 7.9% for 24 h (Zhang et al., 2011b). In the viewpoint of global environment and the reduction of CO2, it is desirable to synthesize DMC from CO2 under mild reaction conditions.
In this context, the conversion of CO2 and methanol to DMC under milder conditions was studied and the mechanism was investigated by X-ray absorption near-edge structure (XANES). Methanol could be synthesized photocatalytically from CO2 ((Ahmed et al., 2011, 2012); Morikawa et al. under review). In future, the DMC synthesis reported in this work could be combined with photocatalysis to synthesize DMC from CO2 as a single starting material.
Materials and Methods
Preparation of CeO2
Cerium oxide samples were prepared from cerium nitrate hexahydrate (Wako Pure Chemical, >98.0%). It was dissolved in deionized water (<0.06 μS cm−1) to make the concentration to 0.2 mol L−1. A 5% ammonia aqueous solution (Wako Pure Chemical) was added to the solution to reach the pH 10. Obtained yellow precipitate was filtered using a polytetrafluoroethene-based membrane filter (Omnipore JGWP04700, Millipore) with a pore size of 0.2 μm and washed several times with deionized water. The obtained powder was calcined in air at 673 K for 4 h. Then, the powder was connected to a vacuum system using rotary and diffusion pumps (10−6 Pa) and the temperature was elevated at a ramping rate of 5 K min−1 from 290 to 673 K and kept at 673 K for 1 h.
A part of freshly-prepared CeO2 above was reduced under 25 kPa of hydrogen. The temperature was elevated from 290 to 673 K with the ramping rate of 10 K min−1 and maintained at 673 K for 1 h.
Preparation of Cu–−CeO2
3.8–950 mg of copper nitrate trihydrate (Wako Pure Chemical, >99.9%) was dissolved in 10 mL of deionized water. The Cu2+ solution was added to 1.0 g of CeO2 powder prepared in section Preparation of CeO2. Then, 25% of ammonia aqueous solution was added to the suspension until the pH reached 9.5. The mixture was reacted at 290 K for 1 h with magnetically stirred at a rate of 300 rpm. The color of precipitate was yellow, yellow green, and dark brown when the Cu content was 0.1, 1, and 20 wt%, respectively. The precipitate was filtered using JGWP04700 membrane and washed by several times with deionized water. The obtained powder was dried at 353 K for 12 h and denoted Cu–CeO2. The Cu–CeO2 samples were treated under H2 (25 kPa). The temperature was elevated from 290 to 673 K with the ramping rate of 10 K min−1, maintained at 673 K for 1 h, and evacuated (10−6 Pa) at 673 K for 30 min.
Characterization
Nitrogen adsorption isotherm measurements were performed at 77 K within the pressure range 1.0–90 kPa in a vacuum system connected to diffusion and rotary pumps (10−6 Pa) and equipped with a capacitance manometer (Models CCMT-1000A and GM-2001, ULVAC). The Brunauer-Emmett-Teller (BET) surface area (SBET) was calculated on the basis of eight-point measurements between 10 and 46 kPa (P/P0 = 0.10–0.45) on the adsorption isotherm. The sample was evacuated at 423 K for 90 min before the measurements.
The electronic state of cerium in catalysts was investigated by the synchrotron X-ray measurements. The catalyst powder samples were prepared in vacuum (10−6 Pa) and transferred directly to a Pyrex glass cell equipped with 50 μm-thick Kapton (Dupont) windows on both sides. The samples in N2 (60 kPa) or CO2 gas (60 kPa) were sealed with fire and transported to beamline.
Ce L3-edge X-ray absorption fine structure (XAFS) spectra were measured at 290 K in a transmission mode in the Photon Factory at the High-Energy Accelerator Research Organization (Tsukuba, Japan) on beamline 9C and also in SPring-8 (Sayo, Japan) on beamline 01B1. The X-ray energy was calibrated at the first intense peak top energy (5731.1 eV) for the Ce L3-edge spectrum of CeO2. The XAFS data were analyzed with a software package XDAP version 2.2.7 (Vaarkamp et al., 2006).
DMC Synthesis Tests
An autoclave (Taiatsu Glass Kogyo, inner volume 120 mL; Model TVS–N2–120) was used for the DMC synthesis tests using CeO2 and Cu–CeO2 catalysts. The inner space of autoclave was purged with argon gas (>99.998%) at a rate of 300 mL min−1. 10 mL of dehydrated methanol (Kanto Chemical, 99.8%) and 100 mg of catalyst were introduced in the autoclave with the flow of Ar, not in contact with air. Next, CO2 was flowed at a rate of 300 mL min−1 for 15 min. The outlet valve of reactor was closed and the interior pressure was increased to 2.0, 1.0, 0.50, 0.10 MPa at 290 K. Then, the temperature of the reactor was elevated from 290 to 393, 373, or 353 K with the ramping rate of 4 K min−1, and maintained at the destination temperature for 2–6 h. As a comparison, 10 mL of dehydrated methanol, 50 mg of catalyst, and 3.6 MPa of CO2 were introduced in the autoclave in similar way at 290 K. Then, the temperature of reactor was elevated from 290 to 403 K with the ramping rate of 4 K min−1, and maintained at 403 K for 2 h. The reaction suspension was filtered using a JGWP04700 membrane. The filtrate was analyzed by gas chromatograph equipped with frame ionization detector (Model GC-18A, Shimadzu) equipped with a capillary column Ultra ALLOY-5 (Frontier Laboratories; inner diameter 250 μm, length 30 m). The conversion (%) of methanol to DMC was defined as
Results
DMC Synthesis from CO2 and Methanol
Pretreatment effects in H2
In the test under the 2.8 MPa CO2 (initial pressure) at 393 K for 6 h, DMC formation rate using incipient CeO2 catalyst was 0.44 mmol h−1 g−1cat and that for reduced CeO2 was 0.70 mmol h−1 g−1cat (Table 1A). Total initial pressure of CO2 and methanol was 3.5 MPa at 393 K. By the pretreatment effect in H2 at 673 K, the synthesis rate increased by a factor of 1.6 times. The conversion (%) of methanol to DMC was improved to 0.33% (Table 1A).
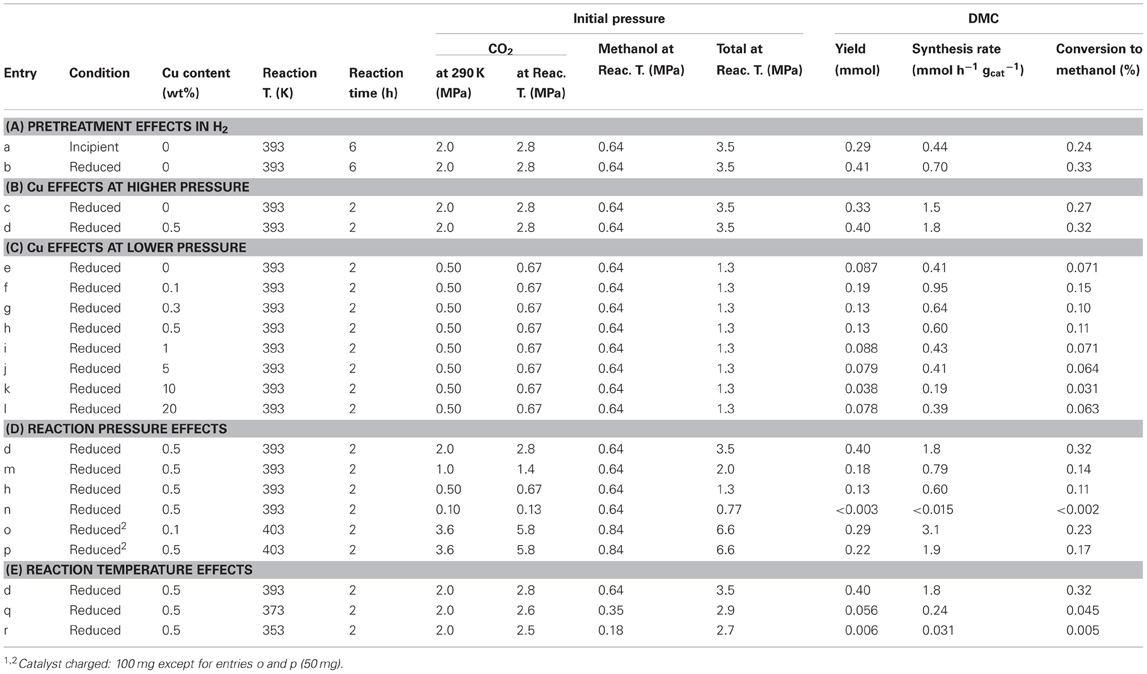
Table 1. Conditions and results of DMC synthesis from methanol and CO2 over Ce oxide and Cu–promoted Ce oxide catalysts1.
Effects of the Cu addition
The DMC synthesis rate using reduced Cu–CeO2 (0.5 wt% Cu) was compared to that using reduced CeO2 at 393 K for 2 h. The rates were 1.8 and 1.5 mmol h−1 g−1cat, respectively (Table 1B). By the inclusion of 0.5 wt% of Cu in the catalyst, the rate increased by a factor of 1.2 times. The conversion of methanol to DMC was improved to 0.32% (Table 1B).
Next, the effects of Cu addition were investigated by progressively changing the Cu content between 0 and 20 wt% under lower initial pressure (0.67 MPa) of CO2 at 393 K. By the inclusion of 0.1 wt% of Cu in Cu–CeO2 catalyst, the DMC synthesis rate increased 2.3-fold higher: from 0.41 mmol h−1 g−1cat (reduced CeO2) to 0.95 mmol h−1 g−1cat (Table 1C; Figure 1). However, further increase of Cu content between 0.3 and 1 wt% in Cu–CeO2 catalysts was not effective compared to the test results for the Cu–CeO2 catalyst, 0.1 wt% of Cu (Table 1C). When the Cu content was between 1 and 20 wt%, the synthesis rates gradually approached to constant, similar to the one for undoped CeO2 (0.41 mmol h−1 g−1cat) (Table 1C; Figure 1).
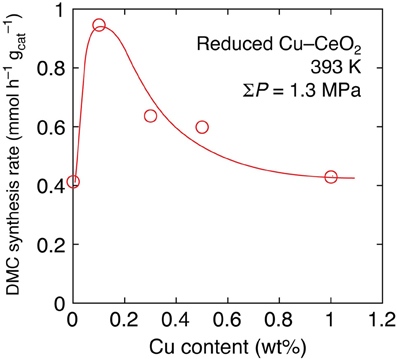
Figure 1. The dependence of DMC synthesis rates on the copper content in reduced Cu–CeO2 catalysts under total initial pressure of 1.3 MPa at 393 K.
Reaction pressure effects
In the reaction tests at 393 K using reduced Cu–CeO2 catalyst (0.5 wt% Cu), partial pressure of CO2 introduced at 290 K was varied between 2.0 and 0.10 MPa. The partial (initial) pressure of CO2 increased to between 2.8 and 0.13 MPa at reaction temperature of 393 K (Table 1D). The DMC synthesis rates were plotted as a function of initial pressure of CO2 and initial total pressure of CO2+ methanol at 393 K (Figure 2). The DMC synthesis was possible under the total pressure of 1.3 MPa, but the amount of produced DMC was below detection limit (3 μmol) under the total pressure of 0.77 MPa at 393 K (Table 1D). The DMC synthesis rates were proportional to partial pressure of CO2 (Figure 2).
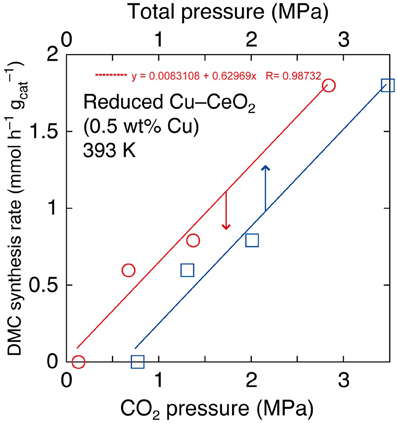
Figure 2. The dependence of DMC synthesis rates on the partial pressure of CO2 and total pressure using Cu–CeO2 catalyst (0.5 wt% of Cu) at 393 K.
The reaction pressure effects were also tested under severer reaction conditions: at 403 K and 6.6 MPa using Cu–CeO2 catalysts (0.1 and 0.5 wt% Cu) (Table 1o,p). The synthesis rates (3.1–1.9 mmol h−1 g−1cat) were higher by a factor of 3.3–3.2 times compared to corresponding test results at 393 K and 1.3 MPa (Table 1C,D). Thus, major reason of relatively low DMC synthesis rates in this paper was mild reaction conditions at 393–353 K and 3.5–1.3 MPa.
Reaction temperature effects
Further, the reaction temperature was varied between 393 and 353 K using reduced Cu–CeO2 catalyst (0.5 wt% Cu) under the CO2 partial pressure of 2.0 MPa introduced at 290 K. The CO2 pressure increased to between 2.8 and 2.5 MPa at reaction temperatures of 393–353 K (Table 1E). Under the total pressure of 2.7 MPa, DMC synthesis was possible as low as 353 K: 0.031 mmol h−1 g−1cat (Table 1E; Figure 3). The apparent activation energy was estimated to 120 kJ mol−1 based on the Arrhenius plot (Figure 3, inset).
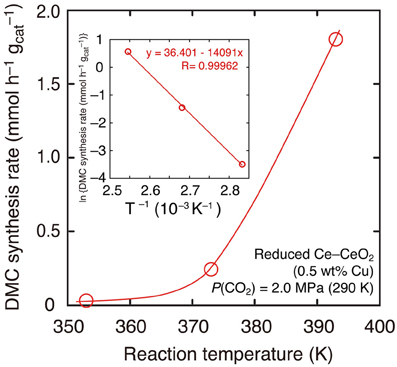
Figure 3. DMC synthesis rates over Cu–CeO2 catalyst (0.5 wt% of Cu) at 393–353 K. Initial partial pressure of CO2 was 2.0 MPa at 290 K. (Inset) The associated Arrhenius plot and the fit to the equation.
Bet Surface Area and Ce L3-Edge Xanes
The BET surface area was 78 and 94 m2 g−1cat for Cu–CeO2 samples consisting of 0.1 and 0.5 wt% Cu, respectively (Table 2).

Table 2. BET surface area of Cu–promoted Ce oxide catalysts.
Ce L3-edge XANES spectra taken for Ce–based catalysts and also standard Ce compounds were depicted in Figure 4. Twin peaks appeared at 5731 and 5738 eV in the XANES spectrum for as-synthesized CeO2 (spectrum a), indicating that valence state of Ce4+ was predominant (Zhang et al., 2004).
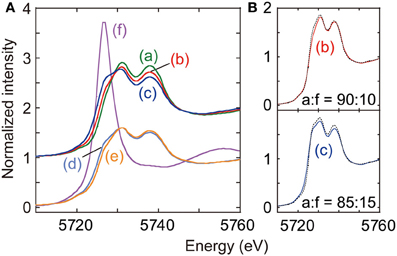
Figure 4. (A) Ce L3-edge XANES for as-synthesized CeO2 (a), CeO2 reduced at 673 K (b), Cu–CeO2 (0.5 wt% Cu) reduced at 673 K (c), Cu–CeO2 (0.1 wt% Cu) under N2 after reduction at 673 K (d), Cu–CeO2 (0.1 wt% Cu) under CO2 after reduction at 673 K (e), and Ce(NO3)3·6H2O (f). (B) Fits to spectra (b) and (c) with the combination of spectra (a) and (f). Best-fit results were shown by changing the mixing ratio of data (a) and (f).
When the CeO2 was reduced under hydrogen at 673 K (spectrum b), a shoulder peak gradually grew at 5727 eV, on the lower side of peak at 5731 eV. As an intense whiteline peak appeared at 5726.7 eV in the spectrum for CeIII(NO2)3·6H2O (Figure 4A-f), the shoulder peak for spectrum b suggested the partial reduction of initial Ce4+ to Ce3+. The partial reduction of CeO2 is traditionally known to promote electron-donating catalysis, e.g., ammonia synthesis (Izumi et al., 1996; Aika et al., 1997). The spectrum b was fitted with the spectrum a for fresh CeO2 and spectrum f for Ce(NO2)3·6H2O by changing the mixing ratio of standard spectra (Izumi and Nagamori, 2000; Izumi et al., 2007). Spectra a and f were used as models of Ce4+ and Ce3+ states, respectively. The goodness of fit was evaluated based on the residual-factor (Rf)
The spectrum b was best fitted with the mixing ratio of Ce4+:Ce3+ = 90:10 (Figure 4B-b).
The shoulder peak at 5727 eV also appeared in the XANES spectrum for reduced Cu–CeO2 (0.5 wt% Cu; spectrum c), and the intensity was greater than that in spectrum b for reduced CeO2. The spectrum c was also fitted with the spectrum a (Ce4+) and spectrum f (Ce3+) by changing the mixing ratio. The best fit was realized with the mixing ratio was Ce4+:Ce3+ = 85:15 (Figure 4B-c).
The Ce L3-edge XANES spectrum for reduced Cu–CeO2 catalyst (0.1 wt% Cu; spectrum d) was essentially identical with that for reduced CeO2 catalyst (spectrum b). The best-fit ratio with the mixing standard spectrum component of Ce4+ and Ce3+ was 90 and 10%. However, the shoulder peak at 5727 eV in the spectrum significantly weakened when 60 kPa of CO2 was introduced to the reduced Cu–CeO2 (0.1 wt% Cu) at 290 K (spectrum e). The best-fit ratio with the mixing standard spectrum component of Ce4+ and Ce3+ was 95 and 5%. The decrease of shoulder peak intensity at 5727 eV suggested re-oxidation of Ce3+ to Ce4+ by the reaction with CO2.
Discussion
DMC Synthesis Under Milder Conditions
DMC synthesis from CO2 and methanol was reported at a synthesis rate of 1.8–5.1 mmol h−1 g−1cat using CeO2 at 403 K and 8.7 MPa for 2–4 h (Yoshida et al., 2006). DMC synthesis rate from CO2 and methanol in this work using reduced CeO2 at 393 K and 3.5 MPa for 6 h was lower: 0.70 mmol h−1 g−1cat (Table 1A) due to lower reaction temperature and lower pressure. Because the forward reaction reduces the molar amount of materials in system from three to two and is uphill reaction (Pacheco and Marshall, 1997) (Equation 1), reaction conditions of lower reaction temperature and lower pressure are disadvantageous for the DMC synthesis reaction. The synthesis rate was enhanced by a factor of 1.6 times by the pre-reduction in H2 for CeO2 (Table 1A).
The disadvantage of moderate reaction conditions was compensated by the Cu addition to CeO2 catalysts. At 393 K and 3.5 MPa for 2 h, the DMC synthesis rates increased to 1.8 mmol h−1 g−1cat by the addition of 0.5 wt% of Cu (Table 1B).
The effects of Cu addition to the DMC synthesis rates were compared at even milder reaction conditions: at 393 K and 1.3 MPa for 2 h (Table 1C). Under the reaction conditions, the 0.1 wt% of Cu was most effective and it promoted the synthesis rate by a factor of 2.3 times (Figure 1). One of the plausible explanations is that the positive effects to induce the Ce4+ site reduction to facilitate H2 dissociation and spillover on the catalyst surface and negative effects to block (cover) the surface active sites for DMC synthesis, e.g., the adsorption/activation sites for methanol, compromised to make a synthesis rate maximum at the Cu amount of 0.1 wt%.
The maximal DMC synthesis rate using Cu–CeO2 catalyst (0.1 wt% Cu) at 393 K and 1.3 MPa was 0.95 mmol h−1 g−1cat (Table 1f), but the rate at 403 K and 6.6 MPa was quite higher (3.1 mmol h−1 g−1cat), nearly equivalent to those in literature using CeO2 (1.8–5.1 mmol h−1 g−1cat) at even severe conditions (403 K and 8.7 MPa) (Yoshida et al., 2006), demonstrating the effects of pre-reduction and/or the Cu addition to CeO2 found in this work. The SBET values (78–94 m2 g−1cat; Table 2) for Cu–CeO2 catalysts (0.1–0.5 wt% Cu) were also similar to those for CeO2 catalyst reported (80 m2 g−1cat) (Yoshida et al., 2006).
The effects of ZrO2 mixed with CeO2 (Tomishige et al., 2001; Zhang et al., 2011b) were also interpreted to enhance the redox chemistry between Ce4+ and Ce3+. In this sense, the redox of Cu+ and Cu2+ may enhance the redox between Ce4+ and Ce3+. We tested Co–CeO2 catalyst under the reaction condition of Table 1d. The conversion of methanol to DMC was 0.23% (not listed), slightly inferior to Cu–CeO2 catalyst. Furthermore, in our preliminary results, the conversions to DMC using Fe–CeO2 and Ni–CeO2 catalysts were nearly equivalent to that using Co–CeO2 catalyst. Thus, the effects of hydrogen activation and/or redox of added metal (Fe, Co, Ni, or Cu) to CeO2 may work in similar way: enhancing effects of mixed metal ions and/or activating effects of hydrogen during pretreatment.
The reactions at lower reaction temperatures were tested (Table 1E). At 2.7 MPa using Cu–CeO2 catalyst (0.5 wt% of Cu), DMC was formed at as low as 353 K (Table 1E). The temperature dependence of DMC synthesis rates nicely followed the Arrhenius equation to give the apparent activation energy: 120 kJ mol−1 (Figure 3, inset). Similar range of apparent activation energy (107 kJ mol−1) was obtained using homogeneous Sn catalysts in the temperature range of 357–403 K (Kalhor et al., 2011). The dependences of DMC synthesis rates on the CO2 pressure and total reactant pressure were also investigated at 393 K using Cu–CeO2 catalyst (0.5 wt% of Cu; Table 1D). DMC was synthesized as low as 1.3 MPa.
The progress of catalysts to synthesize DMC from CO2 is quite fast, especially under relatively mild conditions (see the Introduction section). The Cu–CeO2 catalysts in this study are one of the good catalysts to work at relatively mild conditions. The dependence of synthesis rates on pressure and temperature (Table 1) was interpreted based on X-ray spectroscopy in next section by monitoring the oxygen defect sites and Ce3+.
Active Sites of DMC Synthesis
The dependence of DMC synthesis on the CO2 pressure (previous section) was proportional (Figure 2). This fact suggested that the key reaction step of DMC synthesis depended linearly on the CO2 concentration.
To provide the insight into the surface reaction mechanism, the electronic state and structure for Ce sites were investigated using Ce L3-edge XANES. 10–15% of the Ce4+ sites of as-prepared CeO2 or Cu–CeO2 (0.5 wt% of Cu) were reduced to Ce3+ based on the shoulder peak intensity at 5727 eV (Figure 4b,c). Similarly, 10% of the Ce4+ sites of as-prepared Cu–CeO2 (0.1 wt% of Cu) were reduced to Ce3+ under H2 at 673 K, but a half of the Ce3+ sites were re-oxidized to Ce4+ by the introduction of CO2 at 290 K (Figure 4d,e). These changes in the XANES spectra suggested the adsorption of CO2 at the surface defect sites over the catalyst and the associated, neighboring Ce3+ sites to the defects were re-oxidized to Ce4+. This reduction and re-oxidation mechanism was already reported on CeO2 layers grown over Cu(111) surface (Staudt et al., 2010).
The proposed reaction mechanism was shown in Figure 5. Based on the dependence of DMC synthesis rates on the CO2 pressure and the change of a shoulder peak at 5727 eV in the Ce L3-edge XANES spectra, O vacancy was assumed as defect site and worked to adsorb CO2. The population of O vacancy should increase by the reduction in H2 and/or by the presence of Cu sites in catalysts. In order to synthesize DMC, surface Lewis base sites are required to subtract H atom from methanol (Figure 5). If each H atom was subtracted at the Lewis base site from two methanol molecules, DMC and water molecules are formed to restore an O vacancy site.
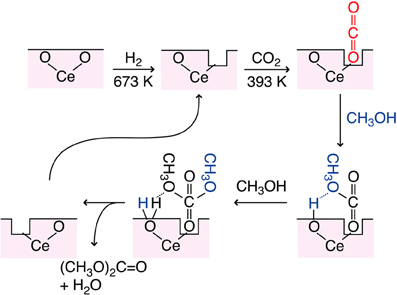
Figure 5. Proposed reaction mechanism of CO2 conversion to DMC using partially-reduced Ce oxide surface.
Conclusions
Reduction of CeO2 and Cu–promoted CeO2 catalysts in hydrogen at 673 K was effective to enhance the DMC synthesis from CO2 and methanol by a factor of 1.6–1.2 times. Added Cu worked cooperatively with CeO2 catalysts as it accelerated the partial reduction of Ce4+ sites to Ce3+. At the same time, doped Cu sites may block surface active sites. As a compromise, the DMC synthesis rate was maximal: 0.95 mmol h−1 g−1cat at 393 K and 1.3 MPa (total pressure) in 2 h when the Cu amount was 0.1 wt% for reduced Cu–CeO2 catalyst. The DMC synthesis was possible at the reaction temperature as low as 353 K (2.7 MPa) using the reduced Cu–CeO2 catalyst. The apparent activation energy was calculated to be 120 kJ mol−1. Based on the Ce L3-edge XANES, 10% of Ce sites were reduced to Ce3+ by the reduction in H2 for Cu–CeO2 (0.1 wt% of Cu) while half of them were re-oxidized to Ce4+ by the introduction of CO2 at 290 K. A linear rate dependence on CO2 pressure and the re-oxidation in CO2 suggest that the adsorption of CO2 might be the key step in DMC synthesis. H subtraction from methanol needs to occur at the neighboring sites of adsorbed CO2. Two methoxy groups and adsorbed CO2 combine then to form DMC and water and restores surface O vacancy (defect site).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful for the financial support received from the Asahi Glass Foundation (2009–2010). The X-ray absorption experiments were performed with the approval of the Photon Factory Review Committee (No. 2012G683, 2011G083, 2011G033) and the grant of the Priority Program for Disaster-Affected Quantum Beam Facilities (2011A1977, 2011A1978, SPring-8 & KEK). The authors also thank Yusuke Yoshida and Yuta Ogura at Chiba University, Graduate School of Science, for the discussion of reaction conditions.
References
Ahmed, N., Morikawa, M., and Izumi, Y. (2012). Photocatalytic conversion of carbon dioxide into methanol using optimized layered double hydroxide catalysts. Catal. Today 185, 263–269. doi: 10.1016/j.cattod.2011.08.010
Ahmed, N., Shibata, Y., Tanniguchi, T., and Izumi, Y. (2011). Photocatalytic conversion of carbon dioxide into methanol using zinc–copper–M(III) (M = aluminum, gallium) layered double hydroxide. J. Catal. 279, 123–135. doi: 10.1016/j.jcat.2011.01.004
Aika, K., Kubota, J., Kadowaki, Y., Niwa, Y., and Izumi, Y. (1997). Molecular sensing techniques for the characterization and design of new ammonia catalysts. Appl. Surf. Sci. 121/122, 488–491. doi: 10.1016/S0169-4332(97)00343-7
Aouissi, A., Apblett, A. W., AL-Othman, Z. A., and Al-Amro, A. (2010). Direct synthesis of dimethyl carbonate from methanol and carbon dioxide using heteropolyoxometalates: the effects of cation and addenda atoms. Transit. Met. Chem. 35, 927–931. doi: 10.1007/s11243-010-9413-7
Ballivet-Tkatchenko, D., Dos Santos, J. H. Z., Philippot, K., and Vasireddy, S. (2011). Carbon dioxide conversion to dimethyl carbonate: the effect of silica as support for SnO2 and ZrO2 catalysts. Comp. Rend. Chim. 14, 780–785. doi: 10.1016/j.crci.2010.08.003
Bian, J., Xiao, M., Wang, S. J., Lu, Y. X., and Meng, Y. Z. (2009a). Novel application of thermally expanded graphite as the support of catalysts for direct synthesis of DMC from CH3OH and CO2. J. Colloid Interface Sci. 334, 50–57. doi: 10.1016/j.jcis.2009.03.009
Bian, J., Xiao, M., Wang, S., Lu, Y., and Meng, Y. (2009b). Direct synthesis of DMC from CH3OH and CO2 over V-doped Cu–Ni/AC catalysts. Catal. Commun. 10, 1142–1145. doi: 10.1016/j.catcom.2008.12.008
Chen, Y., Xiao, M., Wang, S., Han, D., Lu, Y., and Meng, Y. (2012). Porous diatomite-immobilized Cu-Ni bimetallic nanocatalysts for direct synthesis of dimethyl carbonate. J. Nanomater. 2012, 610410. doi: 10.1155/2012/610410
Izumi, Y. (2013). Recent advances in the photocatalytic conversion of carbon dioxide to fuels with water and/or hydrogen using solar energy and beyond. Coord. Chem. Rev. 257, 171–186. doi: 10.1016/j.ccr.2012.04.018
Izumi, Y., Iwata, Y., and Aika, K. (1996). Catalysis on ruthenium clusters supported on CeO2 or Ni–doped CeO2: adsorption behavior of H2 and ammonia synthesis. J. Phys. Chem. 100, 9421–9428. doi: 10.1021/jp952602o
Izumi, Y., Konishi, K., Obaid, D., Miyajima, T., and Yoshitake, H. (2007). X-ray absorption fine structure combined with X-ray fluorescence spectroscopy. Monitoring of vanadium sites in mesoporous titania, excited under visible light by selective detection of vanadium Kβ5, 2 fluorescence. Anal. Chem. 79, 6933–6940. doi: 10.1021/ac070427p
Izumi, Y., and Nagamori, H. (2000). Site-selective X-ray absorption fine structure (XAFS) spectroscopy (2). XAFS spectra tuned to surface active sites of Cu/ZnO and Cr/SiO2 catalysts. Bull. Chem. Soc. Jpn. 73, 1581–1587. doi: 10.1246/bcsj.73.1581
Kalhor, M. P., Chermette, H., Chambrey, S., and Ballivet-Tkatchenko, D. (2011). From CO2 to dimethyl carbonate with dialkyldimethoxystannanes: the key role of monomeric species. Phys. Chem. Chem. Phys. 13, 2401–2408. doi: 10.1039/c0cp02089c
La, K. W., Jung, J. C., Kim, H., Baeck, S. H., and Song, I. K. (2007). Effect of acid-base properties of H3PW12O40/CexTi1−xO2 catalysts on the direct synthesis of dimethyl carbonate from methanol and carbon dioxide: a TPD study of H3PW12O40/CexTi1−xO2 catalysts. J. Mol. Catal. A 269, 41–45. doi: 10.1016/j.molcata.2007.01.006
Lee, H. J., Park, S., Song, I. K., and Jung, J. C. (2011). Direct synthesis of dimethyl carbonate from methanol and carbon dioxide over Ga2O3/Ce0.6Zr0.4O2 catalysts: effect of acidity and basicity of the catalysts. Catal. Lett. 141, 531–537. doi: 10.1007/s10562-010-0544-4
Li, C. F., and Zhong, S. H. (2003). Study on application of membrane reactor in direct synthesis DMC from CO2 and CH3OH over Cu–KF/MgSiO catalyst. Catal. Today 82, 83–90. doi: 10.1016/S0920-5861(03)00205-0
Ono, Y. (1997). Catalysis in the production and reactions of dimethyl carbonate, an environmentally benign building block. Appl. Catal. A 155, 133–166. doi: 10.1016/S0926-860X(96)00402-4
Pacheco, M. A., and Marshall, C. L. (1997). Review of dimethyl carbonate (DMC) manufacture and its characteristics as a fuel additive. Energy Fuels 11, 2–29. doi: 10.1021/ef9600974
Sakakura, T., Choi, J. C., Saito, Y., Masuda, T., Sako, T., and Oriyama, T. (1999). Metal-catalyzed dimethyl carbonate synthesis from carbon dixoide and acetals. J. Org. Chem. 64, 4506–4508. doi: 10.1021/jo990155t
Sakakura, T., Choi, J. C., Saito, Y., and Sako, T. (2000). Synthesis of dimethyl carbonate from carbon dioxide: catalysis and mechanism. Polyhedron 19, 573–576. doi: 10.1016/S0277-5387(99)00411-8
Sakakura, T., Saito, Y., Okano, M., Choi, J. C., and Sako, T. (1998). Selective conversion of carbon dioxide to dimethyl carbonate by molecular catalysts. J. Org. Chem. 63, 7095–7096. doi: 10.1021/jo980460z
Shi, Y., Jin, X., Hu, Y., Wnag, S., Li, J., and Shi, Z. (2013). Synthesis and characterization of bis[2-(1H-benzimidazol-2-yl)benzoate]nickel(II), and its use for preparation of dimethyl carbonate from methanol and CO2. Res. Chem. Intermed. doi: 10.1007/s11164-013-1030-6. [Epub ahead of print].
Staudt, T., Lykhach, Y., Tsud, N., Skála, T., Prince, K. C., Matolín, V., et al. (2010). Ceria reoxidation by CO2: a model study. J. Catal. 275, 181–185. doi: 10.1016/j.jcat.2010.07.032
Tomishige, K., Furusawa, Y., Ikeda, Y., Asadullah, M., and Fujimoto, K. (2001). CeO2–ZrO2 solid solution catalyst for selective synthesis of dimethyl carbonate from methanol and carbon dioxide. Catal. Lett. 76, 71–74. doi: 10.1023/A:1016711722721
Tomishige, K., Sakaihori, T., Ikeda, Y., and Fujimoto, K. (1999). A novel method of direct synthesis of dimethyl carbonate from methanol and carbon dioxide catalyzed by zirconia. Catal. Lett. 58, 225–229. doi: 10.1023/A:1019098405444
Vaarkamp, M., Linders, H., and Koningsberger, D. (2006). Software package XDAP version 2.2.7. Woudenberg: XAFS Services International.
Wang, X. J., Xiao, M., Wang, S. J., Lu, Y. X., and Meng, Y. Z. (2007). Direct synthesis of dimethyl carbonate from carbon dioxide and methanol using supported copper (Ni, V, O) catalyst with photo-assistance. J. Mol. Catal. A 278, 92–96. doi: 10.1016/j.molcata.2007.08.028
Wu, X. L., Meng, Y. Z., Xiao, M., and Lu, Y. X. (2006). Direct synthesis of dimethyl carbonate (DMC) using Cu–Ni/VSO as catalyst. J. Mol. Catal. A 249, 93–97. doi: 10.1016/j.molcata.2006.01.007
Yoshida, Y., Arai, Y., Kado, S., Kunimori, K., and Tomishige, K. (2006). Direct synthesis of organic carbonates from the reaction of CO2 with methanol and ethanol over CeO2 catalysts. Catal. Today 115, 95–101. doi: 10.1016/j.cattod.2006.02.027
Zhang, F., Wang, P., Koberstein, J., Khalid, S., and Chan, S. W. (2004). Cerium oxidation state in ceria nanoparticles studied with X-ray photoelectron spectroscopy and absorption near edge spectroscopy. Surf. Sci. 563, 74–82. doi: 10.1016/j.susc.2004.05.138
Zhang, Z., Wang, R., and Ma, X. (2011a). Direct synthesis of DMC from CO2 and CH3OH over CexZr1−x−0.1Y0.1O2 catalysts. Adv. Mater. Res. 287–290, 1632–1635. doi: 10.4028/www.scientific.net/AMR.287-290.1632
Keywords: CO2, dimethyl carbonate, cerium oxide, partial reduction, X-ray absorption near-edge structure (XANES), oxygen vacancy, hydrogen subtraction, environmental catalyst
Citation: Wada S, Oka K, Watanabe K and Izumi Y (2013) Catalytic conversion of carbon dioxide into dimethyl carbonate using reduced copper-cerium oxide catalysts as low as 353 K and 1.3 MPa and the reaction mechanism. Front. Chem. 1:8. doi: 10.3389/fchem.2013.00008
Received: 15 March 2013; Paper pending published: 19 April 2013;
Accepted: 06 June 2013; Published online: 26 June 2013.
Edited by:
Viswanathan Balasubramanian, Indian Institute of Technology, IndiaReviewed by:
Kannan Srinivasan, CSIR-Central Salt and Marine Chemicals Research Institute, IndiaKrishnamurthy K. Ramaswamy, Indian Institute of Technology, India
Copyright © 2013 Wada, Oka, Watanabe and Izumi. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Yasuo Izumi, Department of Chemistry, Graduate School of Science, Chiba University, Yayoi 1–33, Inage-ku, Chiba 263–8522, Japan e-mail: yizumi@faculty.chiba-u.jp