Selectivity and self-diffusion of CO2 and H2 in a mixture on a graphite surface
 Thuat T. Trinh
Thuat T. Trinh Thijs J. H. Vlugt
Thijs J. H. Vlugt May-Britt Hägg
May-Britt Hägg Dick Bedeaux
Dick Bedeaux Signe Kjelstrup
Signe Kjelstrup- 1Department of Chemistry, Norwegian University of Science and Technology, Trondheim, Norway
- 2Department of Process and Energy, Delft University of Technology, Delft, Netherlands
- 3Department of Chemical Engineering, Norwegian University of Science and Technology, Trondheim, Norway
We performed classical molecular dynamics (MD) simulations to understand the mechanism of adsorption from a gas mixture of CO2 and H2 (mole fraction of CO2 = 0.30) and diffusion along a graphite surface, with the aim to help enrich industrial off-gases in CO2, separating out H2. The temperature of the system in the simulation covered typical industrial conditions for off-gas treatment (250–550 K). The interaction energy of single molecules CO2 or H2 on graphite surface was calculated with classical force fields (FFs) and with Density Functional Theory (DFT). The results were in good agreement. The binding energy of CO2 on graphite surface is three times larger than that of H2. At lower temperatures, the selectivity of CO2 over H2 is five times larger than at higher temperatures. The position of the dividing surface was used to explain how the adsorption varies with pore size. In the temperature range studied, the self-diffusion coefficient of CO2 is always smaller than of H2. The temperature variation of the selectivities and the self-diffusion coefficient imply that the carbon molecular sieve membrane can be used for gas enrichment of CO2.
Introduction
The production of cheap membranes for CO2 gas separation purposes is of primary importance for the realization of carbon capture and sequestration technologies (He et al., 2009; He and Hägg, 2011, 2012). One of the important applications of membranes is to separate CO2 from a mixture of gases (Bernardo et al., 2009). Pressure swing adsorption (PSA) is one of the most common techniques to capture CO2 from a mixture of CO2 and H2. This process requires large pressures, being different in the adsorption and desorption steps (Bernardo et al., 2009). In the adsorption step, CO2 absorbs strongly into the carbon material at high pressure. Then in the later step, CO2 desorbs at a much lower pressure. The energy costs depend on the manner this is performed; in one or more steps, with or without heat integrated. By using a molecular sieve membrane the separation can be performed as a continuous process, where the CO2 is removed both by adsorption and diffusion from the high pressure side (feed side) to the low pressure side (permeate side). To provide an energy efficient design, we will need knowledge of molecular behavior, in particular of the selectivity and of transport properties at selected process conditions. Although there is a lot of recent progress in the modification of graphene material for adsorption and separation application of CO2 and H2 these material are not cheap (Kim et al., 2013; Li et al., 2013). Nano-porous, fibrous, carbonaceous materials are promising candidates from an economic point of view. In order to make further progress and produce molecular sieve membranes, better knowledge of several issues is needed. Central for membrane functionality are pore size, surface binding, surface wall transport, pore inlet control, carbon structure and composition. This work aims to provide such knowledge for a simplified, graphitic membrane, laying the grounds for more realistic future studies.
There are several experimental works and simulations devoted to understand adsorption of single component CO2 and H2 on carbon based material such as activated carbon and graphite (Guo et al., 2006; Haas et al., 2009; Levesque and Lamari, 2009; Jin et al., 2011; Saha et al., 2011). The experimentally obtained adsorption isotherm of CO2 on active carbon is well-described by several models such as Langmuir (Jin et al., 2011), Tóth (Himeno et al., 2005), Dubinin-Astakhov (D-A) (Saha et al., 2011; Sevilla and Fuertes, 2012). Reported values for the enthalpy of adsorption depend on the type of adsorbent. Saha et al. reported that heats of adsorption of CO2 in Maxsorb II and ACF (A-20) material are around −20 kJ/mol (Saha et al., 2011), while the untreated activated carbon C3345 material has a heat of adsorption −14 kJ/mol (Jin et al., 2011). Guo et al. reported that the heat of adsorption varied in the range −10 to −28 kJ/mol depending on the modification of the activated carbon material (Guo et al., 2006). Himeno et al. reported adsorption enthalpies in the range −16 to −25 kJ/mol for pure CO2 on five different commercial activated carbons (Himeno et al., 2005).
Several simulation studies have given the adsorption isotherms for CO2 on planar and pore-like graphite surfaces. Lim et al. (2010) reported data using a Langmuir adsorption model, and provided the self-diffusion coefficient (Ds= 10−8−10−9 m2/s) of CO2 in a narrow pore (width 0.65–0.75 nm) for temperatures T = 298−318 K. Zhou et al. reported results for a wider range of pore sizes (0.7–3.4 nm) (Zhou and Wang, 2000). Their values are comparable with those of Lim et al. The authors reported that CO2 could form double layers. Both layers had a typical liquid-like structure (Zhou and Wang, 2000). Levesque et al. calculated the heat of CO2 adsorption on activated carbon using Monte-Carlo simulations (Levesque and Lamari, 2009). The authors discussed how the adsorption enthalpy depended on the distribution of pore sizes.
Adsorption and diffusion of single component H2 on graphite have recently been measured (Haas et al., 2009; Simon et al., 2010). The self-diffusion coefficient of H2 on a graphite surface was found, using quasielastic neutron scattering (QENS) (Haas et al., 2009), to be in the range 10−6−10−7 m2/s. Simulations found that pure H2 on the graphite surface had a high lateral mobility (Simon et al., 2010).
Few computational results are reported on the selective adsorption of a mixture of CO2 and H2 on a graphite surface. Cao et al. described the graphite surface selectivity of the mixture at bulk compositions 50:50 and 20:80 at three different temperatures, slit pore sizes up to 3.0 nm and pressures up to 10 atm, using Monte Carlo simulations (Cao and Wu, 2005). The selectivity of CO2 over H2 depended on the pore size and the temperature. More recently, Kumar et al. (Vasanth Kumar and Rodríguez-Reinoso, 2012) reported results for CO2/H2 mixtures for molar ratios 10:90 and 20:80 on different graphite structures (nanotube, slit pores, or computer generated) at room temperature 298 K. It was shown that mixture separation was best with nanotubes. There are few studies on diffusion of CO2 and H2 on the graphite surface.
These studies give a motivation for the present work. We want to add to the knowledge of adsorption isotherms for a mixture CO2 and H2 at a typical syngas compositions (Rostrup-Nielsen and Christiansen, 2011) (mole fraction of CO2 = 0.30) on a graphite surface, find the selectivity and self-diffusion coefficient for the components along the surface, and study these properties for a wide range of temperatures (T = 250, 550 K). Molecular dynamics (MD) simulations are well-suited to determine such properties. A snapshot of the gas mixture in equilibrium with the graphite is shown in Figure 1.
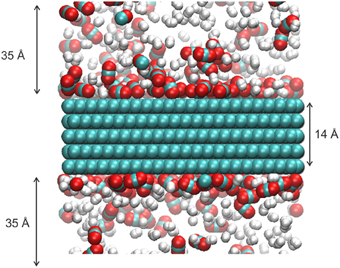
Figure 1. Typical snapshot of the gas mixture of CO2 and H2 in equilibrium with a graphite surface. The temperature is T = 300 K and the number of particles is N = 700. The green, red, white are represented carbon, oxygen, and hydrogen atom, respectively.
Model
Isotherm Adsorption
The reaction between the gas phase and the adsorbed phase on the surface can be written for each component:
At equilibrium, the ideal gas chemical potential is equal to the surface chemical potential for each component:
where μ0g is the standard chemical potential of the gas phase, i.e., the ideal chemical potential at the reference pressure p0.
The chemical potential for the surface is
where μ*s is the standard state chemical potential, γ is the activity coefficient, and Γ and Γ * are the surface adsorption and standard surface adsorption, respectively. The surface adsorption is an excess quantity (see Equation 14 below for the definition).
Simulation Details
To model the CO2 adsorption and transport on the graphite surface, we performed classical MD simulations using the DL_POLY classic version 2.18 package (Smith et al., 2002). The system consisted of a graphite crystal and a mixture of CO2 and H2 molecules, ratio 30:70, an example of a syngas mixture (Rostrup-Nielsen and Christiansen, 2011). The graphite had hexagonal structure with P63/mmc without any defects. The graphite contained 4284 carbon atoms and was constructed from 5 sheets of graphene which represented the property of graphite well (Boukhvalov et al., 2008). We oriented the graphene sheets in our simulation box such that the surfaces of the sheets were perpendicular to the z direction. The size of the simulation box was 42 × 51 × 84 Å3. In the z direction, the system covers a pore size of 70 Å and a graphite width of 14 Å (Figure 1). Periodic boundary conditions are used in all directions. At least 10 systems with different total number (N) of molecules, where 10 < N < 7.0 were simulated. For each N, simulations were performed at 8 different temperatures in the range 250–550 K.
The MD simulation had time steps of 1fs. The initial configuration was constructed by randomly distributing the CO2/H2 mixture over the graphite surface. The system was stabilized during 1000 ps by NVT runs with the Nosé-Hoover thermostat (Martyna et al., 1992). When the system was in the thermal equilibrium, we performed another 1000 ps run with microcanonical ensemble conditions (NVE) to study adsorption and transport properties. The average values of temperature and pressure in NVE simulation were within 1% of expected values. In total 2 × 106 MD steps was performed and this is sufficiently long to get good statistics and consistent trajectories. Each trajectory was printed every 100 time step and stored for further analysis.
Potential Energy Interaction
We fixed the graphite layer and used the rigid body model of Transferable Potentials for Phase Equilibria (TraPPE) for CO2 and H2 molecule. This potential can describe well the vapor-liquid and the liquid-solid equilibria of CO2 (Potoff and Siepmann, 2001). The intermolecular potential contained long range Coulombic interactions, and a shifted and truncated 12-6 Lennard-Jones (LJ) potential (Allen and Tildesley, 1989).
where rij is the distance between atoms i and j, εij, and σij are LJ potential parameters, and rc is the cutoff radius. The LJ interaction parameters between the different types of atoms were calculated from the Lorentz-Berthlot mixing rules (Allen and Tildesley, 1989)
The Coulombic interactions were:
where qi, qj are the charges on atoms i, j, and ε0 is the dielectric constant. In our work, we use the Smoothed Particle Mesh Ewald technique implemented in the DL_POLY package for the electrostatic interactions, see Essmann et al. (1995) for more details. The parameters, taken from the DREIDING (Mayo et al., 1990) and TraPPE (Potoff and Siepmann, 2001) force fields (FFs), are listed in Table 1.
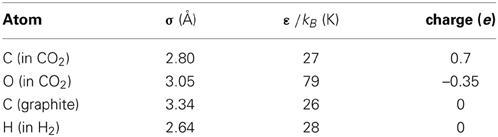
Table 1. Interaction potential parameters used in simulation.
Density Functional Theory (DFT) Calculations
To evaluate the results using classical FFs, we also performed DFT to calculate the binding energy of CO2 and H2 on graphite surface. For the ab-initio simulations, DFT optimization and single energy were performed using Quickstep (Vandevondele et al., 2005) which is part of the CP2K program package (http://cp2k.berlios.de, 2011). Quickstep employs the Gaussian and plane waves (GPW) method (Lippert et al., 1997) which makes efficient and accurate density-functional calculations of large systems possible. We used the Goedecker-Teter-Hutter (GTH) pseudopotentials (Goedecker et al., 1996; Hartwigsen et al., 1998) to describe atomic cores and the PBE exchange-correlation functional (Perdew et al., 1996). One-electron wave functions were developed under the DZVP-MOLOPT (DZPM) basis set, offering a double-zeta valence complemented with polarization functions (Vandevondele and Hutter, 2007). An energy cut-off of 400 Ry was selected for the additional plane wave basis sets. To describe the van der Waals interactions, an empirical dispersion correction of Grimme's type was applied (Grimme, 2006).
DFT is a computationally expensive method for a large system. Hence we used a much smaller model than with the FF method. Five sheets of 32 carbon atom each was used to construct the graphite surface. The graphite geometry was chosen similarly to the FF simulation. The system was fully optimized, and then CO2 and H2 molecules were fixed at selected distance from the surface for single point energy calculations.
We used the DFT method to calculate the interaction energy between each component (CO2, H2) and graphite surface.
For X = CO2 or H2:EiX, EGraphite−X, EGraphite, EX are the interaction energy, potential energy of graphite-X system, potential energy of graphite and potential energy of X, respectively.
The optimum distance of adsorption is the distance between molecule and graphite surface where the interaction energy profile has a minimum.
Results and Discussion
Interactions Between CO2/H2 and the Graphite Surface
Figure 2 shows the adsorption energy profile of a single CO2 or H2 molecule on the graphite surface calculated with the FF and DFT methods for the optimum molecule-surface distance. This distance and the adsorption energy are given in Table 2 for both methods. CO2 is favorably adsorbed at the bridge site and is parallel to the surface, while H2 prefers the hollow site and is perpendicular to the surface. Our finding is supported by Rubes et al. (2010). The profiles of the plots in Figure 2 are very similar, meaning that the FF results can be seen as a good representation of the DFT calculations. The values of the interaction energy of CO2 and H2 on the surface (Table 2) are typical for physisorption. For CO2 on surface, the interaction energy EiCO2 = −15 and −17 kJ/mol for FF and DFT, respectively. The optimum distance of CO2 and surface is around 3.20–3.30 Å (Table 2). The interaction energy in the case of H2 on the surface is smaller than in the case of CO2 on the surface (EiH2 = −6 and −5 kJ/mol for FF and DFT, compare Figures 2B to 2A) while the optimum distance of H2 and the surface is around 3.0 Å (Figure 2B). Furthermore, the interaction energies are in good agreement with other DFT calculations (Montoya et al., 2003; Rubes et al., 2010), meaning that our results for binding energies and distances are robust.
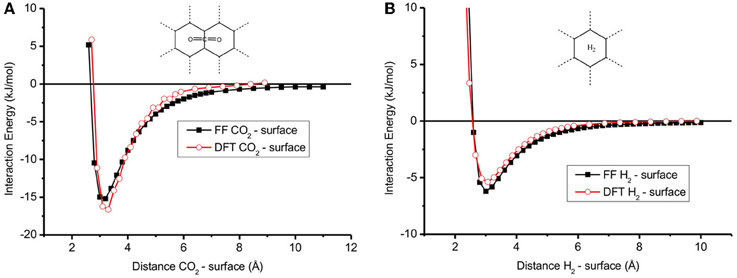
Figure 2. Interaction energy between (A) CO2—graphite surface and (B) H2—graphite surface calculated with DFT and Force Field method. The result of FF method is comparable with DFT method.
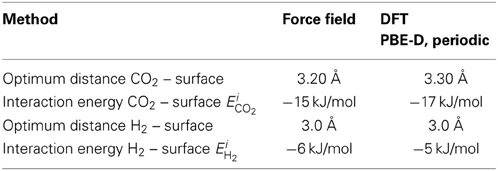
Table 2. Optimum distance for adsorption and corresponding interaction energies for the FF and DFT methods used.
The Structure of Mixture on Surface
Typical snapshots of mixtures adsorbed on graphite surfaces at different temperatures and total number of particles are depicted in Figure 3. With the same number of total molecules, there are more molecules adsorbed on the surface at lower temperature (compare Figures 3A,B) than at higher temperatures (Compare Figures 3C,D). The ratio CO2/H2 on the graphite surface is also larger in the low temperature range than at high temperatures. H2 and CO2 appears randomly distributed on the surface when surface has low loading (Figures 3A–C). But when the surface has high loading and CO2 is preferred at the surface, the H2 molecules seem to occupy the voids between the CO2 molecules (Figure 3D).
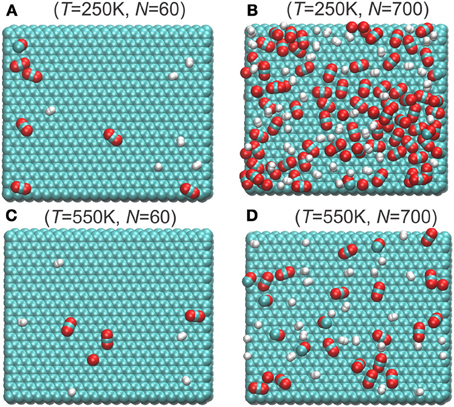
Figure 3. Typical snaphosts of top view of molecules adsorbed on the graphite surface at different temperatures and total number of particles (A) T = 250 K, N = 60; (B) T = 250 K, N = 700; (C) T = 550 K, N = 60; (D) T = 550 K, N = 700, cf. Figure 6. Only the adsorbed layers below the dividing surface are shown (cf. Equation 14). The green, red, white represent carbon, oxygen, and hydrogen atoms, respectively.
Surface Excess Densities
In the thermodynamic description, we use the surface excess concentration (adsorption) Γ, as defined originally by Gibbs, see Kjelstrup and Bedeaux (2008) for a detailed description. The interface is defined as the thin layer between phases where densities deviate from the densities in the adjacent phases. We restrict ourselves to surfaces parallel to the graphite surface, so
where Γ is the adsorption, and C, Cgas are the total concentration of molecules and the concentration in the gas, respectively (Figure 4). The Heaviside function, θ, is by definition unity, when the argument is positive, and zero when argument is negative. The extension of the surface can differ from molecule to molecule, as illustrated for the two molecules in question in Figure 4, and a choice must be made. The positions αH2 and αCO2 are defined as the positions where the concentrations of H2 and CO2 are 5% above the bulk value. For the CO2/H2 mixture, we choose the dividing surface α = αCO2 as given in the figure for the integral in Equation (14). Adsorption isotherms (Figures 6, 7 below) were obtained by plotting the surface excess concentration provided by Equation (14) for both components using this position, vs. the gas pressure. The gas pressures of CO2 and H2 were obtained by separate calculations where the simulation box contained only CO2 or H2 at different temperatures and concentrations. The total gas pressure is the sum of partial pressure of CO2 and H2.
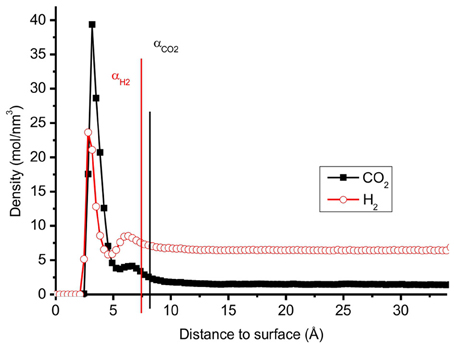
Figure 4. The distribution of pure CO2 and pure H2 molecules perpendicular to the surface in a mixture with N = 700 at temperature T = 300 K. One can distinguish two regions for each molecule; 0-α: adsorbed layer, above α: gas phase.
The distributions of CO2 and H2 molecules perpendicular to the surface, have two peaks, see Figure 4. The first peak of CO2 is located around 3.2 Å and the first of H2 is located around 3.0 Å. These peaks correspond to the optimum distances of adsorption of the gas molecules as described in the previous section. The radial distribution functions (RDF) of CO2–H2 and CO2–CO2 molecules of the different layers across the surface are reported in Figure 5. The RDF of CO2–CO2 in the adsorbed phase has a liquid-like behavior and this agrees with other simulations of pure CO2 on graphite surface (Zhou and Wang, 2000). The RDF of CO2–CO2 in the gas phase is less ordered, showing a homogenous behavior. The positions of maximum RDF of the adsorbed layers and the gas of CO2 are comparable. The RDF of CO2–H2 has a typical gas-like behavior, which indicates that CO2–H2 is near an ideal mixture. The interactions between the gas components are not as important as the interactions between the gas and the graphite surface.
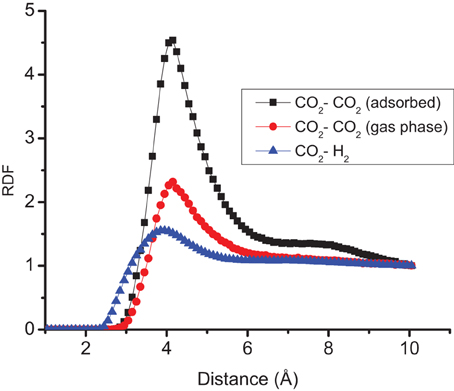
Figure 5. Radial distribution function RDF of CO2–H2, CO2–CO2 in the adsorbed layer and CO2–CO2 in the gas phase at system T = 300 K, N = 700, and x CO2 = 0.30.
The Adsorption on A Graphite Surface
The adsorption of CO2 and H2 at different temperatures are presented in Figure 6. When the temperature increases, the adsorption decreases, as expected. This behavior was also observed with CO2/H2 mixtures of molar ratios (20:80 and 10:90) on a different carbon pore structure, using Monte Carlo simulations (Vasanth Kumar and Rodríguez-Reinoso, 2012). The adsorption of hydrogen, ΓH2, is much lower than the value of Γ CO2. We explain the preference of CO2 to H2 on the surface by its stronger interaction with graphite (Table 2). The H2 adsorbs less than CO2 and prefers the gas phase. The total adsorption of both CO2 and H2 is shown in Figure 7. When the temperature increases the mixture adsorbs less.
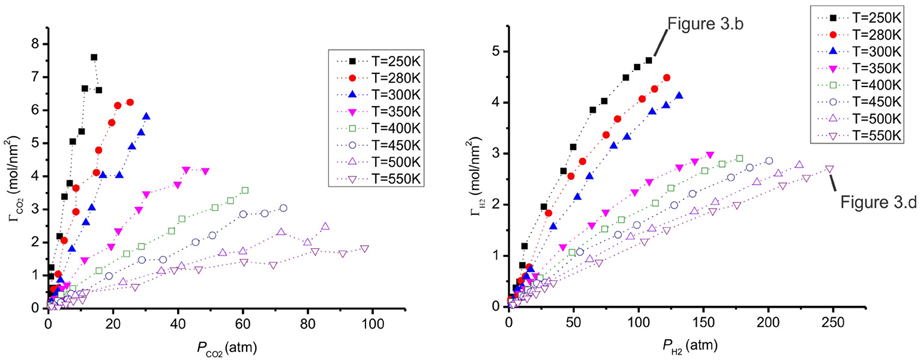
Figure 6. The adsorption of CO2 and H2 as a function of partial pressure of CO2 (left) and H2 (right) at different temperatures. The states used for the snapshots in Figures 3B,D are shown.
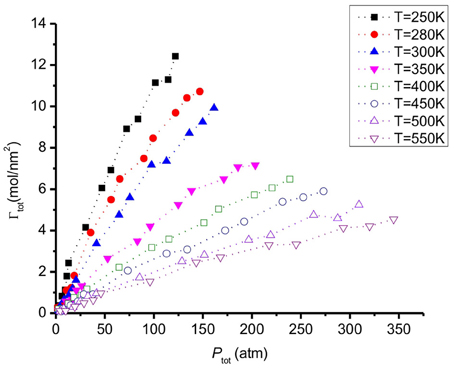
Figure 7. Total amount of CO2/H2 adsorbed on a graphite surface as a function of gas pressure at different temperatures.
Separation of mixtures
The separation ratio S (selectivity) of CO2/H2 mixture is defined as:
The selectivity is commonly used to define the efficiency of a (membrane) material to separate CO2 from a mixture of CO2, H2. For activated carbon, S depends on the mixture composition and on the pore sizes (Vasanth Kumar and Rodríguez-Reinoso, 2012). Our surface model can be seen as a graphite crystal with an extra-large slit pore, of 7 nm diameter. The results obtained for S are presented in Figure 8. At low pressures (<25 atm), the accuracy in S is small (within ±30%) because the small number of molecules leads to poor statistics. At higher pressures (>25 atm), the values of S vary around a constant average value within ±10% for each temperature.
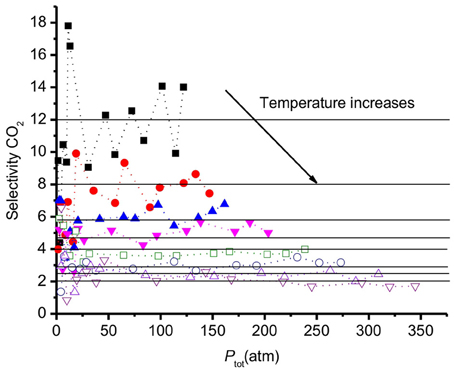
Figure 8. Selectivity of CO2 relative to H2 vs. the total pressure of the system. Results refer to a graphite surface with pore size 7 nm at different temperatures from 250 to 550 K. The selectivity decreases with increasing temperatures.
Monte Carlo simulations by Kumar et al. (Vasanth Kumar and Rodríguez-Reinoso, 2012) showed that the selectivity increases with increasing pressure with a slit pore geometry. Cao et al. (Cao and Wu, 2005) reported that the selectivity of CO2 over H2 size decreased when the pressure increased in the low pressure region, using Monte Carlo simulations. The results of Cao et al. can be understood from the selectivity of CO2 over H2 being maximum for carbon pores around 15 Å (Cao and Wu, 2005), being the double of the surface extension shown in Figure 4. The number of molecules adsorbed larger, relatively speaking, for pore sizes below 2α, leading to high selectivities for such pores or pore distributions. By finding the extension of the surface, one can thus decide on the optimal pore size of the material.
The data in Figure 8 show that the selectivity is essentially invariant of the pressure for pressures above 25 atm. The results indicate that the selectivity goes down below 25 atm. All values S are in the range 2–18 and decreases when the temperature increases. At 250K, the average selectivity is 12. At the highest temperature, 550 K, S reduces to the average value 2. At high temperatures the two gases have similar adsorption behavior, CO2 does not adsorb much stronger than H2. This changes at lower temperatures. The trend of S is in good agreement with other simulations of CO2/H2 mixtures in slit pores with smaller pore sizes, using the GCMC technique (Cao and Wu, 2005).
Surface Self-Diffusion
The self-diffusion of CO2 and H2 along the surface was studied. The self-diffusion coefficient of molecule was obtained from:
where d is the dimensionality (for surface d = 2), N is the total molecules, ri (t) and ri (0) are the projections of the position of molecules on the surface at time t and time 0.
All molecules were included in the mean-squared displacement calculations as described in previous studies (Haas et al., 2009). By plotting the logarithm of the diffusion coefficients found vs. the inverse of temperature, we obtained an Arrhenius plot. This was used to estimate the temperature dependence of the diffusion coefficient according to
where D0 is the pre-exponential factor, R is the gas constant, and Eact is the activation energy.
Figure 9 shows an example of mean-squared displacement of CO2 and H2 parallel to the graphite surface at temperature 300 K. It is clearly shown that H2 diffuses much faster than CO2. We observe a similar trend for all cases: H2 always has a higher self-diffusion coefficient than CO2, because H2 is lighter than CO2.
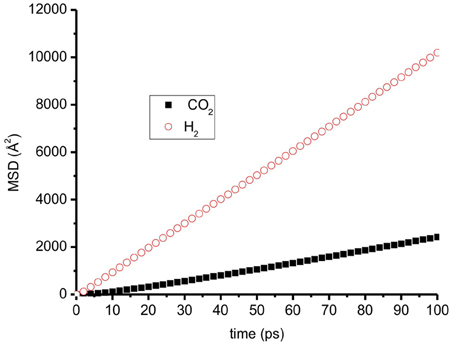
Figure 9. The mean-squared displacement (MSD) of CO2 and H2 on a graphite surface at T = 300 K, N = 700, mole fraction of CO2 = 0.3 H2 has much higher MSD than CO2.
The activation barrier for self-diffusion was obtained by calculating the slope of the linear relationship between the natural logarithm of the self-diffusion coefficient and 1/T (Figure 10). We found that activation barriers for self-diffusion of CO2 varying in the range EactCO2 = 3.5–4.3 kJ/mol. These values are smaller than the values reported by Lim et al. (2010), giving energy barriers in the range 5.77–6.08 kJ/mol for CO2 diffusion. The pores were smaller than 1.0 nm in this case, however. The self-diffusion coefficient of CO2 along the graphite surface is higher than values obtained from simulations with smaller pore sizes (<1 nm) (Zhou and Wang, 2000; Lim et al., 2010). This is because larger pores allow surface CO2 more space to diffuse, and less interaction between CO2 and carbon atoms of graphite. Under the conditions used here with pore size ~7 nm, CO2 will diffuse relatively faster and with a smaller diffusion barrier than inside a 1 nm slit pore. This adds to the knowledge on CO2 diffusion on graphite surfaces.
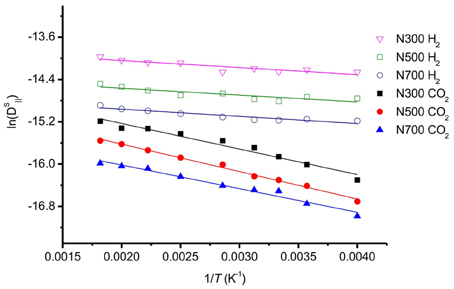
Figure 10. Natural logarithm of the self-diffusion coefficients as a function of the inverse temperature of selected system.
For H2 on graphite surface, we found self-diffusion barriers in the range EactH2 = 1.0–1.1 kJ/mol. The self-diffusion barrier for H2 is good agreement with experimental data for H2 transport on graphite surface. QENS gave 1.0–1.2 kJ/mol, Haas et al. (2009). The D0H2 in our study was also in a very good agreement with other experimental values (Table 3).
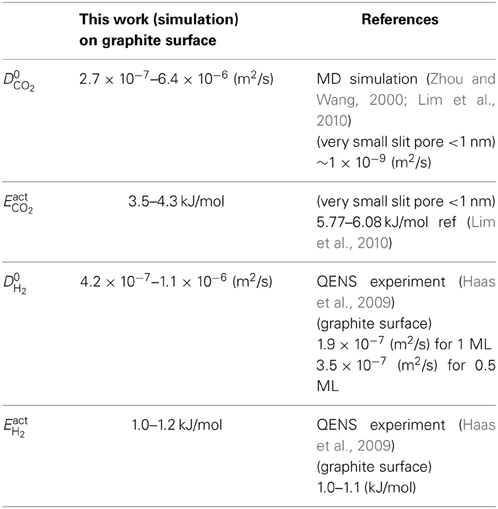
Table 3. Summary of results on self-diffusion parameters cf. Equation (17) for a mixture of CO2 and H2 on a graphite surface.
The diffusion coefficient of H2 is larger than that of CO2 (Figure 11). The barrier to self-diffusion of CO2 is four times larger than that of H2 on a graphite surface. The CO2 diffusion depends much more on the temperature than that of H2. Hence at high temperature, CO2, and H2 will have similar diffusion coefficients.
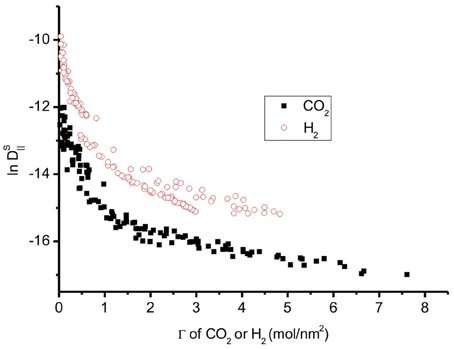
Figure 11. Natural logarithm of all self-diffusion coefficients vs. adsorption of CO2 and H2. H2 has higher diffusion coefficients than CO2.
The results (Figure 8) have shown that the selectivity is more or less invariant to the total pressure above 25 atm, but highly dependent on the temperature. The permeation of a gas through a membrane is a product of diffusion and adsorption, and the main driving force for separation is given by the partial pressure difference over the membrane. This means that separation of CO2 from a mixture with H2 at any pressure can best occur at low temperatures, when graphite as an adsorbing material is most effective for CO2. The adsorbed CO2 will then induce pore size reduction, hence hindering H2 to permeate, and hence CO2 can be selectively permeated. At high temperature, the permeated gas will be enriched in H2 since adsorbed CO2 will no longer be blocking the pores, and there will hardly be any selectivity between the two. However, if the pore size can be tailored to the range of 3–4 Å, one may achieve a diffusional selectivity in favor of H2 at high temperatures (Figure 10).
The PSA process is based on adsorption at high pressures and desorption at low pressures. By combining PSA with low-high temperature (TSA-process), further enrichment of CO2 could be obtained by repeating these equilibrium adsorption steps on activated carbon. By modifying the structure of a graphite surface, one may enhance the separation of CO2 out of mixture with H2; both when considering a PSA-TSA process as well as for carbon molecular sieve membranes.
Conclusion
In this work, we have used Equilibrium MD to study the adsorption, selectivity, and self-diffusion of a mixture of CO2 and H2 (overall mole fraction 0.30 of CO2) adsorbed on a slit graphite surface. The results show that there is a preferential adsorption of CO2 to H2 in the adsorbed layer, which depends on the temperature. CO2 adsorbs stronger than H2 at low temperatures, while at high temperatures there is little preferential adsorption of CO2 over H2. The sorption selectivity of CO2 over H2 on the graphite surface is invariant to pressure above 25 atm, but reduces when temperature increases. The self-diffusion of CO2 on graphite surface is the order of magnitude ~10−8 m2/s. This is larger than for CO2 confined in small slit pore by orders of magnitude. The self-diffusion of H2 on graphite is in very good agreement with available experimental data (Haas et al., 2009). CO2 has a higher energy barrier of diffusion than H2. These results of the equilibrium system are useful for process enrichment studies of CO2.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge The Research Council of Norway NFR project no 209337 and The Faculty of Natural Science and Technology, Norwegian University of Science and Technology (NTNU) for financial support. The calculation power is granted by The Norwegian Metacenter for Computational Science (NOTUR).
References
Allen, M. P., and Tildesley, D. J. (1989). Computer Simulation of Liquids. Oxford: Oxford university press.
Bernardo, P., Drioli, E., and Golemme, G. (2009). Membrane gas separation: a review/state of the art. Ind. Eng. Chem. Res. 48, 4638–4663. doi: 10.1021/ie8019032
Boukhvalov, D. W., Katsnelson, M. I., and Lichtenstein, A. I. (2008). Hydrogen on graphene: electronic structure, total energy, structural distortions and magnetism from first-principles calculations. Phys. Rev. B 77:035427. doi: 10.1103/PhysRevB.77.035427
Cao, D. P., and Wu, J. Z. (2005). Modeling the selectivity of activated carbons for efficient separation of hydrogen and carbon dioxide. Carbon 43, 1364–1370. doi: 10.1016/j.carbon.2005.01.004
Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H., and Pedersen, L. G. (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. doi: 10.1063/1.470117
Goedecker, S., Teter, M., and Hutter, J. (1996). Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 54:1703. doi: 10.1103/PhysRevB.54.1703
Grimme, S. (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799. doi: 10.1002/jcc.20495
Guo, B., Chang, L., and Xie, K. (2006). Adsorption of carbon dioxide on activated carbon. J. Nat. Gas Chem. 15, 223–229. doi: 10.1016/S1003-9953(06)60030-3
Haas, O.-E., Simon, J. M., and Kjelstrup, S. (2009). Surface self-diffusion and mean displacement of hydrogen on graphite and a PEM fuel cell catalyst support. J. Phys. Chem. C 113, 20281–20289. doi: 10.1021/jp902491s
Hartwigsen, C., Goedecker, S., and Hutter, J. (1998). Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 58:3641. doi: 10.1103/PhysRevB.58.3641
He, X., Arvid Lie, J., Sheridan, E., and Hägg, M.-B. (2009). CO2 capture by hollow fibre carbon membranes: experiments and process simulations. Energy Proc. 1, 261–268. doi: 10.1016/j.egypro.2009.01.037
He, X., and Hägg, M.-B. (2011). Hollow fiber carbon membranes: investigations for CO2 capture. J. Membr. Sci. 378, 1–9. doi: 10.1016/j.memsci.2010.10.070
He, X., and Hägg, M.-B. (2012). Structural, kinetic and performance characterization of hollow fiber carbon membranes. J. Membr. Sci. 390–391, 23–31. doi: 10.1016/j.memsci.2011.10.052
Himeno, S., Komatsu, T., and Fujita, S. (2005). High-pressure adsorption equilibria of methane and carbon dioxide on several activated carbons. J. Chem. Eng. Data 50, 369–376. doi: 10.1021/je049786x
http://cp2k.berlios.de (2011). Branch 2.2 Edn.
Jin, Y., Lee, D., Lee, S., Moon, W., and Jeon, S. (2011). Gravimetric analysis of CO2 adsorption on activated carbon at various pressures and temperatures using piezoelectric microcantilevers. Anal. Chem. 83, 7194–7197. doi: 10.1021/ac201786n
Kim, H. W., Yoon, H. W., Yoon, S.-M., Yoo, B. M., Ahn, B. K., Cho, Y. H., et al. (2013). Selective gas transport through few-layered graphene and graphene oxide membranes. Science 342, 91–95. doi: 10.1126/science.1236098
Kjelstrup, S., and Bedeaux, D. (2008). Non-Equilibrium Thermodynamics of Heterogeneous Systems. Singapore: World Scientific Singapore.
Levesque, D., and Lamari, F. D. (2009). Pore geometry and isosteric heat: an analysis of carbon dioxide adsorption on activated carbon. Mol. Phys. 107, 591–597. doi: 10.1080/00268970902905802
Li, H., Song, Z., Zhang, X., Huang, Y., Li, S., Mao, Y., et al. (2013). Ultrathin, molecular-sieving graphene oxide membranes for selective hydrogen separation. Science 342, 95–98. doi: 10.1126/science.1236686
Lim, Y.-I., Bhatia, S. K., Nguyen, T. X., and Nicholson, D. (2010). Prediction of carbon dioxide permeability in carbon slit pores. J. Membr. Sci. 355, 186–199. doi: 10.1016/j.memsci.2010.03.030
Lippert, G., Hutter, J., and Parrinello, M. (1997). A hybrid Gaussian and plane wave density functional scheme. Mol. Phys. 92, 477–488. doi: 10.1080/002689797170220
Martyna, G. J., Klein, M. L., and Tuckerman, M. (1992). Nosé–Hoover chains: the canonical ensemble via continuous dynamics. J. Chem. Phys. 97, 2635. doi: 10.1063/1.463940
Mayo, S. L., Olafson, B. D., and Goddard, W. A. (1990). DREIDING: a generic force field for molecular simulations. J. Phys. Chem. 94, 8897–8909. doi: 10.1021/j100389a010
Montoya, A., Mondragón, F., and Truong, T. N. (2003). CO2 adsorption on carbonaceous surfaces: a combined experimental and theoretical study. Carbon 41, 29–39. doi: 10.1016/S0008-6223(02)00249-X
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Potoff, J. J., and Siepmann, J. I. (2001). Vapor–liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 47, 1676–1682. doi: 10.1002/aic.690470719
Rostrup-Nielsen, J. R., and Christiansen, L. J. (2011). Concepts in Syngas Manufacture. Singapore: World Scientific.
Rubes, M., Kysilka, J., Nachtigall, P., and Bludsky, O. (2010). DFT/CC investigation of physical adsorption on a graphite (0001) surface. Phys. Chem. Chem. Phys. 12, 6438–6444. doi: 10.1039/c001155j
Saha, B. B., Jribi, S., Koyama, S., and E-Sharkawy, I. I. (2011). Carbon dioxide adsorption isotherms on activated carbons. J. Chem. Eng. Data 56, 1974–1981. doi: 10.1021/je100973t
Sevilla, M., and Fuertes, A. B. (2012). CO2 adsorption by activated templated carbons. J. Colloid Interface Sci. 366, 147–154. doi: 10.1016/j.jcis.2011.09.038
Simon, J. M., Haas, O. E., and Kjelstrup, S. (2010). Adsorption and desorption of H2 on graphite by molecular dynamics simulations. J. Phys. Chem. C 114, 10212–10220. doi: 10.1021/jp1011022
Smith, W., Yong, C., and Rodger, P. (2002). DL_POLY: application to molecular simulation. Mol. Simul. 28, 385–471. doi: 10.1080/08927020290018769
Vandevondele, J., and Hutter, J. (2007). Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105. doi: 10.1063/1.2770708
Vandevondele, J., Krack, M., Mohamed, F., Parrinello, M., Chassaing, T., and Hutter, J. (2005). QUICKSTEP: fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 167, 103–128. doi: 10.1016/j.cpc.2004.12.014
Vasanth Kumar, K., and Rodríguez-Reinoso, F. (2012). Effect of pore structure on the selectivity of carbon materials for the separation of CO2/H2 mixtures: new insights from molecular simulation. RSC Adv. 2, 9671. doi: 10.1039/c2ra20775c
Keywords: CO2−H2 mixture, adsorption, diffusion, molecular dynamics simulation, graphite
Citation: Trinh TT, Vlugt TJH, Hägg M-B, Bedeaux D and Kjelstrup S (2013) Selectivity and self-diffusion of CO2 and H2 in a mixture on a graphite surface. Front. Chem. 1:38. doi: 10.3389/fchem.2013.00038
Received: 04 September 2013; Accepted: 12 December 2013;
Published online: 24 December 2013.
Edited by:
Doo Soo Chung, Seoul National University, South KoreaReviewed by:
Giancarlo Franzese, Universitat de Barcelona, SpainYun Hee Jang, Gwangju Institute of Science and Technology, South Korea
Copyright © 2013 Trinh, Vlugt, Hägg, Bedeaux and Kjelstrup. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Signe Kjelstrup, Department of Chemistry, Norwegian University of Science and Technology, Realfagbygget, Høgskoleringen 5, NO 7491 Trondheim, Norway e-mail: signe.kjelstrup@ntnu.no