S-nitrosylation of the thioredoxin-like domains of protein disulfide isomerase and its role in neurodegenerative conditions
 Myra E. Conway
Myra E. Conway Matthew Harris
Matthew Harris- Department of Applied Sciences, University of the West of England, Bristol, UK
Correct protein folding and inhibition of protein aggregation is facilitated by a cellular “quality control system” that engages a network of protein interactions including molecular chaperones and the ubiquitin proteasome system. Key chaperones involved in these regulatory mechanisms are the protein disulfide isomerases (PDI) and their homologs, predominantly expressed in the endoplasmic reticulum of most tissues. Redox changes that disrupt ER homeostasis can lead to modification of these enzymes or chaperones with the loss of their proposed neuroprotective role resulting in an increase in protein misfolding. Misfolded protein aggregates have been observed in several disease states and are considered to play a pivotal role in the pathogenesis of neurodegenerative conditions such as Alzheimer's disease, Parkinson's disease, and Amyotrophic Lateral sclerosis. This review will focus on the importance of the thioredoxin-like CGHC active site of PDI and how our understanding of this structural motif will play a key role in unraveling the pathogenic mechanisms that underpin these neurodegenerative conditions.
Introduction
Protein disulfide isomerases (EC 5.3.4.1) are key metabolic proteins which primarily reside in the endoplasmic reticulum (ER) and operate in synergy with other chaperones, such as glucose-regulated protein (78 kDa) (GRP78) that controls the processing and correct folding of proteins (Wilkinson and Gilbert, 2004). Expression of PDI has also been reported outside the ER, in the mitochondria, nucleus, cytosol and cell surface (Yoshimori et al., 1990; Koehler et al., 2006; El Hindy et al., 2014). PDI forms part of the thioredoxin superfamily, which catalyze the formation and breakage of disulfide bonds, through thiol:disulfide exchange, facilitating correct refolding of the protein by rearranging the configuration of disulfide bonds (Figure 1) (Tu et al., 2000). These proteins have also associated roles as chaperones in refolding without enzymatic involvement.
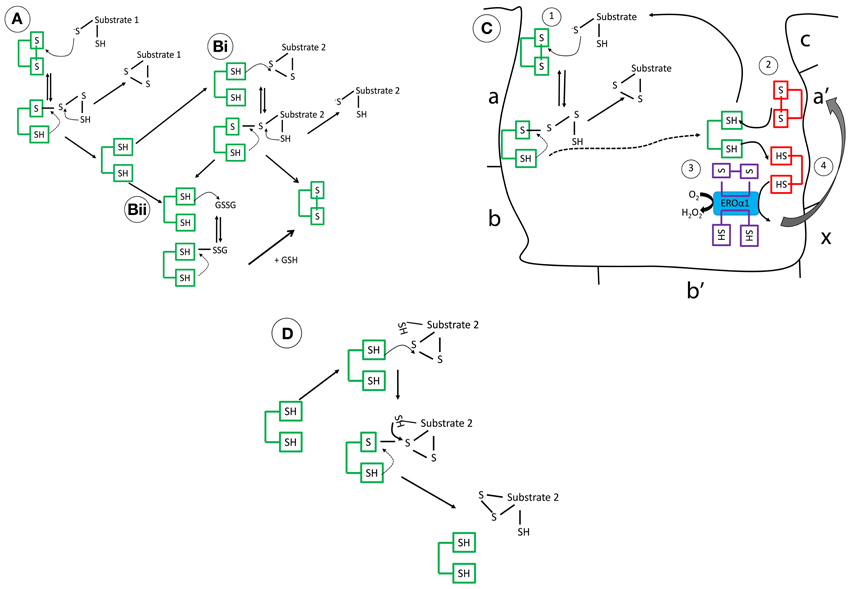
Figure 1. The mechanistic details of PDI as (A) an oxidase (B) a reductase (C) interaction of reduced PDI with ERO1 (D) an isomerase. These mechanisms are complex, governed in part by its intriguing structure but also the pKa of the involved cysteine residues and the redox microenvironment. (A) Operating as an oxidase, forming new disulfide bonds, PDI in its oxidized form, is targeted through the nucleophilic cysteine of the substrate, forming a mixed disulphide. The substrate is then released oxidized after donating its reducing equivalents to PDI. (Bi) As a reductase, the N-terminal cysteine of PDI, which has a low pKa (approximately 4.5), targets its substrate disulfide bond through a nucleophilic attack, forming a mixed disulfide. Now, the substrate thiol is free to interact with another disulfide bond. This results in the oxidation of PDI. (Bii) Alternatively, reduced PDI can be re-oxidized by GSSG or an oxidoreductase, the ER flavo-oxidase Ero1 (its preferred substrate), but peroxiredoxins are also efficient. The kinetic rates of reaction of PDI in vitro are dependent on the substrate and the GSH/GSSG ratio. (C) In brief, reduced PDI, generated after reaction 1(dotted line), is oxidized by the a′ domain (reaction 2), finally, reoxidation of the a′ domain is catalyzed by Ero1 and channels the electrons to final acceptors such as molecular oxygen (reaction 3/4) (D) Rearranging of disulfide bonds is less understood but incorrect disulfide bonding undergoes isomerization creating the correct pattern of disulfide bond formation.
The functional diversity of the PDI family depends on its metabolic substrates, the redox environment, but more importantly their characteristic thioredoxin domain-like structures, which govern the redox thiol-disulfide isomerase and chaperone activity. Depletion of PDI in mammalian cells results in a delay in disulfide bond formation of secretory proteins, highlighting its vital role in oxidative protein folding (Rutkevich et al., 2010). Neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Amyotrophic lateral sclerosis (ALS) share several common pathological characteristics, in particular, the abnormal accumulation of misfolded proteins forming inclusions, where a common role for PDI has been assigned (Kim et al., 2000; Muchowski, 2002; Tuite and Melki, 2007). This review will focus on the function of PDI in protein folding and the impact of increased cellular stress on its proposed neuroprotective role.
Structure of Human PDI Dictates its Functional Diversity
The PDI family has 21 members, all of which have thioredoxin-like fold structures, with most including 1–3 redox-active -CXXC- motifs (Ellgaard and Ruddock, 2005; Appenzeller-Herzog and Ellgard, 2008; Hatahet and Ruddock, 2009). The individual domains of human PDI [Figure 1C (a, a′, b, and b′)] have been resolved using NMR (PDB codes; 1Mek, 1X5C, 3BJ5, and 2K18) (Kemmink et al., 1996, 1999; Denisov et al., 2007; Nguyen et al., 2008). However, it was not until the recent crystallization of the three-dimensional structure of yeast PDI [4°C (PDB:2B5E) and 22°C (PDB:3BOA)] and human PDI (hPDI) [oxidized (PDB:4EL1) and reduced (PDB:4EKZ)] that insight into how these domains operate cooperatively was detailed (Tian et al., 2006; Wang et al., 2013). hPDI has four thioredoxin-like domains (a, a′, b, and b′), where two of these (a and a′) have a catalytic redox-active –CGHC- motif (Wang et al., 2013). In yeast (the structure resolved at 4°C) and hPDI, the a/a′ domains face each other in a “U-shaped” structure and are solvent exposed allowing interaction with their respective substrates (Tian et al., 2006; Wang et al., 2013). The b domains confer substrate specificity and form a hydrophobic rigid base connecting the two flexible a domains. The a′ and b′ domain are connected through a 19 amino acid inter-domain link, called the x-linker, which is predicted to promote flexibility, influencing substrate binding (Wang et al., 2010). Another key feature is the highly-acidic C-terminus, which is involved in calcium binding and contains the ER-retrieval motif KDEL. In hPDI, the folds of the domains show conformational differences in the reduced and oxidized form, where the distance between the two active site -CXXC- regions increases from 27.6 to 40.3 Å, offering a more open structure exposing the hydrophobic areas of the b domains when oxidized, supporting a role for redox in chaperone PDI activity (Wang et al., 2013). Reported by several groups, homo-dimerization of the bb′ domain, regulated by the x-linker region, is suggested to block substrate access, regulating PDI activity (Byrne et al., 2009; Wallis et al., 2009; Bastos-Aristizabal et al., 2014). Molecular dynamic simulation of three-dimensional structures suggested that in solution hPDI may adopt more rigid structures facilitated by inter-domain salt-bridges, encouraging inter-domain disulfide bond formation (Yang et al., 2014). These exciting advances in structural analysis have expanded our knowledge of this highly complex protein and have accelerated our move toward understanding the functional diversity of PDI (The mechanistic details of PDI are summarized in Figure 1).
Molecular Chaperones, PDI, and Cellular Stress
The cell responds to an increase in immature or misfolded proteins through a concerted protein network response, involving the upregulation of several molecular chaperones that prevent protein aggregation, termed the unfolded protein response (UPR), followed by ER-associated degradation (ERAD), which specifically recognizes terminally misfolded proteins. Molecular chaperones such as the heat shock proteins (e.g., HSP70 and HSP90) are ubiquitously expressed in all cells and in all subcellular compartments. They represent a diverse group of proteins and are characterized based on their dependence on ATP, holdases, are ATP dependent and foldases, are independent of ATP, the former considered the first line of defense (reviewed by Niforou et al., 2014). Bip/GPR78 and PDI, among other chaperones, function in the ER stress response as neuro-protectors by alleviating the level of misfolded proteins. However, by mechanisms which involve increased cellular stress, these “quality control systems” fail leading to the aggregation and deposition of mis-folded proteins, which not only result in loss of protein function but also lead to cell toxicity and ultimately cell death, a pathway linked to many neurodegenerative diseases.
Excess reactive oxygen and nitrogen species (ROS/RNS) generated through oxidative metabolism, or triggered from environmental toxins and also normal aging can cause an imbalance between the production of ROS/RNS and the cell's defense systems, including anti-oxidant enzymes (such as the glutaredoxin system), glutathione, and molecular chaperones (Nakamura and Lipton, 2009). Neurons are particularly vulnerable due to their comparatively low levels of certain antioxidants and reliance on energy from ROS/RNS-generating mitochondrial metabolism (Mattson et al., 2002). Pertinent to this review is the modification of PDI through S-nitrosylation, the addition of a nitric oxide (NO) moiety to a reactive thiol (-SH) group (SNO-PDI). Under normal physiological conditions, S-nitrosylation modulates the function of target proteins, playing a dynamic role in numerous biological processes. In contrast, under pathological conditions, stress induced S-nitrosylation of certain proteins, including PDI, can alter protein function triggering a cascade of events resulting in neuronal cell death that ultimately results in neurodegeneration (Nakamura and Lipton, 2007; Nakamura et al., 2013). Increased PDI expression has been reported in neurodegenerative diseases, to ease ER stress from the accumulation of misfolded proteins, however, the neuroprotective effect of its disulfide-isomerase and chaperone activity can be inhibited by S-nitrosylation (Uehara et al., 2006; Andreu et al., 2012), discussed below with respect to AD, PD, and ALS (summarized in Figure 2).
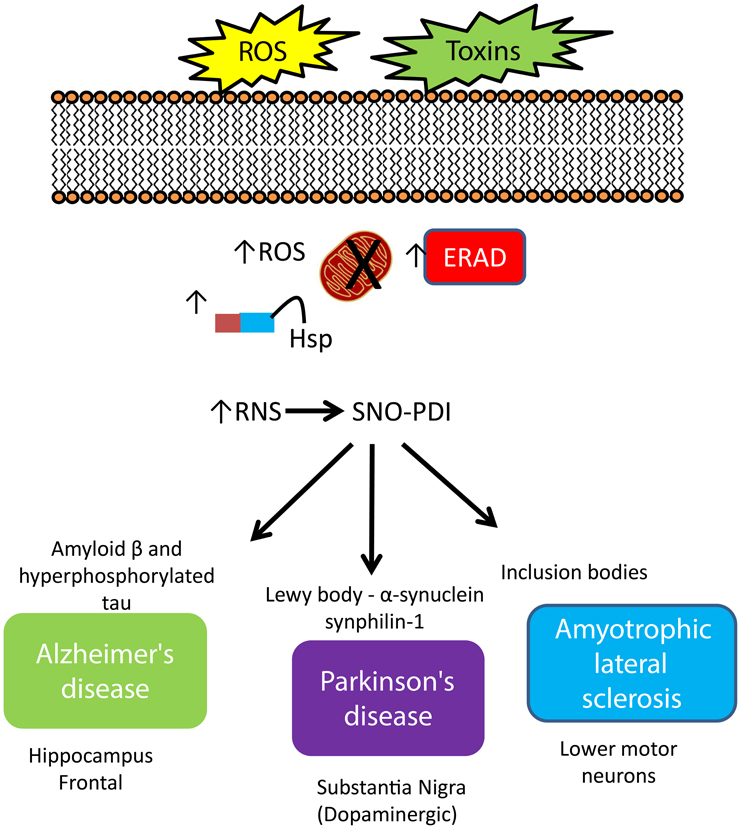
Figure 2. Increased expression of PDI and regulation of protein misfolding in neurodegenerative disease. An increase in cellular stress originating from both environmental and intracellular sources initiates a cascade of defenses to protect the cell against damage, including increased expression of molecular chaperones, initiation of the ER-stress response, where the role of PDI strongly features. The response of PDI generates a signature pattern across all neurodegenerative conditions indicating a key role for this protein as a neuroprotector against cellular stress. However, S-nitrosylation impacts activity and compromises this protective role leading to an increase in misfold proteins, creating aggregates, contributing to cell death.
SNO-PDI Colocalizing with Amyloid β and Tau: Implications in Alzheimer's Disease
The key morphology of AD includes cerebral atrophy, β-amyloid (Aβ) deposits, and neuritic changes such as neurofibrillary tangles (NFT) (reviewed in Hardy, 2003), which manifests clinically as a progressive loss in cognition, mood changes, and memory loss (Alzheimer, 1907). These NFTs contain hyperphosphorylated, microtubule-associated tau and are linked to the clinical progressive stages of AD (Braak and Braak, 1991; Morishima-Kawashima and Ihara, 2002). Several metabolic mechanisms also contribute to disease progression, including glutamate toxicity, calcium overload, an increase in cellular stress linked with mitochondrial and ER dysfunction, protein misfolding and a dysregulation of autophagy (Sattler and Tymianski, 2000; Calabrese et al., 2006; Nakamura and Lipton, 2007; Nixon, 2007; Nakamura and Lipton, 2008).
The accumulation of misfolded proteins and influx of cellular Ca2+, associated with AD, can cause ER stress in neurons and are considered to contribute to AD (Schroder, 2008). A role for PDI in contributing to AD pathology has been supported through evidence of its co-localization with NFT and the dystrophic neurites of senile plaques in AD patient brain tissues, particularly in the hippocampus and frontal lobe, areas most affected by AD (Honjo et al., 2010, 2012). Co-localization of PDI with tau to the ER was also reported in the SH-SY5Y cell line, a model of AD pathology (Xu et al., 2013). Interestingly, a small number of NFTs immuno-positive for PDI was found in normal control brains, but these were few and scattered (Honjo et al., 2010). However, it may indicate that in normal aging brains ER stress can result in misfolded proteins, where a role for PDI is likely.
In AD, an increase in cellular ROS/RNS is associated with alterations in Aβ metabolism (Dalle-Donne et al., 2006; Mangialasche et al., 2009). These changes in cellular stress also targets susceptible proteins and damages mitochondria (related to Aβ production). ROS-damaged mitochondria results in the oxidation of mitochondrial DNA (genes of oxidative phosphorylation are reduced), lipids [increased 4-hydroxy-2-nonenal (HNE)] (Hardas et al., 2013) and proteins (nitration of enolase) leading to mitochondrial dysfunction and impaired cellular respiration (Castegna et al., 2002). Moreover, mitochondria-derived ROS can also enhance Aβ formation, contributing to cell death (Leuner et al., 2012). Together with the presentation of specific mutations in the presenilin-1 gene, overt mitochondrial damage, where a role for cellular ROS predominates, is considered to factor strongly in the pathogenesis of AD (reviewed in García-Escudero et al., 2013).
Important to this review is the impact of increased cellular ROS on PDI activity and its role in AD. In AD brain an increased expression of PDI was reported, however, pathophysiological relevant amounts of S-nitrosylated PDI was also noted (Uehara et al., 2006; Hoffstrom et al., 2010; Andreu et al., 2012). In these studies, S-nitrosylation of PDI, through the redox active thiols of the a and a′ domain (Figure 1C), inhibited enzyme activity and resulted in the accumulation of poly-ubiquitinated proteins, an increase in ER stress and induction of apoptosis (Uehara et al., 2006; Nakamura and Lipton, 2009). ER stress can initiate a cascade of events including activation of the UPR, which aims to slow down protein synthesis and increase protein folding capacity (reviewed in Salminen et al., 2009). For reasons not entirely understood this system becomes overwhelmed resulting in the increase in protein aggregation, a contributing factor to several neurodegenerative conditions. S-glutathionylation of PDI, induced through RNS, may also factor in this mechanism (Townsend et al., 2009). The accrual of unfolded proteins in the ER lumen is in fact sufficient to produce ROS/RNS, suggesting a vicious cycle of ER stress and local oxidative stress finally leading to cell death when unresolved (Malhotra and Kaufman, 2007). The role of PDI in preventing aggregates, such as Aβ, extends also to NFT, where PDI inhibited nucleation and elongation of Tau244−372 fibrillization in SH-SY5Y cells, again illustrating its role as a neuroprotector (Xu et al., 2013).
A homolog of PDI, protein disulfide isomerase P5 (P5), was found to also co-localize with tau in NFTs (Honjo et al., 2014). Knock-down of PDI or P5 in SH-SY5Y cells resulted in decreased cell viability under ER stress (Honjo et al., 2014). Like PDI, the P5 protein was also shown to be S-nitrosylated in AD patient brains, however, unlike that reported for PDI, expression levels of P5 were significantly decreased (Honjo et al., 2014). ERp57, another PDI family protein, has been reported in the CSF of AD patients physically associated with amyloid-β (Erickson et al., 2005), suggesting a potential role as a carrier protein that prevents aggregation of Aβ. Together, these studies clearly indicate a direct association between SNO-PDI, and protein misfolding, understanding of which will identify potential new therapeutic targets for AD.
α-Synuclein Aggregation Regulated by SNO-PDI in Parkinson's Disease
Build-up of protein aggregates strongly features in PD pathology. Here, cytoplasmic Lewy bodies, which stain for aggregated α-synuclein in the substantia nigra, results in the clinical presentation of bradykinesia, rest tremor, rigidity, gait, and postural abnormalities (Spillantini et al., 1997; Baba et al., 1998; Kawamata et al., 2001; Neystat et al., 2002). This is also a heterogeneous disease where protein aggregation and increased cellular stress, among others, contribute to disease pathology (reviewed in Nakamura and Lipton, 2011). Pharmacological models of PD in vitro have illustrated that enhanced production of ROS is the main cause of ER stress, where a significant up-regulation of PDI and ERp57 are linked to a global ER stress response (Ryu et al., 2002; Holtz and O'Malley, 2003; Holtz et al., 2006). These findings have also been confirmed in α-synuclein transgenic mouse models, as well as in brain tissue derived from PD patients (Conn et al., 2004; Colla et al., 2012). Although clinically distinct from AD, S-nitrosylated PDI was also reported in PD brain tissue, where S-nitrosylation of PDI inhibited its enzymatic activity, resulting in the accumulation of polyubiquinated proteins, indicating similarities in mechanisms underlying different neurodegenerative conditions (Uehara et al., 2006).
In vitro studies have shown that PDI colocalized and interacted with α-synuclein in dopaminergic MES cells, and under normal physiological conditions, forms a complex with α-synuclein, in the process, preventing abnormal aggregation (Honjo et al., 2011a; Xu et al., 2014). However, when PDI was S-nitrosylated, this interaction was disturbed, resulting in α-synuclein oligomerization, indicating a key role for the a and a′ domains in this mechanism (Wu et al., 2014). The authors concluded that PDI can relieve neurodegeneration by decreasing α-synuclein aggregation, but when S-nitrosylated the consequential dysfunction of PDI results in α-synuclein aggregation. Chemical modification of PDI promotes the aggregation and build-up of the minor Parkinsonian-specific biomarker synphilin-1 in a NO dependent manner (Uehara et al., 2006; Uehara, 2007). However, over-expression of wild-type PDI (non-SNO-PDI) reduced synphilin-1-containing aggregates in SHSY-5Y cells (Uehara et al., 2006). A recent paper examining the effect of SNO-PDI formation on α-synuclein aggregation and Lewy-like neurite formation, found that in addition to SNO-PDI formation activating α-synuclein aggregation, it was also seen to provoke co-localization and in parallel the formation of α-synuclein:synphilin-1-containing Lewy-body-like aggregates, supporting the role of SNO-PDI in PD pathogenesis (Kabiraj et al., 2014). Although the functional contribution of PDIs to the pathogenesis of PD in vivo requires further validation, collectively these studies illustrate that up-regulation of PDI represents a protective response to abnormal protein aggregation, contributing to the re-establishment of protein homeostasis, whereas S-nitrosylation clearly impacts protein aggregation and deposition.
SNO-PDI, SOD1, NOX, and Amyotrophic Lateral Sclerosis
ALS is characterized by the accumulation of cytoplasmic aggregated ubiquitinated proteins in motor neurons and surrounding oligodendrocytes, which clinically presents as progressive muscle weakness, typically with a 3–5 year survival (reviewed in Blokuis et al., 2013). Mutations in 16 genes have been identified to be associated with classical ALS, including mutations in superoxide dismutase (SOD1), TARDBP, fused in sarcoma (FUS), UBQLN2, profiling, and C9ORF72, but these are rare (Ferraiuolo et al., 2011; Rademakers and Blitterswijk, 2012; Chen et al., 2013). Approximately 90% of all ALS cases are sporadic, with the second most frequent, familial ALS, attributed to dominantly inherited mutations in Cu/Zn SOD1 (Rosen, 1993). The major inclusions associated with both familial and sporadic ALS are the TDP-43-positive ubiquitinated protein aggregates that accumulate in the cytoplasm, where PDI was also shown to reside (Honjo et al., 2011b). Another inclusion associated with ALS, FUS is normally located in the nucleus but in ALS is redistributed to the cytoplasm (Dormann et al., 2010). In NSC-34 cells, mutant FUS expression triggers ER stress and co-localizes with PDI (Farg et al., 2012). Moreover, PDI was shown to co-localize with FUS-positive ubiquitinated inclusions, in human ALS patient spinal cords, supporting the in vitro cell models.
A significant increase in PDI expression, alongside other ER stress markers such as ERp57, was reported in ALS spinal cord and CSF compared to controls and also in the G93A SOD1 mouse model of ALS (Atkin et al., 2006, 2008; Massignan et al., 2007; Honjo et al., 2011a). This increase in PDI expression is not just limited to neurons, but has also been observed in the microglia and astrocytes of murine models (Jaronen et al., 2013). However, in ALS, PDI is also S-nitrosylated impacting its neuroprotective role, again sharing common mechanistic traits to AD and PD (Walker et al., 2010; Jeon et al., 2014). In ALS mouse and cell models, overexpression of PDI decreases both the accumulation of SOD1 aggregates and neuronal cell death, whereas inhibition of PDI using bacitracin or siRNA knockdown increases the formation of aggregates (Atkin et al., 2006, 2008; Walker et al., 2010; Jeon et al., 2014). Furthermore, when production of NO generation via NOS was blocked using N-nitro-L-arginine, the formation of SNO-PDI and mutant SOD1 aggregates significantly decreased (Chen et al., 2013), but interestingly studies using the PDI redox-mutant suggest that it is the chaperone activity of PDI that was responsible (Jeon et al., 2014). The therapeutic effects of Reticulon-4A, found to have a protective role against disease onset and progression in an ALS mouse model, were associated with intracellular redistribution of PDI, supporting the role of PDI as a therapeutic target (Yang et al., 2009).
In vivo ALS studies have indicated that NADPH oxidase (NOX) activation and superoxide production are elevated in microglia and may contribute to motorneuron death, through increased cellular S-nitrosylation (Wu et al., 2006). In vitro studies showed that PDI acts as an important regulator of NOX activation and subsequent ROS production in microglial cells, suggesting that an up-regulation of PDI may not just have a protective role in protein aggregation but also in regulating UPR-triggered NOX activation, in a cell-type specific manner (Jaronen et al., 2013). Proteomic screening identified PDI and ERp57 as biomarkers for ALS, with ERp57 highlighted for its potential to monitor disease progression (Nardo et al., 2011). Due to the key role PDI appears to play in ALS, a genome association study examined the effects of two specific single-nucleotide polymorphisms (SNPs) in the P4HB gene, which codes for PDI, which showed a significant association with familial ALS, suggesting that P4HB is a modifier gene in ALS susceptibility and may present a potential therapeutic target (Kwok et al., 2013). The same study also identified two genotypes and one diplotype that modify disease survival. These findings are consistent with the view that PDI expression is clearly linked to ALS pathology and either de-nitrosylation of PDI or up-regulation of total PDI may provide a therapeutic benefit in ALS.
Conclusion
An increase in the generation of ROS/RNS through external and internal cellular mechanisms can overwhelm the anti-oxidant and chaperone systems contributing to an increase in cellular calcium, mitochondrial dysfunction, protein misfolding/aggregation leading to cell death. This pathogenic pathway shares common features across all neurodegenerative conditions despite clear genetic links that differentiate between them. A role for PDI in protein misfolding has emerged as a key link to understanding how these aggregates accumulate in the brain with similar patterns of colocalization of PDI with disease specific aggregates. Despite clear evidence that protein aggregation in central to the pathology of neurodegenerative conditions, many questions remain unanswered. How do these models of protein misfolding translate in vivo? Is it possible to generate a therapeutic translation to mimic increased PDI expression that is not subject to S-nitrosylation? Or will increased expression of PDI stimulate unfavorable metabolic reactions, attenuating its potential role as a target therapeutic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is funded through a Research Fellowship from BRACE, Bristol, UK
References
Alzheimer, A. (1907). Über eine eigenartige Erkrankung der Hirninde. Z. Psychiatr. Psychis. Gerichtl. Med. 64, 146–148.
Andreu, C., Woehlbier, U., Torres, M., and Hetz, C. (2012). Protein disulphide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett. 586, 2826–2834. doi: 10.1016/j.febslet.2012.07.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Appenzeller-Herzog, C., and Ellgard, L. (2008). The human PDI family: versatility packed into a single fold. Biochim. Biophys. Acta 86, 535–548. doi: 10.1016/j.bbamcr.2007.11.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Atkin, J., Farg, M., Turner, B., Tomas, D., Lysaght, J., Nunan, J., et al. (2006). Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 281, 30152–30165. doi: 10.1074/jbc.M603393200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Atkin, J., Farg, M., Walker, A., McLean, C., Tomas, D., and Horne, M. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baba, M., Nakajo, S., Tu, P., Tomita, T., Nakaya, K., Lee, V., et al. (1998). Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884.
Bastos-Aristizabal, S., Kozlov, G., and Gehring, K. (2014). Structural insight into the dimerization of human protein disulfide isomerase. Protein Sci. 23, 618–626. doi: 10.1002/pro.2444
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Blokuis, A., Groen, E., Koppers, M., van den Berg, L., and Pasterkamp, R. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794. doi: 10.1007/s00401-013-1125-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Byrne, L. J., Sidhu, A., Wallis, A. K., Ruddock, L. W., Freedman, R. B., Howard, M. J., et al. (2009). Mapping of the ligand-binding site on the b′ domain of human PDI: interaction with peptide ligands and the x-linker region. Biochem. J. 423, 209–217. doi: 10.1042/BJ20090565
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Calabrese, V., Sultana, R., Scapagnini, G., Guagliano, E., Sapienza, M., Bella, R., et al. (2006). Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer's disease. Antioxid. Redox Signal. 8, 1975–1986. doi: 10.1089/ars.2006.8.1975
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Castegna, A., Aksenov, M., Thongboonkerd, V., Klein, J. B., Pierce, W. M., Booze, R., et al. (2002). Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem. 82, 1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, X., Zhang, X., Li, C., Guan, T., Shang, H., Cui, L., et al. (2013). S-nitrosylated protein disulphide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J. Neurochem. 124, 45–58. doi: 10.1111/jnc.12046
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colla, E., Coune, P., Liu, Y., Pletnikova, O., and Troncoso, J. (2012). Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Conn, K., Gao, W., McKee, A., Lan, M., Ullman, M., Eisenhauer, P., et al. (2004). Identification of the protein disulphide isomerase family member PDIp in experimental Parkinson's disease and Lewy body pathology. Brain Res. 1022, 164–172. doi: 10.1016/j.brainres.2004.07.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dalle-Donne, I., Rossi, R., Colombo, R., Giustarini, D., and Milzani, A. (2006). Biomarkers of oxidative damage in human disease. Clin. Chem. 52, 601–623. doi: 10.1373/clinchem.2005.061408
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Denisov, A. Y., Maattanen, P., Sprules, T., Thomas, D. Y., and Gehring, K. (2007). 1H, 13C and 15N resonance assignments of the bb′ domains of human protein disulfide isomerase. Biomol. NMR Assign. 1, 129–130. doi: 10.1007/s12104-007-9035-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dormann, D., Rodde, R., Edbauer, D., Bentmann, E., Fischer, I., Hruscha, A., et al. (2010). ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 29, 2841–2857. doi: 10.1038/emboj.2010.143
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
El Hindy, M., Hezwani, M., Corry, D., Hull, J., El Amraoui, F., Harris, M., et al. (2014). The branched-chain aminotransferase proteins: novel redox chaperones for protein disulphide isomerase – Implications in Alzheimer's Disease. Antioxid. Redox Signal. 20, 2497–2513. doi: 10.1089/ars.2012.4869
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ellgaard, L., and Ruddock, L. W. (2005). The human protein disulphide isomerase family:substrate interactions and functional properties. EMBO Rep. 6, 28–32 doi: 10.1038/sj.embor.7400311
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Erickson, R., Dunning, L., Olson, D., Cohen, S., Davis, A., Wood, W., et al. (2005). In cerebrospinal fluid ER chaperones ERp57 and calreticulin bind beta-amyloid. Biochem. Biophys. Res. Commun. 332, 50–57. doi: 10.1016/j.bbrc.2005.04.090
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Farg, M., Soo, K., Walker, A., Pham, H., Orian, J., Horne, M., et al. (2012). Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulphide-isomerase. Neurobiol. Aging 33, 2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ferraiuolo, L., Higginbottom, A., Heath, P., Barber, S., Greenald, D., Kirby, J., et al. (2011). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134, 2627–2641. doi: 10.1093/brain/awr193
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
García-Escudero, V., Martín-Maestro, P., Perry, G., and Avila, J. (2013). Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell. Longev. 2013:162152. doi: 10.1155/2013/162152
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hardas, S. S., Sultana, R., Clark, A. M., Beckett, T. L., Szweda, L. I., Murphy, M. P., and Butterfield, D. A. (2013). Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 30, 80–85. doi: 10.1016/j.redox.2013.01.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hardy, J. (2003). The relationship between amyloid and tau. J. Mol. Neurosci. 20, 203–206. doi: 10.1385/JMN:20:2:203
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hatahet, F., and Ruddock, L. W. (2009). Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850. doi: 10.1089/ars.2009.2466
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hoffstrom, B., Kaplan, A., Letso, R., Schmid, R., Turmel, G., Lo, D., et al. (2010). Inhibitors of protein disulphide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 6, 900–906. doi: 10.1038/nchembio.467
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holtz, W., and O'Malley, K. (2003). Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J. Biol. Chem. 278, 19367–19377. doi: 10.1074/jbc.M211821200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holtz, W., Turetzky, J., Jong, Y., and O'Malley, K. (2006). Oxidative stress-triggered unfolded protein response is upstream of intrinsic cell death evoked by parkinsonian mimetics. J. Neurochem. 99, 54–69. doi: 10.1111/j.1471-4159.2006.04025.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Honjo, Y., Horibe, T., Torisawa, A., Ito, H., Nakanishi, A., Mori, H., et al. (2014). Protein disulphide isomerase P5-immunopositive inclusions in patients with Alzheimer's disease. J. Alzheimers Dis. 38, 601–609. doi: 10.3233/JAD-130632
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Honjo, Y., Ito, H., Horibe, T., Shimada, H., Nakanishi, A., Mori, H., et al. (2012). Derlin-1-immunopositive inclusions in patients with Alzheimer's disease. Neuroreport 23, 611–615. doi: 10.1097/WNR.0b013e3283552a75
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Honjo, Y., Ito, H., Horibe, T., Takahashi, R., and Kawakami, K. (2010). Protein disulphide isomerase-immunopositive inclusions in patients with Alzheimer disease. Brain Res. 1349, 90–96. doi: 10.1016/j.brainres.2010.06.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Honjo, Y., Ito, H., Horibe, T., Takahashi, R., and Kawakami, K. (2011b). Protein disulphide isomerase immunopositive glial cytoplasmic inclusions in patients with multiple system atrophy. Int. J. Neurosci. 121, 543–550. doi: 10.3109/00207454.2011.585440
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Honjo, Y., Kaneko, S., Ito, H., Horibe, T., Nagashima, M., Nakamura, M., et al. (2011a). Protein disulphide isomerase-immunopositive inclusions in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 12, 444–450. doi: 10.3109/17482968.2011.594055
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jaronen, M., Vehvilainen, P., Malm, T., Keksa-Goldsteine, V., Pollari, E., Valonen, P., et al. (2013). Protein disulphide isomerase in ALS mouse glia links protein misfolding with NADPH oxidase-catalyzed superoxide production. Hum. Mol. Genet. 22, 646–655. doi: 10.1093/hmg/dds472
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jeon, G., Nakamura, T., Lee, J., Choi, W., Ahn, S., Lee, K., et al. (2014). Potential effect of S-nitrosylated protein disulphide isomerase on mutant SOD1 aggregation and neuronal cell death in amyotrophic lateral sclerosis. Mol. Neurobiol. 49, 796–807. doi: 10.1007/s12035-013-8562-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kabiraj, P., Marin, J., Varela-Ramirez, A., Zubia, E., and Narayan, M. (2014). Ellagic acid mitigates SNO-PDI induced aggregation of Parkinsonian biomarkers. ACS Chem. Neurosci. 5, 1209–1220. doi: 10.1021/cn500214k
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kawamata, H., McLean, P., Sharma, N., and Hyman, B. (2001). Interaction of alpha-synuclein and synphilin-1: effect of Parkinson's disease-associated mutations. J. Neurochem. 77, 929–934. doi: 10.1046/j.1471-4159.2001.00301.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kemmink, J., Darby, N. J., Dijkstra, K., Nilges, M., and Creighton, T. E. (1996). Structure determination of the N-terminal thioredoxin-like domain of protein disulphide isomerase using multidimensional heteronuclear 13C/15N NMR spectroscopy. Biochemistry 35, 7684–7691. doi: 10.1021/bi960335m
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kemmink, J., Dijkstra, K., Mariani, M., Scheek, R. M., Penka, E., Nilges, M., et al. (1999). The structure in solution of the b domain of protein disulfide isomerase. J. Biomol. NMR 13, 357–368. doi: 10.1023/A:1008341820489
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, H., Russell, R., Raina, A., Harris, P., Siedlak, S., Zhu, X., et al. (2000). Protein disulphide isomerase in Alzheimer disease. Antioxid. Redox Signal. 2, 485–489. doi: 10.1089/15230860050192260
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koehler, C. M., Beverly, K. N., and Leverich, E. P. (2006). Redox pathways of the mitochondrion. Antioxid. Redox Signal. 8, 813–822. doi: 10.1089/ars.2006.8.813
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kwok, C., Morris, A., Frampton, J., Smith, B., Shaw, C., and de Belleroche, J. (2013). Association studies indicate that protein disulphide isomerase is a risk factor in amyotrophic lateral sclerosis. Free Radic. Biol. Med. 58, 81–86. doi: 10.1016/j.freeradbiomed.2013.01.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leuner, K., Schütt, T., Kurz, C., Eckert, S. H., Schiller, C., Occhipinti, A., et al. (2012). Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal. 16, 1421–1433. doi: 10.1089/ars.2011.4173
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Malhotra, J., and Kaufman, R. (2007). The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18, 716–731. doi: 10.1016/j.semcdb.2007.09.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mangialasche, F., Polidori, M., Monastero, R., Ercolani, S., Camarda, C., Cecchetti, R., et al. (2009). Biomarkers of oxidative and nitrosative damage in Alzheimer's disease and mild cognitive impairment. Ageing Res. Rev. 8, 285–305. doi: 10.1016/j.arr.2009.04.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Massignan, T., Casoni, F., Basso, M., Stefanazzi, P., Biasini, E., Tortarolo, M., et al. (2007). Proteomic analysis of spinal cord of presymptomatic amyotrophic lateral sclerosis G93A SOD1 mouse. Biochem. Biophys. Res. Commun. 353, 719–725. doi: 10.1016/j.bbrc.2006.12.075
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mattson, M., Chan, S., and Duan, W. (2002). Modification of brain aging and neurodegenerative disorders by genes, diet, and behaviour. Physiol. Rev. 82, 637–672.
Morishima-Kawashima, M., and Ihara, Y. (2002). Alzheimer's disease: β-amyloid protein and tau. J. Neurosci. 70, 392–401.
Muchowski, P. (2002). Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron 35, 9–12. doi: 10.1016/S0896-6273(02)00761-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakamura, T., and Lipton, S. (2007). Molecular mechanisms of nitrosative stress-mediated protein misfolding in neurodegenerative diseases. Cell Mol. Life Sci. 64, 1609–1620. doi: 10.1007/s00018-007-6525-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakamura, T., and Lipton, S. (2008). Emerging roles of S-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox Signal. 10, 87–101. doi: 10.1089/ars.2007.1858
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakamura, T., and Lipton, S. (2009). Cell death:protein misfolding and neurodegenerative diseases. Apoptosis 14, 455–468. doi: 10.1007/s10495-008-0301-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakamura, T., and Lipton, S. (2011). Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 18, 1478–1486. doi: 10.1038/cdd.2011.65
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakamura, T., Tu, S., Akhtar, M. W., Sunico, C., Okamoto, S., and Lipton, S. (2013). Aberrant protein S-nitrosylation in neurodegenerative disease. Neuron 78, 596–614. doi: 10.1016/j.neuron.2013.05.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nardo, G., Pozzi, S., Pignataro, M., Lauranzano, E., Spano, G., Garbelli, S., et al. (2011). Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 6:e25545. doi: 10.1371/journal.pone.0025545
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neystat, M., Rzhetskaya, M., Kholodilov, N., and Burke, R. (2002). Analysis of synphilin-1 and synuclein interactions by yeast two-hybrid beta-galactosidase liquid assay. Neurosci. Lett. 325, 119–123. doi: 10.1016/S0304-3940(02)00253-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nguyen, V. D., Wallis, K., Howard, M. J., Haapalainen, A. M., Salo, K. E., Saaranen, M. J., et al. (2008). Alternative conformations of the x region of human protein disulphide-isomerase modulate exposure of the substrate binding b′ domain. J. Mol. Biol. 383, 1144–1155. doi: 10.1016/j.jmb.2008.08.085
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Niforou, K., Chemondou, C., and Trougakos, I. (2014). Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2, 323–332. doi: 10.1016/j.redox.2014.01.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nixon, R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120, 4081–4091. doi: 10.1242/jcs.019265
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rademakers, R., and Blitterswijk, M. (2012). Motor neuron disease in 2012: novel causal genes and disease modifiers. Nat. Rev. Neurol. 9, 63–64. doi: 10.1038/nrneurol.2012.276
Rosen, D. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364, 362. doi: 10.1038/364362c0
Rutkevich, L., Cohen-Doyle, M., Brockmeier, U., and Williams, D. (2010). Functional relationship between protein disulphide isomerase family members during the oxidative folding of human secretory proteins. Mol. Biol. Cell. 21, 3093–3105. doi: 10.1091/mbc.E10-04-0356
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ryu, E., Harding, H., Angelastro, J., Vitolo, O., Ron, D., and Greene, L. (2002). Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J. Neurosci. 22, 10690–10698.
Salminen, A., Kauppinen, A., Suuronen, T., Kaarniranta, K., and Ojala, J. (2009). ER stress in Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's pathology. J. Neuroinflammation 26:41. doi: 10.1186/1742-2094-6-41
Sattler, R., and Tymianski, M. (2000). Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 78, 3–13. doi: 10.1007/s001090000077
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schroder, M. (2008). Endoplasmic reticulum stress responses. Cell. Mol. Life Sci. 65, 862–894. doi: 10.1007/s00018-007-7383-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Spillantini, M., Schmidt, M., Lee, V., Trojanowski, J., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tian, G., Xiang, S., Noiva, R., Lennarz, W., and Schindelin, H. (2006). The crystal structure of yeast protein disulphide isomerase suggests cooperatively between its active sites. Cell 124, 61–73. doi: 10.1016/j.cell.2005.10.044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Townsend, D. M., Manevich, Y., He, L., Xiong, Y., Bowers, R. R. Jr., Hutchens, S., et al. (2009). Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 69, 7626–7634. doi: 10.1158/0008-5472.CAN-09-0493
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tu, B. P., Ho-Schleyer, S. C., Travers, K. J., and Weissman, J. S. (2000). Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 290, 1571–1574. doi: 10.1126/science.290.5496.1571
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tuite, M. F., and Melki, R. (2007). Protein misfolding and aggregation in ageing and disease: molecular processes and therapeutic perspectives. Prion 1, 116–120. doi: 10.4161/pri.1.2.4651
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Uehara, T. (2007). Accumulation of misfolded protein through nitrosative stress linked to neurodegenerative disorders. Antioxid. Redox Signal. 9, 597–601. doi: 10.1089/ars.2006.1517
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Uehara, T., Nakamura, T., Yao, D., Shi, Z., Gu, Z., Ma, Y., et al. (2006). S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517. doi: 10.1038/nature04782
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walker, A., Farg, M., Bye, C., McLean, C., Horne, M., and Atkin, J. (2010). Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133, 105–116. doi: 10.1093/brain/awp267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wallis, A. K., Sidhu, A., Byrne, L. J., Howard, M. J., Ruddock, L. W., Williamson, R. A., et al. (2009). The ligand-binding b′ domain of human protein disulphide-isomerase mediates homodimerization. Protein Sci. 18, 2569–2577. doi: 10.1002/pro.270
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, C., Chen, S., Wang, X., Wang, L., Wallis, A. K., Freedman, R. B., et al. (2010). Plasticity of human protein disulfide isomerase: evidence for mobility around the X-linker region and its functional significance. J. Biol. Chem. 285, 26788–26797. doi: 10.1074/jbc.M110.107839
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, C., Li, W., Ren, J., Fang, J., Ke, H., Gong, W., et al. (2013). Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 19, 36–45. doi: 10.1089/ars.2012.4630
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wilkinson, B., and Gilbert, H. F. (2004). Protein disulfide isomerase. Biochim. Biophys. Acta 1699, 35–44. doi: 10.1016/j.bbapap.2004.02.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, D., Re, D., Nagai, M., Ischiropoulos, H., and Przedborski, S. (2006). The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. U.S.A. 103, 12132–12137. doi: 10.1073/pnas.0603670103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, X., Wang, A., Chen, L., Huang, E., Xie, W., Liu, C., et al. (2014). S-nitrosylating protein disulphide isomerase mediates α-synuclein aggregation caused by methamphetamine exposure in PC12 cells. Toxicol. Lett. 230, 19–27. doi: 10.1016/j.toxlet.2014.07.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, B., Jin, C., Deng, Y., Liu, W., Yang, T., Feng, S., et al. (2014). Alpha-synuclein oligomerization in manganese-induced nerve cell injury in brain slices: a role of NO-mediated S-nitrosylation of protein disulphide isomerase. Mol. Neurobiol. 50, 1098–1110. doi: 10.1007/s12035-014-8711-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, L., Liu, X., Chen, J., and Liang, Y. (2013). Protein disulphide isomerase interacts with Tau protein and inhibits its fibrillization. PLoS ONE 8:e76657. doi: 10.1371/journal.pone.0076657
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, S., Wang, X., Cui, L., Ding, X., Niu, L., Yang, F., et al. (2014). Compact conformations of human protein disulfide isomerase. PLoS ONE 9:e103472. doi: 10.1371/journal.pone.0103472
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, Y., Harel, N., and Strittmatter, S. (2009). Reticulon-4S (Nogo-A) redistributes protein disulphide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. J. Neurosci. 29, 13850–13859. doi: 10.1523/JNEUROSCI.2312-09.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: PDI, thioredoxin-like -CXXC- motifs, protein folding, S-nitrosylation, neurodegeneration
Citation: Conway ME and Harris M (2015) S-nitrosylation of the thioredoxin-like domains of protein disulfide isomerase and its role in neurodegenerative conditions. Front. Chem. 3:27. doi: 10.3389/fchem.2015.00027
Received: 30 January 2015; Accepted: 30 March 2015;
Published: 16 April 2015.
Edited by:
Bulent Mutus, University of Windsor, CanadaReviewed by:
Eleonora Napoli, University of California, Davis, USAMay Khanna, University of Arizona, USA
Copyright © 2015 Conway and Harris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Myra E. Conway, Department of Applied Sciences, University of the West of England, Coldharbour Lane, Bristol BS16 1QY, UK myra.conway@uwe.ac.uk