The immunomodulatory properties of Helicobacter pylori confer protection against allergic and chronic inflammatory disorders
- Institute of Molecular Cancer Research, University of Zürich, Zürich, Switzerland
Chronic infection with the gastric bacterial pathogen Helicobacter pylori causes gastritis and predisposes carriers to a high risk of developing gastric and duodenal ulcers, gastric cancer, and gastric lymphoma, but has also recently been shown to protect against certain allergic and chronic inflammatory disorders. The immunomodulatory properties that allow the bacteria to persist for decades in infected individuals in the face of a vigorous, yet ultimately non-protective, innate, and adaptive immune response may at the same time confer protection against allergies, asthma, and inflammatory bowel diseases. Experimental evidence from mouse models suggests that H. pylori has evolved to skew the adaptive immune response toward immune tolerance rather than immunity, which promotes persistent infection on the one hand, and inhibits auto-aggressive and allergic T-cell responses on the other. Regulatory T-cells mediating peripheral immune tolerance have emerged as key cellular players in facilitating persistent infection as well as protection from allergies, in both observational studies in humans and experimental work in mice. Recent data suggest that H. pylori actively targets dendritic cells to promote tolerance induction. The findings discussed in this review raise the possibility of harnessing the immunomodulatory properties of H. pylori for the prevention and treatment of allergic and auto-immune diseases, and also provide new insights relevant for H. pylori-specific vaccine development.
Introduction
Helicobacter pylori is the most common bacterial infection in humans worldwide. The bacteria possess the remarkable ability to persist in infected individuals for many decades and have intimately co-existed with humans at least since they first migrated out of East Africa approximately 60000 years ago (Linz et al., 2007). During this long period of co-evolution, the bacteria have acquired traits that allow them to evade and subvert both innate and adaptive branches of the immune system to ensure their persistence in the face of a vigorous, yet ultimately non-protective, local and systemic immune response (Muller et al., 2011). The evolutionary costs possibly associated with the gastric pathology induced by chronic H. pylori infection have been proposed to be offset by potential benefits and a selective advantage for human carriers (Blaser and Atherton, 2004). Indeed, epidemiological and experimental data now point to a strong protective effect of H. pylori infection on the development of many extra-gastric diseases, including gastroesophageal reflux disease and its associated sequels (Vaezi et al., 2000; Islami and Kamangar, 2008; Whiteman et al., 2010), childhood asthma and allergy (Chen and Blaser, 2008; Amberbir et al., 2011), and certain metabolic disorders (Osawa et al., 2005; Mera et al., 2006). The epidemiological and experimental evidence for possible protective properties of H. pylori infection, as well as the mechanistic basis underlying these effects, are the subject of this review.
H. pylori Protects Against Allergic Asthma and Other Atopic Diseases
The Pathogenesis of Asthma
In allergic asthma, genetically susceptible individuals respond to environmental allergens with inappropriate T-cell-mediated immune responses, leading to chronic obstruction of the airways. Allergic asthma is characterized by the accumulation of inflammatory infiltrates in the lung, airway hyper-responsiveness to a variety of specific and non-specific stimuli, increased serum immunoglobulin E (IgE) levels, and mucus hypersecretion. Chronic inflammation further leads to structural changes (airway remodeling) with collagen deposits, hyperplasia, and thickening of the airway wall. In asthmatic patients, allergic episodes trigger the bronchoalveolar infiltration of various immune cell populations, mostly eosinophils, mast cells, and activated CD4+ T-cells. Effector T-helper 2 (Th2) cells play a central role in orchestrating the immune response to allergens by releasing cytokines that trigger the predominant features of asthma (Robinson et al., 1992): the secretion of IL-4 and IL-13 contributes to B-cell production of IgE (Wills-Karp et al., 1998; Bacharier and Geha, 2000), the release of IL-5 drives eosinophilic inflammation (Wang et al., 1989; Rosenberg et al., 2007), and IL-9 stimulates mast cell proliferation (Renauld et al., 1995). The action of IL-4 and IL-13 on lung epithelia further induces goblet cell metaplasia, whereas IL-13 acting on smooth muscle cells promotes the development of airway hyper-responsiveness (Wills-Karp et al., 1998). Recently, other subsets of T-helper cells have been linked to asthma pathogenesis, including Th9 (Shimbara et al., 2000; Erpenbeck et al., 2003), Th25 (Tamachi et al., 2006; Ballantyne et al., 2007), and Th22 cells (Nakagome et al., 2011). A subset of lung-infiltrating T-cells known as Th17 cells has been described to account for neutrophilic airway inflammation, but also for enhanced Th2-cell-mediated eosinophilic airway inflammation (Wakashin et al., 2008). IL-17 secretion by both Th17 cells and eosinophils was further found to be increased in asthmatic patients (Molet et al., 2001). Although asthma is generally considered to be an adaptive immune disorder, the innate arm of the immune system also contributes to the pathology through the production of pro-inflammatory mediators by bronchial epithelial cells (Kauffman et al., 2006), mast cells (Amin et al., 2005), basophils (Ono et al., 2010), natural killer T (NKT) cells (Akbari et al., 2003), and dendritic cells (DC; Lambrecht et al., 2000).
There is now ample evidence that a variety of suppressive and regulatory mechanisms are crucially involved in preventing the activation of potentially harmful effector responses in the lungs of healthy individuals (Ray et al., 2010). This task is predominantly accomplished by CD4+CD25+ regulatory T-cells (Tregs) that either develop in the thymus (natural Tregs) or are induced in the periphery in response to specific antigens (adaptive or induced Tregs). Tregs can suppress pathogenic T-cell responses through direct contact with their target cells or via the release of anti-inflammatory cytokines such as IL-10 and transforming growth factor beta (TGF-β). Both CD25hi (Curotto de Lafaille et al., 2008) and IL-10-producing Tregs (Akdis et al., 2004) have been implicated in preventing Th2 responses to allergens. Peripheral blood-derived CD4+CD25+ Treg subsets of healthy individuals indeed inhibit the proliferation and cytokine production of their allergen-responsive CD4+CD25− effector counterparts in vitro. In contrast, this ability was reduced in Treg subsets of allergic individuals, suggesting that effective suppression of pathogenic Th2 responses might be defective or overridden in patients with allergic diseases (Robinson, 2009). In bronchoalveolar lavage fluid (BALF) from asthmatic children, both the percentage and the suppressive capacity of Tregs were reduced compared to healthy control individuals (Kay, 2001). In a mouse model of ovalbumin-induced airway inflammation, the transfer of ovalbumin-specific Tregs could prevent or reverse the development of airway hyper-responsiveness and Th2 immune responses in an IL-10-dependent manner (Boudousquie et al., 2009). Overall, these findings suggest that functional Tregs of healthy individuals shift allergen-specific immune responses toward tolerance, thereby preventing the development of asthma and other allergic disorders.
Helicobacter Infection is Inversely Correlated with Allergic Asthma in Humans
Many atopic individuals never develop allergic disease manifestations in their lifetime, suggesting that the genetic background acts synergistically with environmental factors to determine individual allergy susceptibility. The influence of environmental factors is drastically illustrated by the alarming increase of asthma and associated allergic disease incidence in Western societies over the last decades, especially among children (Eder et al., 2006). A plausible explanation has been formulated in the “hygiene hypothesis,” which postulates a causal inverse relationship between allergies and pediatric infectious diseases (Strachan, 1989). At the immunological level, this hypothesis proposes that early life exposure to microbial antigens is required for the normal maturation of the immune system and the generation of protective regulatory T-cell responses. This notion has recently been revisited by Blaser and Falkow (2009), who suggest that the specific loss of our ancestral indigenous microbiota due to modern hygienic practices and the widespread use of antibiotics, rather than a general decline in arbitrary childhood infections, is causally associated with the epidemic increase of asthma and other allergic diseases.
In experimental models of airway inflammation, several viral and parasitic pathogens, including influenza viruses and helminths, have been implicated in protection against asthma and allergy (Wilson et al., 2005; Kitagaki et al., 2006). In addition, the role of H. pylori as a protective agent against atopic disorders has been suggested by numerous cross-sectional (Matricardi et al., 2000; Kosunen et al., 2002; Linneberg et al., 2003; Jarvis et al., 2004; Radon et al., 2004; von Hertzen et al., 2006; Herbarth et al., 2007; Janson et al., 2007; Shiotani et al., 2008) and case–control studies (Bodner et al., 2000; Matricardi et al., 2000; Tsang et al., 2000; Jun et al., 2005). McCune and colleges have reported that individuals carrying H. pylori were 30% less likely to have concomitant allergic conditions, including asthma, eczema, and allergic rhinitis (McCune et al., 2003). Several independent studies have further suggested more pronounced protective effects of H. pylori in children and in individuals with early onset asthma (Chen and Blaser, 2007, 2008; Amberbir et al., 2011) and in CagA-seropositive individuals (Chen and Blaser, 2007; Reibman et al., 2008).
Asthma Protection Conferred by H. pylori is Mediated by Regulatory T-Cells
We have recently reported that experimental H. pylori infection prevents allergic asthma in a mouse model of ovalbumin- or house dust mite-induced airway inflammation (Arnold et al., 2011a). Infected mice were efficiently protected against allergen-induced airway hyper-responsiveness, tissue inflammation, and goblet cell metaplasia, and exhibited reduced pulmonary and bronchoalveolar infiltration with eosinophils, Th2 cells, and Th17 cells (Arnold et al., 2011a). The protection against asthma could be attributed to H. pylori-induced, highly suppressive Tregs, which accumulate in the lungs of infected mice and block pathogenic effector T-cell responses (Figure 1). The depletion of CD4+CD25+ Tregs by systemic administration of a CD25-neutralizing antibody led to enhanced pulmonary inflammation and abrogated the characteristic T-cell hypo-responsiveness to ovalbumin in infected mice. The adoptive transfer of Tregs purified from the mesenteric lymph nodes of infected but not naïve donors was further sufficient to transfer protection to uninfected recipients (Arnold et al., 2011a). In addition, H. pylori-infected animals were characterized by lung-infiltrating semi-mature DC, which might suggest that Tregs generated during infection have the ability to influence extra-gastric immune responses by retaining DCs in a semi-mature state (Figure 1). An analogous mechanism was recently proposed by Onishi et al. (2008) who found that FoxP3+ Tregs form aggregates on DCs to actively down-regulate their co-stimulatory molecules CD80 and CD86, thus competing with naïve T-cells for access to DCs and limiting their ability to activate effector T-cell responses. Our finding of the accumulation of Tregs and semi-mature DCs in the lungs of infected, but not asthmatic mice (Arnold et al., 2011a) is in line with this model of asthma prevention (summarized schematically in Figure 1).
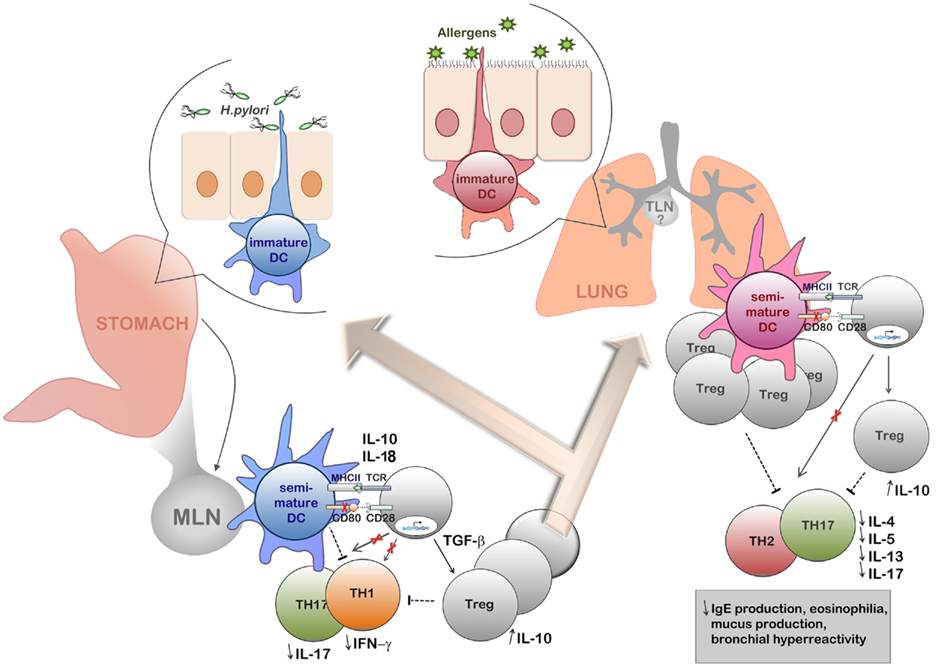
Figure 1. Schematic representation of the current model of H. pylori-induced immune tolerance and asthma protection. Tolerogenic dendritic cells and H. pylori-induced regulatory T-cells act in concert to prevent adaptive Th1/Th17-driven immunity to the infection and to inhibit allergen-specific Th2 responses. In chronically infected humans, H. pylori resides exclusively in the gastric mucosa, where it is presumably encountered and detected by tissue-resident DC populations extending dendrites into the gastric lumen. H. pylori-experienced DCs migrate to the gut-draining mesenteric lymph nodes, where they act as potent inducers of TGF-β-dependent FoxP3+ regulatory T-cells, but fail to prime H. pylori-specific Th1 and Th17 responses. Induced Tregs may further perpetuate the tolerogenic effects of H. pylori-experienced DCs by retaining mesenteric lymph node DCs in a semi-mature state and by directly suppressing H. pylori-specific gastric Th1 and Th17 responses, thereby protecting the host from excessive gastric immunopathology. Newly induced Tregs further migrate to the lung, where they suppress allergen-specific Th2 and Th17 responses involved in the pathogenesis of asthma. The generation of allergic T-cell responses may be blocked either through the tolerogenic effects of Tregs on DCs (retaining DCs in a semi-mature state) or directly through suppression of Th2 and Th17 responses via Treg/T-effector cell contact or via soluble cytokines, in particular IL-10. The ultimate outcome of gastric H. pylori infection on the allergen-challenged lung is reduced eosinophilia, mucus production and airway hyper-responsiveness. The involvement of the tracheal lymph nodes in H. pylori-induced asthma suppression is likely, but currently not well understood.
Asthma Protection Conferred by H. pylori is Linked to the Expression of Specific Virulence Factors and is Most Pronounced in Young Individuals
Although a negative association between CagA-positive H. pylori infection status and allergic disease manifestations has been suggested (Chen and Blaser, 2007), protection conferred by H. pylori infection in our model was not linked to the expression of a functional type IV secretion system (Arnold et al., 2011a). Interestingly, the mucosal or systemic administration of the H. pylori neutrophil-activating protein (HP-NAP) in a therapeutic model of asthma was shown to inhibit bronchial inflammation through agonistic ligation of toll-like receptor 2 (TLR2; Codolo et al., 2008). HP-NAP delivery reduced lung eosinophilia in response to repeated ovalbumin challenge and decreased the production of IL-4, IL-5, and GM–CSF in the bronchioalveolar fluid (Codolo et al., 2008). In H. pylori-infected individuals, stimulation of lamina propria lymphocytes with HP-NAP further increased IL-10 production compared to uninfected controls, while reducing the proliferative and IFN-γ responses of stimulated PBMC (Windle et al., 2005). Because IL-10 is a key cytokine in the resolution of asthmatic inflammation (Xystrakis et al., 2006; Ogawa et al., 2008), this raises the possibility that specific H. pylori virulence factors promote protection against allergic diseases by stimulating the Treg-specific production of IL-10.
Early onset asthma in children and adolescents is rare in the H. pylori-infected population (Chen and Blaser, 2008). H. pylori-infected children are known to preferentially launch Treg responses to the pathogen (Harris et al., 2008), which may account for the particularly beneficial effects of H. pylori in this population. Similarly, experimental H. pylori infection induces a continuum of protection against airway inflammation that is negatively correlated with age at the time of infection. Protection was most evident in neonatally infected mice, which develop H. pylori-specific immunological tolerance mediated by long-lived, inducible Tregs (Arnold et al., 2011b). Because the neonatal period of life is a unique developmental stage in which immune responses are highly plastic and inherently biased toward tolerance, Tregs generated during the first weeks and months of life are thought to differ qualitatively in their suppressive potential compared to their adult counterparts. Indeed, murine neonatal CD4+ T-cells were shown to intrinsically differentiate into CD4+FoxP3+ Tregs in response to TCR stimulation and TGF-β signals (Wang et al., 2010). They further stably express high levels of FoxP3, which renders them particularly suppressive. It is therefore tempting to speculate that the observed inverse correlation between H. pylori colonization and allergic asthma is mechanistically linked to neonatally acquired immune tolerance to the bacterium (see The Beneficial Effects of H. pylori on Allergic and Chronic Inflammatory Disorders are Mediated by Tregs and Tolerogenic DCs).
H. pylori Protects Against Inflammatory Bowel Disease
The Pathogenesis of Inflammatory Bowel Disease
Other immune system disorders for which inverse associations with H. pylori have been examined are the inflammatory bowel diseases (IBDs), chronic inflammatory conditions of unknown etiology of the gastrointestinal (GI) tract. Two main types of IBDs–Crohn’s disease and ulcerative colitis are distinguished based on the affected GI region and transmural involvement. Crohn’s disease is characterized by transmural inflammation of the bowel preferentially involving the terminal ileum and right colon. Ulcerative colitis manifests as a chronic inflammatory condition of the colonic mucosa; in its most limited form it may be restricted to the distal rectum, while the entire colon is involved in its most advanced form. The main symptoms of both IBDs are diarrhea, abdominal pain, and weight loss. Standard medications for both IBDs include salicylates, corticosteroids, and other immunomodulators; surgery is required for the treatment of bowel stenosis, abscesses, and internal fistulas in Crohn’s disease patients. Curative treatment is currently not possible as the etiology of both IBDs is unclear.
In healthy individuals, intestinal mucosal homeostasis is controlled by FoxP3+CD4+ Tregs, which efficiently suppress pathogenic T-cell responses through IL-10 and TGF-β (Maloy et al., 2003; Kamanaka et al., 2006; Li et al., 2007a). The failure of regulatory networks to control excessive T-cell responses breaks this equilibrium and leads to chronic inflammation. In patients with ulcerative colitis, the intestinal production of IL-4 and IL-13 cytokines is reminiscent of an atypical Th2 adaptive response (Fuss et al., 1996), whereas Crohn’s disease patients present a Th1-polarized cytokine profile, with production of interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), and IL-12 (Fuss et al., 1996). In addition, both types of diseases are characterized by the accumulation of IL-17-producing CD4+ T-cells, termed Th17 cells (Annunziato et al., 2007). These cells differentiate from naïve T-cells through the synergistic effects of IL-6 and TGF-β, IL-1, and IL-21 (Veldhoen et al., 2006; Korn et al., 2007) and require IL-23 for their maintenance and expansion (Ouyang et al., 2008). Recent work has shown that IL-23 drives chronic intestinal inflammation in a mouse model of colitis by directly targeting T-cells, and induces their proliferation and accumulation in the colon (Hue et al., 2006). IL-23 further favors the emergence of an immunogenic IL-17A+IFN-γ+ double-positive CD4+ T-cell subset (Hue et al., 2006; Ahern et al., 2010) and inhibits the differentiation of FoxP3+ Treg cells (Kullberg et al., 2006; Ahern et al., 2010), suggesting that the IL-23/Th17 axis is a major determinant in the pathogenesis of IBD. The key role of IL-23 is also supported in humans, as increased expression of IL-23 is detected in IBD patients and mutations in the IL23R gene increases susceptibility to both Crohn’s disease and ulcerative colitis (Ahern et al., 2010). IL-23 can also drive inflammation in the absence of T-cells. In lymphocyte-deficient Rag−/− mice, the development of colitis following Helicobacter hepaticus infection or treatment with agonistic anti-CD40 antibody depended on IL-23 (Hue et al., 2006; Yen et al., 2006). This inflammation was further attributed to an IL-23-responsive innate lymphoid population that expresses IL23R, Thy1 and RORγt, and secretes IL-17 and IFN-γ (Buonocore et al., 2010). The detection of a similar innate lymphoid cell population in the inflamed intestine of patients provided evidence for a functional role of IL-23-responsive innate cells in the pathogenesis of IBD (Buonocore et al., 2010).
Helicobacter Infection is Inversely Correlated with IBD
Several epidemiological studies have examined a possible inverse correlation between IBDs and Helicobacter infection. A Hungarian study investigating 133 IBD patients (both Crohn’s and colitis patients) and similar numbers of controls found a significant inverse association with H. pylori infection (Pronai et al., 2004); whereas only 13% of IBD patients carried H. pylori, the rates ranged from 39 to 67% in various control groups (Pronai et al., 2004). This result was confirmed in a Polish study examining 94 pediatric IBD patients (both types of IBD), which revealed a lower H. pylori colonization rate in patients compared to healthy controls (9.6 vs. 38.4%, p < 0.0001; Sladek et al., 2007). A recent meta-analysis conducted by Luther et al. (2010) of 30 articles examining such a possible link confirmed that H. pylori infection may indeed confer some level of protection against IBD, with only 27% of IBD patients showing evidence of H. pylori infection compared to 41% of patients in the control group. The authors caution, however, that the heterogeneity among examined studies and the possibility of publication bias may limit the certainty of their findings. It is therefore all the more interesting that the same group has provided experimental evidence of protective effects of H. pylori infection on Salmonella typhimurium-induced colitis. Upon co-infection of both bacteria, H. pylori suppressed Salmonella-specific Th17 responses in the cecum, and reduced cecal inflammation caused by Salmonella infection (Higgins et al., 2011). The protective effects were linked to increased levels of IL-10 in the mesenteric lymph nodes of co-infected over Salmonella-only infected mice, suggesting that this regulatory cytokine modulates the differentiation and/or activity of Th17 cells (Higgins et al., 2011). The same group has attributed the protective effects of H. pylori on colitis to the bacteria’s chromosomal DNA, which appears to exhibit a high ratio of immunoregulatory to immunostimulatory sequences (Luther et al., 2011) and is by itself sufficient to prevent sodium dextran sulfate-induced colitis. In their experimental protocol, Luther et al. (2011) treated mice with one orally administered dose of 20–50 μg H. pylori DNA prior to subjecting them to an acute and a chronic protocol of colitis induction; in both models, administration of the DNA reduced the pathology and also attenuated other parameters of DSS-induced colitis such as bleeding and weight. The protective properties of H. pylori DNA were attributed to inhibition of cytokine production by DC, which upon addition of the DNA failed to produce type I interferon and IL-12 in response to E. coli DNA (Luther et al., 2011). Whether H. pylori DNA is indeed the relevant factor conferring protection against IBD in humans or mice remains to be elucidated in more detail; in fact, data showing protection by live infection in IBD models other than acute Salmonella-induced colitis are currently not available.
H. pylori May Protect Against Other T-Cell-Driven Auto-Immune Diseases
Given the documented protective effects of H. pylori infection on asthma and other allergic disease manifestations on the one hand, and IBD on the other, the possibility has been raised that the presence or absence of this infection may also influence the risk of developing additional T-cell-driven immunological or metabolic disorders (Blaser and Falkow, 2009). The incidence of auto-immune diseases caused by the aberrant activation of aggressive autoreactive T-cells, such as multiple sclerosis (MS) and type I diabetes mellitus, has increased sharply in the second half of the twentieth century (Bach, 2002), i.e., in the time frame in which H. pylori has begun to disappear – at least in recent birth cohorts – from human populations in most developed countries (Blaser and Falkow, 2009). Socioeconomic conditions favoring lower H. pylori transmission and infection rates, such as frequent use of antibiotics in childhood, small family size, and non-crowded housing (Blaser and Falkow, 2009) have all also been found to be associated with a higher prevalence of allergic and auto-immune diseases (Bach, 2002). While a possible inverse correlation between H. pylori infection and auto-immune diseases remains largely speculative at this point in time, it is tempting to postulate that the same immunomodulatory and immunoregulatory mechanisms protecting infected individuals from allergic asthma may also be operative against excessive T-cell-driven auto-immune activation. The pathogenesis of auto-immune diseases has been studied most thoroughly in MS, an auto-immune disorder directed against the myelin sheath of neuronal axons, causing demyelination and a broad spectrum of CNS symptoms. In MS, as in IBD (see H. pylori Protects Against Inflammatory Bowel Disease), the Th17 subset of helper T-cells is thought to be the driving force behind the chronic (neuro-) inflammation causing disease symptoms. It is now widely accepted that Th17 cells, not Th1 cells as believed previously (Gutcher and Becher, 2007), are the main encephalitogenic population in auto-immune neuro-inflammation in experimental auto-immune encephalomyelitis (EAE), the standard mouse model of MS. Pathogenic auto-immune Th17 cells are characterized by the secretion of the cytokines IL-22, IL-21, IL-17A, IL-17F, and GM–CSF (Littman and Rudensky, 2010). A recent study has reported a dominant role for Th17-derived GM–CSF in auto-immune CNS inflammation based on the evidence that autoreactive helper T-cells specifically lacking GM–CSF failed to initiate neuro-inflammation despite expression of IL-17A and IFN-γ, whereas GM–CSF secretion by Ifng−/−Il17a −/− helper T-cells was sufficient to induce EAE (Codarri et al., 2011).
Very little solid epidemiological data is available to date to support a protective effect of H. pylori on the development of MS. One study has found an inverse correlation of H. pylori infection with MS in the Japanese population (Li et al., 2007b), and some studies point to a higher prevalence of MS in adults with a record of having been afflicted with asthma in childhood (Ponsonby et al., 2006). In the study by Li et al. (2007b) examining 105 MS patients and 85 healthy controls, H. pylori seropositivity was significantly lower in patients with conventional MS (22.6%) relative to the healthy controls (42.4%). This result obviously remains to be confirmed in larger patient cohorts, and should also be experimentally examined in the EAE model of MS. In EAE, CNS inflammation and progressive paralysis of the tail and hind limbs is entirely driven by myelin-specific auto-aggressive T-cells (Codarri et al., 2011), and should therefore be susceptible to H. pylori-induced, Treg-mediated immunoregulation. Other experimental models of auto-immune disease, such as the non-obese diabetic model of type I diabetes (Chaparro et al., 2006) may provide informative results as well.
The Beneficial Effects of H. pylori on Allergic and Chronic Inflammatory Disorders are Mediated by Tregs and Tolerogenic DCs
Tregs Promote H. pylori Persistence, Limit Gastric Infection-Associated Immunopathology and Prevent Airway Hyper-Responsiveness in Mice
Numerous recent reports have implicated Tregs and DCs with tolerogenic activity in mediating the systemic immunomodulatory effects of H. pylori infection, both in human carriers, and in experimentally infected animals. In a seminal study examining human gastric T-cell responses to H. pylori infection, Robinson et al. (2008) showed that patients with peptic ulcer disease exhibited stronger Th1 and Th2 responses to H. pylori than asymptomatic carriers; conversely, the latter group predominantly mounted Treg responses to the infection. IL-10-expressing Tregs were particularly abundant in the gastric mucosa of the normal carriers compared to the peptic ulcer disease patients; interestingly, mucosal IL-10 levels were directly correlated with bacterial densities, with asymptomatic carriers showing high IL-10 expression receiving the highest colonization scores and peptic ulcer disease patients with low IL-10 expression receiving comparatively low colonization scores (Robinson et al., 2008). In a similar study conducted by Harris et al. (2008) the relatively mild gastritis typical of H. pylori-infected children could also be linked to Treg-predominant T-cell responses. Children with a mild form of gastritis exhibited higher gastric mucosal Treg numbers and higher levels of the regulatory cytokines IL-10 and TGF-β than adults with more severe gastritis (Harris et al., 2008). Evidence for a functional role for Tregs and Treg-derived cytokines in promoting H. pylori persistence on the one hand, and in mediating H. pylori-induced immunomodulation on the other has been provided in experimental infection models. The earliest such evidence came from IL-10−/− mice, which are able to spontaneously clear Helicobacter infections, but suffer from – at least temporarily – strongly enhanced Th1 mediated gastritis (Ismail et al., 2003). The depletion of Tregs in mice infected with Helicobacter at 6 weeks of age using a CD25-specific antibody resulted in a strong reduction in colonization, and in accelerated and enhanced gastritis (Sayi et al., 2011). The depletion of Tregs in a genetic mouse model in which the diphtheria toxin receptor is expressed under the Treg-specific foxp3 promoter (foxp3-DTR-transgenic mouse) also resulted in clearance of the infection and in severe gastritis, even accompanied by the development of preneoplastic gastric lesions (Arnold et al., 2011b). Mice whose CD4+ T-cells cannot respond to TGF-β due to transgenic expression of a dominant-negative form of TGF-β receptor II also develop strongly enhanced pathology and spontaneously reduce bacterial burdens (Arnold et al., 2011b), indicating that TGF-β-dependent inducible Tregs, but not TGF-β-independent natural Tregs, are predominantly involved in maintaining persistence and in mediating H. pylori-specific immunomodulation. Interestingly, the systemic depletion of Tregs in the foxp3-DTR-transgenic model improved the clearance of H. pylori by vaccinated mice, suggesting that the efficacy of an H. pylori vaccine is significantly hampered by the Treg-mediated suppression of protective effector T-cell responses (Hitzler et al., 2011). The latest evidence for an important role of Tregs in the immunomodulation conferring protection against asthma was provided by the finding that the depletion of Tregs abrogates asthma protection (Arnold et al., 2011a). Conversely, as mentioned above, purified Tregs alone were sufficient to transfer protection from H. pylori-infected donors to uninfected recipients (Arnold et al., 2011a). Whereas as few as 100’000 Tregs isolated from neonatally infected donors were suppressive in the asthma model, Tregs from uninfected or adult-infected mice failed to confer protection (Arnold et al., 2011a). The selective suppressivity of Tregs from neonatally infected mice can be attributed to the fact that neonatal exposure to H. pylori induces immune tolerance to the bacteria (Arnold et al., 2011b).
H. pylori Re-Program DCs Toward Tolerogenicity
In contrast to natural Tregs, which originate from the thymus, inducible Tregs are generated in the periphery. Certain populations of poorly immunogenic DCs are believed to initiate and maintain peripheral immune tolerance through the induction of anergy, deletion of autoreactive T-cells and the instruction and differentiation of inducible Tregs (Maldonado and von Andrian, 2010). Such tolerogenic DCs function by converting naive T-cells into FoxP3+ Tregs through antigen presentation in the absence of co-stimulatory signals or cytokines, either alone or in combination with the production of soluble and membrane-bound tolerogenic factors such as IL-10, TGF-β, retinoic acid, and programmed death ligands (PD-Ls; Kretschmer et al., 2005; Maldonado and von Andrian, 2010). DCs appear to indeed play a central role in the induction and maintenance of H. pylori-specific immune tolerance (Figure 2). The depletion of DCs impairs vaccine-induced protective immunity to a similar degree as the depletion of Tregs in the same model (Hitzler et al., 2011). In experimental infection models using adult-infected mice, the depletion of DCs improves the control of the infection and strongly enhances gastric T-cell infiltration and chronic gastritis (Hitzler et al., 2011). DCs isolated from the mesenteric lymph nodes of neonatally infected, tolerant mice exhibit tolerogenic properties ex vivo, i.e., they act as poor inducers of Th1 or Th17 cells, and excellent inducers of Tregs (Oertli et al., 2012). The depletion of DCs from neonatally infected, tolerant mice is consequently sufficient to break tolerance (Oertli et al., 2012). Whereas a functional role for DCs in balancing tolerance and immunity in Helicobacter infection is thus well established, the underlying mechanism is much less thoroughly understood. Evidence from in vitro infection of bone-marrow-derived DCs suggests that H. pylori possesses the ability to profoundly re-program DCs toward tolerogenicity (Figure 2; Kao et al., 2010; Oertli et al., 2012). DCs that have been exposed to H. pylori fail to undergo maturation upon stimulation with E. coli LPS; the IL-12 secretion and up-regulation of the co-stimulatory molecules CD80, CD86, and CD40 that are hallmarks of LPS-matured DCs are prevented by the infection (Figure 2; Oertli et al., 2012). H. pylori-exposed DCs further efficiently induce FoxP3 expression in co-cultured naive T-cells in a TGF-β-dependent manner, but fail to prime Th1 or Th17 cells (Kao et al., 2010; Oertli et al., 2012). The ability of H. pylori to re-program DCs in such a manner requires direct contact (Oertli et al., 2012), but is independent of the virulence factor CagA (Kao et al., 2010).
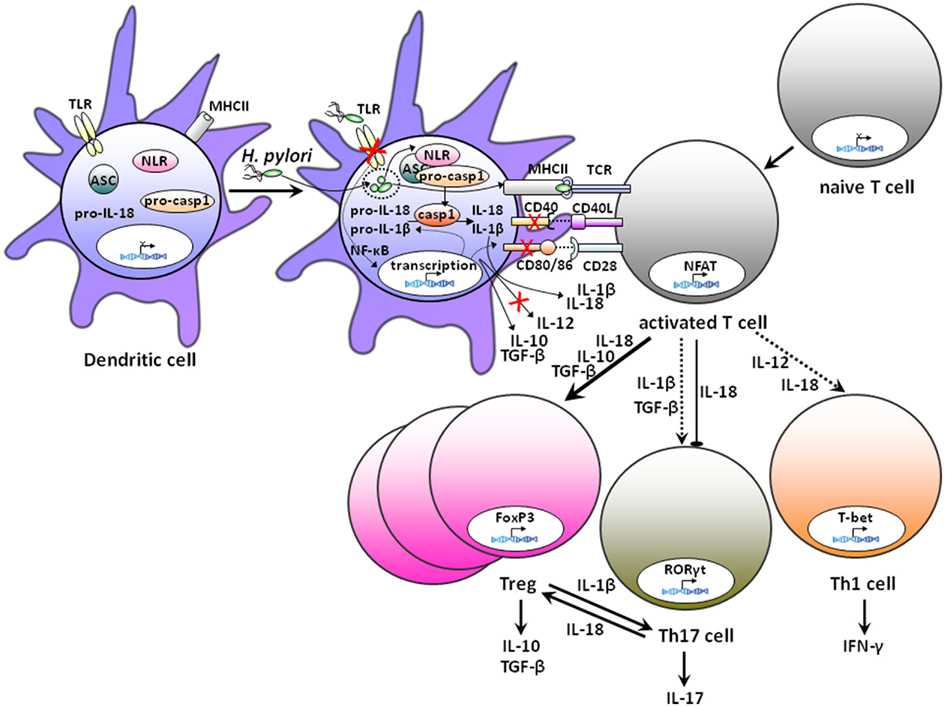
Figure 2. Schematic representation of the effects of H. pylori exposure on DCs and the DC/T-cell interaction. Exposure to H. pylori induces semi-mature DCs with high expression of MHC class II, but only low to moderate expression of the co-stimulatory molecules CD40, CD80, and CD86, and of the cytokine IL-12. In contrast, IL-10 is made in large quantities by H. pylori-experienced DCs. Inflammasome activation by H. pylori through as yet uncharacterized cytoplasmic nod-like receptors (NLRs) leads to caspase-1 activation and the processing and secretion of IL-1β and IL-18. IL-1β promotes Th17 differentiation, whereas IL-18 is required for Th1 and Treg differentiation. H. pylori-experienced DCs actively induce the conversion of naive T-cells to FoxP3+ Tregs in a process that requires IL-18, TGF-β, and possibly IL-10. In contrast, H. pylori-experienced DCs are poor inducers of Th17 and Th1 differentiation. The documented lack of H. pylori TLR ligands in conjunction with efficient inflammasome activation by the bacteria suggests that the relative availability of pro-IL-1β (low level expression due to lack of transcriptional activation) and pro-IL-18 (high levels due to constitutive expression) for caspase-1 processing may dictate the outcome of the DC/T-cell interaction.
The Tolerogenicity of DCs Requires the Synthesis, Processing, and Secretion of Interleukin-18
The tolerogenic activity of DCs requires the DC-intrinsic expression and processing of IL-18 (Figure 2), as demonstrated by the inability of IL-18−/− DCs to induce FoxP3 expression in co-cultured T-cells and the failure of IL-18 receptor-deficient (IL-18R−/−) T-cells to convert to FoxP3+ Tregs upon co-culturing with H. pylori-infected wild type DCs (Oertli et al., 2012). Indeed, IL-18−/− as well as IL-18R−/− mice exhibit significantly lower Treg numbers in their mesenteric lymph nodes than wild type mice under conditions of Helicobacter infection, and generate stronger Th17 responses and develop more severe infection-associated immunopathology (Oertli et al., 2012). CD4+CD25+ cells isolated from infected IL-18−/− or IL-18R−/− donors fail to prevent allergen-induced asthma, indicating that not only the differentiation, but also the suppressive activity of Tregs depends on IL-18 signaling (Oertli et al., 2012). The current model thus assumes that the availability of IL-18 dictates whether naive T-cells co-cultured with DCs differentiate into Th1, Th17, or Treg cells; whereas Th1 and Treg differentiation depend crucially on IL-18, Th17 cells develop under conditions where IL-18 is lacking (Figure 2). The results obtained in the H. pylori model are reminiscent of the hypersusceptibility of IL-18−/− animals toward experimentally induced colitis, which has been attributed to the lack of Nlrp6 inflammasome activation (Elinav et al., 2011). Which nod-like receptors are involved in the innate immune recognition of H. pylori leading to caspase-1 activation and IL-18 processing remains to be determined. It is interesting to note in this context that H. pylori lacks many TLR ligands shared by other gram-negative, pathogenic bacteria. H. pylori flagellin is a poor ligand of TLR5 (Gewirtz et al., 2004) due to mutations in the TLR5 recognition site of the N-terminal D1 domain of flagellin (Andersen-Nissen et al., 2005). The bacterium’s LPS consists predominantly of the tetra-acylated lipid A variety, which is known to exhibit 1000-fold reduced bioactivity as compared to E. coli LPS (Moran et al., 1997). While H. pylori harbors TLR2 ligands (Rad et al., 2009; Sayi et al., 2011), these exhibit predominantly anti-inflammatory properties in vivo (Sayi et al., 2011). The combined results imply that the lack of (pro-inflammatory) TLR signaling in conjunction with high level inflammasome activation and IL-18 secretion may favor Treg over Th17 differentiation during H. pylori infection. Our recent finding that addition of E. coli LPS can reverse the tolerogenic effects of H. pylori on DCs (Oertli et al., 2012) lends further support to this model. LPS is a strong inducer of IL-1β, which in turn is required for Th17 polarization (Figure 2). The relative availability of IL-1β and IL-18, which is influenced by TLR-/Myd88- and NF-κB-dependent transcriptional activation of IL-1β expression (IL-18, in contrast, is preformed and stored in granules, and not subject to extensive transcriptional regulation), thus dictates whether Tregs or Th17 cells are preferentially induced. In the context of H. pylori exposure of DCs, IL-18 is produced in copious amounts due to efficient inflammasome activation; in contrast, due to the concomitant lack of TLR-mediated transcriptional activation, IL-1β is not available for caspase-1-mediated processing, leading to the preferential differentiation of naive T-cells into Tregs as opposed to Th17 cells (Figure 2).
As Th17 cells have been implicated in adaptive immunity to H. pylori infection (DeLyria et al., 2009; Velin et al., 2009; Hitzler et al., 2011), the preferential induction of Tregs over Th17 cells may have conferred a selective advantage to the bacteria in the 60000 years of co-evolution of H. pylori with its human host (Moodley et al., 2009) and explains why infected individuals with strong gastric Treg, but weak T-effector responses show the highest levels of colonization and the least severe gastric pathology (Robinson et al., 2008). An only recently recognized bystander effect of the strong H. pylori-specific Treg induction is cross-suppression of allergen- or autoantigen-specific T-cell responses (Arnold et al., 2011a), which results in the above-discussed protective effects against asthma and other allergies, as well as against IBDs.
Conclusion and Perspectives
Two avenues of active research in the Helicobacter field will likely benefit most from integrating the concepts outlined here into current models and strategies; on the one hand, it is obvious that H. pylori – or at least its tolerance-promoting properties – should be harnessed for the development of new preventive or therapeutic strategies in the treatment of asthma and other allergies, and of IBDs. Whether an infection-independent strategy sometimes referred to as “tolerizing vaccination” will work in this context remains to be seen. On the other hand, vaccine development efforts directed at eradicating H. pylori (therapeutically or prophylactically) must take into account the need to overcome the immunomodulatory properties of this infection if sterilizing immunity is to be achieved. Finally, it will be interesting to compare the strategies that H. pylori has evolved to establish and maintain persistent infection to those exploited by other chronic bacteria such as mycobacteria and Salmonella. Overriding the immune evasion and – modulation strategies of persistent bacterial infections is a crucial first step in breaking the asymptomatic carrier state and in successfully interrupting the transmission cycle that perpetuates the worldwide public health problems associated with these persistent infections.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the laboratory of Anne Müller is supported by the University of Zurich Research Priority Program in Systems Biology, the Swiss National Science Foundation, and the Swiss and Zurich Cantonal Cancer Leagues.
References
Ahern, P. P., Schiering, C., Buonocore, S., McGeachy, M. J., Cua, D. J., Maloy, K. J., and Powrie, F. (2010). Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 33, 279–288.
Akbari, O., Stock, P., Meyer, E., Kronenberg, M., Sidobre, S., Nakayama, T., Taniguchi, M., Grusby, M. J., DeKruyff, R. H., and Umetsu, D. T. (2003). Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat. Med. 9, 582–588.
Akdis, M., Verhagen, J., Taylor, A., Karamloo, F., Karagiannidis, C., Crameri, R., Thunberg, S., Deniz, G., Valenta, R., Fiebig, H., Kegel, C., Disch, R., Schmidt-Weber, C. B., Blaser, K., and Akdis, C. A. (2004). Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J. Exp. Med. 199, 1567–1575.
Amberbir, A., Medhin, G., Erku, W., Alem, A., Simms, R., Robinson, K., Fogarty, A., Britton, J., Venn, A., and Davey, G. (2011). Effects of Helicobacter pylori, geohelminth infection and selected commensal bacteria on the risk of allergic disease and sensitization in 3-year-old Ethiopian children. Clin. Exp. Allergy 41, 1422–1430.
Amin, K., Janson, C., Boman, G., and Venge, P. (2005). The extracellular deposition of mast cell products is increased in hypertrophic airways smooth muscles in allergic asthma but not in nonallergic asthma. Allergy 60, 1241–1247.
Andersen-Nissen, E., Smith, K. D., Strobe, K. L., Barrett, S. L., Cookson, B. T., Logan, S. M., and Aderem, A. (2005). Evasion of Toll-like receptor 5 by flagellated bacteria. Proc. Natl. Acad. Sci. U.S.A. 102, 9247–9252.
Annunziato, F., Cosmi, L., Santarlasci, V., Maggi, L., Liotta, F., Mazzinghi, B., Parente, E., Fili, L., Ferri, S., Frosali, F., Giudici, F., Romagnani, P., Parronchi, P., Tonelli, F., Maggi, E., and Romagnani, S. (2007). Phenotypic and functional features of human Th17 cells. J. Exp. Med. 204, 1849–1861.
Arnold, I. C., Dehzad, N., Reuter, S., Martin, H., Becher, B., Taube, C., and Muller, A. (2011a). Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J. Clin. Invest. 121, 3088–3093.
Arnold, I. C., Lee, J. Y., Amieva, M. R., Roers, A., Flavell, R. A., Sparwasser, T., and Muller, A. (2011b). Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140, 199–209.
Bach, J. F. (2002). The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 347, 911–920.
Bacharier, L. B., and Geha, R. S. (2000). Molecular mechanisms of IgE regulation. J. Allergy Clin. Immunol. 105, S547–S558.
Ballantyne, S. J., Barlow, J. L., Jolin, H. E., Nath, P., Williams, A. S., Chung, K. F., Sturton, G., Wong, S. H., and McKenzie, A. N. (2007). Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J. Allergy Clin. Immunol. 120, 1324–1331.
Blaser, M. J., and Atherton, J. C. (2004). Helicobacter pylori persistence: biology and disease. J. Clin. Invest. 113, 321–333.
Blaser, M. J., and Falkow, S. (2009). What are the consequences of the disappearing human microbiota? Nat. Rev. Microbiol. 7, 887–894.
Bodner, C., Anderson, W. J., Reid, T. S., and Godden, D. J. (2000). Childhood exposure to infection and risk of adult onset wheeze and atopy. Thorax 55, 383–387.
Boudousquie, C., Pellaton, C., Barbier, N., and Spertini, F. (2009). CD4+CD25+ T cell depletion impairs tolerance induction in a murine model of asthma. Clin. Exp. Allergy 39, 1415–1426.
Buonocore, S., Ahern, P. P., Uhlig, H. H., Ivanov, I. I., Littman, D. R., Maloy, K. J., and Powrie, F. (2010). Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375.
Chaparro, R. J., Konigshofer, Y., Beilhack, G. F., Shizuru, J. A., McDevitt, H. O., and Chien, Y. H. (2006). Nonobese diabetic mice express aspects of both type 1 and type 2 diabetes. Proc. Natl. Acad. Sci. U.S.A. 103, 12475–12480.
Chen, Y., and Blaser, M. J. (2007). Inverse associations of Helicobacter pylori with asthma and allergy. Arch. Intern. Med. 167, 821–827.
Chen, Y., and Blaser, M. J. (2008). Helicobacter pylori colonization is inversely associated with childhood asthma. J. Infect. Dis. 198, 553–560.
Codarri, L., Gyulveszi, G., Tosevski, V., Hesske, L., Fontana, A., Magnenat, L., Suter, T., and Becher, B. (2011). RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 12, 560–567.
Codolo, G., Mazzi, P., Amedei, A., Del Prete, G., Berton, G., D’Elios, M. M., and de Bernard, M. (2008). The neutrophil-activating protein of Helicobacter pylori down-modulates Th2 inflammation in ovalbumin-induced allergic asthma. Cell. Microbiol. 10, 2355–2363.
Curotto de Lafaille, M. A., Kutchukhidze, N., Shen, S., Ding, Y., Yee, H., and Lafaille, J. J. (2008). Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity 29, 114–126.
DeLyria, E. S., Redline, R. W., and Blanchard, T. G. (2009). Vaccination of mice against H pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology 136, 247–256.
Eder, W., Ege, M. J., and von Mutius, E. (2006). The asthma epidemic. N. Engl. J. Med. 355, 2226–2235.
Elinav, E., Strowig, T., Kau, A. L., Henao-Mejia, J., Thaiss, C. A., Booth, C. J., Peaper, D. R., Bertin, J., Eisenbarth, S. C., Gordon, J. I., and Flavell, R. A. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757.
Erpenbeck, V. J., Hohlfeld, J. M., Volkmann, B., Hagenberg, A., Geldmacher, H., Braun, A., and Krug, N. (2003). Segmental allergen challenge in patients with atopic asthma leads to increased IL-9 expression in bronchoalveolar lavage fluid lymphocytes. J. Allergy Clin. Immunol. 111, 1319–1327.
Fuss, I. J., Neurath, M., Boirivant, M., Klein, J. S., de la Motte, C., Strong, S. A., Fiocchi, C., and Strober, W. (1996). Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 157, 1261–1270.
Gewirtz, A. T., Yu, Y., Krishna, U. S., Israel, D. A., Lyons, S. L., and Peek, R. M. Jr. (2004). Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J. Infect. Dis. 189, 1914–1920.
Gutcher, I., and Becher, B. (2007). APC-derived cytokines and T cell polarization in autoimmune inflammation. J. Clin. Invest. 117, 1119–1127.
Harris, P. R., Wright, S. W., Serrano, C., Riera, F., Duarte, I., Torres, J., Pena, A., Rollan, A., Viviani, P., Guiraldes, E., Schmitz, J. M., Lorenz, R. G., Novak, L., Smythies, L. E., and Smith, P. D. (2008). Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology 134, 491–499.
Herbarth, O., Bauer, M., Fritz, G. J., Herbarth, P., Rolle-Kampczyk, U., Krumbiegel, P., Richter, M., and Richter, T. (2007). Helicobacter pylori colonisation and eczema. J. Epidemiol. Community. Health 61, 638–640.
Higgins, P. D., Johnson, L. A., Luther, J., Zhang, M., Sauder, K. L., Blanco, L. P., and Kao, J. Y. (2011). Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm. Bowel Dis. 17, 1398–1408.
Hitzler, I., Oertli, M., Becher, B., Agger, E. M., and Muller, A. (2011). Dendritic cells prevent rather than promote immunity conferred by a Helicobacter vaccine using a mycobacterial adjuvant. Gastroenterology 141, 186–196.
Hue, S., Ahern, P., Buonocore, S., Kullberg, M. C., Cua, D. J., McKenzie, B. S., Powrie, F., and Maloy, K. J. (2006). Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483.
Islami, F., and Kamangar, F. (2008). Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev. Res. (Phila) 1, 329–338.
Ismail, H. F., Fick, P., Zhang, J., Lynch, R. G., and Berg, D. J. (2003). Depletion of neutrophils in IL-10(-/-) mice delays clearance of gastric Helicobacter infection and decreases the Th1 immune response to Helicobacter. J. Immunol. 170, 3782–3789.
Janson, C., Asbjornsdottir, H., Birgisdottir, A., Sigurjonsdottir, R. B., Gunnbjornsdottir, M., Gislason, D., Olafsson, I., Cook, E., Jogi, R., Gislason, T., and Thjodleifsson, B. (2007). The effect of infectious burden on the prevalence of atopy and respiratory allergies in Iceland, Estonia, and Sweden. J. Allergy Clin. Immunol. 120, 673–679.
Jarvis, D., Luczynska, C., Chinn, S., and Burney, P. (2004). The association of hepatitis A and Helicobacter pylori with sensitization to common allergens, asthma and hay fever in a population of young British adults. Allergy 59, 1063–1067.
Jun, Z. J., Lei, Y., Shimizu, Y., Dobashi, K., and Mori, M. (2005). Helicobacter pylori seroprevalence in patients with mild asthma. Tohoku J. Exp. Med. 207, 287–291.
Kamanaka, M., Kim, S. T., Wan, Y. Y., Sutterwala, F. S., Lara-Tejero, M., Galan, J. E., Harhaj, E., and Flavell, R. A. (2006). Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 25, 941–952.
Kao, J. Y., Zhang, M., Miller, M. J., Mills, J. C., Wang, B., Liu, M., Eaton, K. A., Zou, W., Berndt, B. E., Cole, T. S., Takeuchi, T., Owyang, S. Y., and Luther, J. (2010). Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138, 1046–1054.
Kauffman, H. F., Tamm, M., Timmerman, J. A., and Borger, P. (2006). House dust mite major allergens Der p 1 and Der p 5 activate human airway-derived epithelial cells by protease-dependent and protease-independent mechanisms. Clin. Mol. Allergy 4, 5.
Kitagaki, K., Businga, T. R., Racila, D., Elliott, D. E., Weinstock, J. V., and Kline, J. N. (2006). Intestinal helminths protect in a murine model of asthma. J. Immunol. 177, 1628–1635.
Korn, T., Bettelli, E., Gao, W., Awasthi, A., Jager, A., Strom, T. B., Oukka, M., and Kuchroo, V. K. (2007). IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448, 484–487.
Kosunen, T. U., Hook-Nikanne, J., Salomaa, A., Sarna, S., Aromaa, A., and Haahtela, T. (2002). Increase of allergen-specific immunoglobulin E antibodies from 1973 to 1994 in a Finnish population and a possible relationship to Helicobacter pylori infections. Clin. Exp. Allergy 32, 373–378.
Kretschmer, K., Apostolou, I., Hawiger, D., Khazaie, K., Nussenzweig, M. C., and von Boehmer, H. (2005). Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 6, 1219–1227.
Kullberg, M. C., Jankovic, D., Feng, C. G., Hue, S., Gorelick, P. L., McKenzie, B. S., Cua, D. J., Powrie, F., Cheever, A. W., Maloy, K. J., and Sher, A. (2006). IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 203, 2485–2494.
Lambrecht, B. N., De Veerman, M., Coyle, A. J., Gutierrez-Ramos, J. C., Thielemans, K., and Pauwels, R. A. (2000). Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J. Clin. Invest. 106, 551–559.
Li, M. O., Wan, Y. Y., and Flavell, R. A. (2007a). T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26, 579–591.
Li, W., Minohara, M., Su, J. J., Matsuoka, T., Osoegawa, M., Ishizu, T., and Kira, J. (2007b). Helicobacter pylori infection is a potential protective factor against conventional multiple sclerosis in the Japanese population. J. Neuroimmunol. 184, 227–231.
Linneberg, A., Ostergaard, C., Tvede, M., Andersen, L. P., Nielsen, N. H., Madsen, F., Frolund, L., Dirksen, A., and Jorgensen, T. (2003). IgG antibodies against microorganisms and atopic disease in Danish adults: the Copenhagen allergy study. J. Allergy Clin. Immunol. 111, 847–853.
Linz, B., Balloux, F., Moodley, Y., Manica, A., Liu, H., Roumagnac, P., Falush, D., Stamer, C., Prugnolle, F., van der Merwe, S. W., Yamaoka, Y., Graham, D. Y., Perez-Trallero, E., Wadstrom, T., Suerbaum, S., and Achtman, M. (2007). An African origin for the intimate association between humans and Helicobacter pylori. Nature 445, 915–918.
Littman, D. R., and Rudensky, A. Y. (2010). Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140, 845–858.
Luther, J., Dave, M., Higgins, P. D., and Kao, J. Y. (2010). Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm. Bowel Dis. 16, 1077–1084.
Luther, J., Owyang, S. Y., Takeuchi, T., Cole, T. S., Zhang, M., Liu, M., Erb-Downward, J., Rubenstein, J. H., Chen, C. C., Pierzchala, A. V., Paul, J. A., and Kao, J. Y. (2011). Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut 60, 1479–1486.
Maldonado, R. A., and von Andrian, U. H. (2010). How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol. 108, 111–165.
Maloy, K. J., Salaun, L., Cahill, R., Dougan, G., Saunders, N. J., and Powrie, F. (2003). CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 197, 111–119.
Matricardi, P. M., Rosmini, F., Riondino, S., Fortini, M., Ferrigno, L., Rapicetta, M., and Bonini, S. (2000). Exposure to foodborne and orofecal microbes versus airborne viruses in relation to atopy and allergic asthma: epidemiological study. BMJ 320, 412–417.
McCune, A., Lane, A., Murray, L., Harvey, I., Nair, P., Donovan, J., and Harvey, R. (2003). Reduced risk of atopic disorders in adults with Helicobacter pylori infection. Eur. J. Gastroenterol. Hepatol. 15, 637–640.
Mera, R. M., Correa, P., Fontham, E. E., Reina, J. C., Pradilla, A., Alzate, A., and Bravo, L. E. (2006). Effects of a new Helicobacter pylori infection on height and weight in Colombian children. Ann. Epidemiol. 16, 347–351.
Molet, S., Hamid, Q., Davoine, F., Nutku, E., Taha, R., Page, N., Olivenstein, R., Elias, J., and Chakir, J. (2001). IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J. Allergy Clin. Immunol. 108, 430–438.
Moodley, Y., Linz, B., Yamaoka, Y., Windsor, H. M., Breurec, S., Wu, J. Y., Maady, A., Bernhoft, S., Thiberge, J. M., Phuanukoonnon, S., Jobb, G., Siba, P., Graham, D. Y., Marshall, B. J., and Achtman, M. (2009). The peopling of the Pacific from a bacterial perspective. Science 323, 527–530.
Moran, A. P., Lindner, B., and Walsh, E. J. (1997). Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J. Bacteriol. 179, 6453–6463.
Muller, A., Oertli, M., and Arnold, I. C. (2011). H. pylori exploits and manipulates innate and adaptive immune cell signaling pathways to establish persistent infection. Cell Commun. Signal 9, 25.
Nakagome, K., Imamura, M., Kawahata, K., Harada, H., Okunishi, K., Matsumoto, T., Sasaki, O., Tanaka, R., Kano, M. R., Chang, H., Hanawa, H., Miyazaki, J., Yamamoto, K., and Dohi, M. (2011). High expression of IL-22 suppresses antigen-induced immune responses and eosinophilic airway inflammation via an IL-10-associated mechanism. J. Immunol. 187, 5077–5089.
Oertli, M., Sundquist, M., Hitzler, I., Engler, D. B., Arnold, I. C., Reuter, S., Maxeiner, J., Hansson, M., Taube, C., Quiding-Järbrink, M., and Müller, A. (2012). DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori – specific immune tolerance, and asthma protection. J. Clin. Invest. doi: 10.1172/JCI61029
Ogawa, Y., Duru, E. A., and Ameredes, B. T. (2008). Role of IL-10 in the resolution of airway inflammation. Curr. Mol. Med. 8, 437–445.
Onishi, Y., Fehervari, Z., Yamaguchi, T., and Sakaguchi, S. (2008). Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. U.S.A. 105, 10113–10118.
Ono, E., Taniguchi, M., Higashi, N., Mita, H., Kajiwara, K., Yamaguchi, H., Tatsuno, S., Fukutomi, Y., Tanimoto, H., Sekiya, K., Oshikata, C., Tsuburai, T., Tsurikisawa, N., Otomo, M., Maeda, Y., Hasegawa, M., Miyazaki, E., Kumamoto, T., and Akiyama, K. (2010). CD203c expression on human basophils is associated with asthma exacerbation. J. Allergy Clin. Immunol. 125, 483–489, e483.
Osawa, H., Nakazato, M., Date, Y., Kita, H., Ohnishi, H., Ueno, H., Shiiya, T., Satoh, K., Ishino, Y., and Sugano, K. (2005). Impaired production of gastric ghrelin in chronic gastritis associated with Helicobacter pylori. J. Clin. Endocrinol. Metab. 90, 10–16.
Ouyang, W., Kolls, J. K., and Zheng, Y. (2008). The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28, 454–467.
Ponsonby, A. L., Dwyer, T., van der Mei, I., Kemp, A., Blizzard, L., Taylor, B., Kilpatrick, T., and Simmons, R. (2006). Asthma onset prior to multiple sclerosis and the contribution of sibling exposure in early life. Clin. Exp. Immunol. 146, 463–470.
Pronai, L., Schandl, L., Orosz, Z., Magyar, P., and Tulassay, Z. (2004). Lower prevalence of Helicobacter pylori infection in patients with inflammatory bowel disease but not with chronic obstructive pulmonary disease – antibiotic use in the history does not play a significant role. Helicobacter 9, 278–283.
Rad, R., Ballhorn, W., Voland, P., Eisenacher, K., Mages, J., Rad, L., Ferstl, R., Lang, R., Wagner, H., Schmid, R. M., Bauer, S., Prinz, C., Kirschning, C. J., and Krug, A. (2009). Extracellular and intracellular pattern recognition receptors cooperate in the recognition of Helicobacter pylori. Gastroenterology 136, 2247–2257.
Radon, K., Windstetter, D., Eckart, J., Dressel, H., Leitritz, L., Reichert, J., Schmid, M., Praml, G., Schosser, M., von Mutius, E., and Nowak, D. (2004). Farming exposure in childhood, exposure to markers of infections and the development of atopy in rural subjects. Clin. Exp. Allergy 34, 1178–1183.
Ray, A., Khare, A., Krishnamoorthy, N., Qi, Z., and Ray, P. (2010). Regulatory T cells in many flavors control asthma. Mucosal Immunol. 3, 216–229.
Reibman, J., Marmor, M., Filner, J., Fernandez-Beros, M. E., Rogers, L., Perez-Perez, G. I., and Blaser, M. J. (2008). Asthma is inversely associated with Helicobacter pylori status in an urban population. PLoS ONE 3, e4060. doi: 10.1371/journal.pone.0004060
Renauld, J. C., Kermouni, A., Vink, A., Louahed, J., and Van Snick, J. (1995). Interleukin-9 and its receptor: involvement in mast cell differentiation and T cell oncogenesis. J. Leukoc. Biol. 57, 353–360.
Robinson, D. S., Hamid, Q., Ying, S., Tsicopoulos, A., Barkans, J., Bentley, A. M., Corrigan, C., Durham, S. R., and Kay, A. B. (1992). Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N. Engl. J. Med. 326, 298–304.
Robinson, K., Kenefeck, R., Pidgeon, E. L., Shakib, S., Patel, S., Polson, R. J., Zaitoun, A. M., and Atherton, J. C. (2008). Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut 57, 1375–1385.
Rosenberg, H. F., Phipps, S., and Foster, P. S. (2007). Eosinophil trafficking in allergy and asthma. J. Allergy Clin. Immunol. 119, 1303–1310; quiz 1311–1302.
Sayi, A., Kohler, E., Toller, I. M., Flavell, R. A., Muller, W., Roers, A., and Muller, A. (2011). TLR-2-activated B cells suppress Helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J. Immunol. 186, 878–890.
Shimbara, A., Christodoulopoulos, P., Soussi-Gounni, A., Olivenstein, R., Nakamura, Y., Levitt, R. C., Nicolaides, N. C., Holroyd, K. J., Tsicopoulos, A., Lafitte, J. J., Wallaert, B., and Hamid, Q. A. (2000). IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J. Allergy Clin. Immunol. 105, 108–115.
Shiotani, A., Miyanishi, T., Kamada, T., and Haruma, K. (2008). Helicobacter pylori infection and allergic diseases: epidemiological study in Japanese university students. J. Gastroenterol. Hepatol. 23, e29–e33.
Sladek, M., Jedynak-Wasowicz, U., Wedrychowicz, A., Kowalska-Duplaga, K., Pieczarkowski, S., and Fyderek, K. (2007). The low prevalence of Helicobacter pylori gastritis in newly diagnosed inflammatory bowel disease children and adolescent. Prz. Lek. 64(Suppl. 3), 65–67.
Tamachi, T., Maezawa, Y., Ikeda, K., Kagami, S., Hatano, M., Seto, Y., Suto, A., Suzuki, K., Watanabe, N., Saito, Y., Tokuhisa, T., Iwamoto, I., and Nakajima, H. (2006). IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J. Allergy Clin. Immunol. 118, 606–614.
Tsang, K. W., Lam, W. K., Chan, K. N., Hu, W., Wu, A., Kwok, E., Zheng, L., Wong, B. C., and Lam, S. K. (2000). Helicobacter pylori sero-prevalence in asthma. Respir. Med. 94, 756–759.
Vaezi, M. F., Falk, G. W., Peek, R. M., Vicari, J. J., Goldblum, J. R., Perez-Perez, G. I., Rice, T. W., Blaser, M. J., and Richter, J. E. (2000). CagA-positive strains of Helicobacter pylori may protect against Barrett’s esophagus. Am. J. Gastroenterol. 95, 2206–2211.
Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M., and Stockinger, B. (2006). TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189.
Velin, D., Favre, L., Bernasconi, E., Bachmann, D., Pythoud, C., Saiji, E., Bouzourene, H., and Michetti, P. (2009). Interleukin-17 is a critical mediator of vaccine-induced reduction of Helicobacter infection in the mouse model. Gastroenterology 136, 2237–2246, e2231.
von Hertzen, L. C., Laatikainen, T., Makela, M. J., Jousilahti, P., Kosunen, T. U., Petays, T., Pussinen, P. J., Haahtela, T., and Vartiainen, E. (2006). Infectious burden as a determinant of atopy – a comparison between adults in Finnish and Russian Karelia. Int. Arch. Allergy Immunol. 140, 89–95.
Wakashin, H., Hirose, K., Maezawa, Y., Kagami, S., Suto, A., Watanabe, N., Saito, Y., Hatano, M., Tokuhisa, T., Iwakura, Y., Puccetti, P., Iwamoto, I., and Nakajima, H. (2008). IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am. J. Respir. Crit. Care Med. 178, 1023–1032.
Wang, G., Miyahara, Y., Guo, Z., Khattar, M., Stepkowski, S. M., and Chen, W. (2010). “Default” generation of neonatal regulatory T cells. J. Immunol. 185, 71–78.
Wang, J. M., Rambaldi, A., Biondi, A., Chen, Z. G., Sanderson, C. J., and Mantovani, A. (1989). Recombinant human interleukin 5 is a selective eosinophil chemoattractant. Eur. J. Immunol. 19, 701–705.
Whiteman, D. C., Parmar, P., Fahey, P., Moore, S. P., Stark, M., Zhao, Z. Z., Montgomery, G. W., Green, A. C., Hayward, N. K., and Webb, P. M. (2010). Association of Helicobacter pylori infection with reduced risk for esophageal cancer is independent of environmental and genetic modifiers. Gastroenterology 139, 73–83.
Wills-Karp, M., Luyimbazi, J., Xu, X., Schofield, B., Neben, T. Y., Karp, C. L., and Donaldson, D. D. (1998). Interleukin-13: central mediator of allergic asthma. Science 282, 2258–2261.
Wilson, M. S., Taylor, M. D., Balic, A., Finney, C. A., Lamb, J. R., and Maizels, R. M. (2005). Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J. Exp. Med. 202, 1199–1212.
Windle, H. J., Ang, Y. S., Athie-Morales, V., McManus, R., and Kelleher, D. (2005). Human peripheral and gastric lymphocyte responses to Helicobacter pylori NapA and AphC differ in infected and uninfected individuals. Gut 54, 25–32.
Xystrakis, E., Kusumakar, S., Boswell, S., Peek, E., Urry, Z., Richards, D. F., Adikibi, T., Pridgeon, C., Dallman, M., Loke, T. K., Robinson, D. S., Barrat, F. J., O’Garra, A., Lavender, P., Lee, T. H., Corrigan, C., and Hawrylowicz, C. M. (2006). Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J. Clin. Invest. 116, 146–155.
Yen, D., Cheung, J., Scheerens, H., Poulet, F., McClanahan, T., McKenzie, B., Kleinschek, M. A., Owyang, A., Mattson, J., Blumenschein, W., Murphy, E., Sathe, M., Cua, D. J., Kastelein, R. A., and Rennick, D. (2006). IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116, 1310–1316.
Keywords: Helicobacter immunomodulation, asthma and allergies, dendritic cells and regulatory T-cells, immune tolerance
Citation: Arnold IC, Hitzler I and Müller A (2012) The immunomodulatory properties of Helicobacter pylori confer protection against allergic and chronic inflammatory disorders. Front. Cell. Inf. Microbio. 2:10. doi: 10.3389/fcimb.2012.00010
Received: 01 December 2011; Accepted: 30 January 2012;
Published online: 16 February 2012.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
Steve Blanke, University of Illinois Urbana-Champaign, USAHolly Algood, Vanderbilt University, USA
Copyright: © 2012 Arnold, Hitzler and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Anne Müller, Institute of Molecular Cancer Research, University of Zürich, Winterthurerstr. 190, 8057 Zürich, Switzerland. e-mail: mueller@imcr.uzh.ch
†Present address: Isabelle C. Arnold, Sir William Dunn School of Pathology, University of Oxford, Oxford, UK.