Characterization of the mechanism of protection mediated by CS-D7, a monoclonal antibody to Staphylococcus aureus iron regulated surface determinant B (IsdB)
- 1Department of Vaccine Basic Research, Merck Research Labs, Merck/MSD, West Point, PA, USA
- 2Department of Biologics Research, Merck Research Labs, Merck/MSD, West Point, PA, USA
We previously reported the development of a human monoclonal antibody (CS-D7, IgG1) with specificity and affinity for the iron regulated surface determinant B (IsdB) of Staphylococcus aureus. CS-D7 mediates opsonophagocytic killing in vitro and protection in a murine sepsis model. In light of recent data indicating that IsdB specific T cells (CD4+, Th17), not Ab, mediate protection after vaccination with IsdB, it is important to investigate the mechanism of protection mediated by CS-D7. The mAb was examined to determine if it blocked heme binding to IsdB in vitro. The mAb was not found to have heme blocking activity, nor did it prevent bacterial growth under in vivo conditions, in an implanted growth chamber. To assess the role of the mAb Fc a point mutation was introduced at aa 297 (CS-D7·N297A). This point mutation removes Fc effector functions. In vitro analysis of the mutein confirmed that it lacked measurable binding to FcγR, and that it did not fix complement. The mutein had dramatically reduced in vitro opsonic OP activity compared to CS-D7. Nonetheless, the mutein conferred protection equivalent to the wild type mAb in the murine sepsis model. Both wild type and mutein mAbs were efficacious in FcγR deletion mice (including both FcγRII−/− mice and FcγRIII−/− mice), indicating that these receptors were not essential for mAb mediated protection in vivo. Protection mediated by CS-D7 was lost in Balb/c mice depleted of C3 with cobra venom factor (CFV), was lost in mice depleted of superoxide dismutase (SOD) in P47phox deletion mice, and as previously reported, was absent in SCID mice (Joshi et al., 2012). Enhanced clearance of S. aureus in the liver of CS-D7 treated mice and enhanced production of IFN-γ, but not of IL17, may play a role in the mechanism of protection mediated by the mAb. CS-D7 apparently mediates survival in challenged mice through a mechanism involving complement, phagocytes, and lymphocytes, but which does not depend on interaction with FcγR, or on blocking heme uptake.
Introduction
Staphylococcus aureus is a significant cause of hospital acquired bloodstream and catheter infections (Thwaites et al., 2011), and is a leading cause of endocarditis, osteomyelitis, and skin and soft tissue infections (Lowy, 1998a). Although it has been extensively investigated, natural protective immunity against S. aureus is poorly understood. Acute infection with S. aureus does not prevent re-infection with this bacteria (Lee, 1996). Preclinical and clinical data indicate that immunization with intact bacteria induces high serum antibody immune titers to staphylococcus, but does not confer protection against S. aureus infection (Lee, 1996; Schaffer and Lee, 2008). In vivo bacterial clearance is currently believed to be facilitated by antibody (Ab) and complement mediated uptake and killing by phagocytes (Peterson et al., 1978; Leijh et al., 1981; Verbrugh et al., 1982; Gregory et al., 1996; Verdrengh and Tarkowski, 1997; Cunnion et al., 2004). S. aureus is a commensal species that colonizes people transiently or permanently, and therefore, individuals have antibodies to S. aureus. Iron regulated surface determinant B (IsdB) is an antigen expressed on the cell surface of S. aureus in iron limited environments, with a MW of approximately 72 kD. Its function is to capture and import heme iron from hemoglobin (Mazmanian et al., 2003). Due to the low iron environment of mammalian blood and tissue, IsdB is upregulated during infection in vivo (Brown et al., 2009). Humans, as well as mammals examined to date, have pre-existing antibody titers to IsdB (Lowy, 1998b), but it is unknown whether these pre-existing titers offer protection.
We previously reported the development of a fully human monoclonal antibody (CS-D7, IgG1) specific for IsdB of Staphylococcus aureus (Ebert et al., 2010). The mAb was isolated from the Cambridge Antibody Technology (CAT) scFv antibody library and has high affinity and specificity for IsdB. It recognizes a conformational epitope spanning amino acids 50–285 of the antigen. As reported, this mAb had functional activity in vitro (opsonophagocytic killing activity) and significantly enhanced survival in the murine sepsis model in Balb/c mice. In recent work, Joshi and co-authors demonstrated that T cells not B cells were the critical effector cells conferring enhanced survival following S. aureus challenge in the Balb/c murine sepsis model (Joshi et al., 2012). Using IsdB specific lymphocytes adoptively transferred from wild type into SCID mice, enhanced survival was determined to be mediated entirely by IsdB specific CD4+ T cells, with B cells and Ab playing no measurable role. This observation, that IsdB specific Ab does not play a critical role in the model, is in apparent disagreement with previously published data indicating that enhanced survival in the murine sepsis model correlates with anti-IsdB Ab titers (Kuklin et al., 2006) and with data indicating that Ab can confer enhanced survival in the same Balb/c sepsis model (Brown et al., 2009; Kim et al., 2010). We, therefore, sought to investigate the protective mechanism of mAb CS-D7 in the murine sepsis model. Since this model is routinely used to verify the efficacy of vaccine antigens targeting S. aureus (Fattom et al., 1996; Kuklin et al., 2006; Stranger-Jones et al., 2006; Vernachio et al., 2006; Spellberg et al., 2008), it is important to define the meaning of immune mediated enhanced survival in this important animal model.
We found that although CS-D7 mediates enhanced survival in the murine sepsis model, that activity was most likely not dependent on direct inhibition of bacterial growth or survival, or on prevention of the physiological function of IsdB. We investigated the role of the mAb Fc and found that a mutein lacking Fc function conferred equivalent survival to the wild type mAb in the murine sepsis model. This was a surprising finding, as the mechanism of antigen specific mAb mediated bacterial clearance is thought to rely on engagement of the receptors for complement and Fcγ by C3b or iC3b and Ab bound to the bacteria surface. The engagement of both the Fcγ receptor and the CR1 receptor, leads to synergy of bacteria uptake and killing by effector cells (Philippe, 2004; Roozendaal and Carroll, 2006). We investigated the role of other components necessary for bacterial clearance in vivo, and found that complement and phagocytes were necessary for protection mediated by CS-D7 in Balb/c mice. CS-D7 mediated survival in the sepsis model is dependent on the presence of lymphocytes (Joshi et al., 2012), therefore, lymphocytes may contribute essential cytokines for stimulation of a strong phagocyte killing response. It was determined that CS-D7 may enhance survival through a non-classical Ab mediated uptake/killing mechanism. A possible mechanism will be discussed.
Materials and Methods
Recombinant Proteins
Recombinant IsdB was prepared as previously described (Kuklin et al., 2006). Heme-free IsdB was prepared by isolation from recombinant heme containing IsdB following peak separation on a cation binding gel resin. Separation of the two fractions of IsdB was readily made by following the absorbance at 280 nm for the presence of protein, and at 400 nm for the presence of iron containing IsdB. The extracellular domains (ECDs) of FcγRIIA and FcγRIIIA were expressed recombinantly in Pichia pastoris at Merck/MSD (Rahway, NJ) (Li et al., 2006). After filtration, the FcγR's were isolated by Ni2+ affinity columns and further purified by hydrophobic interaction chromatography. SDS-PAGE electrophoresis indicated the proteins were of the correct molecular weight and the ability of the FcγR ECD's to bind mAbs was confirmed by surface plasmon resonance.
To construct the antibody Fc variant CS-D7· N297A, mutagenic primers were designed and synthesized. Mutant clones were generated through PCR and transformation steps by using QuikChange Lightning Site-Directed Mutagenesis kit and QuikChange Lightning Multi Site-Directed Mutagenesis kit (Stratagene, Santa Clara, CA). Commercially available antibodies were acquired and used as supplied by the manufacturer. Additional antibodies were produced by transient transfection in CHO or HEK cells (Sino Biological, Beijing, China) (Ebert et al., 2010). Antibodies were stored in 6% sucrose, 100 mM arginine, 100 mM histidine, pH 6.0.
Bacteria
The following strains were used in this investigation: S. aureus Becker (obtained from Chia Lee, University of Arkansas, MSSA, spa type 715, CC45), SA025 (clinical isolate from pancreatic cyst, MRSA, SCCmec IV, spa type 785, CC22), and 8325-4 spa minus (obtained from Intercell AG). Bacteria were grown on tryptic soy agar (TSA), or tryptic soy broth (TSB) overnight, pelleted and stored as frozen 15% glycerol stocks. Alternatively, bacteria were passaged two to three times to stationary phase in iron restricted medium (<0.1 mg/L), Roswell Park Memorial Institute (RPMI) 1640 medium (Life Technologies, Grand island, NY), pelleted and stored frozen in 15% glycerol. For use in experiments, bacteria were thawed, pelleted, and resuspended in the appropriate buffer or medium.
Mice
Balb/c, P47 phox−/− (B6.129S2-Ncf1tm1shl N14) missing a key component of the superoxide generating system, and FcγRII−/− (Fcgr2b; C129S4(B6)-Fcgr2btm1Ttk/cAnNTac N12; Balb/c background) missing the FcγRIIγ protein; and FcγRI−/− (Fcer1g; C.129P2(B6)-Fcerlgtm1RavN12; Balb/c background) missing the γ chain subunit of the Fc receptors, were obtained from Taconic (Hudson, NY), and housed in micro-isolator cages during experiments.
Growth of S. aureus in Chambers Implanted in Rats
In vivo growth of S. aureus Becker was investigated using growth chambers implanted sub-dermally on the back of rats (n = 12). The growth chambers were made from sterile dialysis tubing, with a MW cutoff of 50 kDa, and with a volume of 1 mL. To investigate whether mAb could inhibit the survival and growth of S. aureus Becker, 1.4 × 106 CFU bacteria (from TSA culture) with or without 1 mg of mAb, was added to growth chambers. It was previously determined that IsdB specific mAb binding to 5 × 106 CFU of IsdB expressing S. aureus Becker is saturated at approximately 5 μg (Brown et al., 2009). Therefore, the bacteria were exposed to a large excess of antibody during growth in the chamber. At designated times, rats (n = 3–4) were sacrificed and growth chambers removed aseptically. Bacteria were harvested and colony counts (CFU/mL) were obtained through serial dilution of the bacterial culture followed by plating on TSA overnight at 37°C.
Heme Binding Uptake Assay
See Supplemental Data.
IgG/Fcγ Receptor Binding Assay
FcγR ELISA binding assays were performed as previously described (Shields et al., 2001) with minor modifications. Human FcγRIIA ECD (1 μg/mL) or human FcγRIIIA ECD (2 μg/mL) diluted in PBS was coated overnight at 4°C in white 96-well Maxisorp plates (Nunc, Rochester, NY). The plates were washed three times (BioTek ELX405) with PBS (phosphate buffered saline) containing 0.05% Tween20. Serial dilutions of the test antibodies in blocking buffer [2% BSA (Jackson ImmunoResearch Laboratories, West Grove, PA) in SuperBlock T20 (Pierce, Rockford, IL)] were added for 90 min with rocking. After washing, bound antibodies were labeled with either 0.1 μg/mL (for FcγRIIA) or 1 μg/mL (for FcγRIIIA) of biotinylated anti-κ light chain F(ab′)2 (Southern Biotech, Birmingham, AL) followed by 100 ng/mL of Streptavidin-Europium (PerkinElmer, Waltham, MA). Time-resolved fluorescence was measured by a Victor3 spectrophotometer (Perkin-Elmer, CityplaceWaltham, StateMA) and the relative fluorescence was plotted versus the log of the antibody concentration.
Complement C3b Deposition Assay
An in vitro cell free C3b deposition assay was developed to measure functional complement activation dependent on IgG-C1q binding. High binding microtiter plates were coated with mAb in PBS (12.5 μg/mL) and incubated at 4°C overnight. Plates were blocked with 0.1% BSA in PBS for 2 h at room temperature, and washed with PBS containing Ca2+ and Mg2+. C1q (Calbiochem, EMD Chemicals, USA) was serially diluted into C1q depleted human serum (Quidel). The mix was added to the mAb coated microtiter plate and incubated 30 min at room temperature. C3b was detected by addition of sheep antihuman C3b-FITC (Accurate Chemical and Scientific Corp., Westbury, NY) to each well for 1 h at room temperature. Assay readout was measured at 535 nm.
Opsonophagocytic (OP) Uptake Assay
The OP assay has been described previously (Cope et al., 2008). This assay measures Ab and complement mediated opsonophagocytosis (uptake) by HL60 phagocytes, but not killing by the phagocytes. Components for the assay were dimethylformamide (DMF) differentiated HL60 cells, baby pig sera as a source of complement, 5′-6′-FAM-SE fluorescently labeled, iron starved S. aureus. In this flow cytometry based assay, non-fluorescent HL60 cells were examined for the presence (via phagocytosis) of opsonized fluorescently labeled S. aureus 8325-4 (spa minus), to determine OP activity of antibodies. The bacteria strain used in the assay was chosen to avoid non-specific binding of antibody to the bacteria surface through binding to Protein A. During assay development, it was determined that a spa positive strain (i.e., Becker) was found to have equivalent activity to the 8325-4 strain, however for simplification of the assay and interpretation of results, strain 8325-4 was routinely used. Briefly, 2.4 × 106 iron restricted bacteria were combined with antibodies. After a short incubation of 30 min, complement was added to the mixture and incubated for an additional 30 min at 37°C. Following this incubation the bacteria were washed and labeled with FAM-SE. Then 2 × 105 HL60 cells were mixed with the bacteria at a ratio of 8 bacteria to 1 HL60 cell. The reaction mix was incubated at 37°C, 170 RPM for 30 min. The HL60 phagocytosis of bacteria was stopped by addition of cold PBS. The number of HL60 cells containing engulfed bacteria was measured on a FACSCalibur (Becton Dickinson).
Passive Immunization and Challenge Models
All animal work was performed in accordance with the Merck Research Laboratories Institutional Animal Care and Use committee guidelines. Antibodies were evaluated for efficacy using passive immunization of mice in the murine sepsis model (Brown et al., 2009). In preliminary experiments, S. aureus SA025 (from TSA culture) was titrated via i.v. injection to determine the LD80 dose for Balb/c mice. Next, mAb CS-D7 was dose titrated via i.p. injection prior to i.v. challenge, to achieve approximately 80% survival at day 4 post challenge (data not shown). The optimal bacterial challenge inoculum was determined to be approximately 2–3 × 108 CFU and the optimal antibody dose was 400 μg. The final model used was as follows; 400 μg of antibody per mouse (n = 5) was injected via the i.p. route 2 h prior to challenge of bacteria strain SA025 (from TSA culture, 2–3 × 108 CFU) via tail vein injection. Mice were monitored for survival for 96 h. Monitoring of survival for 96 h post challenge was chosen because that time period reflected the largest differences between experimental and control groups, and few mice died thereafter. Data from three or more experiments were pooled for survival analysis. To deplete mice of C3, mice were treated with CVF (Cunnion et al., 2004). The amount of CVF was titrated to give essentially no C3 in the murine sera. C3 was measured using a murine ELISA for complement factor 3 (ALPCO™ Diagnostics, Salem, NH).
Murine Cytokine and CFU Measurement
Mice (n = 20–24 per group) were injected i.p. with mAb followed by i.v. S. aureus challenge as described above. Subgroups of 5–6 mice were sacrificed at designated time points and blood and tissue collected. Organs were placed in 2 mL of cold PBS. Blood and tissue reserved for cytokine measurement were immediately frozen by placing on dry ice. Tissues were stored at −80°C until use, at which time they were removed, homogenized and evaluated for cytokines. Murine cytokines were measured using kits from MSD (Meso Scale Discovery, Inc., Gaithersburg, MD), as per the manufacturer's instructions. Tissues reserved for CFU measurement were placed on wet ice and stored at 4°C until homogenized (in a total of 5 mL PBS) and 50 μL of homogenate plated for CFU evaluation. As a control, survival of 10 mice was observed for the duration of the experiment to ensure that the cytokine and/or CFU data reflected conditions of efficacy of the antibody. The experiments were performed two or more times with representative data listed.
Statistical Methods
For comparison of survival in the murine challenge experiments, results of individual survival experiments of mice were pooled and curves were analyzed using the Prism® software (Prism for Windows, version 5.01, GraphPad Software, Inc., La Jolla, CA), and choosing the Log rank, Mantel Cox test statistical method for testing significance. Comparisons of CFU/mL were made using the two tailed unpaired T test with Prism® software. Comparisons of cytokines/mL were made using the two tailed T test with Windows, version 5.01, Excel® software.
Results
mAb CS-D7 does not Directly Inhibit Growth of S. aureus in Implanted Chambers In vivo
It was previously reported that CS-D7 enhanced survival in the murine lethal challenge model. To investigate the mechanism of this activity, the potential for direct inhibition of growth of S. aureus by the antibody was evaluated. Growth chambers implanted sub-dermally on the back of rats were used to measure outgrowth of the bacteria under conditions that induced expression of the antigen, that is, under iron restricted, but nutrient rich, in vivo conditions. This method allowed quantitative and qualitative analysis of the resulting bacteria. In preliminary experiments, it was determined that S. aureus Becker (starting concentration of 1 × 106 CFU) reached stationary phase at approximately 10–12 h (at a concentration of 4–6 × 108 CFU), in these chambers (Supplemental Data, Figure A1). It was also demonstrated that there was rapid expression of IsdB by the bacteria, and >95% of the bacteria expressed the antigen at 8 h after implantation in the chamber (as demonstrated by flow cytometry, Supplemental Data, Figure A1).
To investigate whether CS-D7 inhibited the survival and growth of S. aureus under these conditions, bacteria plus an excess of either mAb CS-D7 or 20C2HA (non-specific isotype matched control mAb) were added to growth chambers (Figure 1). At several time points (3–6 h), it was observed that the bacteria were aggregated in the presence of mAb CS-D7, but not the presence of control mAb 20C2HA. This most likely represents cross linking of bacteria due to CS-D7 binding to IsdB expressed on the bacteria surface. Aggregation may have contributed to an apparently lower CFU/mL at the 6 h time point, due to the difficulty of making a single CFU suspension from aggregated bacteria for serial dilution and plating. By 24 h, there were equivalent numbers of CFU in all three growth chamber conditions. Therefore, although CS-D7 bound to S. aureus, this binding did not significantly inhibit survival or division of the bacteria. Binding of CS-D7 did lead to transient bacteria aggregation.
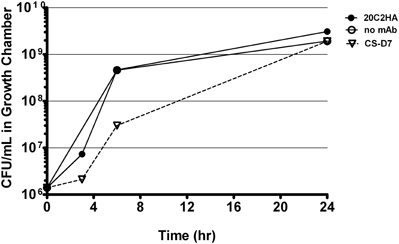
Figure 1. S. aureus growth in the presence of IsdB specific mAb or isotype control mAb. Growth chambers containing 1 × 106 CFU Becker and 1 mg of mAb CS-D7 (inverted triangle), or 20C2HA, (closed circles), or no mAb (open circles) were evaluated for outgrowth of bacteria during a 24 h period. At designated times, a subset of rats were euthanized, chambers removed and bacteria were quantified by serial dilution on TSA.
mAb CS-D7 does not Measurably Inhibit Heme Uptake from Hemoglobin In vitro
IsdB is a bacterial surface protein which functions to capture heme, a source of iron, from hemoglobin for import into the bacteria (Mazmanian et al., 2003). An antibody which could block this function of the antigen might possess enhanced efficacy against the bacteria (Kim et al., 2010). It is known that CS-D7 can bind equivalently to both heme free and heme replete IsdB (unpublished data), indicating that the presence of heme does not adversely affect the epitope which CS-D7 recognizes. To investigate potential mAb interference of heme uptake, an assay was developed to determine if CS-D7 binding to heme free IsdB would prevent heme transfer from hemoglobin to the antigen (See Supplemental Data and Supplemental Table A1). For this evaluation, CS-D7 and control mAb 20C2HA were incubated separately with IsdB− (heme free IsdB) and the complexes were exposed to human hemoglobin immobilized on Sepharose®. Equal amounts of protein samples were combined with the hemoglobin resin (containing a 1:1 molar ratio of antibody to antigen) in each case. As indicated in Table 1, the IsdB− in samples 1 and 2 bound heme after interaction with the hemoglobin-Sepharose®. In addition to CS-D7, a second IsdB specific mAb was evaluated, mAb 2H2 (Brown et al., 2009). The sample containing 2H2 plus IsdB−, and the sample containing CS-D7 plus IsdB−, had equivalent A400/A280 ratios after exposure to hemoglobin (0.24 and 0.22 respectively), as the sample containing the non-specific control mAb 20C2HA plus IsdB− (0.23). This indicated that the presence of the two test mAbs did not prevent IsdB− from binding heme, under these in vitro conditions. This result did not rule out the possibility that CS-D7 and/or 2H2 caused the heme iron to bind at a position other than the NEAT domain (Grigg et al., 2007) of the antigen. Exposure of the hemoglobin-Sepharose® to antibody alone did not result in binding of heme to the mAb.
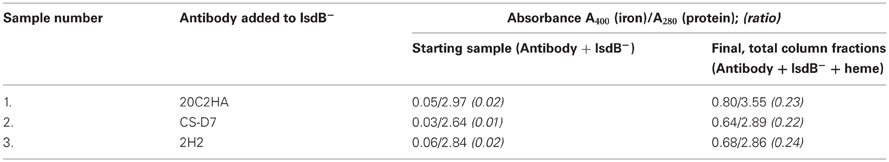
Table 1. Heme transfer from hemoglobin to heme minus IsdB (IsdB−) in the presence of mAbs.
Functionally Fc Defective Mutein CS-D7·N297A was Prepared for Evaluation of mAB OP Activity
Based on the above results, the mechanism of protection mediated by CS-D7 may not involve direct growth suppression of the bacteria, or inhibition of the physiological function of IsdB. Therefore, experiments were devised to determine if CS-D7 enhanced uptake and killing of S. aureusin vivo. To this end, an Fc mutant of CS-D7 (IgG1 isotype) was made. A point mutation was introduced which converted amino acid 297 from asparagine to alanine. Due to this mutation, the Fc cannot be glycosylated and therefore, loses its binding activity for Fcγ receptors and C1q (Lund et al., 1996). To verify that the mutein no longer bound to Fc, an in vitro ELISA to measure Fc receptor binding was employed, as described in Methods. Results from this assay indicated that while CS-D7 had robust binding to the Fc receptors tested, CS-D7·N297A had undetectable binding to either of the two receptors FcγRIIa or FcγRIIIa (Figures 2A,B).
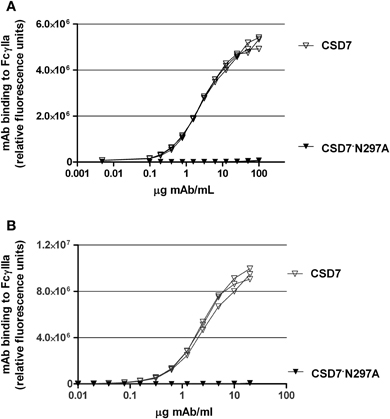
Figure 2. CS-D7 or CS-D7·N297A mAb binding to FcγRIIa or FcγRIIIa. Binding of mAbs to (A) FcγIIa or (B) FcγIIIa was measured as described in Methods. Data shown are individual curves for triplet determinations.
To verify that the mutein CS-D7·N297A had lost its ability to fix complement, an assay to measure C3b deposition was developed. This assay measured the generation of C3b in C1q replenished human serum, when incubated with mAb bound to a microtiter plate surface. When CS-D7 was evaluated for complement activation in this assay, robust generation of C3b was observed (Figure 3), which was similar in quantity to the C3b generated in the presence of licensed mAb Rituxan© (Genentech) (Zhou et al., 2008; Pawluczkowycz et al., 2009). However, in the presence of mutein CS-D7·N297A, generation of C3b was not observed. This indicated that the classical pathway of complement fixation, starting with binding of C1q to the Fc of the mAb, was not activated by CS-D7·N297A.
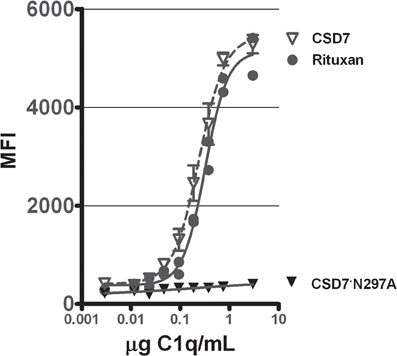
Figure 3. Stimulation of complement C3b formation in human plasma by mAbs CS-D7, CS-D7·N297A, or Rituxan©. Microtiter plates coated with one of three mAbs were exposed to C1q serially diluted into C1q deficient human sera. C3b generation was quantitated as described in Methods. Data shown are the mean ± s.e. n = 3.
OP activity was assessed in a third in vitro evaluation of functionality of CS-D7·N297A since the first two assays used to evaluate CS-D7·N297A were not cell based assays. Therefore, the mutein was compared to wild type CS-D7 in an OP assay, in the presence of HL60 phagocytic cells and fluorescently labeled S. aureus, as described in Methods (Figure 4). In this assay, there was a background level of approximately 20% fluorescent HL60 in the absence of antibody. The wild type CS-D7 had robust OP activity. In the presence of the maximal amount of 10 μg CS-D7mAb/mL, there was an increase of 50% of HL60 containing phagocytosed bacteria, above background levels. At the same concentration, only 10% of HL60 in the presence of the isotype control mAb, and 0% of HL60 in the presence of the mutein contained phagocytosed bacteria above the background level. Both the isotype control and the mutein raised levels of uptake to approximately 20% above background at a 10-fold higher concentration of antibody, but further increases in concentration did not increase uptake above that level. Therefore, in agreement with the analysis above, the mutein had dramatically less functional activity than the wild type mAb.
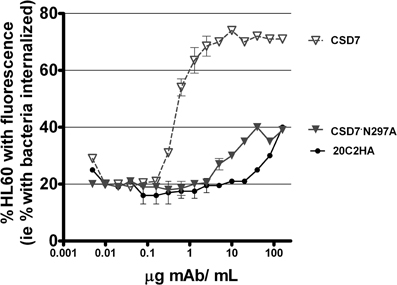
Figure 4. OP activity mediated by mAbs CS-D7, CS-D7·N297A, or isotype control 20C2HA. OP activity was measured by combining differentiated HL60 with fluorescently labeled opsonized S. aureus, incubating the mixture, and measuring HL60 with engulfed bacteria, as described in the Methods section. Data shown are the mean ± s.e. n = 2.
CS-D7 and Mutein CS-D7·N297A Confer Equivalent Protection in the Murine Sepsis Model
After establishing that the mutein had significantly reduced Fc functionality, the mutein and wild type mAbs were compared for efficacy in the murine sepsis model. Balb/c mice were passively immunized with 400 μg of CS-D7, or CS-D7·N297A, or the isotype control 20C2HA. Mice (n = 5) were challenged via the tail vein with S. aureus SA025, and were monitored for survival for 96 h (Table 2). The experiment was repeated several times and data were pooled for analysis. Unexpectedly, CS-D7 and the mutein had equivalent activity in this model (P = 0.4852), and were significantly superior to the isotype control mAb (P < 0.001).
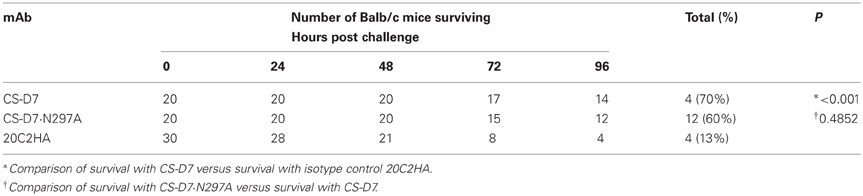
Table 2. Comparison of mAb efficacy in the murine sepsis model.
CS-D7 and CS-D7·N297A Confer Equivalent Protection in FcR−/− mice
Since the mutein was equally active in the murine sepsis model to the wild type CS-D7, it appeared that binding to Fcγ receptors and ability to fix complement were less important for the mechanism of protection in this model. To further determine whether binding to Fc receptors was non-essential for protection, mice genetically deficient for either the activating receptor (Fcγ IIII) or the inhibitory receptor (Fcγ RII) were investigated in the murine sepsis model. Mice (n = 5) were passively immunized i.p. with mAb prior to receiving a lethal dose of S. aureus via the tail vein. The experiment was repeated four to five times and data were combined for analysis. In these experiments, the wild type and the mutein had equivalent activity (Tables 3 and 4), which was significantly higher than for the control mAb 20C2HA. MAb mediated survival was lower in these deletion murine strains than in the wild type Balb/c mice. This is most likely due to increased sensitivity of the mice to S. aureus challenge.

Table 3. Passive immunization and challenge of FcγRIII−/− mice.
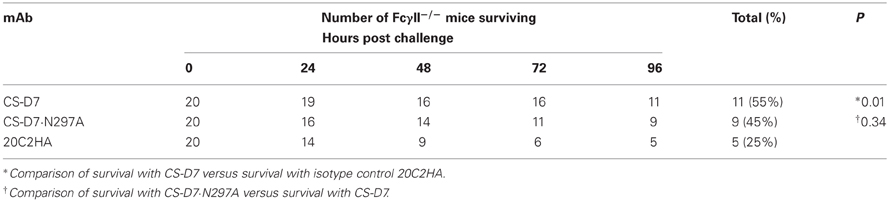
Table 4. Passive immunization and challenge of FcγII−/− mice.
Neither CS-D7 nor CS-D7·N297A Confer Protection in the Absence of an Intact Complement System, or in the Absence of Effector Cells with Functioning Oxidative Burst
To investigate whether the mechanism of CS-D7 efficacy in the murine sepsis model depended on phagocytes and complement, experiments were conducted to remove these components from mice prior to challenge. In the first case, Balb/c mice were depleted of C3 using CVF (Cunnion et al., 2004). CVF activates the alternative pathway and quickly depletes the host of C3, although not the components of the classical pathway. Mice were passively immunized and challenged as above. In mice depleted of C3, the mAbs were not protective (Table 5). To effectively remove neutrophils and other phagocytes with an oxidative burst mechanism from playing a role in mAb mediated survival, P47 phox−/− mice lacking a functional oxidative burst mechanism were employed. These mice were passively immunized and challenged, but they were not significantly protected by the mAbs (Table 6).

Table 5. Passive immunization and challenge of complement deficient mice.

Table 6. Passive immunization and challenge of P47 phox−/− (neutrophil impaired) mice.
mAb CS-D7 Significantly Reduces CFU Burden in Liver, but not in other Organs
To determine if CS-D7 enhanced bacteria clearance in the murine model, Balb/c mice were passively immunized with CS-D7 or isotype control mAb 20C2HA, or PBS alone, and challenged as above. At designated time points, mice were sacrificed and blood and organs were harvested. Due to increasing disease from S. aureus challenge in control mice, mice were not evaluated after 48 h post challenge for CFU burden. Survival was observed until 96 h post challenge in a subgroup of 10 mice (survival rate for CS-D7, 72%; 20C2HA, 28%; PBS, 39%). As shown in Figure 5, there were no significant differences in the blood burden (CFU/0.01mL) of the animals, nor in the bacteria burden in the filter organs, with the exception of a significant reduction in the liver of CS-D7 immunized animals at 1 h (P = 0.007) and 4 h post challenge (P = 0.015). There was a trend of CS-D7 mediating faster clearance in other organs, for example in the kidneys, but this difference did not reach statistical significance (P > 0.05). Interestingly there was an apparent higher level of CFU recovered from the spleens of animals injected with CS-D7 than Ab control animals at 48 h post challenge, however this difference was not statistically significant, and was no different from the PBS control animals.

Figure 5. Bacteria burden in murine tissue post immunization and challenge with S. aureus. Balb/c mice were passively immunized with CS-D7 (circles) or isotype control 20C2HA (squares) or PBS alone (triangles), and 2 h later were challenged via the tail vein with S. aureus SA025. At designated time points, subsets of animals were sacrificed and the blood and organs were harvested. Tissue bacterial burden was evaluated by homogenization of each organ in a total of 5 mL PBS and plating of 50 μL homogenate on TSA. The CFU count per 50 μL is plotted. For blood CFU determination, 10 μL of undiluted blood was plated. Data shown are means and ranges from mice (n = 4) in two experiments.
CS-D7 Mediated Enhanced Survival may Affect cytokine Levels Post Challenge in the Blood and the Organs
CS-D7 mediated efficacy may be reflected in a modulation of cytokine levels. To investigate this possibility, Balb/c mice were passively immunized and challenged as above. At designated time points, groups of mice were sacrificed and blood and tissue harvested. A selection of cytokines, representing T helper subsets, or having pro-inflammatory properties, were evaluated. Cytokines INFγ (Th1), IL17 (Th17), IL5 (Th2), IL6 (pro-inflammatory mediator), and KCGRO (pro-inflammatory chemokine) were evaluated post challenge. IL17 and IL5 concentrations did not change over time for both the blood and the organs, and were equivalent between CS-D7 treated mice and 20C2HA treated mice (data not shown). Of the cytokines IFN-γ, IL6, and KCGRO, there was a trend toward higher IFN-γ in mice treated with CS-D7. In the sera at 24 and 48 h post challenge (P = 0.03) (Table 7), and in the kidney (P = 0.05), liver (P = 0.008), and spleen (P = 0.08) at 24 h post challenge (Table 8), IFN-γ levels were higher in CS-D7 treated mice. There was also a trend toward lower IL6 levels, which was observed at 4 h (P = 0.005), 24 h (P = 0.0004), and 48 h in the sera, and at 1 h in the kidney (P = 0.011). At 24 h post challenge, KCGRO was reduced in the blood of CS-D7 treated mice. However, there was no trend with this cytokine at other times or in other organs.
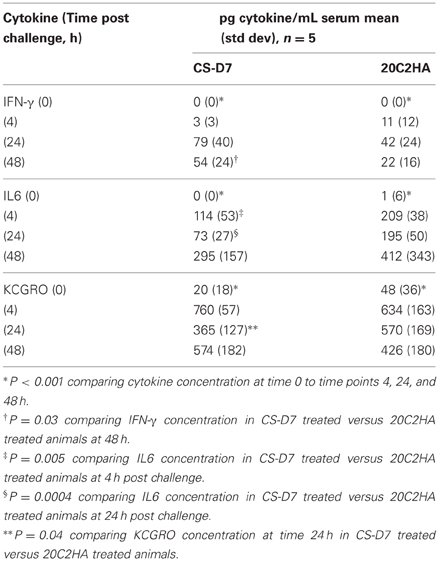
Table 7. Serum cytokine levels after passive immunization with one of two mAbs and challenge with S. aureus SA025.
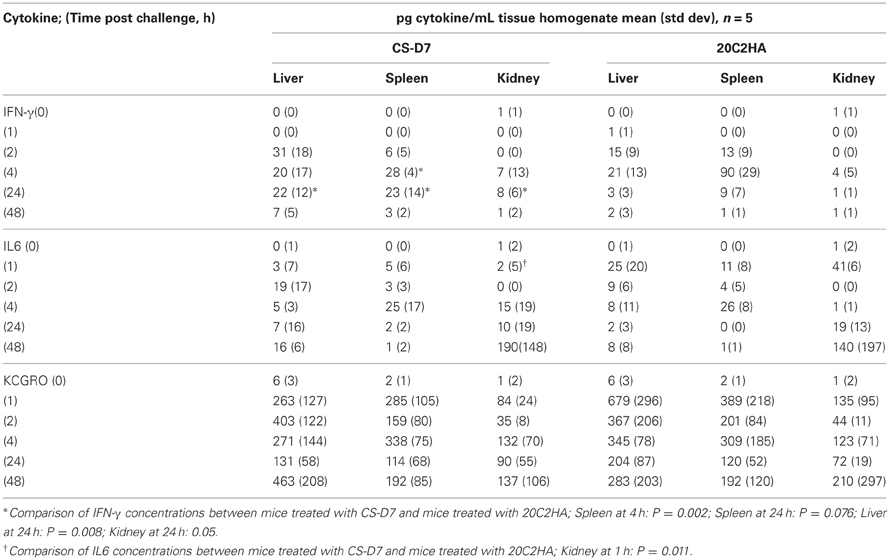
Table 8. Tissue homogenate cytokine levels after passive immunization with one of two mAbs and challenge with S. aureus SA025.
Discussion
Clearance of bacteria from the blood stream is of vital importance in host defense against disseminated infection. Bacteria removal and killing occurs after opsonization with antigen specific antibody and/or complement. Both the innate (e.g., complement) and the adaptive (e.g., antibody) arms of the immune system cooperate in this function, indicating its evolutionary importance. A central method of bacterial removal from the bloodstream is mediated through binding to CR1 on erythrocytes (Edberg et al., 1992; Lindorfer et al., 2001a,b). Complement (C3b) opsonized S. aureus is carried to the liver by binding to CR1 on erythrocytes, where bacteria are taken up and removed by phagocytic cells of the reticuloendothelial system. Kupffer cells express CRIg, which is believed to be the capture receptor of complement opsonized bacteria (Helmy et al., 2006). Opsonized bacteria may be passed from erythrocytes, or captured directly from blood, by Kupffer cells. CRIg binds both C3b and iC3b. Neutrophils are called to the liver by chemokines and cytokines such as IL8 and IL17. Upon arrival at sites of bacterial residence, neutrophils may release DNA nets to capture bacteria (Hickey and Kubes, 2009; Pilsczek et al., 2010). In the presence of antigen specific Ab, bacteria may be phagocytosed by different sets of effector cells, such as neutrophils and tissue macrophages, expressing both the FcγR and complement receptors. Binding of the two types of receptors is synergistic and greatly facilitates uptake and killing (Liszewski and Atkinson, 1993). Therefore, the presence of antigen specific Ab is a great advantage to the host for pathogen clearance. CS-D7 has high specificity and affinity for its target, S. aureus IsdB. IsdB is surface expressed in iron restricted environments, such as found in the human host, and as such has been proposed as a vaccine target (Kuklin et al., 2006; Stranger-Jones et al., 2006). Passively administered anti-IsdB Ab has demonstrated efficacy in rodent models (Ebert et al., 2010; Kim et al., 2010), however, in active immunization with IsdB, Ab response is not involved in protection in the murine sepsis model (Joshi et al., 2012). Therefore, it was of importance to determine the mechanism of protection mediated by the mAb CS-D7.
In an effort to determine if CS-D7 mediates protection by interfering with bacteria growth, S. aureus was cultured under in vivo conditions in the presence of CS-D7. The expression of IsdB is highly upregulated under this optimal in vivo environment with more than 50% of cells expressing antigen by 4 h, and essentially 100% of cells expressing the antigen by 8 h post implantation into rats. We found that during growth in implanted chambers, in which bacterial and host proteases may be present in high concentrations, the antigen remained available for antibody binding for the length of observation, 48 h. Expression of IsdB, in the presence of excess CS-D7, did not impact outgrowth of bacteria, implying that IsdB was not necessary for outgrowth under these conditions, or that the mAb binding to IsdB did not interfere with growth. However, the presence of CS-D7 did lead to transient aggregation of the bacteria under those conditions. It is possible that CS-D7 could mediate protection by interfering with heme uptake by the bacteria. The CS-D7 binding region on IsdB (aa 50–285) spans the domain 1 NEAT motif (aa 140–269) (Ebert et al., 2010). MAb binding to IsdB did not appear to interfere with heme transfer from human hemoglobin to IsdB in vitro. Since heme binding occurs through the domain 2 NEAT motif (aa 337–462), this may not be surprising (Muryoi et al., 2008). Kim and co-authors recently published data indicating that rabbit polyclonal antibodies to IsdB inhibited hemoglobin binding to IsdB. Their observations may relate to the use of polyclonal antibodies which would bind multiple epitopes on IsdB, whereas, CS-D7 recognizes a single epitope which may not obstruct hemoglobin binding.
Next, the functional activity of CS-D7 Fc was examined. Based on the current understanding of bacteria clearance, it was expected that CS-D7 would mediate protection through binding to its cognate antigen, inducing complement fixation through the classical pathway, and enhancing uptake and killing by phagocytes. To test this hypothesis, the mAb was mutated with a point mutation at aa 297. As expected, after mutation, the mutein did not mediate binding to FcγR nor did it fix complement in human serum (as measured by C3b production), whereas the wild type mAb did possess these functions. Activity in the OP assay was also dramatically reduced, essentially equivalent to the isotype control mAb. The mutein was then compared to the wild type mAb for protective efficacy. Unexpectedly, the mutein was as effective as the wild type mAb at enhancing survival in the murine sepsis model.
Further testing was performed to explore the mechanism of action of CS-D7 and the mutein. To confirm that Fc binding to FcγR was not critical for mAb efficacy, passive immunization experiments were conducted in FcγR deficient mice. In both FcγII−/− or FcγRIII−/− mice, both mAbs conferred equivalent protection, which was significantly more than the isotype control mAb. Overall survival in these deletion mice was lower than for wild type Balb/c mice. This may be due to defects in multiple cell types in the deletion mice (e.g., NK, macrophages, neutrophils, mast cells, and basophils in the FcγRIII−/−), amplifying the effects of the challenge dose. Based on the challenge experiments, mAb binding to either the stimulatory or the inhibitory FcγR was not essential for in vivo efficacy. It was demonstrated that mutein CS-D7.N297A did not fix C3b in vitro, in the presence of C1q replenished human sera. To confirm that activation of the classical pathway was not necessary for in vivo mAb efficacy, mouse complement component C3 was depleted using CVF (Liszewski and Atkinson, 1993). It was observed that in the absence of C3, neither CS-D7 nor CS-D7.N297A enhanced survival in the murine sepsis model. Therefore, C3 was a critical component for mAb mediated survival in this model. Since CVF also depletes the host of the alternative complement components, the presence of that pathway may also be important in this model.
Excess CS-D7, but not the isotype control, led to transient aggregation of S. aureus in the in vivo growth chamber, in parallel to an increase in IsdB expression. The aggregates are equivalent to IC. It is known that IC formation stimulates the activation of C3 to C3b and iC3b. In fact, one of the main functions of C3b is to bind to IC and reduce deposits of IC aggregates in small blood vessels (Liszewski and Atkinson, 1993). Therefore, CS-D7 may stimulate formation of C3b and iC3b through cross linking of the bacteria, as opposed to stimulation of the classical complement cascade. However, mere binding of a mAb to IsdB does not necessarily yield efficacy, as demonstrated previously for certain murine IsdB-specific mAbs (Brown et al., 2009). Therefore, the potential to bind IsdB in such a way as to produce cross linking of bacteria and complement activation may be important for efficacy.
Phagocytic cells were essential for mAb efficacy in vivo. When phagocytes (neutrophils and macrophages) were rendered non-functional by effectively deleting the oxidative burst mechanism (in P47 phox−/− mice), efficacy of the mAbs was abrogated. To further investigate bacteria clearance in the presence of CS-D7, blood and organ bacteria burden were measured post immunization and challenge. Differences in bacterial burden between CS-D7 treated and isotype control treated mice were observed in the liver, at 1 and 4 h post challenge. The liver is the primary organ responsible for clearance of blood born S. aureus, therefore, this observation suggests CS-D7 efficacy involves enhanced bacteria killing in the liver. Lack of more obvious differences in bacteria burden in other tissue may have several explanations. The S. aureus inoculum used to challenge mice in the sepsis model does not have IsdB expressed on its surface, due to culture of the bacteria on TSA. The expression of IsdB occurs over the course of several hours in vivo. Thus, antibody binding to IsdB on the bacteria takes place over several hours (Ebert et al., 2010). Additionally, a large challenge dose was necessary to produce sepsis in this model, and individual mice clear bacteria at different rates. Therefore, it may be difficult to capture a time at which expression of IsdB, leading to mAb mediated clearance of the bacteria, can be clearly observed; i.e., at any time, there may be only subtle differences in bacterial burden between CS-D7 and 20C2HA immunized animals. This subtle difference can be observed in the data shown in Figure 5, for example in the kidney. However differences in the CFU burden in these filter organs were not statistically significant. Lastly, although CFU measurements indicate similar numbers of viable bacteria, some or most of these may be sequestered in phagocytic cells in CS-D7 treated animals, potentially rendering them less able to cause pathologic sequelae leading to death of the animal.
Previously it was demonstrated that although CS-D7 confers enhanced survival in Balb/c mice, SCID mice (on a CB-17 background), lacking lymphocytes, were not protected after passive transfer of mAb (Joshi et al., 2012). There is increasing evidence that T cells play an active role in IgG mediated efficacy against pathogens such as Cryptococcus neoformans, Francisella tularensis, and Pseudomonas aeruginosa (Casadevall and Pirofski, 2003). T cell secretion of certain cytokines or chemokines may be essential to IgG mediated protection. IL17 is known to stimulate neutrophils, which is important for neutrophil opsonophagocytosis and killing. It has been demonstrated that IL17 is critical for protection from S. aureus infection in non-immunized mice, as well as for IsdB immunized mice (Ishigame et al., 2009; Cho et al., 2010; Joshi et al., 2012). Therefore, the concentrations of several cytokines including IL17 were evaluated in the blood and organs of passively immunized and challenged mice. In mice treated with CS-D7 there was a higher level of IFN-γ in the sera and organs, observed at 24 h post challenge. T cells are the major source for IFN-γ, thus confirming the observed essential need for T cells in this model. IFN-γ is a potent activator of phagocytic cells, especially macrophages, inducing increased microbicidal activity. This activity could explain the observed enhanced clearance of S. aureus in the liver. An additional possibility for the mechanism of CS-D7 would be to prevent or damp down a cytokine storm post challenge. In that case, pro-inflammatory cytokines should be decreased in blood and organs. Lower levels of IL6, a potent pro-inflammatory cytokine, were measured in the sera and kidneys. The observed reduction of IL6 levels support the existence of a reduced pro-inflammatory or acute phase response in the CS-D7 treated mice. There was a single time point at which reduced KCGRO was observed (at 24 h in the sera), but at all other time points levels in sera and tissues were not significantly different between CS-D7 treated animals and controls. This most likely reflects the importance for neutrophil recruitment and function in this model, in the presence of either CS-D7 or control mAb. Importantly, IL17 levels were not measurably stimulated in either the blood or tissues. Therefore, the role of IL17 is either not important, or is yet undefined for mAb mediated protection in this model. Additionally, the Th2 cytokine IL5 was not measurably stimulated in these animals.
In summary, we have demonstrated that CS-D7 mediated protection in the murine sepsis model is dependent on complement, phagocytes and lymphocytes, but was not dependent on Fc functionality, classical complement activation, or direct inhibition of bacterial growth. The mechanism of CS-D7 mediated protection in this model may be triggered by cross linking, or aggregation, of S. aureusin vivo. Although this mechanism may not seem relevant to physiological infections, it may play a previously un-appreciated role in bacterial clearance under certain circumstances. When bacteria are localized to a site in which replication leads to large numbers, Ab may bind and aggregate the cells, leading to enhanced complement deposition and uptake by neutrophils or tissue macrophages. Additionally, during certain bloodstream infections, bacteria cross-linked by Ab (IC) may become aggregated and enhance the deposition of complement. Therefore, IsdB specific Ab may play a role in the murine sepsis model, and perhaps in human infection, by enhancing complement fixation on invading S. aureus through a non-classical mechanism. Importantly, in vitro assays designed to evaluate the supposed function of Abs may be inadequate or even misleading for predicting in vivo efficacy. Therefore, careful investigation of the mechanism of efficacy should be performed in order to design appropriate in vitro assays for testing Ab potency.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
IsdB, iron regulated surface determinant B; OP, opsonophagocytic uptake activity; CVF, cobra venom factor; IC, immune complexes; ECD, extracellular domains.
References
Brown, M., Kowalski, R., Zorman, J., Wang, X. M., Towne, V., Zhao, Q. J., Secore, S., Finnefrock, A. C., Ebert, T., Pancari, G., Isett, K., Zhang, Y. H., Anderson, A. S., Montgomery, D., Cope, L., and McNeely, T. (2009). Selection and characterization of murine monoclonal antibodies to Staphylococcus aureus iron-regulated surface determinant B with functional activity in vitro and in vivo. Clin. Vaccine Immunol. 16, 1095–1104.
Casadevall, A., and Pirofski, L. A. (2003). Antibody-mediated regulation of cellular immunity and the inflammatory response. Trends Immunol. 24, 474–478.
Cho, J. S., Pietras, E. M., Garcia, N. C., Ramos, R. I., Farzam, D. M., Monroe, H. R., Magorien, J. E., Blauvelt, A., Kolls, J. K., Cheung, A. L., Cheng, G. H., Modlin, R. L., and Miller, L. S. (2010). IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest. 120, 1762–1773.
Cope, L., Fan, H., Wu, X., Ebert, T., Caulfield, M., and McNeely, T. (2008). Development of an assay to measure functional antibodies to Staphylococcus aureus IsdB. International Symposium on Staphylococci and Staphylococcal Infection, 2008.
Cunnion, K. M., Benjamin, D. K., Hester, C. G., and Frank, M. M. (2004). Role of complement receptors 1 and 2 (CD35 and CD21), C3, C4, and C5 in survival by mice of Staphylococcus aureus bacteremia. J. Lab. Clin. Med. 143, 358–365.
Ebert, T., Smith, S., Pancari, G., Clark, D., Hampton, R., Secore, S., Towne, V., Fan, H., Wang, X. M., Wu, X., Ernst, R., Harvey, B. R., Finnefrock, A. C., Wang, F., Tan, C., Durr, E., Cope, L., Anderson, A., An, Z., and McNeely, T. (2010). A fully human monoclonal antibody to Staphylococcus aureus iron regulated surface determinant B (IsdB) with functional activity in vitro and in vivo. Hum. Antibodies 19, 113–128.
Edberg, J. C., Kimberly, R. P., and Taylor, R. P. (1992). Functional-characterization of nonhuman primate erythrocyte immune adherence receptors-implications for the uptake of immune-complexes by the cells of the mononuclear phagocytic system. Eur. J. Immunol. 22, 1333–1339.
Fattom, A. I., Sarwar, J., Ortiz, A., and Naso, R. (1996). A Staphylococcus aureus capsular polysaccharide (CP) vaccine and CP-specific antibodies protect mice against bacterial challenge. Infect. Immun. 64, 1659–1665.
Gregory, S. H., Sagnimeni, A. J., and Wing, E. J. (1996). Bacteria in the bloodstream are trapped in the liver and killed by immigrating neutrophils. J. Immunol. 157, 2514–2520.
Grigg, J. C., Vermeiren, C. L., Heinrichs, D. E., and Murphy, M. E. P. (2007). Haem recognition by a Staphylococcus aureus NEAT domain. Mol. Microbiol. 63, 139–149.
Helmy, K. Y., Katschke, J., Gorgani, N. N., Kljavin, N. M., Elliott, J. M., Diehl, L., Scales, S. J., Ghilardi, N., and Lookeren Campagne, M. (2006). CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 124, 915–927.
Hickey, M. J., and Kubes, P. (2009). Intravascular immunity: the host-pathogen encounter in blood vessels. Nat. Rev. Immunol. 9, 364–375.
Ishigame, H., Kakuta, S., Nagai, T., Kadoki, M., Nambu, A., Komiyama, Y., Fujikado, N., Tanahashi, Y., Akitsu, A., Kotaki, H., Sudo, K., Nakae, S., Sasakawa, C., and Iwakura, Y. (2009). Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30, 108–119.
Joshi, A., Pancari, G., Cope, L., Bowman, E. P., Cua, D. J., Proctor, R. A., and McNeely, T. (2012). Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model. Hum. Vaccin. Immunother. 8, PMID 22327491. [Epub ahead of print].
Kim, H. K., DeDent, A., Cheng, A. G., McAdow, M., Bagnoli, F., Missiakas, D. M., and Schneewind, O. (2010). IsdA and IsdB antibodies protect mice against Staphylococcus aureus abscess formation and lethal challenge. Vaccine 28, 6382–6392.
Kuklin, N. A., Clark, D. J., Secore, S., Cook, J., Cope, L. D., McNeely, T., Noble, L., Brown, M. J., Zorman, J. K., Wang, X. M., Pancari, G., Fan, H. X., Isett, K., Burgess, B., Bryan, J., Brownlow, M., George, H., Meinz, M., Liddell, M. E., Kelly, R., Schultz, L., Montgomery, D., Onishi, J., Losada, M., Martin, M., Ebert, T., Tan, C. Y., Schofield, T. L., Nagy, E., Meineke, A., Joyce, J. G., Kurtz, M. B., Caulfield, M. J., Jansen, K. U., McClements, W., and Anderson, A. S. (2006). A novel Staphylococcus aureus vaccine: iron surface determinant B induces rapid antibody responses in rhesus macaques and specific increased survival in a murine S. aureus sepsis model. Infect. Immun. 74, 2215–2223.
Lee, J. C. (1996). The prospects for developing a vaccine against Staphylococcus aureus. Trends Microbiol. 4, 162–166.
Leijh, P. C. J., Vandenbarselaar, M. T., Daha, M. R., and Vanfurth, R. (1981). Participation of immunoglobulins and complement components in the intracellular killing of Staphylococcus aureus and Escherichia coli by human granulocytes. Infect. Immun. 33, 714–724.
Li, H., Sethuraman, N., Stadheim, T. A., Zha, D., Prinz, B., Ballew, N., Bobrowicz, P., Choi, B. K., Cook, W. J., Cukan, M., Houston-Cummings, N. R., Davidson, R., Gong, B., Hamilton, S. R., Hoopes, J. P., Jiang, Y., Kim, N., Mansfield, R., Nett, J. H., Rios, S., Strawbridge, R., Wildt, S., and Gerngross, T. U. (2006). Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat. Biotechnol. 24, 210–215.
Lindorfer, M. A., Hahn, C. S., Foley, P. L., and Taylor, R. P. (2001a). Heteropolymer-mediated clearance of immune complexes via erythrocyte CR1: mechanisms and applications. Immunol. Rev. 183, 10–24.
Lindorfer, M. A., Nardin, A., Foley, P. L., Solga, M. D., Bankovich, A. J., Martin, E. N., Henderson, A. L., Price, C. W., Gyimesi, E., Wozencraft, C. P., Goldberg, J. B., Sutherland, W. M., and Taylor, R. P. (2001b). Targeting of Pseudomonas aeruginosa in the bloodstream with bispecific monoclonal antibodies. J. Immunol. 167, 2240–2249.
Liszewski, M. K., and Atkinson, J. P. (1993). “The complement System,” in Fundamental Immunology, ed W. E. Paul (New York, NY: Raven Press Ltd.), 917–939.
Lowy, F. D. (1998b). Medical progress-Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532.
Lund, J., Takahashi, N., Pound, J. D., Goodall, M., and Jefferis, R. (1996). Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc gamma receptor I and influence the synthesis of its oligosaccharide chains. J. Immunol. 157, 4963–4969.
Mazmanian, S. K., Skaar, E. P., Gaspar, A. H., Humayun, M., Gornicki, P., Jelenska, J., Joachmiak, A., Missiakas, D. M., and Schneewind, O. (2003). Passage of heme-iron across the envelope of Staphylococcus aureus. Science 299, 906–909.
Muryoi, N., Tiedemann, M. T., Pluym, M., Cheung, J., Heinrichs, D. E., and Stillman, M. J. (2008). Demonstration of the iron-regulated surface determinant (Isd) heme transfer pathway in Staphylococcus aureus. J. Biol. Chem. 283, 28125–28136.
Pawluczkowycz, A. W., Beurskens, F. J., Beum, P. V., Lindorfer, M. A., van de Winkel, J. G., Parren, P. W., and Taylor, R. P. (2009). Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J. Immunol. 183, 749–758.
Peterson, P. K., Wilkinson, B. J., Kim, Y., Schmeling, D., Douglas, S. D., Quie, P. G., and Verhoef, J. (1978). Key role of peptidoglycan in opsonization of Staphylococcus aureus. J. Clin. Invest. 61, 597–609.
Philippe, G. (2004). Complement: a unique innate immune sensor for danger signals. Mol. Immunol. 41, 1089–1098.
Pilsczek, F. H., Salina, D., Poon, K. K., Fahey, C., Yipp, B. G., Sibley, C. D., Robbins, S. M., Green, F. H., Surette, M. G., Sugai, M., Bowden, M., Hussain, M., Zhang, K., and Kubes, P. (2010). A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 185, 7413–7425.
Roozendaal, R., and Carroll, M. C. (2006). Emerging patterns in complement-mediated pathogen recognition. Cell 125, 29–32.
Schaffer, A. C., and Lee, J. C. (2008). Vaccination and passive immunisation against Staphylococcus aureus. Int. J. Antimicrob. Agents 32, S71–S78.
Shields, R. L., Namenuk, A. K., Hong, K., Meng, Y. G., Rae, J., Briggs, J., Xie, D., Lai, J., Stadlen, A., Li, B., Fox, J. A., and Presta, L. G. (2001). High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 276, 6591–6604.
Spellberg, B., Ibrahim, A. S., Yeaman, M. R., Lin, L., Fu, Y., Avanesian, V., Bayer, A. S., Filler, S. G., Lipke, P., Otoo, H., and Edwards, J. E. Jr. (2008). The antifungal vaccine derived from the recombinant N terminus of Als3p protects mice against the bacterium Staphylococcus aureus. Infect. Immun. 76, 4574–4580.
Stranger-Jones, Y. K., Bae, T., and Schneewind, O. (2006). Vaccine assembly from surface proteins of Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 103, 16942–16947.
Thwaites, G. E., Edgeworth, J. D., Gkrania-Klotsas, E., Kirby, A., Tilley, R., Török, M. E., Walker, S., Wertheim, H. F., Wilson, P., and Llewelyn, M. J. (2011). Clinical management of Staphylococcus aureus bacteraemia. Lancet Infect. Dis. 11, 208–222.
Verbrugh, H. A., Peterson, P. K., Nguyen, B. Y. T., Sisson, S. P., and Kim, Y. (1982). Opsonization of encapsulated Staphylococcus aureus the role of specific antibody and complement. J. Immunol. 129, 1681–1687.
Verdrengh, M., and Tarkowski, A. (1997). Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect. Immun. 65, 2517–2521.
Vernachio, J. H., Bayer, A. S., Ames, B., Bryant, D., Prater, B. D., Syribeys, P. J., Gorovits, E. L., and Patti, J. M. (2006). Human immunoglobulin G recognizing fibrinogen-binding surface proteins is protective against both Staphylococcus aureus and Staphylococcus epidermidis infections in vivo. Antimicrob. Agents Chemother. 50, 511–518.
Zhou, X., Hu, W., and Qin, X. (2008). The role of complement in the mechanism of action of rituximab for B-cell lymphoma: implications for therapy. Oncologist 13, 954–966.
Appendix
Heme binding uptake assay
An assay was developed to assess whether IsdB specific mAb CS-D7 could block IsdB from binding to immobilized heme (containing iron). Reagents used in the binding assay included IsdB prepared free of heme (IsdB−), or containing heme, (IsdB+), human hemoglobin prepared from fresh erythrocytes, hemoglobin:Sepharose® (hemoglobin was cross-linked to activated Sepharose® after preparation from fresh human packed red blood cells), test mAb and isotype matched negative control mAb 20C2HA. The antibody and IsdB− were combined, allowing the antibody to bind to the antigen. This combination was then exposed to hemoglobin (via passage over a hemoglobin:Sepharose® column) to determine whether the test antibody could prevent heme transfer from the hemoglobin to the IsdB− antigen. The IsdB− or IsdB−/antibody passing through the hemoglobin column was evaluated for acquisition of heme (iron) by monitoring the antigen complex at 400 nm (the peak of absorbance for iron, no absorbance by protein). Column fractions were also monitored for protein content by measuring absorbance at 280 nm (no iron absorption at this wavelength). The assay procedure was as follows: IsdB− was combined in a 1:1 molar ratio with one of the monoclonal antibodies. After a short incubation, the antigen/antibody complex was applied to the hemoglobin-Sepharose column. Column flow through was collected and monitored for absorbance at 400 nm, and at 280 nm.
In preliminary experiments, IsdB− was evaluated for acquisition of heme iron from human hemoglobin in the absence of Ab. IsdB− was passed over a hemoglobin:Sepharose® column (sample 1, Table A1). The absorbance at 400 nm indicated that the initially heme free IsdB− contained little iron until the protein was exposed to immobilized hemoglobin. It was observed that although a portion of the IsdB− passed through the column (not binding to the matrix), a portion bound tightly to the column (sample 1, Table A1). In further tests, it was determined that 8M urea was required to completely elute the sample from the hemoglobin-Sepharose®. Removal of hemoglobin from the gel matrix by 8M urea was minimal (as determined by SDS PAGE). Urea did completely remove the starting sample (as demonstrated by SDS PAGE). Both the IsdB− flowing through the column, and the urea eluted IsdB− demonstrated an increase in absorbance at 400 nm, indicating acquired iron. Absorbance at A400 for both flow through and elution fractions was essentially at the background level if mAb CS-D7 was applied to the column in the absence of IsdB−.
A fully heme loaded IsdB+ sample was also applied to the hemoglobin column (sample 2, Table A1), and it was observed that this material consisted of two fractions also, one which bound tightly to the column, and one which did not bind. These data indicated that the heme loaded form of IsdB behaved similarly to the heme free antigen with regard to interaction with the immobilized hemoglobin, as did the heme free antigen. For simplification purposes, the two fractions were combined (i.e., for analysis of results, the flow through was combined with the urea eluted sample, see text and Table 1).

Table A1. IsdB interaction with immobilized human hemoglobin.
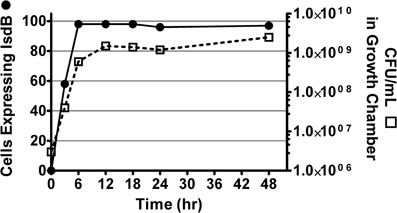
Figure A1. S. aureus cultured in growth chambers in vivo. S. aureus Becker was placed in growth chambers made from dialysis tubing aseptically placed under the skin of rats. At designated times, rats were sacrificed and growth chambers removed for quantitation of CFU (squares) by serial dilution and plating on TSA, and for quantitation of surface expression of IsdB (circles) by flow cytometry (% of cells positive for IsdB).
Keywords: iron regulated surface determinant B (IsdB), Staphylococcus aureus, vaccination, passive immunization, opsonophagocytosis
Citation: Pancari G, Fan H, Smith S, Joshi A, Haimbach R, Clark D, Li Y, Hua J, McKelvey T, Ou Y, Drummond J, Cope L, Montgomery D and McNeely T (2012) Characterization of the mechanism of protection mediated by CS-D7, a monoclonal antibody to Staphylococcus aureus iron regulated surface determinant B (IsdB). Front. Cell. Inf. Microbio. 2:36. doi: 10.3389/fcimb.2012.00036
Received: 13 December 2011; Accepted: 03 March 2012;
Published online: 20 March 2012.
Edited by:
David Heinrichs, University of Western Ontario, CanadaReviewed by:
Olaf Schneewind, University of Chicago, USAJean C. Lee, Brigham and Women's Hospital, USA
Robert S. Daum, University of Chicago, USA
Copyright: © 2012 Pancari, Fan, Smith, Joshi, Haimbach, Clark, Li, Hua, McKelvey, Ou, Drummond, Cope, Montgomery and McNeely. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Tessie McNeely, Department of Vaccine Basic Research, Merck Research Labs, Merck/MSD, WP26-253, West Point, PA 19486, USA. e-mail: tessie_mcneely@merck.com