Evolutionary blueprint for host- and niche-adaptation in Staphylococcus aureus clonal complex CC30
- 1Department of Microbiology, Schulich School of Medicine and Dentistry, Siebens Drake Research Institute and Centre for Human Immunology, University of Western Ontario, London, ON, Canada
- 2Department of Molecular and Cellular Biology, University of Guelph, Guelph, ON, Canada
Staphylococcus aureus clonal complex CC30 has caused infectious epidemics for more than 60 years, and, therefore, provides a model system to evaluate how evolution has influenced the disease potential of closely related strains. In previous multiple genome comparisons, phylogenetic analyses established three major branches that evolved from a common ancestor. Clade 1, comprised of historic pandemic phage type 80/81 methicillin susceptible S. aureus (MSSA), and Clade 2 comprised of contemporary community acquired methicillin resistant S. aureus (CA-MRSA) were hyper-virulent in murine infection models. Conversely, Clade 3 strains comprised of contemporary hospital associated MRSA (HA-MRSA) and clinical MSSA exhibited attenuated virulence, due to common single nucleotide polymorphisms (SNP's) that abrogate production of α-hemolysin Hla, and interfere with signaling of the accessory gene regulator agr. We have now completed additional in silico genome comparisons of 15 additional CC30 genomes in the public domain, to assess the hypothesis that Clade 3 has evolved to favor niche adaptation. In addition to SNP's that influence agr and hla, other common traits of Clade 3 include tryptophan auxotrophy due to a di-nucleotide deletion within trpD, a premature stop codon within isdH encoding an immunogenic cell surface protein involved in iron acquisition, loss of a genomic toxin–antitoxin (TA) addiction module, acquisition of S. aureus pathogenicity islands SaPI4, and SaPI2 encoding toxic shock syndrome toxin tst, and increased copy number of insertion sequence ISSau2, which appears to target transcription terminators. Compared to other Clade 3 MSSA, S. aureus MN8, which is associated with Staphylococcal toxic shock syndrome, exhibited a unique ISSau2 insertion, and enhanced production of toxic shock syndrome toxin encoded by SaPI2. Cumulatively, our data support the notion that Clade 3 strains are following an evolutionary blueprint toward niche-adaptation.
Introduction
Society has become imbued with the Superbug label to define strains of antibiotic resistant bacteria that cause hospital-associated outbreaks of infection (Foster, 2004; Abbott, 2005; Brazier, 2008; Guo et al., 2011). This term, denoting the sudden emergence and spread of new antibiotic resistant strains, could also be applied to an historic global pandemic caused by a penicillin-resistant S. aureus clone known as phage type PT80/81, which emerged in Australia, Great Britain, and North America in the early 1950's (Rountree and Beard, 1958; Williams et al., 1959; Wormald, 1961; Tanimoto, 1962). The initial outbreaks occurred in hospitals, especially among newborns and nursing mothers, but quickly spread to the wider community, causing unusually severe invasive skin infections, and fatal sepsis or necrotizing pneumonia in young and healthy individuals (Hassall and Rountree, 1959). Although the pandemic dissipated after 10 years (∼1953–1963), concomitant with the introduction of methicillin, genetically related contemporary strains are prominent in both the community and health-care settings. These consist of clinical methicillin susceptible S. aureus (MSSA), the epidemic EMRSA-16 lineage of hospital associated MRSA (HA-MRSA) which has the Type II Staphylococcal cassette chromosome SCCmec element, and the hyper-virulent Southwest Pacific (SWP) clone of community associated MRSA (CA-MRSA) which, like other unrelated CA-MRSA, has Type IV SCCmec. All of these strains belong to clonal complex CC30 as determined by multi locus sequence typing (MLST) analysis (Robinson et al., 2005).
To better understand the evolutionary development of CC30, we recently employed comparative genome sequencing to evaluate nine CC30 strains (DeLeo et al., 2011), including the reference genome of MRSA 252, representing the EMRSA-16 clone of HA-MRSA (Holden et al., 2004; Lindsay and Holden, 2004). Phylogenetic analyses based on a contiguous 1.4 Mb region of each genome, or with concatenated nucleotide segments, supported the existence of three major branches that evolved from a common ancestor. Clade I consists of the historic PT80/81 pandemic. This clonal type is typically ST30spa43, as determined by MLST and staphylococcal Protein A (spa) gene typing, and possesses the Panton Valentin Leukotoxin (PVL), that is also characteristic of contemporary CA-MRSA, including the SWP clone, which is ST30spa19 and comprises Clade 2. Although temporally separated by nearly 50 years, Clades 1 and 2 share a number of common traits, which in addition to PVL, include abundant production of α-hemolysin Hla, elevated transcription of RNAIII encoded by the accessory gene regulator agr locus, and a hypervirulent trait in murine infection models (DeLeo et al., 2011).
Clade 3 is comprised of the EMRSA-16 clone of HA-MRSA, which is typically ST36spa16, and contemporary clinical methicillin susceptible S. aureus, which are often ST30spa33. Although Clade 3 strains exhibited attenuated virulence in murine infection models relative to Clades 1 and 2, these are still associated with a high burden of disease. EMRSA-16, which is known in the United States as USA200, has become one of the most successful HA-MRSA clones (Cox et al., 1995; Enright et al., 2000; Johnson et al., 2001; McDougal et al., 2003; Seybold et al., 2006; Fowler et al., 2007). Others defined an association of CC30 MSSA, frequently ST30spa33, with bacteremia, infective endocarditis, and osteomyelitis (Cassat et al., 2005; Fowler et al., 2007; Nienaber et al., 2011). Staphylococcal toxic shock syndrome, which emerged in the late 1970's (Altemeier et al., 1981), is also associated with CC30, and the tst gene encoding toxic shock syndrome toxin has a strong clonal association with CC30 nasal carriage and bacteremia isolates (Holtfreter et al., 2007). MSSA that resemble the EMRSA-16 clone were also commonly associated with asymptomatic nasal carriage in the United States (Kuehnert et al., 2006), and other studies concur that CC30 is a major clonal complex associated with nasal carriage (Feil et al., 2003; Melles et al., 2004; Kuehnert et al., 2006; Fowler et al., 2007; Ko et al., 2008; Melles et al., 2008). Therefore, although Clade 3 exhibits attenuated virulence in murine infection models, we proposed that the high burden of disease associated with these hospital associated strains could be due to the high incidence of colonization, affording more opportunity to cause infection.
Several observations support the contention that Clade 3 evolved to favor enhanced colonization, at the expense of attenuated virulence. Foremost, the genome of MRSA 252 has the highest content of pseudogenes compared to other S. aureus genomes (Holden et al., 2004), and gene decay is a major force in niche-adaptation of microbial pathogens (Moran and Plague, 2004). Most notable among the pseudogenes was a CAG to TAG transition at Gln113 of hla encoding α-hemolysin (Hla), which is a major lethal virulence factor of CA-MRSA (Bubeck Wardenburg et al., 2007). This mutation, which creates a premature stop codon, is broadly disseminated in Clade 3, including HA-MRSA and clinical MSSA (DeLeo et al., 2011). Clade 3 strains also possessed a single nucleotide polymorphism (SNP) in agrC of the accessory gene regulator agr locus, causing a Gly55 >Arg change in the AgrC sensor protein, leading to attenuated transcription of the RNAIII product that is needed to produce secreted virulence factors (DeLeo et al., 2011). Consequently, attenuated transcription of agr, and inability to produce Hla contribute to the attenuated virulence of Clade 3.
Other defining traits of genomes that are in transition toward niche adaptation include acquisition of mobile genetic elements, and amplification of insertion sequence (IS) elements (Moran and Plague, 2004). However, a common limitation of conducting multiple genome comparisons by mapping short sequence reads from multiple strains on to a known reference genome is that it may not detect large insertions or deletions that differentiate one or more strains from the reference genome. Accordingly, although tst encoding toxic shock syndrome toxin is associated with different S. aureus pathogenicity island (SaPI) structures, and has a strong clonal association with CC30, tst was not present in the MRSA 252 reference genome, and we failed to identify the relevant SaPI through multiple genome comparisons. Herein, we present a detailed analysis of the hypothesis that Clade 3 strains have evolved in favor of niche adaptation, by conducting in silico comparisons of 15 additional CC30 genomes that are available in the public domain. Our analysis of pseudogenes, SaPI and IS elements, and gene deletion events, support the hypothesis that Clade 3 is following an evolutionary blueprint towards host- and niche-adaptation.
Materials and Methods
Bacterial Strains and Growth Conditions
A description of CC30 strains that were used in this study for analysis of secreted proteins and PCR assays is provided in Table 1. In addition, S. aureus RN4220 was obtained from Richard Novick (Novick, 1991). When needed for production of secreted proteins, cultures were grown overnight in tryptic soy broth (TSB; Difco) supplemented with 0.25% glucose, then sub-cultured into 25 ml of fresh TSB in a 125 ml Erlenmeyer flask to an initial optical density of 0.01 (OD600 = 0.01), and grown for 18 h at 37°C on an orbital shaker at 150 rpm. To assess yefM-yoeB addiction module function, cells were grown on brain heart infusion (BHI) agar supplemented with 10 μg/ml erythromycin for plasmid maintenance, and 5 μm cadmium where indicated for induction of the Pcad promoter.
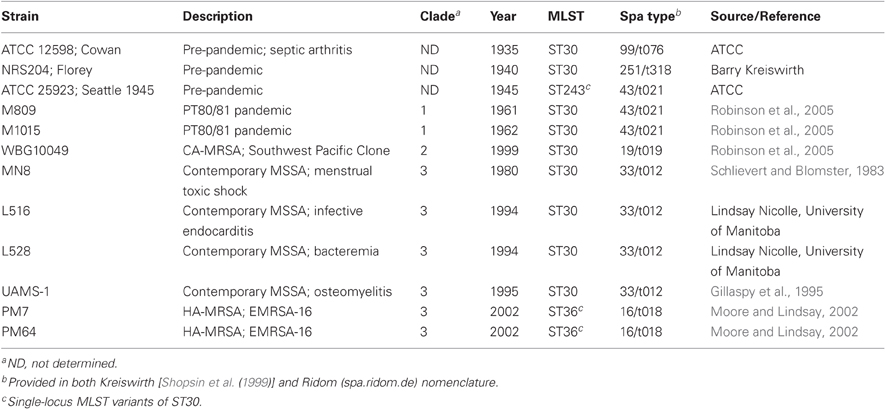
Table 1. Strains used in this study.
Genome Comparisons
Table 2 provides information on 19 CC30 strains for which genome sequence data are available in the public domain. Of these, MRSA 252 (Holden et al., 2004) was used as a reference genome for SNP analysis of multiple CC30 genomes (DeLeo et al., 2011), which in addition to MRSA 252 included three other strains listed in Table 2; M1015, WBG10049, and MN8. Of the genomes referred to in Table 2, those of TCH60 and MRSA 252 are assembled as a single nucleotide sequence, and all others are in assembly phase. Sequence coverage ranged from 10.5× (WW2703/97) to 36× (MN8). Unpublished genome data from strain UAMS-1 was provided by Dr.'s Mark Smeltzer and Jacques Schrenzel. The Basic Local Alignment Search Tool (BLAST) was used to query these genomes with segments of the annotated genome of MRSA 252 (Holden et al., 2004). Query segments were selected on the basis of SNP's, indels, or mobile genetic elements previously noted in the genome of MRSA 252, that were of discriminatory value in assigning evolutionary variants of CC30 (DeLeo et al., 2011). Genomes were also queried with the integrase (int) gene of known S. aureus pathogenicity islands (SaPI), to identify contigs that contained SaPI structures. Genome segments containing SaPI's and other mobile genetic elements or genes of interest were analyzed using MacVector version 7.2.3 software (Accelerys).
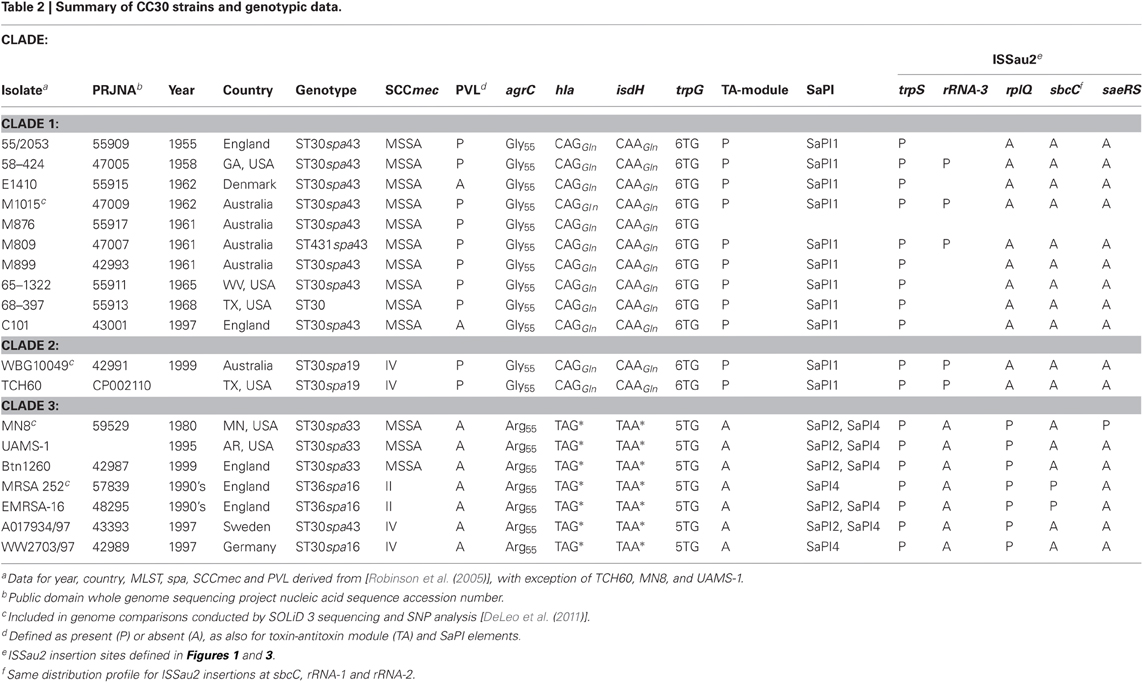
Table 2. Summary of CC30 strains and genotypic data.
PCR was employed to assess presence or absence of two unique ISSau2 insertions in different strains. One insertion adjacent to the saeRS regulatory locus was detected with primers SAR0758_For 5′-CAATATCGAACGCCACTTGAGC-3′, and SAR0757_Rev 5′-CAGCTATGATTGCAGGTTACCAGC-3′. Another insertion adjacent to the 5S-rRNA-3 site (Figure 1) was detected with primers SAR2148_For 5′-TTTCCCTCAACGTCCAGGTGC-3′ and 5S-rRNA-Rev 5′-GCCGAACACAGAAGTTAAGCTCC-3′. PCR was conducted with Roche AmpliTaq Gold DNA polymerase.
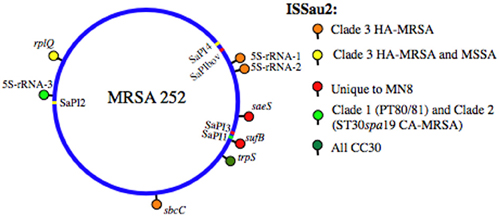
Figure 1. SaPI and ISSau2 distribution in CC30. The location of each element is mapped on the reference genome of MRSA 252 [Holden et al. (2004)]. SaPIs are indicated by colored bars, on the circular blue genome, and are labeled on the interior of the circular genome. SaPI2 and SaPI4 (yellow) are unique to Clade 3. SaPI1 (light green) is present in PT80/81 (Clade 1) and ST30spa19 CA-MRSA (Clade 2). SaPI3 and SaPIbov (red) are not in CC30, but their location is shown for reference, based on location of attS sites. ISSau2 insertions, indicated by colored lollipops above the circular genome, are named after the gene that is adjacent to each insertion. Three different 16S-23S-5S-rRNA loci are abbreviated as 5S-rRNA-1, 5S-rRNA-2, and 5S-rRNA-3.
Analysis of Secreted Proteins
Isolates representing different CC30 genotypes were cultured for 18 h in TSB, after which proteins in the cell-free culture supernatant were precipitated with trichloroacetic acid, and subjected to SDS-PAGE as described previously (Nickerson et al., 2010). For detection of Hla, Western blot assays were conducted with PVDF membrane (Pall Corporation), and rabbit anti-Staphylococcal α-toxin primary antibody (Sigma). The secondary antibody was donkey anti-rabbit IgG IR800 conjugate (Rockland Immunochemicals Inc.), and blots were visualized on an Odyssey Infrared Imager from LiCor Biosciences.
Identification of Coomassie-Blue stained proteins was conducted at the London Regional Proteomics Centre at the University of Western Ontario. Protein bands were excised using an Ettan™ Spot Picker, and processed for mass spectrometry using a Waters MASSPrep Automated Digestor as described (Gyenis et al., 2011). Processed samples were spotted on MALDI plates and analyzed on an Applied Biosystems 4700 Proteomics Analyzer. Data were acquired and processed using 4000 Series Explorer and Data Explorer (Applied Biosystems), and the peptide fingerprints were compared to the NCBInr database for Gram-positive bacteria, using the MASCOT search engine.
Cloning and Expression of yefM-YoeB Toxin–Antitoxin TA-Module
Gene segments were amplified by PCR, and cloned in pCN51 (Charpentier et al., 2004), for expression of the yefM-yoeB antitoxin-toxin genes together, or yoeB toxin on its own, from the cadmium inducible Pcad promoter. PCR was conducted using template DNA from S. aureus strain M1015, with primers YefM_For 5′-cgcggatccgttaactaattaaCAAAGGAGGGTTTATATGATTATC-3′, and YoeB_Rev 5′-ttggcgcgccTTAATCATAATGTGACCATGCCG-3′, generating a 538 nt product containing yefM-yoeB. The lower case nucleotides incorporate BamHI (ggatcc) or AscI (ggcgcgcc) restriction sites, and in YefM_For also add TAA stop codons in all three open reading frames prior to the AGGAGG ribosome binding site of yefM. For yoeB, primers YoeB_For 5′-cgcggatccgttaactaattaacaaaggagggtttatATGAGCAATTACACGGTTAAG-3′, and YoeB_Rev were used to generate a 266 nt product. The underlined lower case nucleotides in YoeB_For incorporate the ribosome binding site that precedes yefM, such that both constructs have identical Pcad promoter and translation initiation signals. PCR products were digested with BamHI and AscI, and ligated into pCN51 that had been digested with the same enzymes. The ligated plasmids were electroporated into restriction deficient S. aureus RN4220, and transformants were selected for growth on BHI agar containing 10 μg/ml erythromycin for maintenance of pCN51.
Results
CC30 Strains are Differentiated by Conserved Pseudogenes
Our present analysis reveals that previously emphasized defects in hla and agrC (DeLeo et al., 2011), co-associate with lesions in isdH and trpD (Table 2). IsdH is a cell surface protein that is highly expressed under iron-limiting conditions, and immunization with IsdH protects against S. aureus nasal carriage and bovine mastitis (Clarke et al., 2006; Pilpa et al., 2006; Ster et al., 2010). In EMRSA-16 and ST30spa33 MSSA, isdH has a SNP that converts CAAGln into a TAA stop codon (Table 2). An observation that many TSST producing strains are auxotrophic for tryptophan (Kreiswirth et al., 1989; Leung et al., 1993) is accounted for by a TG deletion in a 6×TG segment of trpD, in the trpEGDCFBA locus. Therefore, agrC, hla, isdH, and trpD are defective in contemporary MSSA and EMRSA-16 comprising Clade 3, but are functional in PT80/81 and ST30spa19 CA-MRSA comprising Clades 1 and 2 (Table 2). These latter strains also have PVL, which together with Type IV SCCmec is a trait that Clade 2 CA-MRSA shares with other unrelated CA-MRSA, such as USA300 and USA400. Other CC30 strains A01734/94 (ST30spa43), and WW2703/97 (ST30spa16) in Clade 3 are defined as CA-MRSA due to Type IV SCCmec, but they lack PVL, and have the “pseudogene package” (agrC, hla, isdH, and trpD) (Table 2), which we refer to as a niche-adapted trait.
S. aureus Pathogenicity Island (SaPI) Content
Figure 1 summarizes the distribution of insertion sequence ISSau2, and SaPI's in CC30. Irrespective of gene composition or genetic background, a SaPI is defined by a specific integrase int that recognizes an attS site (Lindsay and Holden, 2004; Novick and Subedi, 2007). Our analysis reveals that the attS sites are always located at the 3′-end of a gene, often in association with an operon, and these structures are illustrated in Figure 2. As noted previously (DeLeo et al., 2011), SaPI-4 differentiates Clade 3 from Clades 1 and 2. The attS of SaPI4 is at the 3′-end of the rpsF-ssb-rpsR operon, encoding ribosomal protein S6, a single stranded RNA binding protein, and ribosomal protein S18 (Figure 2A). SaPI4 does not have any known virulence factors, but SAR0385 encodes a protein with a signal peptide, identical to ORF011 of S. aureus phage ϕ1028 (Kwan et al., 2005). Outside of CC30, SaPI4 is restricted to ovine adapted S. aureus strains 011 and 046 (Le Marechal et al., 2011), which have an ortholog of SAR0385 (Figure 2A). In bovine adapted S. aureus ET3-1 (Herron-Olson et al., 2007), an ortholog of SAR0385 is on SaPIbov, where it is flanked by tst, sec, and sel encoding superantigen toxins (Figure 2A). The attS of SaPIbov spans the 3′- end of the xpt-pbuX-guaB-guaA operon, encoding genes for transport and metabolism of purine nucleotides. The rpsR and guaA genes, which contain the attS sites for SaPI4 and SaPIbov, respectively, are in close proximity (Figure 1), and segments of SaPIbov exhibit high similarity to ϕ1028, and SaPI4 (Figure 2A), but there are no genomes that have both SaPI's.
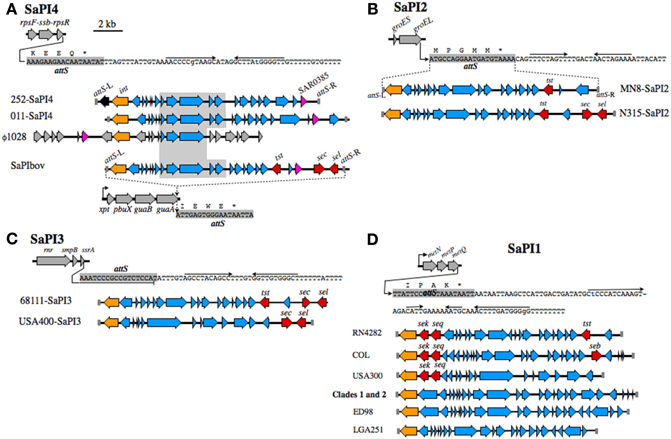
Figure 2. SaPI structures and insertion sites. (A) SaPI4, with an illustration of the rpsF-ssb-rpsR operon, and nucleotide sequence extending from the 3′-end of rpsR. The attS of SaPI4 shaded gray, overlaps the 3′-end of the rpsR open reading frame, and letters above the sequence correspond to the C-terminus of RpsR protein. Arrows above the nucleotide sequence indicate inverted repeats, likely comprising a transcriptional terminator. Illustrations below the sequence compare SaPI4 of S. aureus MRSA 252 with that of ovine adapted strain 011. The duplicated left and right attS sequences are shaded gray, int genes are colored orange, while SAR0385 and orthologous genes, encoding a putative secreted protein, are magenta. A central outlined and shaded segment of SaPI4 is highly conserved in S. aureus phage ϕ1028, and SaPIbov of bovine adapted S. aureus ET3-1. Genes in SaPIbov encoding superantigen toxins are shaded red. The int of SaPIbov is shaded orange to emphasize function, although each SaPI family has a distinct int and attS. Structures of SaPI2 (B), SaPI3 (C), SaPI1 (D), and their genomic insertion sites. Features are labeled as in A. SaPI2 of ST30spa33 strain MN8 (B), is compared to SaPI2 of a non-related MRSA strain N315. SaPI3 (C) is not present in CC30. Structures shown are from strain USA400 [Baba et al. (2002)], which is a CA-MRSA, and strain 68111, which is a triple locus MLST variant of ST30 [Li et al. (2011)]. A clonal complex is comprised of isolates that differ from the ancestral sequence type (ST) at no more than two of seven MLST alleles. Therefore, S. aureus 68111 is distantly related to ST30, but is not within CC30. (D) shows SaPI1 structures from different strains, in comparison to SaPI1 in Clade 1 PT80/81 strains, and Clade 2 ST30spa19 CA-MRSA.
In CC30, the toxic shock syndrome toxin tst is on SaPI2, where attS spans the 3′-end of groES-groEL (Figure 2B). In CC30, tst is the only toxin on SaPI2, but in unrelated HA-MRSA strains N315 and Mu50 (Kuroda et al., 2001), SaPI2 has additional superantigen toxins sec and sel (Figure 2B). In another non-CC30 strain, tst co-associates with sec and sel in SaPI3 (Li et al., 2011), where attS overlaps with the 3′-end of rnr-smpB-ssrA (Figure 2C). This operon encodes a ribonuclease (rnr), a non-translated RNA ssrA, and its cognate binding protein smpB. In the USA400 CA-MRSA, SaPI3 has superantigen toxins sec and sel, but not tst. However, CC30 genomes do not have SaPI3.
The first SaPI identified in S. aureus was SaPI1 in strain RN4282 (Lindsay et al., 1998; Ruzin et al., 2001), which has superantigen enterotoxins sek and seq at the 5′-end, and tst at the 3′-end (Figure 2D). The genome of RN4282 was not sequenced, but the flanking attS sequences of SaPI1 establish the integration site at the 3′-end of metNPQ, encoding a putative methionine transporter. Similar SaPI1 structures are in S. aureus COL and USA300, although these lack tst (Figure 2D). In CC30 SaPI1 is present in Clade 1 and 2 strains (Table 2), but does not have any obvious virulence factors. Similar SaPI1 structures that lack known virulence factors are in S. aureus ED98 (Lowder et al., 2009), which has undergone a recent evolutionary transition from human to poultry host, and in bovine adapted strain LGA251 (Garcia-Alvarez et al., 2011). In summary, although tst has been identified on SaPI1, SaPI2, SaPI3, and SaPIbov, depending on the genetic background, our data indicate that in CC30 it exclusively resides on SaPI2, where unlike other SaPI2 structures, tst does not co-associate with other superantigen toxins.
Analysis of ISSau2 Content
CC30 strains are distinguished by their profiles of ISSau2 (Figure 1), which is a member of the IS3 family (www-is.biotoul.fr/is.html). In E. coli, IS3 is flanked by imperfect inverted 39 nucleotide repeats with terminal 5′-TG and CA-3′ dinucleotides, and has two overlapping reading frames orfA and orfB, which when produced by default, prohibit transposition (Timmerman and Tu, 1985; Sekine et al., 1997). A –1 translational frame-shift within a poly-A tract in the region of overlap between orfA and orfB produces a single protein that catalyzes transposition (Prere et al., 1990; Sekine et al., 1994), with duplication of a three base pair target site (Sekine et al., 1994, 1997). ISSau2 has similar features (Figure 3A), and targets inverted repeats (Figures 3B–G), likely comprising rho-independent transcription terminators. An ancestral insertion common to CC30 genomes occurs in the intergenic segment separating trpS from the oppAFDBC oligopeptide permease operon (Figure 3B). ST30spa19 CA-MRSA and PT80/81 have one additional insertion, adjacent to a 16S-23S-5S rRNA operon (Figures 1 and 3C). Two other 16S-23S-5S-rRNA loci are targeted in EMRSA-16, which has a third unique insertion adjacent to sbcDC (Figures 1, and 3D,F). A fourth insertion adjacent to rplQ is also present in ST30spa33 MSSA (Figures 1 and 3E), including strain MN8, a prototypic menstrual toxic shock strain (Schlievert and Blomster, 1983). MN8 is distinguished from other ST30spa33 strains by two unique insertions (Figure 1), one of which is adjacent to saeRS (Figure 3G), encoding a two-component sensor signal transduction system that is a major regulator of virulence.
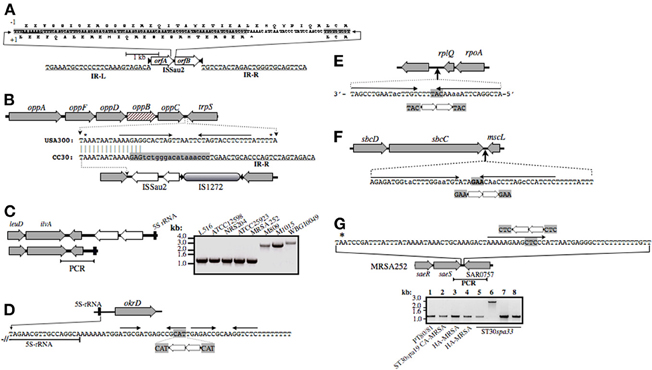
Figure 3. Illustration of ISSau2 (A) and its insertion sites (B–G) in CC30. ISSau2 is comprised of orfA and orfB(A), flanked by 39 nt inverted repeats IR-L and IR-R, with terminal 5′-TG and 3′-CA dinucleotides (A). The sequence above the illustration spans the 3′-end of orfA and 5′-end of orfB. The +1 translation of orfA, terminating at a stop codon, is shown below the sequence. Above the sequence is a translation that would result from a −1 frame-shift within AAAAAAG. The −1 translation from this point onwards continues throug to the end of orfB, and would produce a single 1569 nt trans-frame protein. (B) Illustration of oppAFBDC and trpS genome segment of non-CC30 strain USA300. Beneath this is shown the oppC-trpS intergenic sequence of USA300, aligned to that MRSA 252. Asterisks above the USA300 sequence indicate stop codons of oppC and trpS. Convergent arrows indicate inverted repeats, likely comprising a rho-independent transcription terminator stem-loop structure. In all CC30 strains, the left arm of the stem is disrupted by a flanking repeat of IS1272 (shaded gray), which in turn is disrupted by ISSau2. In MRSA 252, oppB (cross-hatched) has an in-frame internal deletion that is unique to the EMRSA-16 lineage. (C) ISSau2 insertion in the genome of S. aureus TCH60 (top), which is ST30spa19 CA-MRSA (Clade 2), and corresponding segment of MRSA 252 (bottom). PCR with primers spanning the 3′-end of the 5S rRNA and flanking ilvA (right panel) reveal that this insertion is in Clade 1 strains (M809, M1015) and another Clade 2 CA-MRSA (WBG10049), but not other CC30, including strains that pre-date the Clade 1 pandemic (ATCC12598, NRS204, ATCC25923). (D) ISSau2 insertion adjacent to a 16S-23S-5S rRNA operon, and flanking okrD gene, which is unique to Clade 3 HA-MRSA. The sequence below the illustration shows the end of the 5S rRNA transcript, and adjacent rho-independent transcriptional terminator, comprised of tandem stem-loops followed by a poly-T segment. ISSau2 disrupts the right arm of the first stem-loop, with duplication of the CAT target site. (E and F) show similar disruption of a putative stem-loop structure downstream of rplQ, and a likely transcription terminator of the sbcDC operon. (G) Unique ISSau2 insertion in ST30spa33 strain MN8 disrupts stem-loop structure adjacent to saeRS regulator. The saeRS genome segment of MRSA 252 is shown for reference, and the nucleotide sequence downstream of saeS is shown above the illustration. In strain MN8, ISSau2 inserts into the left arm of a putative stem-loop structure, with duplication of the CTC target site. This is confirmed by PCR of a genomic segment spanning saeS and adjacent SAR0757, producing a 2.8 kb amplicon in MN8 (Lane 6), and a 1.2 kb product in all other strains including PT80/81 strain M1015 (Lane 1), ST30spa19 CA-MRSA strain WBG10049 (Lane 2), ST36spa16 HA-MRSA strains PM7 and PM64 (Lanes 3 and 4), and additional ST30spa33 strains UAMS-1 (Lane 5), L516 (Lane 7), and L528 (Lane 8).
Outside of CC30, ISSau2 is restricted to animal adapted S. aureus. Ovine adapted ED133 has seven copies (Guinane et al., 2010), bovine strain LGA251 has three (Garcia-Alvarez et al., 2011), and porcine adapted ST398 has one (Schijffelen et al., 2010). The unassembled genomes of ovine strains 011 and 046 also have at least one copy (Le Marechal et al., 2011). In these animal adapted strains, the integration sites for ISSau2 are mutually exclusive of those in CC30.
Loss of a Toxin–Antitoxin Addiction Module in CC30 Evolution
Toxin-antitoxin (TA) modules encode a stable bactericidal or bacteriostatic toxin, and an unstable antitoxin that forms an inhibitory complex with the toxin. These were first termed addiction modules when discovered on plasmids, since loss of the plasmid during cell division leads to rapid degradation of the unstable antitoxin, followed by activation of the toxin and death of the daughter cells (Meinhart et al., 2003). Most free-living bacteria also have one or more genomic TA modules (Pandey and Gerdes, 2005). An example in S. aureus is mazEF, where MazF is an RNA'se that induces cell stasis by degradation of mRNA (Fu et al., 2007), and MazE is the antitoxin. Most other S. aureus genomes have another uncharacterized TA module, similar to yefM-yoeB in E. coli, where YoeB is a stasis-inducing RNA'se and YefM is the antitoxin (Kamada and Hanaoka, 2005). In ST30spa19 CA-MRSA and PT80/81, yefM-yoeB is flanked by frvX encoding an M42 metallopeptidase/endoglucanase protein family member, and a predicted glutamate synthase, gltS (Figure 4A). In MRSA 252, the flanking genes frvX (SAR2545) and gltS (SAR2547) are present, but not yefM-yoeB, and this is also characteristic of contemporary CC30 MSSA. Other genomes with this trait are restricted to ST398 porcine adapted S. aureus, ovine adapted strains 011 and 046, and as yet undefined strains A9635 and 21200.
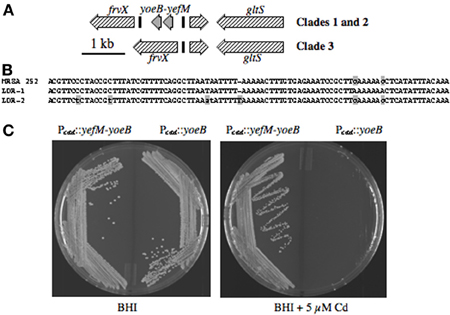
Figure 4. (A) Illustration of yefM-yoeB and flanking DNA in CC30. yefM-yoeB are flanked by repeats LDR-1 and LDR-2 (black bars) in PT80/81 and ST30spa19 CA-MRSA, whereas HA-MRSA and contemporary MSSA have only one LDR and lack yefM-yoeB. (B) Nucleotide sequence of LDR-1 and LDR-2 from strain M1015, and the single LDR of MRSA 252. The lower-case gray shaded nucleotides differentiate LDR-1 and LDR-2. The single LDR of MRSA 252 is a hybrid of LDR-1 and LDR-2, which is suggestive of a recombination event. (C) Growth of S. aureus RN4220 harboring Pcad::yefM-yoeB or Pcad::yoeB, on BHI agar, or BHI supplemented with 5 μM cadmium, to induce Pcad.
Although not annotated, yefM-yoeB is flanked by long direct repeats LDR-1 and LDR-2 (Figure 4B). CC30 genomes that lack yefM-yoeB have a single repeat identical to LDR-1, except for two SNP's at the 3′-end that match LDR-2, suggesting that deletion occurred by recombination between LDR-1 and LDR-2. To assess the function of yoeB, it was cloned by itself, or paired with yefM, in plasmid pCN51 under transcriptional control of Pcad. Cells of S. aureus RN4220 with either plasmid grew well on BHI agar, but on induction with cadmium, cells with yoeB alone did not grow (Figure 4C), confirming its function as a toxin, likely through degradation of mRNA to induce cellular stasis.
Production of Secreted Proteins
Clade 3 MSSA (ST30spa33) are recovered from a spectrum of conditions, including osteomyelitis (Cassat et al., 2005), infective endocarditis (Nienaber et al., 2011), bacteremia (Xiong et al., 2009), and menstrual toxic shock (Lin et al., 2011). Although these all have the same premature stop codon in hla, strains associated with menstrual toxic shock were reported to retain the ability to produce a small amount of Hla (Lin et al., 2011). Further, our data establish that MN8, which is a prototypic menstrual toxic shock strain (Altemeier et al., 1981), is differentiated from other Clade 3 MSSA by unique ISSau2 insertions (Figures 1,3 and Table 2), suggesting that it could also have unique phenotypic traits. We, therefore, evaluated production of secreted proteins in the major CC30 clonal types, including PT80/81 (Clade 1), ST30spa19 CA-MRSA (Clade 2), HA-MRSA (Clade 3), and Clade 3 MSSA recovered from menstrual toxic shock (MN8), osteomyelitis (UAMS-1), infective endocarditis (L516), and bacteremia (L528). Compared to other Clade 3 MSSA, MN8 exhibited more abundant production of secreted proteins (Figure 5A), and when compared to UAMS-1 on three separate occasions, it always produced more secreted protein (data not shown). However, irrespective of this difference, Hla was not detected in any of the ST30spa33 MSSA in a Western blot (Figure 5B).
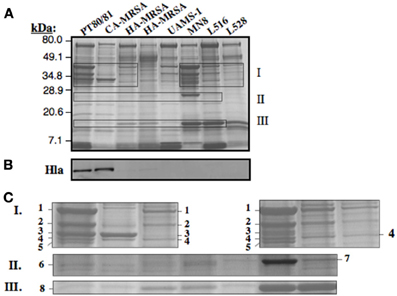
Figure 5. Assessment of CC30 secreted proteins by SDS-PAGE (A) and Western blot for detection of Hla (B). The individual strains are the same as defined in Figure 3G. For visualization of secreted proteins by Coomassie Blue staining (A), a total of 3.0 OD600 units of cell-free culture supernatant was applied to each lane, while 0.02 OD units was applied in Figure 3B. Zones I, II, and III as outlined in the SDS-PAGE gel (A) are enlarged in Figure 3C. The numbered protein bands were excised from the gel, followed by trypsin digestion and mass spectrometry. The identity of proteins in each band is provided in Table 3.
Proteins from zones I, II, and III on the SDS-PAGE (Figure 5A) were selected for trypsin digestion and mass spectrometry (Figure 5C and Table 3). HA-MRSA and ST30spa33 MSSA secreted toxic shock syndrome toxin TSST (Zone II, band 7), encoded by tst on SaPI2, and except for UAMS-1, they also secreted the SAR0385 gene product encoded on SaPI4 (Zone III, band 8). PT80/81 and CA-MRSA produced Hla (Zone I, band 3 and some carry-over in band 4), but Hla was not identified in the co-migrating protein bands from strain MN8. The PT80/81 strain was unique in abundant production of the LukF component of PVL (Zone I, band 2). All strains produced mature glycerol ester hydrolase/lipase (Zone I, band 1), γ-hemolysin components HglC and HglB (band 2 and carry-over in 3), and glycerolphosphoryl diester phosphodiesterase (band 4). With the exception of CA-MRSA, band 4 also contained the HglA component of γ-hemolysin. Therefore, although our data support the contention that Clade 3 MSSA associated with Staphylococcal toxic shock syndrome may be more virulent due to elevated production of secreted proteins, including γ-hemolysin, we found no evidence to support their production of Hla.
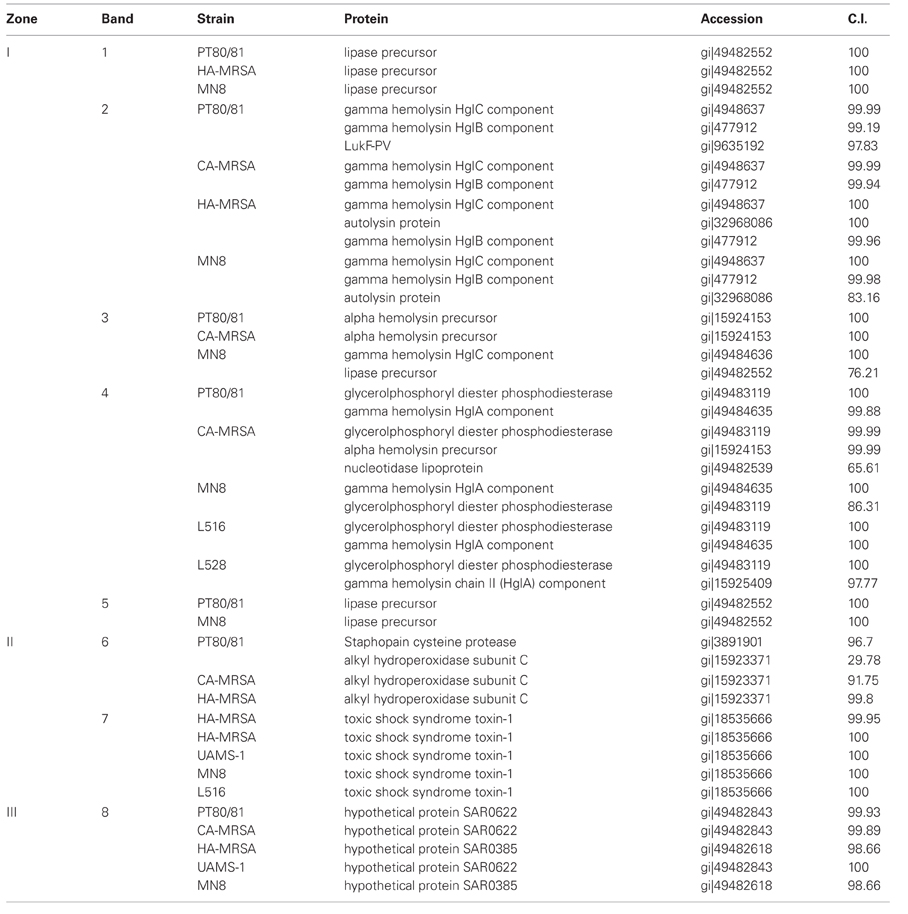
Table 3. Identity of proteins excised from SDS-PAGE (Figures. 5A and C).
Discussion
Gene gain, gene loss, and gene change are major forces in bacterial genome dynamics (Moran and Plague, 2004; Pallen and Wren, 2007), and we have evaluated these processes in S. aureus clonal complex CC30. It was previously established that MRSA 252, representing the EMRSA-16 lineage, has the highest content of pseudogenes compared to other S. aureus genomes (Holden et al., 2004; Lindsay and Holden, 2004). A bovine adapted strain ET3-1 was a close second, and several of the accumulated pseudogenes would eliminate production of a number of cell surface proteins and iron acquisition pathways (Herron-Olson et al., 2007). Importantly, the most robust examples of gene decay come from recently emerged pathogens that have changed lifestyle, usually to live in a simpler host-associated niche (Moran and Plague, 2004). As summarized in Table 2, defining traits of CC30 Clade 3 include (1), a premature stop codon in hla encoding α-Hemolysin; (2), a SNP that causes an Gly55>Arg substitution in the AgrC membrane sensor protein, leading to attenuated transcription of the regulatory RNAIII that is needed to produce secreted virulence factors; (3), isdH and trpD pseudogenes; (4), acquisition of SaPI4, and in most isolates also SaPI2, which possesses the tst gene encoding toxic shock syndrome toxin, and (5), an increase in copy number of ISSau2 relative to Clades 1 and 2. Our analyses have revealed several features that were not previously identified through comparative genome sequencing (DeLeo et al., 2011). These include the presence of SaPII and a functional TA genomic addiction module in Clades 1 and 2, the identification of SaPI2 in Clade 3, and the occurrence of unique ISSau2 insertions in each of the three major Clades. Cumulatively, these findings are concordant with niche adaptation in CC30 Clade 3.
It has long been known that 80% of TSST producing S. aureus strains are auxotrophic for tryptophan (Chu et al., 1985; Leung et al., 1993), which we now attribute to a TG deletion in trpD. It is believed that amino acid auxotrophy contributes to niche adaptation, because auxotrophic bacteria are restricted to a niche where the appropriate amino acid can be obtained. Lactococcus lactis recovered from dairy products are auxotrophic for histidine due to frame-shift mutations in hisC, hisG, and hisH, while strains from non-dairy sources are prototrophic (Delorme et al., 1993). S. aureus ET3-1, which is a predominant clonal type associated with bovine mastitis, also has a frame-shift mutation in hisC (SAB2553), which does not occur in other S. aureus genomes. The trpD gene, which has a TG insertion in CC30 Clade 3, is part of the trpEGDCFBA transcriptional unit, of which trpBA encode the subunits of tryptophan synthase, and trpEGDCF, encode enzymes necessary for synthesis of indole precursor. Chlamydia trachomatis uniformly lack the genes needed to produce indole, but strains that cause ocular vs. genital infections can be differentiated on the basis of the latter being able to produce a functional tryptophan synthase, and it is postulated that tryptophan can be produced by condensation of serine with exogenous indole produced by microflora in the female genital tract (Fehlner-Gardiner et al., 2002; McClarty et al., 2007). Consequently, tryptophan auxotrophy may contribute to tropism of TSST producing CC30 strains for the vaginal mucosa.
Another important factor in evolution of niche adapted strains is an increase in copy number of IS elements, leading to genome deletions and inversions through recombination between adjacent IS elements. An interesting example relevant to our analysis is a reduction in the numbers of operons encoding 16S-23S rRNA in microbial endosymbionts of insect cells (Andersson and Andersson, 1999; Itoh et al., 2002). This was attributed to IS integration within operons encoding 16S-23S rRNA, followed by recombination to generate deletions (Dale et al., 2003). It is, therefore, striking that all three 16S-23S-5S rRNA loci in the CC30 genome are targeted by ISSau2, with an insertion at rRNA-3 being unique to Clade 1, while insertions at rRNA-1 and rRNA-2 are unique to HA-MRSA in Clade 3 (Figure 1 and Table 2). Our analysis suggests that the insertions adjacent to rRNA operons is due to the propensity of ISSau2 to target inverted repeats, which likely comprise rho-independent transcription terminators. ISSau2 is a member of the IS30 family, and two unusual members of the IS30 family in Mycoplasma fermentans and M. bovis, which are obligate intracellular parasites, also target rho-independent transcription terminators, which remain intact and are partially duplicated on transposition (Calcutt et al., 1999; Lysnyansky et al., 2009). Conversely, our data suggest that ISSau2 either disrupts or weakens stem loop structures, as illustrated in Figures 3D–G.
Depending on the orientation of ISSau2 with respect to the adjacent gene, this could have important consequences with respect to control of transposition. It is widely accepted that transposition must be maintained at a low level, a commonly cited reason being that excessive transposition is detrimental to the stability of the host genome (Doolittle et al., 1984). Therefore, endogenous transposase promoters are generally weak, and often partially located in the inverted flanking repeats, such that strong promoters can only be created by juxtaposition of inverted repeats due to formation of head-to tail dimers, or circular copies of the IS as noted for the IS30 family (Dalrymple, 1987). IS elements also have mechanisms to attenuate their activation by impinging transcription, following insertion into active host genes. Impinging transcription across the inverted flanking repeats can either sequester translation initiation signals, or disrupt complex formation between the transposase and inverted repeats. These considerations may help to explain the insertion of ISSau2 adjacent to highly transcribed genes, including all three rRNA operons in the CC30 genome, and adjacent to rplQ encoding the 50S ribosomal subunit protein L17 (Figures 1 and 3). In these situations, ISSau2 is oriented in the antisense orientation with respect to the adjacent gene, such that impinging transcription would also generate antisense RNA to the transposase genes.
In an example that is unique to HA-MRSA in Clade 3 (Figure 1 and Table 2), ISSau2 is inserted in the sense orientation adjacent to sbcDC (Figure 3F), disrupting the predicted transcriptional terminator. The sbcDC genes encode a protein complex that recognizes and cleaves hairpin structures in DNA, has a major role in promoting genome stability and repair of breaks in double stranded DNA, and is induced by the SOS stress response in S. aureus (Connelly et al., 1998; Mascarenhas et al., 2006; Chen et al., 2007; Eykelenboom et al., 2008; Darmon et al., 2010). It is noteworthy in this respect that loss of DNA recombinational repair occurs in the initial stages of genome degeneration, as bacteria undergo a transition from an autonomous free-living state to permanent intracellular existence (Dale et al., 2003). This leads to active expansion of IS elements, which in turn promotes deletion or inversion of genome segments via IS-mediated recombination. Therefore, based on established evolutionary trends, the ISSau2 insertion adjacent to sbcDC may represent an early stage in the pathway toward genome destabilization.
Whether ISSau2 influences expression of adjacent genes is unknown. However, an insertion that is unique to strain MN8, from a case of menstrual Staphylococcal toxic shock, is adjacent to the saeRS two-component sensory signal transducer. SaeRS is a major regulator of virulence in S. aureus (Geiger et al., 2008; Voyich et al., 2009; Nygaard et al., 2010), and although there is as yet no evidence that ISSau2 influences expression of saeRS, we find that relative to ST30spa33 strains that lack this insertion, MN8 exhibits more abundant production of secreted proteins, including the HglA, HglB, and HglC components of γ-hemolysin, as well as TSST and SAR0385 gene product encoded on SaPI2 and SaPI4, respectively. This is consistent with a recent finding that, with the exception of strain MN8, other ST30spa33 strains and EMRSA-16 exhibited strongly attenuated transcription of the RNAIII effector component of the agr global regulator, due to a common SNP in agrC (DeLeo et al., 2011). Although this SNP is also present in strain MN8, transcription of RNAIII was not influenced to the same extent as other strains, suggesting that there was a compensatory mechanism in this strain. Additional work is warranted to determine if this is related to the ISSau2 insertion adjacent to saeRS.
The absence of the yefM-yoeB TA module (toxM) in Clade 3 is also consistent with established evolutionary pathways toward niche adaptation. Most free-living bacteria have multiple genomic TA loci, which are thought to help cope with nutritional stress by inducing a reversible state of dormancy during periods of nutrient depletion (Pedersen et al., 2002; Gerdes et al., 2005), although this has been disputed in E. coli (Tsilibaris et al., 2007). However, obligate intracellular pathogens and symbionts experience a less variable environment, and do not have TA modules (Pandey and Gerdes, 2005). Only a few free-living bacteria lack TA modules, the most notable being Lactococcus lactis, which is niche-adapted in its association with dairy products. Intriguingly, outside of CC30, the only other S. aureus genomes that lack this TA module thus far are restricted to ST398 porcine adapted S. aureus, ovine adapted strains 011 and 046, and as yet undefined strains A9635 and 21200. Although we cannot exclude the possibility that this TA module represents a gene acquisition in Clades 1 and 2, rather than a gene deletion in Clade 3, the broad distribution of this element in other S. aureus genomes supports the contention that this element is a component of the core genome that is lost in evolutionary development of some strains. However, more work is needed to confirm this hypothesis.
In conclusion, our findings support the notion that Clade 3 is following an evolutionary blueprint toward niche-adaptation, while Clade 2 strains consisting of ST30spa19 CA-MRSA retain the feral nature of the historic PT80/81 Clade 1. It is important to note that CA-MRSA are defined by Type IV SCCmec, which in CC30 is also associated with ST30spa43 and ST30spa16 strains in Clade 3 (Table 2). Therefore, strains of CA-MRSA, which are typically associated with hyper-virulence, are emerging with the niche-adapted trait. Given that CA-MRSA must evolve from MSSA through acquisition of Type IV SCCmec, this suggests that the niche-adapted trait is widely disseminated in the human population, and this is supported by several key observations. First, MSSA that resemble the EMRSA-16 clone of HA-MRSA (Clade 3) were the most common clonal type associated with asymptomatic nasal carriage in the United States (Kuehnert et al., 2006), and several other studies concur that CC30 is a major clonal complex associated with nasal carriage (Feil et al., 2003; Melles et al., 2004, 2008; Kuehnert et al., 2006; Fowler et al., 2007; Ko et al., 2008). Second, S. aureus infections are usually caused by the same strain that is associated with nasal carriage, and in our analysis of a panel of 172 CC30 clinical isolates, the occurrence of the hla pseudogene, the agrC SNP, and tst encoded by SaPI2, which are key markers of Clade 3 (Table 2), was 70.9%, 72.1%, and 75.6%, respectively, (DeLeo et al., 2011). Third, in a study that assessed nasal carriage isolates from 107 healthy blood donors, 27% were CC30, and 62% of these CC30 strains possessed tst (Holtfreter et al., 2007), which is a marker of Clade 3. Moreover, in the same study, tst was present in 90% of CC30 bacteremia isolates. In this context, although our studies indicate that the niche adapted trait is associated with attenuated virulence in murine infection models (Holtfreter et al., 2007), a benefit to Clade 3 in having premature stop codons in Hla and IsdH (Table 2) is that both proteins are considered as potential vaccine antigens (Clarke et al., 2006; Wardenburg and Schneewind, 2008; Kennedy et al., 2010; Ster et al., 2010), and Clade 3 strains would be immune to this vaccine strategy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by a grant to Martin J. McGavin from the Natural Sciences and Engineering Research Council of Canada.
References
Altemeier, W. A., Lewis, S., Schlievert, P. M., and Bjornson, H. S. (1981). Studies of the Staphylococcal causation of toxic shock syndrome. Surg. Gynecol. Obstet. 153, 481–485.
Andersson, J. O., and Andersson, S. G. (1999). Insights into the evolutionary process of genome degradation. Curr. Opin. Genet. Dev. 9, 664–671.
Baba, T., Takeuchi, F., Kuroda, M., Yuzawa, H., Aoki, K., Oguchi, A., Nagai, Y., Iwama, N., Asano, K., Naimi, T., Kuroda, H., Cui, L., Yamamoto, K., and Hiramatsu, K. (2002). Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359, 1819–1827.
Brazier, J. S. (2008). Clostridium difficile: from obscurity to superbug. Br. J. Biomed. Sci. 65, 39–44.
Bubeck Wardenburg, J., Bae, T., Otto, M., Deleo, F. R., and Schneewind, O. (2007). Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 13, 1405–1406.
Calcutt, M. J., Lavrrar, J. L., and Wise, K. S. (1999). IS1630 of Mycoplasma fermentans, a novel IS30-type insertion element that targets and duplicates inverted repeats of variable length and sequence during insertion. J. Bacteriol. 181, 7597–7607.
Cassat, J. E., Dunman, P. M., McAleese, F., Murphy, E., Projan, S. J., and Smeltzer, M. S. (2005). Comparative genomics of Staphylococcus aureus Musculoskeletal isolates. J. Bacteriol. 187, 576–592.
Charpentier, E., Anton, A. I., Barry, P., Alfonso, B., Fang, Y., and Novick, R. P. (2004). Novel cassette-based shuttle vector system for Gram-positive bacteria. Appl. Environ. Microbiol. 70, 6076–6085.
Chen, Z., Luong, T. T., and Lee, C. Y. (2007). The sbcDC locus mediates repression of type 5 capsule production as part of the SOS response in Staphylococcus aureus. J. Bacteriol. 189, 7343–7350.
Chu, M. C., Melish, M. E., and James, J. F. (1985). Tryptophan auxotypy associated with Staphylococcus aureus that produce toxic-shock-syndrome toxin. J. Infect. Dis. 151, 1157–1158.
Clarke, S. R., Brummell, K. J., Horsburgh, M. J., McDowell, P. W., Mohamad, S. A., Stapleton, M. R., Acevedo, J., Read, R. C., Day, N. P., Peacock, S. J., Mond, J. J., Kokai-Kun, J. F., and Foster, S. J. (2006). Identification of in vivo-expressed antigens of Staphylococcus aureus and their use in vaccinations for protection against nasal carriage. J. Infect. Dis. 193, 1098–1108.
Connelly, J. C., Kirkham, L. A., and Leach, D. R. (1998). The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc. Natl. Acad. Sci. U.S.A. 95, 7969–7974.
Cox, R. A., Conquest, C., Mallaghan, C., and Marples, R. R. (1995). A major outbreak of methicillin-resistant Staphylococcus aureus caused by a new phage-type (EMRSA-16). J. Hosp. Infect. 29, 87–106.
Dale, C., Wang, B., Moran, N., and Ochman, H. (2003). Loss of DNA recombinational repair enzymes in the initial stages of genome degeneration. Mol. Biol. Evol. 20, 1188–1194.
Dalrymple, B. (1987). Novel rearrangements of IS30 carrying plasmids leading to the reactivation of gene expression. Mol. Gen. Genet. 207, 413–420.
Darmon, E., Eykelenboom, J. K., Lincker, F., Jones, L. H., White, M., Okely, E., Blackwood, J. K., and Leach, D. R. (2010). E. coli SbcCD and RecA control chromosomal rearrangement induced by an interrupted palindrome. Mol. Cell 39, 59–70.
DeLeo, F. R., Kennedy, A. D., Chen, L., Bubeck Wardenburg, J., Kobayashi, S. D., Mathema, B., Braughton, K. R., Whitney, A. R., Villaruz, A. E., Martens, C. A., Porcella, S. F., McGavin, M. J., Otto, M., Musser, J. M., and Kreiswirth, B. N. (2011). Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 108, 18091–18096.
Delorme, C., Godon, J. J., Ehrlich, S. D., and Renault, P. (1993). Gene inactivation in Lactococcus lactis: histidine biosynthesis. J. Bacteriol. 175, 4391–4399.
Doolittle, W. F., Kirkwood, T. B., and Dempster, M. A. (1984). Selfish DNAs with self-restraint. Nature 307, 501–502.
Enright, M. C., Day, N. P., Davies, C. E., Peacock, S. J., and Spratt, B. G. (2000). Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38, 1008–1015.
Eykelenboom, J. K., Blackwood, J. K., Okely, E., and Leach, D. R. (2008). SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol. Cell 29, 644–651.
Fehlner-Gardiner, C., Roshick, C., Carlson, J. H., Hughes, S., Belland, R. J., Caldwell, H. D., and McClarty, G. (2002). Molecular basis defining human Chlamydia trachomatis tissue tropism. A possible role for tryptophan synthase. J. Biol. Chem. 277, 26893–26903.
Feil, E. J., Cooper, J. E., Grundmann, H., Robinson, D. A., Enright, M. C., Berendt, T., Peacock, S. J., Smith, J. M., Murphy, M., Spratt, B. G., Moore, C. E., and Day, N. P. (2003). How clonal is Staphylococcus aureus? J. Bacteriol. 185, 3307–3316.
Fowler, V. G. Jr., Nelson, C. L., McIntyre, L. M., Kreiswirth, B. N., Monk, A., Archer, G. L., Federspiel, J., Naidich, S., Remortel, B., Rude, T., Brown, P., Reller, L. B., Corey, G. R., and Gill, S. R. (2007). Potential associations between hematogenous complications and bacterial genotype in Staphylococcus aureus infection. J. Infect. Dis. 196, 738–747.
Fu, Z., Donegan, N. P., Memmi, G., and Cheung, A. L. (2007). Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189, 8871–8879.
Garcia-Alvarez, L., Holden, M. T., Lindsay, H., Webb, C. R., Brown, D. F., Curran, M. D., Walpole, E., Brooks, K., Pickard, D. J., Teale, C., Parkhill, J., Bentley, S. D., Edwards, G. F., Girvan, E. K., Kearns, A. M., Pichon, B., Hill, R. L., Larsen, A. R., Skov, R. L., Peacock, S. J., Maskell, D. J., and Holmes, M. A. (2011). Meticillin-resistant Staphylococcus aureus with a novel mecA homologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect. Dis. 11, 595–603.
Geiger, T., Goerke, C., Mainiero, M., Kraus, D., and Wolz, C. (2008). The virulence regulator Sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J. Bacteriol. 190, 3419–3428.
Gerdes, K., Christensen, S. K., and Lobner-Olesen, A. (2005). Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3, 371–382.
Gillaspy, A. F., Hickmon, S. G., Skinner, R. A., Thomas, J. R., Nelson, C. L., and Smeltzer, M. S. (1995). Role of the accessory gene regulator (agr) in pathogenesis of Staphylococcal osteomyelitis. Infect. Immun. 63, 3373–3380.
Guinane, C. M., Ben Zakour, N. L., Tormo-Mas, M. A., Weinert, L. A., Lowder, B. V., Cartwright, R. A., Smyth, D. S., Smyth, C. J., Lindsay, J. A., Gould, K. A., Witney, A., Hinds, J., Bollback, J. P., Rambaut, A., Penades, J. R., and Fitzgerald, J. R. (2010). Evolutionary genomics of Staphylococcus aureus reveals insights into the origin and molecular basis of ruminant host adaptation. Genome Biol. Evol. 2, 454–466.
Guo, Y., Wang, J., Niu, G., Shui, W., Sun, Y., Zhou, H., Zhang, Y., Yang, C., Lou, Z., and Rao, Z. (2011). A structural view of the antibiotic degradation enzyme NDM-1 from a superbug. Protein Cell 2, 384–394.
Gyenis, L., Duncan, J. S., Turowec, J. P., Bretner, M., and Litchfield, D. W. (2011). Unbiased functional proteomics strategy for protein kinase inhibitor validation and identification of bona fide protein kinase substrates: application to identification of as a substrate for CK2. J. Proteome Res. 10, 4887–4901.
Herron-Olson, L., Fitzgerald, J. R., Musser, J. M., and Kapur, V. (2007). Molecular correlates of host Specialization in Staphylococcus aureus. PLoS One 2:e1120. doi: 10.1371/journal.pone.0001120
Holden, M. T., Feil, E. J., Lindsay, J. A., Peacock, S. J., Day, N. P., Enright, M. C., Foster, T. J., Moore, C. E., Hurst, L., Atkin, R., Barron, A., Bason, N., Bentley, S. D., Chillingworth, C., Chillingworth, T., Churcher, C., Clark, L., Corton, C., Cronin, A., Doggett, J., Dowd, L., Feltwell, T., Hance, Z., Harris, B., Hauser, H., Holroyd, S., Jagels, K., James, K. D., Lennard, N., Line, A., Mayes, R., Moule, S., Mungall, K., Ormond, D., Quail, M. A., Rabbinowitsch, E., Rutherford, K., Sanders, M., Sharp, S., Simmonds, M., Stevens, K., Whitehead, S., Barrell, B. G., Spratt, B. G., and Parkhill, J. (2004). Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. U.S.A. 101, 9786–9791.
Holtfreter, S., Grumann, D., Schmudde, M., Nguyen, H. T., Eichler, P., Strommenger, B., Kopron, K., Kolata, J., Giedrys-Kalemba, S., Steinmetz, I., Witte, W., and Broker, B. M. (2007). Clonal distribution of superantigen genes in clinical Staphylococcus aureus isolates. J. Clin. Microbiol. 45, 2669–2680.
Itoh, T., Martin, W., and Nei, M. (2002). Acceleration of genomic evolution caused by enhanced mutation rate in endocellular symbionts. Proc. Natl. Acad. Sci. U.S.A. 99, 12944–12948.
Johnson, A. P., Aucken, H. M., Cavendish, S., Ganner, M., Wale, M. C., Warner, M., Livermore, D. M., and Cookson, B. D. (2001). Dominance of EMRSA-15 and -16 among MRSA causing nosocomial bacteraemia in the UK: analysis of isolates from the European Antimicrobial Resistance Surveillance System (EARSS). J. Antimicrob. Chemother. 48, 143–144.
Kamada, K., and Hanaoka, F. (2005). Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell 19, 497–509.
Kennedy, A. D., Bubeck Wardenburg, J., Gardner, D. J., Long, D., Whitney, A. R., Braughton, K. R., Schneewind, O., and Deleo, F. R. (2010). Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 202, 1050–1058.
Ko, K. S., Lee, J. Y., Baek, J. Y., Peck, K. R., Rhee, J. Y., Kwon, K. T., Heo, S. T., Ahn, K. M., and Song, J. H. (2008). Characterization of Staphylococcus aureus nasal carriage from children attending an outpatient clinic in Seoul, Korea. Microb. Drug Resist. 14, 37–44.
Kreiswirth, B. N., Projan, S. J., Schlievert, P. M., and Novick, R. P. (1989). Toxic shock syndrome toxin 1 is encoded by a variable genetic element. Rev. Infect. Dis. 11(Suppl. 1), S83–S88. discussion S88–S89.
Kuehnert, M. J., Kruszon-Moran, D., Hill, H. A., McQuillan, G., McAllister, S. K., Fosheim, G., McDougal, L. K., Chaitram, J., Jensen, B., Fridkin, S. K., Killgore, G., and Tenover, F. C. (2006). Prevalence of Staphylococcus aureus nasal colonization in the United States, 2001–2002. J. Infect. Dis. 193, 172–179.
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., Cui, L., Oguchi, A., Aoki, K., Nagai, Y., Lian, J., Ito, T., Kanamori, M., Matsumaru, H., Maruyama, A., Murakami, H., Hosoyama, A., Mizutani-Ui, Y., Takahashi, N. K., Sawano, T., Inoue, R., Kaito, C., Sekimizu, K., Hirakawa, H., Kuhara, S., Goto, S., Yabuzaki, J., Kanehisa, M., Yamashita, A., Oshima, K., Furuya, K., Yoshino, C., Shiba, T., Hattori, M., Ogasawara, N., Hayashi, H., and Hiramatsu, K. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240.
Kwan, T., Liu, J., Dubow, M., Gros, P., and Pelletier, J. (2005). The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. U.S.A. 102, 5174–5179.
Le Marechal, C., Hernandez, D., Schrenzel, J., Even, S., Berkova, N., Thiery, R., Vautor, E., Fitzgerald, J. R., Francois, P., and Le Loir, Y. (2011). Genome sequences of two Staphylococcus aureus ovine strains that induce severe (strain O11) and mild (strain O46) mastitis. J. Bacteriol. 193, 2353–2354.
Leung, D. Y., Meissner, H. C., Fulton, D. R., Murray, D. L., Kotzin, B. L., and Schlievert, P. M. (1993). Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet 342, 1385–1388.
Li, Z., Stevens, D. L., Hamilton, S. M., Parimon, T., Ma, Y., Kearns, A. M., Ellis, R. W., and Bryant, A. E. (2011). Fatal S. aureus hemorrhagic pneumonia: genetic analysis of a unique clinical isolate producing both PVL and TSST-1. PLoS One 6:e27246. doi: 10.1371/journal.pone.0027246
Lin, Y. C., Anderson, M. J., Kohler, P. L., Strandberg, K. L., Olson, M. E., Horswill, A. R., Schlievert, P. M., and Peterson, M. L. (2011). Proinflammatory exoprotein characterization of toxic shock syndrome Staphylococcus aureus. Biochemistry 50, 7157–7167.
Lindsay, J. A., and Holden, M. T. (2004). Staphylococcus aureus: superbug, super genome? Trends Microbiol. 12, 378–385.
Lindsay, J. A., Ruzin, A., Ross, H. F., Kurepina, N., and Novick, R. P. (1998). The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 29, 527–543.
Lowder, B. V., Guinane, C. M., Ben Zakour, N. L., Weinert, L. A., Conway-Morris, A., Cartwright, R. A., Simpson, A. J., Rambaut, A., Nubel, U., and Fitzgerald, J. R. (2009). Recent human-to-poultry host jump, adaptation, and pandemic spread of Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 106, 19545–19550.
Lysnyansky, I., Calcutt, M. J., Ben-Barak, I., Ron, Y., Levisohn, S., Methe, B. A., and Yogev, D. (2009). Molecular characterization of newly identified IS3, IS4 and IS30 insertion sequence-like elements in Mycoplasma bovis and their possible roles in genome plasticity. FEMS Microbiol. Lett. 294, 172–182.
Mascarenhas, J., Sanchez, H., Tadesse, S., Kidane, D., Krisnamurthy, M., Alonso, J. C., and Graumann, P. L. (2006). Bacillus subtilis SbcC protein plays an important role in DNA inter-strand cross-link repair. BMC Mol. Biol. 7, 20.
McClarty, G., Caldwell, H. D., and Nelson, D. E. (2007). Chlamydial interferon gamma immune evasion influences infection tropism. Curr. Opin. Microbiol. 10, 47–51.
McDougal, L. K., Steward, C. D., Killgore, G. E., Chaitram, J. M., McAllister, S. K., and Tenover, F. C. (2003). Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J. Clin. Microbiol. 41, 5113–5120.
Meinhart, A., Alonso, J. C., Strater, N., and Saenger, W. (2003). Crystal structure of the plasmid maintenance system epsilon/zeta: functional mechanism of toxin zeta and inactivation by epsilon 2 zeta 2 complex formation. Proc. Natl. Acad. Sci. U.S.A. 100, 1661–1666.
Melles, D. C., Gorkink, R. F., Boelens, H. A., Snijders, S. V., Peeters, J. K., Moorhouse, M. J., van der Spek, P. J., van Leeuwen, W. B., Simons, G., Verbrugh, H. A., and van Belkum, A. (2004). Natural population dynamics and expansion of pathogenic clones of Staphylococcus aureus. J. Clin. Invest 114, 1732–1740.
Melles, D. C., Tenover, F. C., Kuehnert, M. J., Witsenboer, H., Peeters, J. K., Verbrugh, H. A., and van Belkum, A. (2008). Overlapping population structures of nasal isolates of Staphylococcus aureus from healthy Dutch and American individuals. J. Clin. Microbiol. 46, 235–241.
Moore, P. C., and Lindsay, J. A. (2002). Molecular characterisation of the dominant UK methicillin-resistant Staphylococcus aureus strains, EMRSA-15 and EMRSA-16. J. Med. Microbiol. 51, 516–521.
Moran, N. A., and Plague, G. R. (2004). Genomic changes following host restriction in bacteria. Curr. Opin. Genet. Dev. 14, 627–633.
Nickerson, N., Ip, J., Passos, D. T., and McGavin, M. J. (2010). Comparison of Staphopain A (ScpA) and B (SspB) precursor activation mechanisms reveals unique secretion kinetics of proSspB (Staphopain B), and a different interaction with its cognate Staphostatin, SspC. Mol. Microbiol. 75, 161–177.
Nienaber, J. J., Sharma Kuinkel, B. K., Clarke-Pearson, M., Lamlertthon, S., Park, L., Rude, T. H., Barriere, S., Woods, C. W., Chu, V. H., Marin, M., Bukovski, S., Garcia, P., Corey, G. R., Korman, T., Doco-Lecompte, T., Murdoch, D. R., Reller, L. B., and Fowler, V. G. Jr. (2011). Methicillin-susceptible Staphylococcus aureus endocarditis isolates are associated with clonal complex 30 genotype and a distinct repertoire of enterotoxins and adhesins. J. Infect. Dis. 204, 704–713.
Novick, R. P., and Subedi, A. (2007). The SaPIs: mobile pathogenicity islands of Staphylococcus. Chem. Immunol. Allergy 93, 42–57.
Nygaard, T. K., Pallister, K. B., Ruzevich, P., Griffith, S., Vuong, C., and Voyich, J. M. (2010). SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J. Infect. Dis. 201, 241–254.
Pandey, D. P., and Gerdes, K. (2005). Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33, 966–976.
Pedersen, K., Christensen, S. K., and Gerdes, K. (2002). Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol. Microbiol. 45, 501–510.
Pilpa, R. M., Fadeev, E. A., Villareal, V. A., Wong, M. L., Phillips, M., and Clubb, R. T. (2006). Solution structure of the NEAT (NEAr Transporter) domain from IsdH/HarA: the human hemoglobin receptor in Staphylococcus aureus. J. Mol. Biol. 360, 435–447.
Prere, M. F., Chandler, M., and Fayet, O. (1990). Transposition in Shigella dysenteriae: isolation and analysis of IS911, a new member of the IS3 group of insertion sequences. J. Bacteriol. 172, 4090–4099.
Robinson, D. A., Kearns, A. M., Holmes, A., Morrison, D., Grundmann, H., Edwards, G., O'Brien, F. G., Tenover, F. C., McDougal, L. K., Monk, A. B., and Enright, M. C. (2005). Re-emergence of early pandemic Staphylococcus aureus as a community-acquired meticillin-resistant clone. Lancet 365, 1256–1258.
Rountree, P. M., and Beard, M. A. (1958). Further observations on infection with phage type 80 Staphylococci in Australia. Med. J. Aust. 45, 789–795.
Ruzin, A., Lindsay, J., and Novick, R. P. (2001). Molecular genetics of SaPI1–a mobile pathogenicity island in Staphylococcus aureus. Mol. Microbiol. 41, 365–377.
Schijffelen, M. J., Boel, C. H., van Strijp, J. A., and Fluit, A. C. (2010). Whole genome analysis of a livestock-associated methicillin-resistant Staphylococcus aureus ST398 isolate from a case of human endocarditis. BMC Genomics 11, 376.
Schlievert, P. M., and Blomster, D. A. (1983). Production of Staphylococcal pyrogenic exotoxin type C: influence of physical and chemical factors. J. Infect. Dis. 147, 236–242.
Sekine, Y., Eisaki, N., and Ohtsubo, E. (1994). Translational control in production of transposase and in transposition of insertion sequence IS3. J. Mol. Biol. 235, 1406–1420.
Sekine, Y., Izumi, K., Mizuno, T., and Ohtsubo, E. (1997). Inhibition of transpositional recombination by OrfA and OrfB proteins encoded by insertion sequence IS3. Genes Cells 2, 547–557.
Seybold, U., Kourbatova, E. V., Johnson, J. G., Halvosa, S. J., Wang, Y. F., King, M. D., Ray, S. M., and Blumberg, H. M. (2006). Emergence of community-associated methicillin-resistant Staphylococcus aureus USA300 genotype as a major cause of health care-associated blood stream infections. Clin. Infect. Dis. 42, 647–656.
Shopsin, B., Gomez, M., Montgomery, S. O., Smith, D. H., Waddington, M., Dodge, D. E., Bost, D. A., Riehman, M., Naidich, S., and Kreiswirth, B. N. (1999). Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J. Clin. Microbiol. 37, 3556–3563.
Ster, C., Beaudoin, F., Diarra, M. S., Jacques, M., Malouin, F., and Lacasse, P. (2010). Evaluation of some Staphylococcus aureus iron-regulated proteins as vaccine targets. Vet. Immunol. Immunopathol. 136, 311–318.
Tanimoto, R. H. (1962). Observations on Staphylococcus aureus phage-type 80/81 Hawaii and its resistance to antibiotics. Hawaii Med. J. 21, 262–265.
Tsilibaris, V., Maenhaut-Michel, G., Mine, N., and van Melderen, L. (2007). What is the benefit to Escherichia coli of having multiple toxin-antitoxin systems in its genome? J. Bacteriol. 189, 6101–6108.
Voyich, J. M., Vuong, C., Dewald, M., Nygaard, T. K., Kocianova, S., Griffith, S., Jones, J., Iverson, C., Sturdevant, D. E., Braughton, K. R., Whitney, A. R., Otto, M., and Deleo, F. R. (2009). The SaeR/S gene regulatory system is essential for innate immune evasion by Staphylococcus aureus. J. Infect. Dis. 199, 1698–1706.
Wardenburg, J. B., and Schneewind, O. (2008). Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205, 287–294.
Williams, J. R., Talbot, E. C., and Maughan, E. (1959). Hospital outbreak of cross-infection due to Staphylococcus pyogenes phage type 80. Br. Med. J. 1, 1374–1378.
Wormald, P. J. (1961). Multiple endemic substrains of Staphylococcus pyogenes phage type 80. Monatsschr. Unfallheilkd. Versicherungsmed. 20, 59–62.
Xiong, Y. Q., Fowler, V. G., Yeaman, M. R., Perdreau-Remington, F., Kreiswirth, B. N., and Bayer, A. S. (2009). Phenotypic and genotypic characteristics of persistent methicillin-resistant Staphylococcus aureus bacteremia in vitro and in an experimental endocarditis model. J. Infect. Dis. 199, 201–208.
Keywords: Staphylococcus aureus, evolution, pseudogene, pathogenicity island, insertion sequence, toxin-antitoxin addiction module, pathoadaptation, virulence
Citation: McGavin MJ, Arsic B and Nickerson NN (2012) Evolutionary blueprint for host- and niche-adaptation in Staphylococcus aureus clonal complex CC30. Front. Cell. Inf. Microbio. 2:48. doi: 10.3389/fcimb.2012.00048
Received: 23 January 2012; Accepted: 20 March 2012;
Published online: 09 April 2012.
Edited by:
David Heinrichs, University of Western Ontario, CanadaReviewed by:
Steven Gill, University of Rochester School of Medicine and Dentistry, USAJodi A. Lindsay, St George's University of London, UK
Copyright: © 2012 McGavin, Arsic and Nickerson. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Martin J. McGavin, Department of Microbiology, Schulich School of Medicine and Dentistry, and Centre for Human Immunology, Western University, London, ON N6A 5C1, Canada. e-mail: martin.mcgavin@schulich.uwo.ca