A framework for assessing the concordance of molecular typing methods and the true strain phylogeny of Campylobacter jejuni and C. coli using draft genome sequence data
- 1 Bureau of Microbial Hazards, Food Directorate, Health Canada, Ottawa, ON, Canada
- 2 Laboratory for Foodborne Zoonoses, Public Health Agency of Canada, Lethbridge, AB, Canada
- 3 National Microbiology Laboratory, Public Health Agency of Canada, Winnipeg, MB, Canada
Tracking of sources of sporadic cases of campylobacteriosis remains challenging, as commonly used molecular typing methods have limited ability to unambiguously link genetically related strains. Genomics has become increasingly prominent in the public health response to enteric pathogens as methods enable characterization of pathogens at an unprecedented level of resolution. However, the cost of sequencing and expertise required for bioinformatic analyses remains prohibitive, and these comprehensive analyses are limited to a few priority strains. Although several molecular typing methods are currently widely used for epidemiological analysis of campylobacters, it is not clear how accurately these methods reflect true strain relationships. To address this, we have developed a framework and associated computational tools to rapidly analyze draft genome sequence data for the assessment of molecular typing methods against a “gold standard” based on the phylogenetic analysis of highly conserved core (HCC) genes with high sequence quality. We analyzed 104 publicly available whole genome sequences (WGS) of C. jejuni and C. coli. In addition to in silico determination of multi-locus sequence typing (MLST), flaA, and porA type, as well as comparative genomic fingerprinting (CGF) type, we inferred a “reference” phylogeny based on 389 HCC genes. Molecular typing data were compared to the reference phylogeny for concordance using the adjusted Wallace coefficient (AWC) with confidence intervals. Although MLST targets the sequence variability in core genes and CGF targets insertions/deletions of accessory genes, both methods are based on multi-locus analysis and provided better estimates of true phylogeny than methods based on single loci (porA, flaA). A more comprehensive WGS dataset including additional genetically related strains, both epidemiologically linked and unlinked, will be necessary to more comprehensively assess the performance of subtyping methods for outbreak investigations and surveillance activities. Analyses of the strengths and weaknesses of widely used typing methodologies in inferring true strain relationships will provide guidance in the interpretation of this data for epidemiological purposes.
Introduction
Campylobacter spp. are the most common cause of bacterial gastroenteritis in Canada (Public Health Agency of Canada, 2009), as around the world, with most cases (>95%) attributed to infection with C. jejuni and C. coli – at a ratio of ∼6:1, respectively. Yet, despite important public health and socioeconomic impacts of this organism (Thomas et al., 2008), limited progress has been made in defining routes of infection and reducing associated illness. This is in part due to the sporadic distribution of the majority of cases of campylobacteriosis (Government of Canada, 2007, 2010), and the associated difficulties in identifying sources of infection. Furthermore, due to the widespread occurrence of this organism in the intestinal tracts of animals and in the environment, there are many possible sources of exposure.
Molecular epidemiology of C. jejuni and C. coli remains challenging due to the nature of the genome evolution in these organisms and the extensive genomic and phenotypic diversity within these species. Genome evolution in C. jejuni and C. coli is largely driven by frequent genomic rearrangements and interstrain genetic exchange (de Boer et al., 2002; Ridley et al., 2008; Wilson et al., 2009a). For these species, recombination appears to affect population structure more rapidly than de novo mutation (Dingle et al., 2001; Biggs et al., 2011). To further complicate matters, there is evidence of stability for some clones (Nielsen et al., 2001), whereas in other cases differences in genetic profiles are observed within a single passage of the organism through an animal host (de Boer et al., 2002). Competition for resources within their gastrointestinal niche likely drives this high rate of evolution through selection of any change that may offer a competitive advantage in this microbe-rich environment (Lefébure and Stanhope, 2009). The rapid evolution of the C. jejuni and C. coli genomes has important consequences for interpretation of molecular typing information. Outbreak isolates may be missed in cases where small genomic changes result in changes of molecular profiles (Hanninen et al., 1999; Nuijten et al., 2000; Sails et al., 2003; Barton et al., 2007). Conversely, some strains will appear to be clonal, and will be linked by typing methods despite true differences in gene content between isolates and absence of epidemiological linkage (Taboada et al., 2008; Biggs et al., 2011).
Several molecular subtyping schemes have been developed for use in characterization of C. jejuni and C. coli isolates for epidemiological investigation (reviewed in Klena and Konkel, 2005). Of these, multi-locus sequence typing (MLST), based on DNA sequence analysis of seven housekeeping genes, is currently the leading method, in part due to the ease of comparison of nucleotide sequence-based typing among labs worldwide (Dingle et al., 2001). This typing scheme has greatly contributed to an improved understanding of Campylobacter epidemiology. Similarly, DNA sequencing of the flagellin gene short variable region (flaA-SVR; Meinersmann et al., 1997, 2005) and the porA gene (Clark et al., 2007) is also routinely used. Alternative methods which incorporate analysis of the accessory genome include comparative genomic fingerprinting (CGF), a low cost, high throughput, and high resolution method that is based on the detection of 40 genes using multiplex PCR (Taboada et al., 2012). Current analyses suggest that CGF is highly concordant with MLST, but with a better discriminatory power (Clark et al., 2012; Taboada et al., 2012). Much like sequence-based methodologies, CGF types can be easily compared among laboratories. With the advent of high throughput next generation sequencing (NGS) technologies, whole genome sequence (WGS) analysis has begun to play an increasing role in microbial epidemiology, particularly in high profile outbreak situations (Gilmour et al., 2010; Chin et al., 2011; Rohde et al., 2011). Unfortunately, current costs limit the use of full genome analysis to a few priority strains (e.g., Parkhill et al., 2000; Pearson et al., 2007). While all of these methods have played an important role in improving our understanding of transmission of campylobacters to human hosts, the range of available typing methods leads to difficulties in meta-analysis of study data.
In order to move toward a common, standard molecular typing methodology suitable for most epidemiological studies, robust evaluation of existing typing schemes is needed. High quality, whole genome sequence (WGS) is the true gold standard for molecular characterization of microbes as all of the information necessary to determine molecular types is encoded within the genome. Analysis of this data can be highly discriminatory among closely related strains (Biggs et al., 2011), but can also be used to infer evolutionary relationship for distantly related organisms (Lefébure and Stanhope, 2009; Lefébure et al., 2010). In the near future, WGS will likely become the method of choice for characterization of microbes; however, use of WGS for surveillance activities is currently not feasible for most laboratories. Nonetheless, there is a growing number of full genomes available for analysis. This data can be used to rigorously assess existing typing schemes to help identify those that would work most effectively for public health activities, and to select improved targets for next generation typing schemes. Furthermore, an improved understanding of the performance of each method will assist in the interpretation of existing studies.
We have used publicly available C. jejuni and C. coli WGS data in the development of a framework to assess performance of MLST, flaA, porA, and CGF typing schemes compared to the inferred “reference” phylogeny based on conserved core genome elements. Such a framework will provide a basis for future, more expansive molecular typing method evaluation based on WGS data.
Materials and Methods
Strains Used in Analysis
A total of 104 strains were included in this study (Table S1 in Supplementary Material). Of these, 24 complete or draft C. jejuni and C. coli sequences were retrieved from GenBank: C. jejuni subsp. jejuni [NCTC 11168 (NC_002163), 81–116 (NC_009839), 81–176 (NC_008787), 84–25 (NZ_AANT00000000), CF93-6 (NZ_AANJ00000000), HB93-13 (NZ_AANQ00000000), CG8421 (NZ_ABGQ00000000), CG8486 (NZ_AASY00000000), 260.94 (NZ_AANK00000000), IA3902 (CP001876.1), ICDCCJ07001 (NC_014802), M1 (CP001900.1), S3 (CP001960.1), 1336 (NZ_ADGL00000000), 305 (ADHL00000000.1), 327 (ADHM00000000.1), 414 (NZ_ADGM00000000), DFVF1099 (ADHK00000000.1), D2600 (AGTF00000000.1), NW(AGTE00000000.1)], C. jejuni subsp. doylei 269.97(NC_009707), C. jejuni RM1221 (NC_003912), and C. coli [RM2228 (AAFL00000000), JV20 (NZ_AEER00000000)]. Sequence data from 39 strains of C. jejuni and 41 strains of C. coli were retrieved from the Short Read Archive under accession numbers SRP001790 and SRA010929, respectively (Lefébure et al., 2010).
Sequence Assembly and Annotation
Illumina traces from 80 of the C. jejuni and C. coli genomes sequenced by Lefébure et al. (2010) were assembled using Velvet (version 1.1.06; Zerbino and Birney, 2008) using a hash length of 25 as this was found to give optimal assemblies. The order of the contigs was inferred by comparison with the C. jejuni NCTC 11168 reference genome using ABACAS (Assefa et al., 2009). Prediction of coding sequences and annotation was completed using the rapid annotation using subsystem technology (RAST; Aziz et al., 2008).
Assessment of WGS Data Quality
In order to generate a measure of quality of WGS data, we examined the C. jejuni genomes [closed reference sequence (RefSeq) genomes (n = 9), draft RefSeq genomes (n = 8), draft 454 genomes (n = 3), and draft Illumina genomes (n = 41)] using a two-step process to examine truncations in core genes predicted in each genome. In the first step, a set of “core genes” for C. jejuni was identified based on a preliminary comparative genomic survey using a subset of RefSeq annotated genomes. Whole genome pair-wise homology searching using BLAST+ (version 2.2.25; Camacho et al., 2009) was performed at the ORF level using the program BLASTP using the strain NCTC 11168 as a reference. Genes were considered “core” if conserved across all of the genomes analyzed, yielding a set of 1,314 genes. In the second step, the 1,314 genes from strain NCTC 11168 were queried against the predicted ORFs for the set of 61 C. jejuni genomes using BLASTP. Alignment lengths were used to identify truncations if shorter than the length of the RefSeq. A one-tailed unpaired t-test was performed using GraphPad Prism version 5.04 for Windows (GraphPad Software, San Diego) to determine statistical significance of increase in number of truncations observed in draft quality genome sequences compared to closed RefSeq.
Core/Accessory Genome Phylogenetic Analysis
A semi-automated approach was developed to rapidly infer a core genome phylogeny for the dataset. In the first step, a robust set of “highly conserved core” (HCC) genes for C. jejuni and C. coli was identified based on a preliminary comparative genomic survey using a subset of RefSeq annotated genomes. Whole genome pair-wise homology searching using BLAST+ (version 2.2.25; Camacho et al., 2009) was performed at the ORF level using the program BLASTP. Genes were considered “core” if conserved across all of the genomes analyzed. A 90% sequence identity cut-off was used to identify HCC genes, yielding a set of 389 genes (Table S3 in Supplementary Material). In the second step, the program CONCATENATOR (Kruczkiewicz et al., 2011), a program written in C# using the.NET Framework 4.0, was used to: (1) identify the homologous sequences for the set of 389 HCC genes in each genome in the dataset using BLASTN; (2) perform individual alignments for each gene using MUSCLE (Edgar, 2004a,b); and (3) concatenate the alignments to produce a single alignment (i.e., a “concatenome”). The reference core genome phylogeny for the dataset was then estimated based on the concatenome using Sea View (Gouy et al., 2010) using uncorrected distances.
In silico Typing Analysis
The program “microbial in silico typer” (MIST) was used to generate in silico molecular typing results from whole genome sequence data (Kruczkiewicz et al., 2011). MIST derives several kinds of in silico typing data from “raw” genome sequences (i.e., contig assemblies), including MLST (Dingle et al., 2001), porA typing (Clark et al., 2007), flaA typing (Meinersmann et al., 1997, 2005), and CGF (Taboada et al., 2012). The full implementation of MIST, which was written in the C# programming language using the.NET Framework 4.0, will be described in detail elsewhere; functionalities used in this study will be briefly described here. Sequence Typing: the sequence for each of the target genes (i.e., MLST genes: aspA, glnA, gltA, glyA, pgm, tkt, uncA; the porA gene; and the flaA gene) was identified in each of the contig assemblies through homology searching using BLAST+ (version 2.2.25; Camacho et al., 2009). Alleles were inferred for each gene by comparing these sequences against allelic sequences obtained from the C. jejuni PubMLST database1. MLST allelic profiles were used to determine the sequence type (ST) and clonal complex (CC) for each strain. Comparative Genomic Fingerprinting. Presence of targets in the CGF40 scheme (Taboada et al., 2012) was determined by performing a homology search for each target using BLASTN against each WGS and using a sequence identity cut-off of 95% to score the presence/absence of each target. To generate CGF40 clusters, pair-wise profile similarities we computed using the simple matching coefficient and clustered using the unweighted-pair group method using average linkages (UPGMA) in Bionumerics (v.5.1; Applied Maths, Austin, TX, USA), using 100, 95, and 90% fingerprint similarities for cluster definition.
Assessment of SNP and Accessory Gene Content Differences
Calculation of pair-wise SNP rates: to estimate pair-wise SNP rates between strains in the dataset, the sequences from the HCC set were concatenated into a single 395,563 bp multiple sequence alignment. All gapped positions resulting from indels or missing data were removed from the alignment, yielding an alignment of 319,428 bp. Calculation of pair-wise accessory gene content differences: for each pair of strains, the total number of SNPs was computed and the SNP rate expressed as the average number of SNPs per 1,000 bp. To estimate accessory genome content differences, pair-wise differences in conservation profiles between strains in the dataset were calculated for a set of 3,903 accessory genes that were selected based on the following criteria: absence in at least one or more genomes; presence in at least two genomes (i.e., no “strain-specific” genes); and non-redundancy (i.e., a single gene was chosen from each set of orthologs).
Comparison of Molecular Typing Methods
In silico typing results were compared to the reference phylogeny by taking the latter and subdividing it into “phylogenetic clusters” at several levels of resolution targeting a specific average intra-cluster SNP rate (5, 10, and 15 SNPs per 1,000 bp). The adjusted Wallace coefficient (AWC; Severiano et al., 2011) was used to compare the phylogenetic clusters to the genotypic clusters obtained from the various methods using MIST. This and other measures of subtyping method performance (Carriço et al., 2006) were analyzed at the Comparing Partitions server2.
Results and Discussion
Effect of Whole Genome Sequence (WGS) Data Quality on Downstream Data Analysis
The dataset assembled for this study represents a collection of public C. jejuni and C. coli WGS data that includes both closed and unfinished genomes designated as RefSeq by NCBI (Pruitt et al., 2002, updated May 23, 2011), as well as draft quality genome assemblies with various levels of sequence coverage and in various states of “fragmentation” (i.e., multiple contigs). It includes “earlier generation” sequence data generated through Sanger sequencing and NGS data generated using the 454 and Illumina platforms. The heterogeneous nature of these data enabled the examination of the impact of sequence data quality on downstream analyses, including pan-genome (i.e., core/accessory genome) analysis, core genome based phylogenetic analysis and the derivation of in silico typing results. In particular, the high quality closed genome sequence data present a benchmark against which to assess the quality of draft assemblies generated using NGS platforms. The latter not only comprise the bulk of WGS data currently available in public databases but, owing to higher-throughput and lower associated costs of data generation, effectively represent the only kind of WGS data currently being generated in laboratories around the world.
Quality assurance remains a significant challenge for NGS data, and is likely to be impacted by the variability in depth of coverage observed for the C. jejuni and C. coli sequences (Table S1 in Supplementary Material), among other things. Lower coverage may result in erroneous base-calling due to inherent systematic error rates of sequencing platforms (i.e., small insertion/deletions in 454 sequences, miscalls in Illumina sequences; Metzker, 2010). In addition, low coverage may impact assembly of sequence reads into long contigs, resulting in assemblies comprised of larger numbers of short contigs and a concomitant increase in incomplete (i.e., “truncated”) gene sequence data during automated gene prediction and sequence annotation. These not only present the potential for allelic miscalls but may also pose significant problems for downstream phylogenetic analysis.
To assess quality of NGS data included in this study, the number of predicted genes that were truncated relative to core genes in C. jejuni NCTC11168 was determined. Draft genome assemblies included in the dataset differed in the number of partial or truncated genes identified (Table S1 in Supplementary Material; Figure 1). Closed genomes included in NCBI’s RefSeq collection had the lowest number of truncated genes and RefSeq genomes that were not closed had significantly more (p < 0.05) truncated genes. The closed RefSeq C. jejuni genome ICDCCj07001 was not included in the statistical analysis as the quality of the sequence appeared to be much lower than in the other closed genomes (83 truncations), and inclusion of this outlier skewed the results. The 454 draft genomes had very high levels of gene truncation, but only three genomes with low coverage (10× to 20×) were available for analysis. Short read (36 bp) Illumina data from the Lefébure et al. (2010) study were minimally processed beyond assembly and scaffolding prior to analysis. The number of truncations in this dataset was similar to what was observed in non-closed RefSeq genomes, but significantly higher (p = 0.0006) than the “gold standard” closed RefSeq genomes. Note that two new C. jejuni genomes (D2600 and NW) with 70× sequencing coverage on Illumina had gene truncation numbers similar to those observed in the closed genome sequence data.
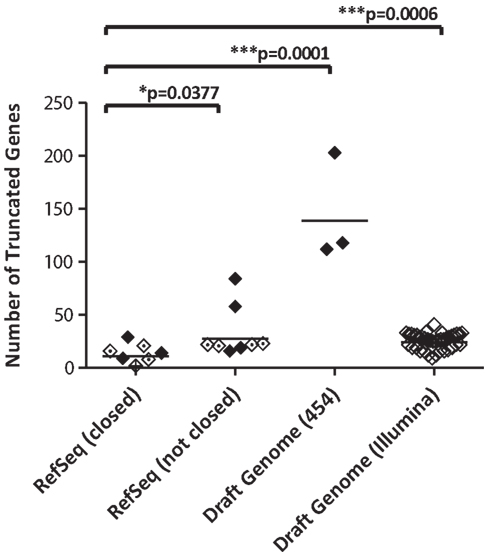
Figure 1. Finishing quality is reflected in the number of truncated predicted genes in C. jejuni whole genome sequences. The C. jejuni genomes used in this analysis were classified according to finishing levels (reference sequence or draft), and/or sequencing platform used [Sanger open diamonds with a central period), 454 (closed diamonds), or Illumina (open diamonds)]. The number of predicted genes with truncations relative to C. jejuni NCTC11168 was calculated for each genome. The mean number of truncations for each group is represented by a line. Closed genomes had significantly fewer truncations than RefSeq genomes that were not closed and draft genomes generated with either 454 or Illumina sequencing technologies.
Our analysis suggests that assessment of truncation in predicted genes relative to a high quality reference genome may be a rapid and informative assessment of overall genome quality for all sequencing platforms included in this analysis (Figure 1). This method was particularly informative in the assessment of quality of reads generated by 454 sequencing technology. Errors in 454 sequencing reads tend to occur in homopolymer repeats (Metzker, 2010), and since Campylobacter genomes have high numbers of homopolymeric adenine and thymine tracts, they would be particularly susceptible to this type of error. Nonetheless, this measure was also found to be effective for both Sanger sequence and Illumina sequence. Draft genomes of similar quality tended to have similar overall numbers of randomly distributed gene truncations. A subset of these, likely in the range of 10–15 truncations observed in the high quality genomes, may represent bona fide allelic variation due to hypervariable homopolymeric tracts that cause premature stop codons in “contingency genes” containing them (Parkhill et al., 2000; Jerome et al., 2011). In contrast, truncations in which sequence breakpoints appear randomly distributed in individual strains across the dataset are more likely to be due to incomplete assembly or poor sequence data. High levels of apparent erroneous truncation were more prevalent in lower quality genome sequence data. Moreover, closed genomes did not necessarily represent the highest quality sequence. For example, the closed RefSeq C. jejuni genome ICDCCJ07001 had an unusually high number of apparently truncated genes compared to other genomes in this category. More extensive analyses of additional species of bacteria in the public database may provide a more complete understanding of how gene truncation may be more generally used as a quality metric.
Despite variability in sequence quality as assessed by gene truncation, most of the sequence typing alleles (i.e., MLST genes, flaA, porA) matched experimentally determined alleles (Table S2 in Supplementary Material, described in detail below). While Illumina sequencing data is known to have a higher error rate than the other technologies (Metzker, 2010; Suzuki et al., 2011), increased coverage greatly reduces false SNP identification. Comparisons of error rates in Illumina and 454 sequencing platforms in sequencing an E. coli strain (Suzuki et al., 2011) found ∼46 false SNPs in this 4.6 Mb genome. If we assume a similar error rate in Campylobacter genomes, we would expect up to 17 false SNPs in each of these smaller genomes. The coverage of the genomes in the Lefébure et al. (2010) strain set was generally much higher than in the Suzuki et al. (2011) study (Table S1 in Supplementary Material); therefore, we would predict a concomitant decrease in error rates in these genomes, as increased coverage is known to decrease these errors (Suzuki et al., 2011). Furthermore, given that this would ultimately represent a small fraction of the inter-isolate SNPs, it is unlikely that this error would substantially impact results of downstream analyses.
From WGS to Core Genome Phylogenetic Analysis
In order to facilitate core genome phylogenetic analysis, we designed a pipeline aimed at identifying a subset of HCC genes with high quality across the dataset and to subsequently infer an estimate of the phylogenetic relationship for the strains used in this analysis. The use of large sets of core genes in phylogenetic analysis represents the best estimate of the “true” phylogenetic relationship of bacterial isolates since it can minimize effects of conflicting signal due to recombination. Moreover, by assessing the quality of core genomes prior to phylogenetic analysis, our approach allowed tolerance of minimally processed draft genome sequence data.
A preliminary comparative genomic survey of annotated genomes for C. jejuni and C. coli was performed to identify a set 389 HCC genes that could be used to derive a reference phylogeny for the dataset. Because differences in the depth of sequence coverage and platform-specific sequencing error bias have the potential to affect the sequence quality of the various draft assemblies, several steps were taken in the automated analysis strategy to maximize the amount of core genome data used while minimizing the potentially adverse effects of erroneous gene sequence data on downstream phylogenetic analysis. For example, although only 215 of the HCC genes had full length predicted gene calls in all 104 genomes analyzed, we identified many cases (n = 279; 166 genes) in which a small (i.e., 1–2 bp) indel led to a frameshift and premature stop codon (i.e., an “indel truncation”). In such cases, the sequence downstream of the indel was retrieved up to the full length of the gene if it could be aligned to the original RefSeq from NCTC 11168. At the same time, because of difficulties in differentiating indels due to sequencing errors from those due to biological causes (bona fide indels, contingency frameshifts) indel positions were ignored in the phylogenetic analysis. In a smaller number of cases (n = 127; 105 genes) we observed the premature truncation of a gene sequence due to proximity to the end of a contig (i.e., a “contig fragmentation”). In these cases, and in cases in which the gene was absent from at least one genome assembly (n = 39; 38 genes), gapped positions due to missing sequence data were also ignored from the analysis. These combined approaches allowed us to make use of a large proportion of the sequence data from the 389 HCC gene set (319,428 out of 395,563 bp).
Overall, the dataset was largely comprised of a genetically diverse set of strains. This presented challenges in terms of assessing the overall inferred phylogeny by standard methods such as bootstrapping and maximum likelihood (Felsenstein, 1989; Schmidt and von Haeseler, 2007). Nonetheless, the resulting phylogeny shows a deep split with significant support between the C. jejuni and C. coli strains (Figure 2). The SNP rates observed for the dataset are consistent with the split since the average interspecies SNP rate among the strains in the dataset was ∼108.6 per 1,000 bp whereas average intraspecies SNP rates were an order of magnitude lower (∼13.9 and 17.3 per 1,000 bp in C. coli and C. jejuni respectively). Although this is in contrast to previous findings based on intraspecies recombinational exchange of MLST alleles (Sheppard et al., 2008) and a 16s rRNA gene phylogeny in which mixing of the two species is observed (data not shown), it is consistent with findings of Lefébure et al. (2010) which suggest that although recombinational exchange between the species occurs, it is of a limited scale and does not remove the dominant phylogenetic signal supporting the species’ split.
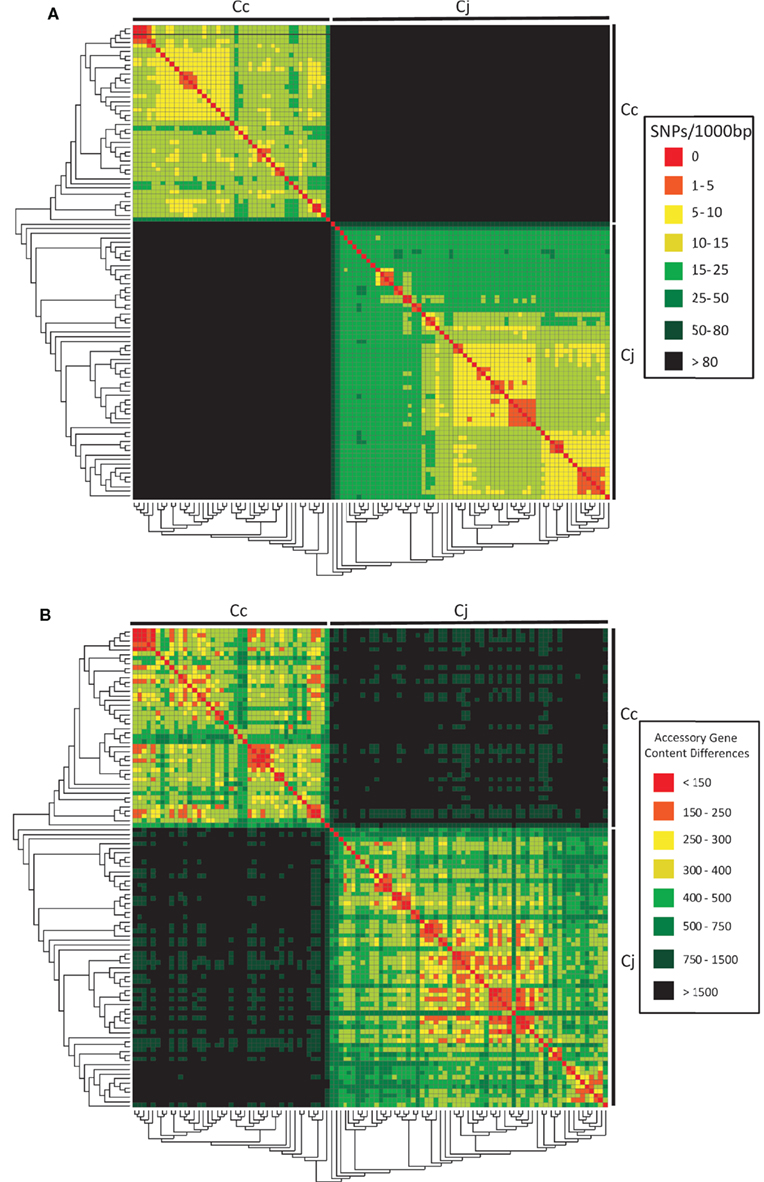
Figure 2. The core genome phylogeny for C. jejuni and C. coli is consistent with underlying genome-wide SNP rates and accessory genome content. The 104 C. jejuni and C. coli WGS were analyzed using an automated pipeline for core genome analysis; a core gene phylogeny derived from 389 core genes is shown here. This phylogeny was compared to the underlying (A) SNP rates and (B) accessory gene content differences. High SNP rates and accessory genome content differences between C. coli and C. jejuni genomes support a deep split between the species. Conversely, small phylogenetic clusters comprised of highly similar strains are supported by lower differences in SNPs and accessory gene content. A high resolution image of this figure is available in the supplementary material.
Within the C. jejuni and C. coli clades, significant support could be found for a small number of highly conserved sub-branches that were well-supported by the underlying SNP distributions (Figure 2A), with strains that form branches with robust bootstrap support sharing significantly lower SNP levels with respect to one another in contrast to unrelated strains in the dataset. At the same time, phylogenetic trees derived for individual core genes do not support the overall consensus phylogeny and this conflicting phylogenetic signal is consistent with significant levels of intraspecific recombinational exchange (results not shown). The HCC gene phylogeny is also compatible with the underlying accessory genome content, with strains within robustly supported branches sharing significantly fewer accessory gene content differences with respect to other strains in the dataset (Figure 2B). It thus appears that the accumulation of differences in accessory genome content is consistent with the accumulation of SNP differences in the core genome (r2 = 0.9703).
Assessment of Automated in silico Typing from WGS Data
For draft genome assemblies it is generally assumed that higher levels of coverage and fewer contigs are indicative of better quality data. As can be seen in Figure 3, a large proportion of sequence typing alleles (MLST genes, flaA, porA) was inferred whether from high quality finished genomes or from minimally processed draft assemblies. Thus, the relationship between quality estimates and their effect on downstream analysis is not always straightforward. For example, although Illumina data generally resulted in assemblies with larger numbers of contigs, the resulting sequence data were of sufficient quality to allow high levels of allele identification from in silico typing analysis. In most cases, allele identification matched the published or publicly available ST types. For example, among 80 strains with known MLST ST (i.e., from Lefébure et al., 2010 or available from the PubMLST database) concordance was found to be 98.4%, or 551/560 alleles. Overall, nine allelic discrepancies in the in silico derived ST were found, affecting data in eight of the genomes. In two of these cases, one of the seven MLST alleles could not be identified, with one of these two missing alleles in the RefSeq for C. coli. In the other seven cases there was a discrepancy at one of the seven loci and it is not possible to ascertain which of the methods gave the incorrect result (Table S2 in Supplementary Material), although in four of these cases the discordant alleles differed by a single SNP; in 3 of the cases alleles were so different (53–70 SNPs) that it is unlikely that sequencing errors were responsible for the observed lack of concordance.
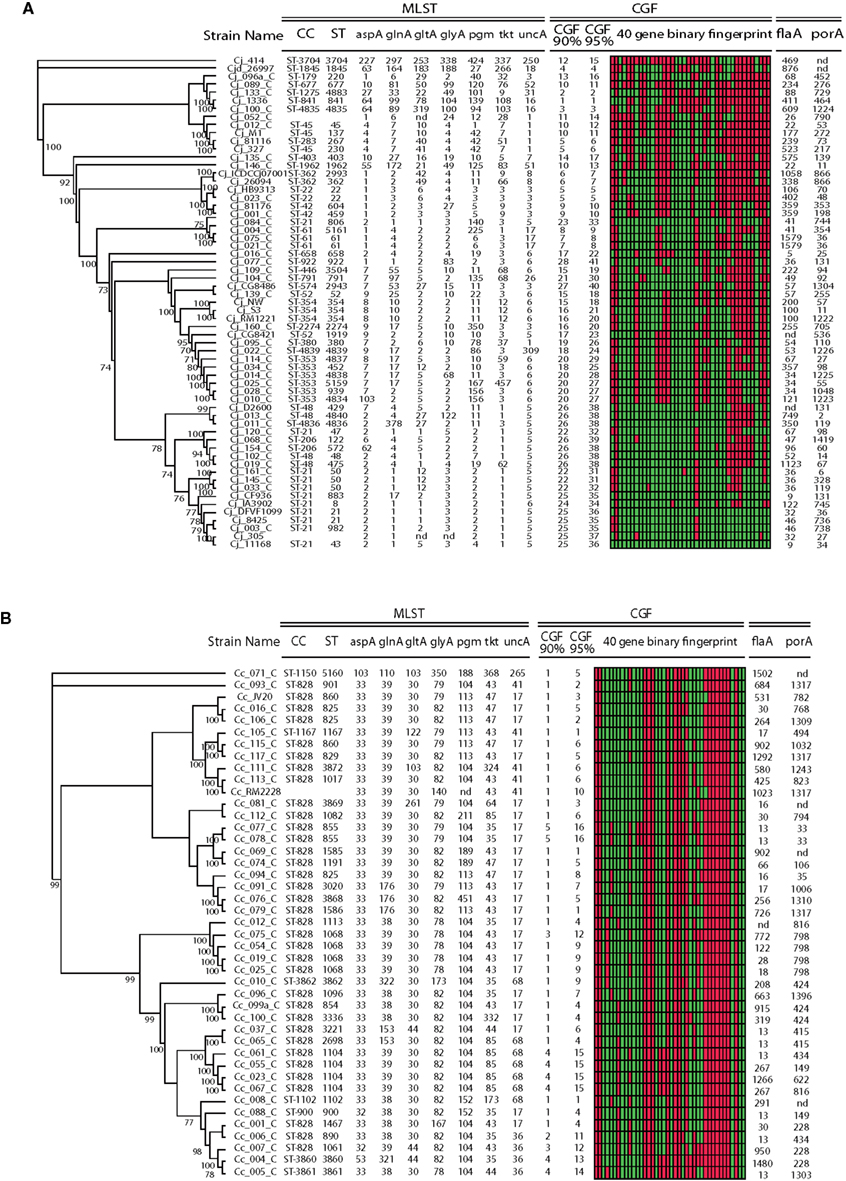
Figure 3. In silico typing data derived from C. jejuni and C. coli and WGS is concordant with core genome phylogeny. Publicly available WGS data for 104 C. jejuni (A) and C. coli (B) strains were used to derive typing profiles using an in silico typing pipeline. Although the dataset is comprised of highly genetically diverse strains, there is concordance between molecular typing and phylogenetic data: strains sharing similar/identical molecular fingerprints were found clustered in the dendrogram and increasing similarity led to shorter branch lengths. CGF cluster numbers are based on 90 and 95% fingerprint identity; green is positive, red is negative. MLST alleles that could not be determined are noted as “nd.”
Multi-Locus Campylobacter Subtyping Methodologies Reflect Core Gene Phylogeny
Molecular fingerprints obtained by subtyping methods represent a low resolution proxy for the full genome complement of a strain. Thus, one possible approach for comparing subtyping data to the underlying core genome phylogenetic data would be to compare the topologies of dendrograms obtained using each method to the reference phylogeny. Nevertheless, because of the relative paucity of data used, most of the topological information encoded in dendrograms from subtyping data lacks robustness and deeper relationships between clusters cannot be reliably inferred. In order to perform the comparison, we deconstructed the reference phylogenetic tree into sets of robust “phylogenetic clusters” reflecting a particular level of genetic similarity (i.e., SNP rate). These could be compared to the clusters obtained by subtyping using measures of concordance that do not rely on overall tree topology. The AWC has recently been proposed as a quantitative measure of congruence of genotypic clusters obtained using different typing methods (Severiano et al., 2011). In order to assess the level of concordance between in silico derived subtyping results and the WGS data, we used the AWC to compare the genotypic clusters obtained from in silico analysis of the WGS data to phylogenetic clusters in the HCC dendrogram reflecting various SNP rates (5, 10, and 15 bp per 1,000 bp).
The number of partitions, or clusters, obtained using the various methods was very high, with the multi-locus typing methods [i.e., CGF40 (100%), ST, and ST-porA] generating unique subtypes for a significant proportion of the strains in the dataset and Simpson’s Index of Diversity values approaching 1 (Table 1). This genetic variability is in large part due to the nature of the strains for which WGS data were publicly available since there is great interest in the scientific community in sequencing strains that may be unique, or that represent lineages that were previously uncharacterized.
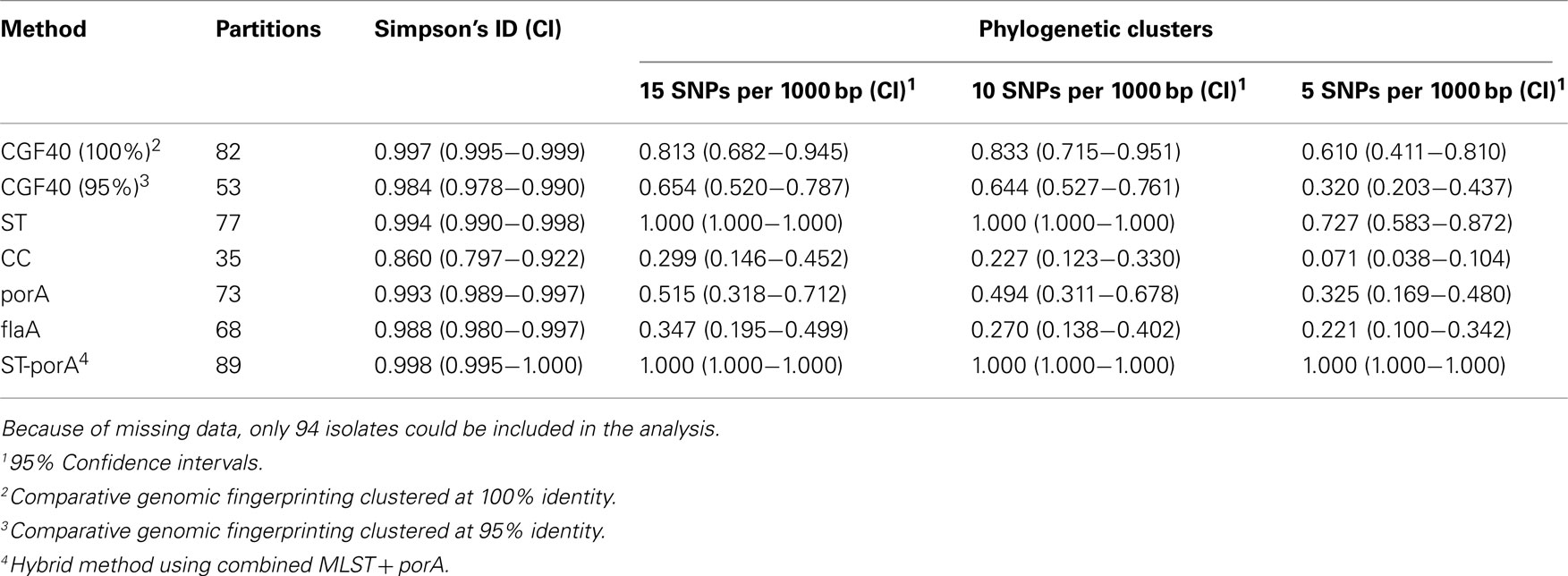
Table 1. Comparison of metrics of subtyping method performance.
In contrast to methods based on single loci (flaA, porA), both multi-locus typing methods (MLST, CGF) were highly congruent with core genome phylogeny (Table 1). These single locus methods are generally used on their own in the context of short term epidemiological analyses, and have been found to be useful for improving discriminatory power of MLST (Dingle et al., 2008; Clark et al., 2012), but perhaps less suitable for examining long term epidemiology; our results are consistent with this view. It is perhaps not entirely unexpected that MLST results provided a very close approximation of core genome phylogeny (Table 1) since the latter is essentially equivalent to a highly extended MLST typing scheme. Indeed, extended typing schemes are being more widely adopted to increase discriminatory power of MLST and to achieve more informative results from such schemes (Dingle et al., 2008; Lang et al., 2010; Zautner et al., 2011). It has been suggested that typing methods that target dispensable genes are better suited to short term epidemiology whereas methods based on core gene sequence such as MLST would more adequately reflect true genetic relationship among strains, and would be more useful for long term epidemiological studies (Wilson et al., 2009b). Given the divergent genomes assessed in this study, which reflect “long term” relationships, the degree to which an accessory genome based binary typing scheme such as CGF40 reflected core genome phylogeny was surprising.
Assessment of Genomic Similarity for Groups of Strains within Genotyping Clusters
The underlying structure of the dataset, which did not produce many multi-strain genotypic clusters, presented challenges to the analysis of congruence of molecular typing methods in a phylogenetic context, particularly at the higher levels of resolution. In order to further assess whether genotypic clusters obtained using the various subtyping methods represent groups of highly genetically related strains, we calculated the average SNP rate per 1,000 bp in the 389 core genes for all sets of strains sharing the same genotypic cluster. The average SNP rate observed for any two strains in the dataset was 61.4 per 1,000 bp and each of the subtyping methods assessed generated genotypic groups with significantly lower SNP rates (i.e., reflecting higher genetic similarity rates), ranging from 2.0 to 15.3 SNPs per 1,000 bp, than the average. Nevertheless, the multi-locus methods generated clusters with consistently lower SNP rates than those observed for the single locus methods (Figure 4A). Moreover, whereas the former had relatively uniform distributions the latter showed significant rate variability. This is consistent with the possibility that due to recombination, single locus methods can in some cases lead to genotypic clusters comprised of strains that are quite genetically different.
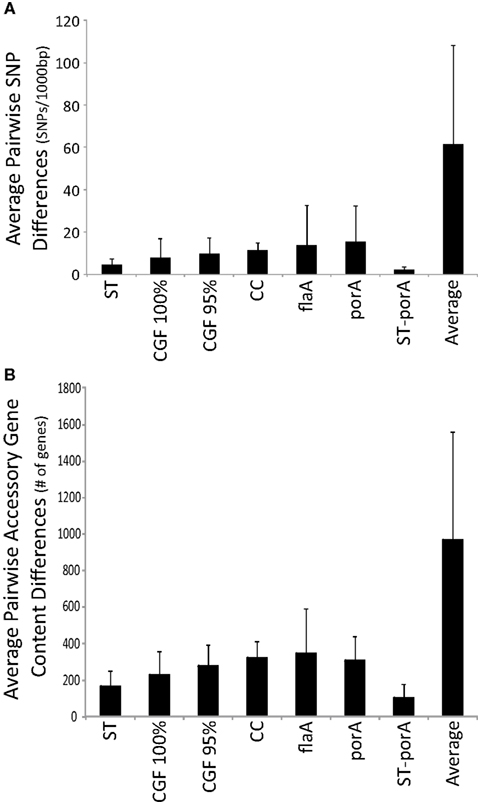
Figure 4. Genotypic clusters are comprised of strains with high genomic similarity. Genotypic clusters obtained using five different molecular typing methods were analyzed for (A) average SNP rates or (B) accessory gene content differences in order to assess intra- and inter-cluster core and accessory genetic similarities, respectively. Although all methods generated genotypic clusters sharing higher core and accessory genetic similarities than expected by chance alone, multi-locus methods led to genotypic clusters with higher genetic similarity.
We next examined the extent to which strains that are indistinguishable based on current subtyping methods differ at the genomic level by assessing the number of conserved genes between pairs of strains in a set of 3,903 genes from the pooled accessory genome among the 104 genomes in the dataset (Figure 2B). As with SNP rates, the average number of accessory gene content differences was consistently lower within genotypic clusters obtained with the various methods (Figure 4B), ranging from n = 103–343, compared to the average rate observed for any two strains (n = 964). Thus, multi-locus methods, whether based on the analysis of core genes (i.e., MLST) or the analysis of accessory genes (i.e., CGF), appear to outperform single locus methods in grouping genetically similar strains.
Although the various subtyping methods generate groups of strains with generally high levels of genetic similarity (i.e., low core gene SNP rates and fewer accessory genome content differences), the WGS data ultimately has the resolution to differentiate the strains within these clusters on the basis of genomic differences in the accessory genome, the core genome or both. Significant genomic differences have been previously observed between strains of the same MLST ST by microarray analysis (Taboada et al., 2008) and more recently by WGS analysis (Biggs et al., 2011). Because genomic differences between clonal strains are likely a reflection of the underlying epidemiology (i.e., separation in time and space), which would allow for the accumulation of such genomic differences, approaches to target such features through laboratory-based assays or to rapidly extract and analyze them from WGS data will become increasingly important in the deployment of genomic-based approaches in an epidemiological context.
Development and Assessment of Next Generation Typing Schemes
The need for a new generation of subtyping methods is underscored by a recent study by Biggs et al. (2011), in which the authors used WGS analysis to show significant genomic variation in two isolates that were indistinguishable by MLST and flaA-SVR typing. This example illustrates how strains that are linked by low resolution subtyping methods may harbor genomic differences consistent with spatial and/or temporal separation and points to the need for higher resolution methods for strain characterization.
Even subtle genomic changes can significantly impact strain characteristics (Carrillo et al., 2004; Jerome et al., 2011) given recent evidence that genomic change in Campylobacter is greatly influenced by positive selection (Lefébure and Stanhope, 2009). Moreover, genomic changes leading to phenotypic traits of public health significance (e.g., antimicrobial resistance, virulence, survival) may significantly impact risk profiles associated with specific genotypes.
Ultimately, whole genome sequence is the gold standard for microbial strain characterization Nonetheless, although rapid high throughput whole genome sequencing is rapidly becoming a feasible option for the investigation of public health events (Gilmour et al., 2010; Chin et al., 2011; Rohde et al., 2011), high throughput lower-resolution methods are still necessary in the context of epidemiological surveillance.
An increasing body of WGS data could be used to inform the development of enhanced subtyping methods and the in silico approach that was used in this study could form the basis for a framework aimed at assessing novel subtyping methods prior to development and experimental implementation. The advantage of such a framework is that it allows for the testing of non-traditional typing targets such that most informative marker combinations could be used to develop enhanced subtyping schemes.
As the cost of sequencing continues to decline, bioinformatics pipelines that enable rapid analysis of draft genome data will enable public health laboratories to not only link WGS data to historical data but to provide optimal strain characterization using “extended MLST” analysis of hundreds of genes comprising core conserved, core variable, and accessory genes. This study demonstrates the feasibility of rapid analysis of minimally processed draft genome sequence data using an automated analytical pipeline.
Conclusion
Full genome sequence data provide the means for the evaluation of novel and existing molecular typing tools. In a post-genomics era, there is the opportunity to devise typing schemes that are based on the selection of informative regions that unambiguously provide evolutionary relationship among strains, but with sufficient resolution to capture subtle genomic changes between related strains that might arise through separation in time/space. A higher level of resolution is necessary to get an adequate representation of the true evolutionary relationship between strains that may otherwise appear to be clonal (Biggs et al., 2011).
This study was limited by the publicly available full genomes. While there is a great deal of genetic diversity captured among the strains for which WGS data are currently available, the dataset did not produce many multi-strain genotypic clusters, which made it difficult to analyze the congruence of molecular typing methods to the core genome phylogeny. The inclusion of additional strains with various degrees of genetic and epidemiologic linkage will be required to address whether current methods are sufficiently discriminatory for distinguishing closely related strains that are temporally or spatially unrelated and the analytical approaches that have been developed in this study will facilitate the assessment of molecular typing methods using a phylogenetic framework. To address this gap, we are currently in the process of sequencing a number of strains collected as part of a large-scale epidemiologic survey and that have been linked by epidemiological data and/or various typing data (Clark et al., 2012). The development of epidemiologically relevant reference panels of strains to be characterized by WGS analysis to be used for the assessment and validation of existing and emerging methods for pathogen characterization would be of great benefit to public health agencies and should be a priority for international collaboration.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Government of Canada through funding by the Genomics Research and Development Initiative. This publication made use of the Campylobacter jejuni Multi-Locus Sequence Typing website (http://pubmlst.org/campylobacter/) developed by Keith Jolley and sited at the University of Oxford (Jolley and Maiden, 2010). The development of this site has been funded by the Wellcome Trust.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Cellular_and_Infection_Microbiology/10.3389/fcimb.2012.00057/abstract
Footnotes
References
Assefa, S., Keane, T. M., Otto, T. D., Newbold, C., and Berriman, M. (2009). ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25, 1968–1969.
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., Formsma, K., Gerdes, S., Glass, E. M., Kubal, M., Meyer, F., Olsen, G. J., Olson, R., Osterman, A. L., Overbeek, R. A., Mcneil, L. K., Paarmann, D., Paczian, T., Parrello, B., Pusch, G. D., Reich, C., Stevens, R., Vassieva, O., Vonstein, V., Wilke, A., and Zagnitko, O. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi:10.1186/1471-2164-9-75
Barton, C., Ng, L. K., Tyler, S. D., and Clark, C. G. (2007). Temperate bacteriophages affect pulsed-field gel electrophoresis patterns of Campylobacter jejuni. J. Clin. Microbiol. 45, 386–391.
Biggs, P. J., Fearnhead, P., Hotter, G., Mohan, V., Collins-Emerson, J., Kwan, E., Besser, T. E., Cookson, A., Carter, P. E., and French, N. P. (2011). Whole-genome comparison of two Campylobacter jejuni isolates of the same sequence type reveals multiple loci of different ancestral lineage. PLoS ONE 6, e27121.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., and Madden, T. L. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. doi:10.1186/1471-2105-10-421
Carriço, J. A., Silva-Costa, C., Melo-Cristino, J., Pinto, F. R., De Lencastre, H., Almeida, J. S., and Ramirez, M. (2006). Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J. Clin. Microbiol. 44, 2524–2532.
Carrillo, C. D., Taboada, E., Nash, J. H., Lanthier, P., Kelly, J., Lau, P. C., Verhulp, R., Mykytczuk, O., Sy, J., Findlay, W. A., Amoako, K., Gomis, S., Willson, P., Austin, J. W., Potter, A., Babiuk, L., Allan, B., and Szymanski, C. M. (2004). Genome-wide expression analyses of Campylobacter jejuni NCTC11168 reveals coordinate regulation of motility and virulence by flhA. J. Biol. Chem. 279, 20327–20338.
Chin, C. S., Sorenson, J., Harris, J. B., Robins, W. P., Charles, R. C., Jean-Charles, R. R., Bullard, J., Webster, D. R., Kasarskis, A., Peluso, P., Paxinos, E. E., Yamaichi, Y., Calderwood, S. B., Mekalanos, J. J., Schadt, E. E., and Waldor, M. K. (2011). The origin of the Haitian cholera outbreak strain. N. Engl. J. Med. 364, 33–42.
Clark, C., Taboada, E., Grant, C., Blakeston, C., Pollari, F., Marshall, B., Rahn, K., Mackinnon, J., Daignault, D., Pillai, D., and Ng, L.-K. (2012). Comparison of molecular typing methods useful for detecting clusters of Campylobacter jejuni and C. coli isolates through routine surveillance. J. Clin. Microbiol. 50, 788–797.
Clark, C. G., Beeston, A., Bryden, L., Wang, G., Barton, C., Cuff, W., Gilmour, M. W., and Ng, L. K. (2007). Phylogenetic relationships of Campylobacter jejuni based on porA sequences. Can. J. Microbiol. 53, 27–38.
de Boer, P., Wagenaar, J. A., Achterberg, R. P., Van Putten, J. P., Schouls, L. M., and Duim, B. (2002). Generation of Campylobacter jejuni genetic diversity in vivo. Mol. Microbiol. 44, 351–359.
Dingle, K. E., Colles, F. M., Wareing, D. R., Ure, R., Fox, A. J., Bolton, F. E., Bootsma, H. J., Willems, R. J., Urwin, R., and Maiden, M. C. (2001). Multilocus sequence typing system for Campylobacter jejuni. J. Clin. Microbiol. 39, 14–23.
Dingle, K. E., Mccarthy, N. D., Cody, A. J., Peto, T. E., and Maiden, M. C. (2008). Extended sequence typing of Campylobacter spp., United Kingdom. Emerg. Infect. Dis. 14, 1620–1622.
Edgar, R. C. (2004a). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113. doi:10.1186/1471-2105-5-113
Edgar, R. C. (2004b). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797.
Gilmour, M. W., Graham, M., Van Domselaar, G., Tyler, S., Kent, H., Trout-Yakel, K. M., Larios, O., Allen, V., Lee, B., and Nadon, C. (2010). High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics 11, 120. doi:10.1186/1471-2164-11-120
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224.
Government of Canada. (2007). Canadian National Enteric Pathogen Surveillance System (C-EnterNet) 2006. Guelph, ON: Public Health Agency of Canada.
Government of Canada. (2010). Canadian National Enteric Pathogen Surveillance System (C-EnterNet) 2008. Guelph, ON: Public Health Agency of Canada.
Hanninen, M. L., Hakkinen, M., and Rautelin, H. (1999). Stability of related human and chicken Campylobacter jejuni genotypes after passage through chick intestine studied by pulsed-field gel electrophoresis. Appl. Environ. Microbiol. 65, 2272–2275.
Jerome, J. P., Bell, J. A., Plovanich-Jones, A. E., Barrick, J. E., Brown, C. T., and Mansfield, L. S. (2011). Standing genetic variation in contingency loci drives the rapid adaptation of Campylobacter jejuni to a novel host. PLoS ONE 6, e16399. doi:10.1371/journal.pone.0016399
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11, 595. doi:10.1186/1471-2105-11-595
Klena, J. D., and Konkel, M. E. (2005). “Methods for epidemiological analysis of Campylobacter jejuni,” in Campylobacter: Molecular and Cellular Biology, eds J. M. Ketley and M. E. Konkel (Wymondham: Horizon Bioscience), 165–179.
Kruczkiewicz, P., Tudor, A., Mutschall, S. K., Buchanan, C. J., Laing, C. R., Thomas, J. E., Gannon, V. P., Clark, C. G., Carrillo, C. D., and Taboada, E. N. (2011). “A bioinformatics toolkit for comparative genomic analysis of Campylobacter jejuni in support of next-generation genotyping methods design,” in 16th International Workshop on Campylobacter, Helicobacter and Related Organisms, Vancouver.
Lang, P., Lefébure, T., Wang, W., Pavinski Bitar, P., Meinersmann, R. J., Kaya, K., and Stanhope, M. J. (2010). Expanded multilocus sequence typing and comparative genomic hybridization of Campylobacter coli isolates from multiple hosts. Appl. Environ. Microbiol. 76, 1913–1925.
Lefébure, T., Bitar, P. D., Suzuki, H., and Stanhope, M. J. (2010). Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol. Evol. 2, 646–655.
Lefébure, T., and Stanhope, M. J. (2009). Pervasive, genome-wide positive selection leading to functional divergence in the bacterial genus Campylobacter. Genome Res. 19, 1224–1232.
Meinersmann, R. J., Helsel, L. O., Fields, P. I., and Hiett, K. L. (1997). Discrimination of Campylobacter jejuni isolates by fla gene sequencing. J. Clin. Microbiol. 35, 2810–2814.
Meinersmann, R. J., Phillips, R. W., Hiett, K. L., and Fedorka-Cray, P. (2005). Differentiation of Campylobacter populations as demonstrated by flagellin short variable region sequences. Appl. Environ. Microbiol. 71, 6368–6374.
Nielsen, E. M., Engberg, J., and Fussing, V. (2001). Genotypic and serotypic stability of Campylobacter jejuni strains during in vitro and in vivo passage. Int. J. Med. Microbiol. 291, 379–385.
Nuijten, P. J., Van Den Berg, A. J., Formentini, I., Van Der Zeijst, B. A., and Jacobs, A. A. (2000). DNA rearrangements in the flagellin locus of an flaA mutant of Campylobacter jejuni during colonization of chicken ceca. Infect. Immun. 68, 7137–7140.
Parkhill, J., Wren, B. W., Mungall, K., Ketley, J. M., Churcher, C., Basham, D., Chillingworth, T., Davies, R. M., Feltwell, T., Holroyd, S., Jagels, K., Karlyshev, A. V., Moule, S., Pallen, M. J., Penn, C. W., Quail, M. A., Rajandream, M. A., Rutherford, K. M., Van Vliet, A. H., Whitehead, S., and Barrell, B. G. (2000). The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403, 665–668.
Pearson, B. M., Gaskin, D. J., Segers, R. P., Wells, J. M., Nuijten, P. J., and Van Vliet, A. H. (2007). The complete genome sequence of Campylobacter jejuni strain 81116 (NCTC11828). J. Bacteriol. 189, 8402–8403.
Pruitt, K., Brown, G., Tatusova, T., and Maglott, D. (2002). “The Reference Sequence (RefSeq) Database,” in The NCBI Handbook, eds J. Mcentyre and J. Ostell (Bethesda, MD: National Center for Biotechnology Information). Available at: http://www.ncbi.nlm.nih.gov/books/NBK21091/
Public Health Agency of Canada. (2009). Canadian Integrated Surveillance Report: Salmonella, Campylobacter, verotoxigenic E. coli and Shigella from 2000-2004. Can. Commun. Dis. Rep. 35, S3.
Ridley, A. M., Toszeghy, M. J., Cawthraw, S. A., Wassenaar, T. M., and Newell, D. G. (2008). Genetic instability is associated with changes in the colonization potential of Campylobacter jejuni in the avian intestine. J. Appl. Microbiol. 105, 95–104.
Rohde, H., Qin, J., Cui, Y., Li, D., Loman, N. J., Hentschke, M., Chen, W., Pu, F., Peng, Y., Li, J., Xi, F., Li, S., Li, Y., Zhang, Z., Yang, X., Zhao, M., Wang, P., Guan, Y., Cen, Z., Zhao, X., Christner, M., Kobbe, R., Loos, S., Oh, J., Yang, L., Danchin, A., Gao, G. F., Song, Y., Yang, H., Wang, J., Xu, J., Pallen, M. J., Aepfelbacher, M., and Yang, R. (2011). Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N. Engl. J. Med. 365, 718–724.
Sails, A. D., Bala, S., and Fields, P. I. (2003). Utility of multilocus sequence typing as an epidemiological tool for investigation of outbreaks of gastroenteritis caused by Campylobacter jejuni. J. Clin. Microbiol. 41, 4733–4739.
Schmidt, H. A., and von Haeseler, A. (2007). Maximum-likelihood analysis using TREE-PUZZLE. Curr. Protoc. Bioinformatics Chapter 6, Unit 6 6.
Severiano, A., Pinto, F. R., Ramirez, M., and Carrico, J. A. (2011). Adjusted Wallace coefficient as a measure of congruence between typing methods. J. Clin. Microbiol. 49, 3997–4000.
Sheppard, S. K., Mccarthy, N. D., Falush, D., and Maiden, M. C. (2008). Convergence of Campylobacter species: implications for bacterial evolution. Science 320, 237–239.
Suzuki, S., Ono, N., Furusawa, C., Ying, B. W., and Yomo, T. (2011). Comparison of sequence reads obtained from three next-generation sequencing platforms. PLoS ONE 6, e19534. doi:10.1371/journal.pone.0019534
Taboada, E. N., Mackinnon, J. M., Luebbert, C. C., Gannon, V. P., Nash, J. H., and Rahn, K. (2008). Comparative genomic assessment of multi-locus sequence typing: rapid accumulation of genomic heterogeneity among clonal isolates of Campylobacter jejuni. BMC Evol. Biol. 8, 229. doi:10.1186/1471-2148-8-229
Taboada, E. N., Ross, S. L., Mutschall, S. K., Mackinnon, J. M., Roberts, M. J., Buchanan, C. J., Kruczkiewicz, P., Jokinen, C., Thomas, J. E., Nash, J. H., Gannon, V. P., Marshall, B., Pollari, F., and Clark, C. G. (2012). Development and validation of a comparative genomic fingerprinting method for high-resolution genotyping of Campylobacter jejuni. J. Clin. Microbiol. 50, 798–809.
Thomas, M. K., Majowicz, S. E., Pollari, F., and Sockett, P. N. (2008). Burden of acute gastrointestinal illness in Canada, 1999-2007: interim summary of NSAGI activities. Can. Commun. Dis. Rep. 34, 8–15.
Wilson, D. J., Gabriel, E., Leatherbarrow, A. J., Cheesbrough, J., Gee, S., Bolton, E., Fox, A., Hart, C. A., Diggle, P. J., and Fearnhead, P. (2009a). Rapid evolution and the importance of recombination to the gastroenteric pathogen Campylobacter jejuni. Mol. Biol. Evol. 26, 385–397.
Wilson, M. K., Lane, A. B., Law, B. F., Miller, W. G., Joens, L. A., Konkel, M. E., and White, B. A. (2009b). Analysis of the Pan Genome of Campylobacter jejuni isolates recovered from poultry by pulsed-field gel electrophoresis, multilocus sequence typing (MLST), and repetitive sequence polymerase chain reaction (rep-PCR) reveals different discriminatory capabilities. Microb. Ecol. 58, 843–855.
Zautner, A. E., Herrmann, S., Corso, J., Tareen, A. M., Alter, T., and Gross, U. (2011). Epidemiological association of different Campylobacter jejuni groups with metabolism-associated genetic markers. Appl. Environ. Microbiol. 77, 2359–2365.
Keywords: Campylobacter spp., genome, MLST, CGF, flaA, porA, molecular epidemiology
Citation: Carrillo CD, Kruczkiewicz P, Mutschall S, Tudor A, Clark C and Taboada EN (2012) A framework for assessing the concordance of molecular typing methods and the true strain phylogeny of Campylobacter jejuni and C. coli using draft genome sequence data. Front. Cell. Inf. Microbio. 2:57. doi: 10.3389/fcimb.2012.00057
Received: 02 December 2011; Accepted: 12 April 2012;
Published online: 01 May 2012.
Edited by:
Alain Stintzi, Ottawa Institute of Systems Biology, CanadaCopyright: © 2012 Carrillo, Kruczkiewicz, Mutschall, Tudor, Clark and Taboada. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Eduardo N. Taboada, Laboratory for Foodborne Zoonoses, Public Health Agency of Canada, c/o Animal Diseases Research Institute, Canadian Food Inspection Agency, Twp Rd. 9-1, Lethbridge, Alberta, T1J 3Z4. e-mail: eduardo.taboada@phac-aspc.gc.ca