MiR-155 induction by microbes/microbial ligands requires NF-κB-dependent de novo protein synthesis
- 1 Molecular, Cellular, and Developmental Biology Program, The Ohio State University, Columbus, OH, USA
- 2 Department of Internal Medicine, The Ohio State University, Columbus, OH, USA
- 3 Integrated Biomedical Science Graduate Program, The Ohio State University, Columbus, OH, USA
- 4 Department of Microbial Infection and Immunity, The Ohio State University, Columbus, OH, USA
- 5 St. Jude Children’s Research Hospital, Memphis, TN, USA
MiR-155 regulates numerous aspects of innate and adaptive immune function. This miR is induced in response to Toll-like receptor ligands, cytokines, and microbial infection. We have previously shown that miR-155 is induced in monocytes/macrophages infected with Francisella tularensis and suppresses expression of the inositol phosphatase SHIP to enhance activation of the PI3K/Akt pathway, which in turn promotes favorable responses for the host. Here we examined how miR-155 expression is regulated during infection. First, our data demonstrate that miR-155 can be induced through soluble factors of bacterial origin and not the host. Second, miR-155 induction is not a direct effect of infection and it requires NF-κB signaling to up-regulate fos/jun transcription factors. Finally, we demonstrate that the requirement for NF-κB-dependent de novo protein synthesis is globally shared by microbial ligands and live bacteria. This study provides new insight into the complex regulation of miR-155 during microbial infection.
Introduction
MicroRNAs (miRs) are potent regulators of gene expression and their dysregulation has been shown to play a critical role in numerous pathogenic conditions (Bushati and Cohen, 2008; van Rooij et al., 2008; Lee and Dutta, 2009; Jiang et al., 2010). Much of the work regarding miR regulation and function has been done in the context of cancer or developmental biology (Bushati and Cohen, 2007); however, less is known about their role in bacterial pathogenesis. Groups studying viral pathogenesis have already embraced the notion that miRs regulate key host cellular processes that influence immune response and microbial persistence (Henke et al., 2008; Hou et al., 2009).
There are a select number of miRs identified to be responsive to inflammatory stimuli and also to regulate different aspects of the immune system (Taganov et al., 2006). One miR that has been of great interest is miR-155, originating from the non-protein-coding gene BIC (Eis et al., 2005), because this miR has been shown to be induced by TLR ligands, interferon and pro-inflammatory cytokines (O’Connell et al., 2007), and intracellular bacteria (Cremer et al., 2009; Izar et al., 2012). Functionally this miR is important for B-cell maturation (Thai et al., 2007; Vigorito et al., 2007), T-cell functions (Rodriguez et al., 2007; Kohlhaas et al., 2009), antigen presentation by dendritic cells (Rodriguez et al., 2007), pro-inflammatory response (Cremer et al., 2009; O’Connell et al., 2010), and oncogenesis (Costinean et al., 2006). Therefore, understanding the regulation and function of miR-155 within the context of microbial infections is of considerable interest.
We recently reported that miR-155 is induced by and regulates host response to the intracellular Gram-negative bacterium Francisella tularensis (Cremer et al., 2009), which is the causative agent of the disease tularemia (Ellis et al., 2002; Santic et al., 2010). There are different subspecies of F. tularensis with varying degrees of virulence in humans. F. tularensis subspecies tularensis (F.t.) is regarded as having the highest virulence in humans and exposure to 10 colony forming units (CFUs) can be lethal if left untreated. F. tularensis subspecies novicida (F.n.) rarely causes disease in humans (Oyston et al., 2004) but has been a useful model for studying tularemia in mice because it remains highly virulent in that organism (Kieffer et al., 2003). Another benefit of studying F.n. is to compare immune response to different subspecies (Butchar et al., 2008), which has yielded new insight potentially accounting for the high virulence of F.t. for example, miR-155 expression is highly induced in human monocytes infected with F.n. but minimally induced by the more virulent F.t. (Cremer et al., 2009).
In an earlier study we reported that miR-155 negatively regulated the phosphatase SHIP, which limited Akt activation (Parsa et al., 2006). In the context of Francisella infection we found that Akt promoted NF-κB activity, the production of pro-inflammatory cytokines (Rajaram et al., 2006), phagosome maturation, inhibition of macrophage cell death (Rajaram et al., 2009), and the survival of mice during in vivo challenge (Rajaram et al., 2006). Thus miR-155 expression is beneficial to the host by promoting the activation of Akt through the inhibition of SHIP. MiR-155 was found to be induced through TLR2 and MyD88, and it required activation of NF-κB (Cremer et al., 2009). In studies using other stimuli, pharmacologic inhibition of either NF-κB or AP-1 resulted in marked reductions of miR-155 expression (Xiao et al., 2009; Dai et al., 2011). However, mutation analysis of the NF-κB and AP-1 sites within the BIC/miR-155 promoter showed that alteration of the AP-1 site led to substantially greater reductions in miR-155 than mutations in either or both of the NF-κB sites (Yin et al., 2008a,b; Xiao et al., 2009). This suggests that, although NF-κB may directly contribute to miR-155 induction by binding its promoter, it also exerts a powerful indirect influence.
In the current study we sought to understand the precise means through which miR-155 was induced during bacterial infection. We report that miR-155 induction by various bacterial ligands and live bacteria requires NF-κB-dependent host cell protein synthesis and thus is not a direct product of TLR stimulation/infection. We further demonstrate that the newly synthesized protein is not a soluble autocrine/paracrine factor but rather the intracellular transcription factor AP-1. To our knowledge this is the first report of the dependence on NF-κB driven de novo protein synthesis for miR-155 induction in response to microbial stimuli.
Results
miR-155 Induction by Francisella Requires New Host Cell Protein Synthesis
To ascertain if miR-155 induction was a direct effect or an indirect effect of infection with F.n. we made use of the eukaryotic protein synthesis inhibitor cycloheximide. Human peripheral blood monocytes (PBM) were infected with F.n. at an MOI of 50 for 6 h in the presence or absence of cycloheximide. We found that cycloheximide significantly impaired miR-155 induction (Figure 1A), which was somewhat surprising since the BIC promoter contains an NF-κB site (Gatto et al., 2008; Yin et al., 2008b). To verify the effectiveness of cycloheximide in blocking protein synthesis we examined the mRNA and protein production of a direct response gene, TNFα. As anticipated TNFα mRNA was induced equally well in the presence or absence of cycloheximide (Figure 1B). However, cycloheximide significantly reduced TNFα protein production (Figure 1C). Of note, cell viability was minimally affected by cycloheximide, as assessed by Trypan blue exclusion.
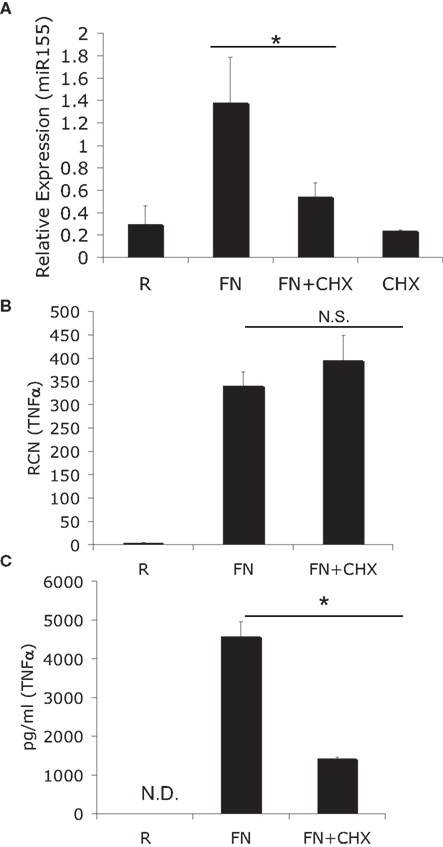
Figure 1. MiR-155 induction requires de novo host-protein synthesis. (A) PBM were infected with F. novicida (FN) at an MOI of 50 for 6 h in the presence or absence of 2 μM cycloheximide (CHX), and then RNA was isolated. “R” stands for resting/uninfected cells. Mature miR-155 expression was assayed by qRT-PCR and data expressed as relative expression from triplicate samples. (B) TNFα mRNA was assayed from the same samples as in (A). (C) Secreted TNFα was measured from the media of the cells in (A) by ELISA. *Designates a p value < 0.05 by Students t-test, while N.S. designates a p value > 0.05. These data are representative of three independent experiments.
MiR-155 Expression Can Be Induced through Soluble Factors
The finding that miR-155 induction requires new host-protein synthesis presents at least two possibilities: miR-155 may be induced through a host autocrine/paracrine-acting factor, or through an internal factor that mediates BIC/miR-155 transcription. To gain further insight into the induction of miR-155 we tested if soluble factors present during the course of infection could mediate the induction of miR-155. For this experiment we infected PBM with F.n. at an MOI of 50 for 24 h. RNA was collected to assay miR-155 expression. Conditioned media was sterile filtered to remove bacteria and then added to new PBM for a 24 h period. The removal of bacteria from the conditioned media was verified by plating filtered media on chocolate agar plates. MiR-155 expression from directly infected cells or cells treated with conditioned media was assayed by qRT-PCR (Figure 2A). Results showed that the filtered, conditioned media is sufficient to mediate partial induction of miR-155. Therefore, although direct contact with Francisella may induce miR-155 (likely through TLR2), it is not required.
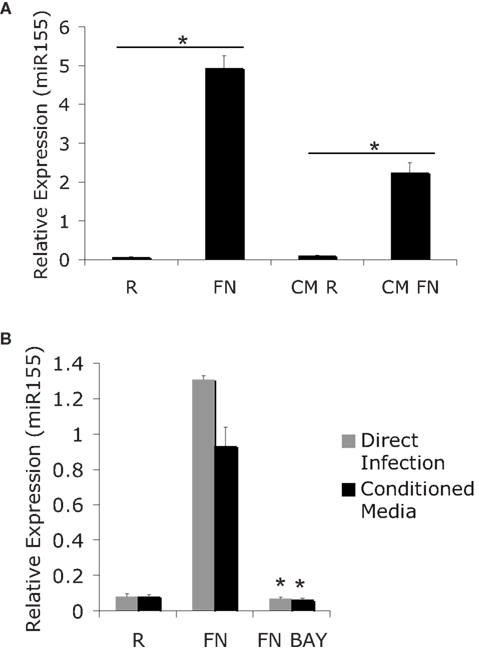
Figure 2. MiR-155 induction can be mediated indirectly though secreted factors. (A) PBM were infected with FN at an MOI of 50 for 24 h. Media from infected cells was sterile filtered to remove bacteria and RNA was isolated from the cells (R and Fn). New PBM were cultured in the conditioned media for 24 h (CM R and CM FN) followed by RNA isolation. Mature miR-155 expression was assayed by qRT-PCR. (B) PBM were infected for 24 h, then the media from the infected cells was sterile filtered to generate conditioned media. New PBM were infected with F.n. at an MOI of 50 for 6 h or treated with conditioned media for 6 h in the presence of BAY-7085-11 (5 μM) or DMSO control. MiR-155 expression was assayed by qRT-PCR. These data are representative of at least three independent experiments. *Designates a p value < 0.05.
We and others have previously reported that miR-155 induction is NF-κB-dependent (Tili et al., 2007; Gatto et al., 2008; Cremer et al., 2009). Having found that miR-155 can be induced through conditioned media from infected cells, we tested whether the induction of miR-155 through conditioned media also required NF-κB. For this experiment we infected PBM at an MOI of 50 of F.n. for 24 h and then sterile filtered the conditioned media. New PBM were infected directly with F.n. at an MOI of 50 for 6 h or cultured in the conditioned media for 6 h. This was done in the presence or absence of the IKK/NF-κB inhibitor BAY-11-7085. These samples were then assayed for miR-155 expression by qRT-PCR (Figure 2B). As in Figure 2A the conditioned media elicited a similar induction of miR-155 to direct infection. Direct infection and conditioned media both required NF-κB activation to induce miR-155 expression.
TNFα and IFNβ are Not Major Mediators of Francisella-Induced miR-155 Expression
We first explored the possibility that miR-155 induction may be mediated by a secreted factor given that TNFα, IFNβ, and IFNγ have all been reported to induce miR-155 (O’Connell et al., 2007). Macrophages have been shown to produce TNFα (Telepnev et al., 2003) and IFNβ (Henry et al., 2007) during Francisella infection. Therefore, we assessed miR-155 induction in the presence or absence of neutralizing antibodies against both cytokines to determine if either cytokine was responsible for miR-155 induction. PBM were infected at an MOI of 50 for 6 h with F.n. alone, F.n. with TNFα neutralizing antibody, or F.n. with isotype control antibody. RNA was collected to assay miR-155 expression by qRT-PCR. Neutralizing TNFα did not impair miR-155 induction (Figure 3A). To examine the ability of the neutralizing antibody to block TNFα an ELISA for TNFα was performed on the media from Figure 3A. The neutralizing antibody impaired the ability of TNFα to be recognized by ELISA, while the isotype control did not (Figure 3B).
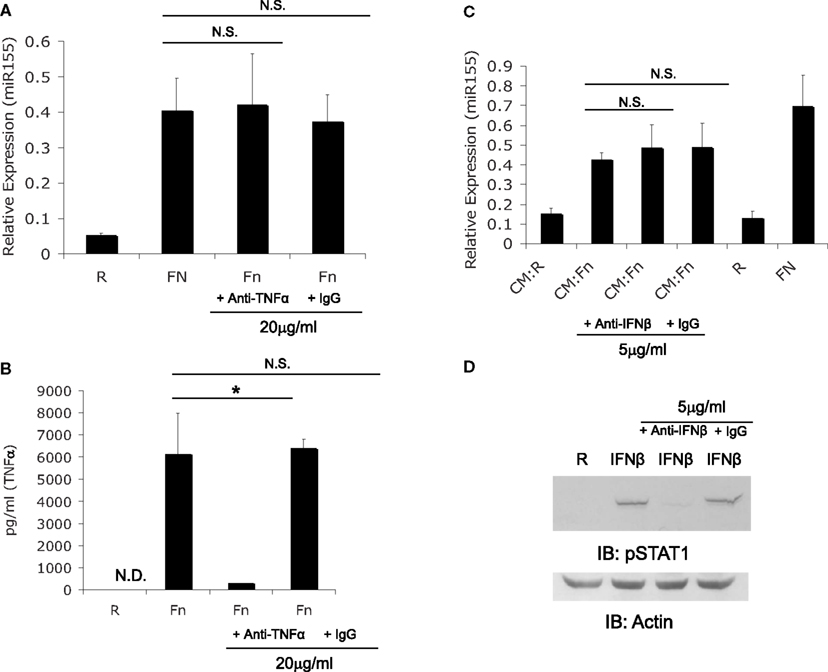
Figure 3. F. novicida induction miR-155 expression is not mediated by TNFα or IFNβ. (A) PBM were treated with TNFα neutralizing antibody or isotype control antibody, then infected with F.n. at an MOI of 50 for 6 h. RNA was collected and assayed for miR-155 expression. (B) The media from the samples in (A) was assayed for human TNFα by ELISA. (C) Conditioned media (CM) was obtained as in Figures 2B. Conditioned media from monocytes uninfected (CM:R) or infected (CM:Fn) with F.n. at an MOI of 50 was sterile filtered and incubated with neutralizing antibody or isotype control antibody at 37°C for 1 h. New PBM were then cultured in the conditioned media conditions, uninfected (R), or direct infection (FN) for 6 h. RNA was collected and miR-155 expression was assayed by qRT-PCR. (D) Effective neutralization of IFNβ was demonstrated by pre-treating media with recombinant IFNβ (500 U/ml) with or without neutralizing antibody or isotype control antibody. Then PBM were cultured in media alone, IFNβ, IFNβ plus neutralizing antibody, or IFNβ plus isotype control for 15 min. Cells were lysed and subject to western blot for phosphorylated STAT1, followed by Actin re-probe.
Next we examined the contribution of IFNβ to F.n. induced miR-155 expression. In order for the neutralizing antibody against IFNβ to be effective, media was pre-incubated with the neutralizing antibody for one hour. PBM were uninfected or infected with F.n. at an MOI of 50 for 24 h, and then sterile filtered to remove bacteria. New PBM then received the conditioned media (CM:R or CM:Fn) or direct infection (R or Fn) for 6 h and were assayed for miR-155 expression. Neutralization of IFNβ did not impair miR-155 induction in PBM (Figure 3C). The effectiveness of the IFNβ neutralizing antibody was shown by treating PBM with recombinant IFNβ for 15 min cell lysates were probed for phosphorylated STAT1, which is activated by IFNβ (Toshchakov et al., 2002). The neutralizing antibody effectively blocked STAT1 phosphorylation (Figure 3D).
Together the data show that while TNFα and IFNβ have the ability to induce miR-155 (O’Connell et al., 2007), F.n.-induced miR-155 expression does not require either cytokine in mediating the effect. We also tested the ability of recombinant TNFα to induce miR-155 expression and find there is only a marginal degree of miR-155 induction relative to the levels produced by F.n. infected monocytes (data not shown). IFNβ has been shown to be produced by murine macrophages (Henry et al., 2007) but not by human monocytes. Recombinant IFNβ had minimal ability to induce miR-155 in primary human monocytes (data not shown).
Cytokine Release is not Required for MiR-155 Induction
Although we excluded TNFα and IFNβ as the soluble secreted factors that induce miR-155 expression, other factors may be involved and they are too numerous to test one-by-one. Hence, we used the exocytosis inhibitor, Brefeldin A, which prevents secretion of cytokines (Zhu et al., 1998). PBM were pre-treated with DMSO vehicle control or Brefeldin A for 30 min, then infected with F.n. at an MOI of 50 for 16 h. Samples were assayed for miR-155 expression by qRT-PCR and results showed that Brefeldin A did not impair miR-155 induction (Figure 4A). We concurrently measured extra- and intra-cellular levels of TNFα and found that Brefeldin A did effectively inhibit secretion (Figures 4B,C).
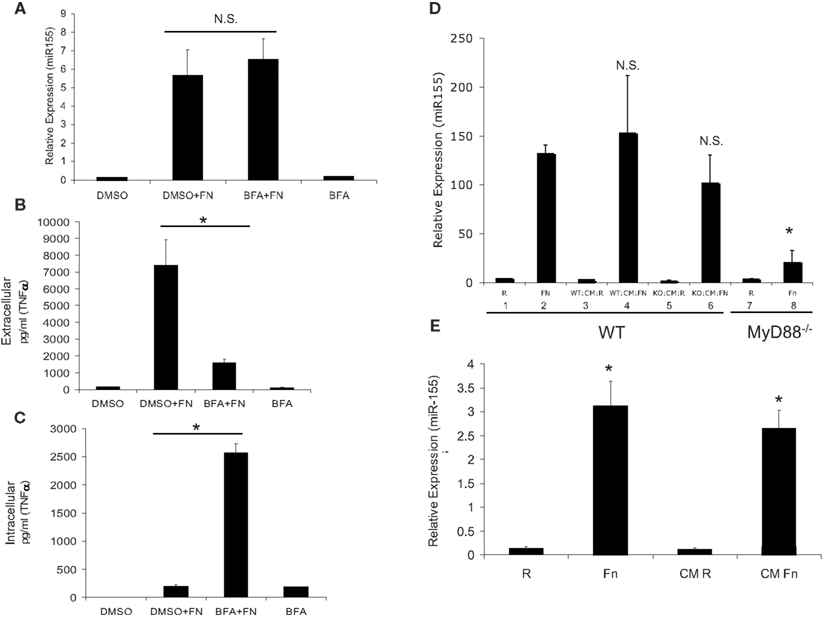
Figure 4. MiR-155 induction is not mediated through host cytokines, but rather by soluble bacterial factors. (A) PBM were infected with F.n. at an MOI of 50 for 16 h in the presence of Brefeldin A (BFA) or DMSO vehicle control. RNA was isolated and mature miR-155 expression was assayed by qRT-PCR. (B) Extracellular TNFα levels were measured by ELISA. (C) Intracellular TNFα levels were measured by ELISA from the cell lysate. (D) WT and MyD88−/− BMMs were infected for 24 h at an MOI of 50 and then sterile filtered to remove bacteria to generate conditioned media (CM). RNA was collected from the infected cells for qRT-PCR (Bars 1, 2, 7, 8). The conditioned media was then placed on new wild-type BMM for 24 h and RNA was subsequently collected for qRT-PCR (Bars 3, 4, 5, 6). These data are representative of three independent experiments. *Designates a p value < 0.05. (E) F.n. was suspended in RPMI-1640 with 10% heat-inactivated fetal bovine serum at a concentration of 3.5 × 108 bacteria/ml (comparable to 5 × 106 PBM infected at MOI 50 in 1 ml) and incubated at 37°C for 24 h. The media was sterile filtered to remove bacteria. PBM were directly infected with F.n. at an MOI of 50 or treated with the filtered media that previously contained F.n. for 16 h. RNA was isolated and miR-155 expression assayed by qRT-PCR. These data are representative of three independent experiments. *Designates a p value < 0.05.
An alternative means to examine the contribution of host cytokines in inducing miR-155 is to test the ability of conditioned media from responsive versus non-responsive cells to induce miR-155. It is known that the production of inflammatory mediators in response to F.n. is TLR2/MyD88-dependent. MyD88−/− macrophages do not produce pro-inflammatory cytokines (Cole et al., 2007; Cremer et al., 2009) or induce miR-155 expression (Cremer et al., 2009). Bone marrow-derived macrophages from wild-type and MyD88−/− mice were uninfected or infected with F.n. at an MOI of 50 for 24 h (Figure 4D). The conditioned media was sterile filtered and used to stimulate new wild-type macrophages. Wild-type macrophage infected with F.n. (Bars 1, 2) or the conditioned media of wild-type macrophages (Bars 3, 4) show a strong induction of miR-155 as expected. However, wild-type macrophages treated with the conditioned media of infected MyD88−/− macrophages also showed strong induction (Bars 5, 6), even though the MyD88−/− macrophages do not have the ability to produce pro-inflammatory cytokines through TLR2 activation (Li et al., 2006; Cole et al., 2007). Finally as expected the MyD88−/− macrophages did not respond to F.n. infection (Bars 7, 8). Therefore, induction of miR-155 can be separated from the production of host inflammatory cytokines.
Although filtering the media removes bacteria, soluble bacterial factors could be preserved and these may play a role in miR-155 induction. To test this we cultured F.n. in RPMI-1640 for 24 h at a density of 3.5 × 108 bacteria/ml (comparable to MOI of 50 for 5 × 106 cells in 1 ml), and then sterile filtered the media. PBM were isolated and then either directly infected with F.n. at an MOI of 50 or cultured with the filtered bacterial media for 24 h (Figure 4E). MiR-155 was highly induced following both treatments. Thus we conclude that miR-155 expression can be induced through filterable factors, yet they are of bacterial and not host origin.
Fos and Jun Transcription Factors are Up-Regulated by NF-κB and Control miR-155 Expression
Our data suggest that miR-155 induction is mediated through soluble bacterial factors and not host-produced factors. However, there is still a requirement for new host-protein synthesis. Since the newly produced protein is not a secreted cytokine, it is likely a transcription factor. Fos and Jun transcription factors act at AP-1 sites (Karin et al., 1997). Their expression is induced by inflammatory stimuli and they are transiently expressed (Hambleton et al., 1996; Hao and Baltimore, 2009). A detailed study of the BIC/miR-155 promoter showed that there are AP-1, NF-κB, and Ets sites but only the AP-1 site is critical for BIC/miR-155 transcription in B cells (Yin et al., 2008b). Fos and Jun can be controlled by NF-κB (Anest et al., 2004) and/or through MAPK (Yin et al., 2008b).
To test if Fos and Jun were responsive to infection, PBM were infected with F.n. at an MOI of 50 for 4 h in the presence or absence of the NF-κB inhibitor (BAY-11-7085). C-Fos (Figure 5A) and c-Jun (Figure 5B) were almost undetectable by Western blot under resting conditions, yet both were strongly up-regulated by infection. Additionally, their up-regulation was NF-κB-dependent. Thus this accounts for the dependence of miR-155 induction on new host-protein synthesis and indirect NF-κB activity. We next examined AP-1 activity by a luciferase reporter. Macrophages were transiently transfected with the reporter and infected with F.n. at an MOI of 50 for 1, 4, 8, or 24 h (Figure 5C). The pattern of AP-1 activity matched that of miR-155 induction over the same time course (Cremer et al., 2009). Lastly we confirmed that AP-1 activity had a dependence on NF-κB. Macrophages expressing the AP-1 reporter were pre-treated with vehicle control, IKK/NF-κB inhibitor (BAY-11-7085), or ERK inhibitor (U0) and then infected with F.n. at an MOI of 50 for 8 h. Inhibition of NF-κB significantly impaired AP-1 activity, and inhibition of ERK disrupted AP-1 activity as expected (Figure 5D). Together the data provide evidence that NF-κB controls AP-1, which is responsible for BIC/miR-155 transcription.
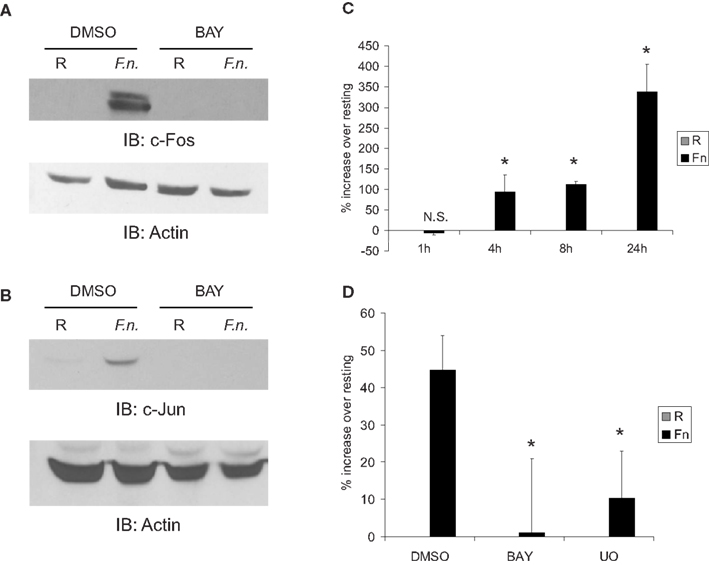
Figure 5. Fos and Jun expression is highly up-regulated by infection in an NF-κB-dependent mechanism to control AP-1 activity. (A) PBM were pre-treated with DMSO or BAY-11-7085 (5 μM; designated as BAY in the figure) for 30 min then infected with F.n. at an MOI of 50 for 4 h. Cells were lysed and subject to western blotting for c-Fos, followed by actin re-probe. (B) Protein-matched lysates were probed for c-Jun and re-probed with actin antibody. (C) RAW264.7 macrophages were transfected with an AP-1 luciferase reporter. 14 h post-transfection cells were uninfected or infected with F.n. at an MOI of 50 for 1, 4, 8, or 24 h. Cells were lysed and luciferase activity was measured by a luminometer in triplicate. Data are represented as % increase over uninfected control. (D) RAW264.7 macrophages were transfected with the AP-1 reporter as in (C). Fourteen hours post-transfection cells were pre-treated with DMSO vehicle control, BAY-11-7085, or U0126 then infected for 8 h. Data are represented as % increase over uninfected samples. These data are representative of three independent experiments. *Designates a p value < 0.05.
Fos Expression is Required for MiR-155 Induction
To directly test the role of Fos/Jun transcription factors as mediators of miR-155 induction during F.n. infection we used siRNA to knock down Fos expression. Macrophages were transfected with siRNA control or siRNA against c-Fos. We had found that Fos and Jun were expressed at very low to undetectable levels basally and induced by F.n. infection. Thus we transfected cells with the siRNA and 30 min post-transfection infected them with F.n. at an MOI of 50 for 4 h. The siRNA was highly effective at knocking down induced c-Fos expression (Figure 6A). We next assayed miR-155 expression in macrophages that had been infected with F.n. at an MOI of 50 for 16 h (Figure 6B). Macrophages transfected with control siRNA displayed a robust induction of miR-155 as expected, whereas in cells where c-Fos had been knocked down miR-155 expression was significantly reduced. Thus, c-Fos is essential for mediating miR-155 induction in response to F.n.
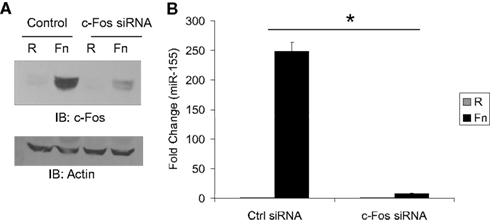
Figure 6. MiR-155 induction is dependent upon c-Fos. (A) RAW 264.7 macrophages were transfected with 4.0 μM siRNA control or siRNA against c-Fos. Thirty minutes post-transfection cells were left uninfected or infected with F.n. at an MOI of 50 for 4 h. Cells were then lysed and subject to western blotting for c-Fos. An antibody against Actin was used to re-probe the blots to ensure equal loading. (B) RAW 264.7 macrophages were transfected as done in part A and infected for 16 h. RNA was collected and miR-155 expression was assayed by qRT-PCR. These data are representative of three independent experiments. *Designates a p value < 0.05.
NF-κB-Dependent de novo Protein Synthesis is Globally Required for miR-155 Induction in Response to Microbial Stimuli
Next, to determine whether the dependence on de novo protein synthesis for miR-155 induction was common to other innate immune stimuli, we tested the responses of monocytes to two TLR agonists and two additional pathogens. Monocytes were incubated with LPS (TLR4), R848 (TLR7/8), B. cenocepacia (B.c.), or M. smegmatis (M.s.). M.s. has been shown to up-regulate miR-155 expression and decrease the expression of the downstream target SHIP in human macrophages (Rajaram et al., 2011). As shown in Figure 7, all stimuli led to significant induction of miR-155 expression, and a concomitant reduction in SHIP expression, as measured by real-time RT-PCR. This suggests a common mechanism of induction, and is in line with a central role of NF-κB.
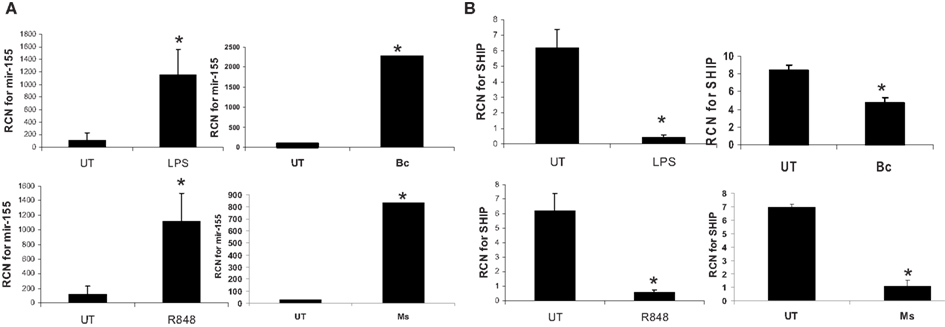
Figure 7. MiR-155 is induced by microbes/microbial ligands with a concomitant decrease in SHIP expression. Human PBM were treated with LPS (500 ng/ml), R848 (1 uM), or infected with B. cenocepacia (designated as Bc in the figure) at MOI 5, or M. smegmatis (designated as Ms in the figure) at MOI 5. mRNA was isolated from the cells 16 h later and assayed by qRT-PCR for (A) miR-155 and (B) SHIP expression. These data are representative of six independent experiments. *Designates a p value < 0.05.
To test directly the role of NF-κB, we treated or infected monocytes as above, but this time with or without pretreatment with the NF-κB inhibitor Bay-11-7085. The inhibitor reduced basal miR-155 and prevented its induction following treatment/infection (Figure 8).
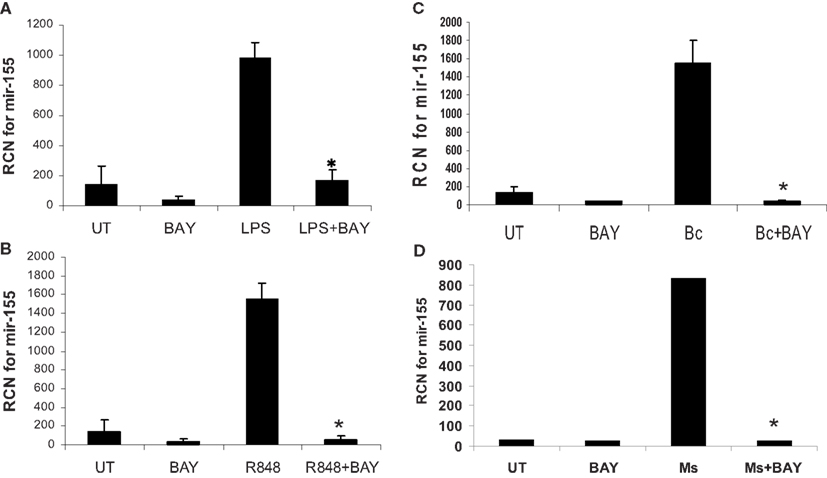
Figure 8. MiR-155 induction by microbes/microbial ligands is NF-κB-dependent. (A–D) Human PBM were pre-treated with or without the NF-κB inhibitor (BAY-11-7085, designated as BAY in the figure), and subsequently treated with (A) LPS (500 ng/ml), (B) R848 (1 uM), or infected with (C) B. cenocepacia (Bc) at MOI 5, or (D) M. smegmatis (Ms) at MOI 5. mRNA was isolated from the cells 16 h later and assayed for miR-155 by qRT-PCR. These data are representative of six independent experiments. *Designates a p value < 0.05.
We then examined the induction of c-Jun and c-Fos following exposure to these agonists and bacteria. Monocytes were incubated with LPS, R848, B.c., or M.s., each with or without pretreatment with the NF-κB inhibitor. As shown in Figure 9, each induced both c-Jun and c-Fos, but NF-κB inhibition very effectively prevented this. Collectively, these results provide evidence that innate immune stimuli lead to c-Jun and c-Fos expression, and that this requires NF-κB.

Figure 9. c-Jun and c-Fos are induced by microbes/microbial ligands in an NF-κB-dependent manner. (A–D) Human PBM were pre-treated with or without the NF-κB inhibitor (BAY-11-7085, designated as BAY in the figure), and subsequently treated with (A) LPS (500 ng/ml), (B) R848 (1 uM), or infected with (C) B. cenocepacia (Bc) at MOI 5, or (D) M. smegmatis (Ms) at MOI 5. Cell lysates were collected 16 h later. Protein-matched lysates were analyzed by Western blotting with c-Jun or c-Fos antibodies. All membranes were re-probed with Actin antibody to ensure equal loading of protein in all lanes. These data are representative of four independent experiments.
Finally, to confirm that it is NF-κB-mediated expression of de novo protein synthesis that drives miR-155, we treated/infected monocytes with or without cycloheximide pretreatment and measured miR-155. Results showed that cycloheximide blocked miR-155 induction (Figure 10). Parallel experiments testing the expression of c-Jun and c-Fos demonstrated that, as expected, cycloheximide treatment inhibited protein expression of these molecules (data not shown).
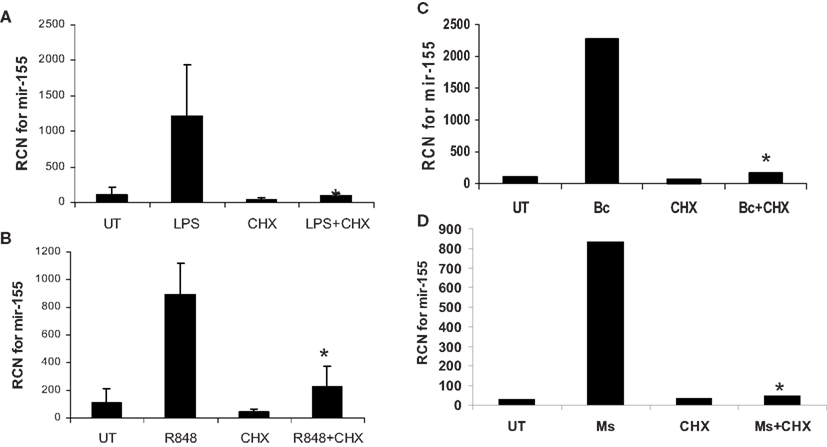
Figure 10. MiR-155 induction by microbes/microbial ligands requires de novo protein synthesis. (A–D) Human PBM were pre-treated with or without the protein synthesis inhibitor (CHX), and subsequently treated with (A) LPS (500 ng/ml), (B) R848 (1 uM), or infected with (C) B. cenocepacia (Bc) at MOI 5, or (D) M. smegmatis (Ms) at MOI 5. mRNA was isolated from the cells 16 h later and assayed for miR-155 by qRT-PCR. These data are representative of six independent experiments. *Designates a p value < 0.05.
Discussion
MiR-155 has been reported to be induced by various factors including TLR ligands, interferon, and pro-inflammatory cytokines (O’Connell et al., 2007). However, the mechanisms of induction are not fully understood. The human BIC promoter contains c-Ets, AP-1 and NF-κB binding sites. Previous studies have demonstrated that NF-κB activation is necessary for miR-155 induction (Gatto et al., 2008; Cremer et al., 2009), however, studies in B lymphocytes demonstrated, by mutational analysis of the BIC promoter, that of the three transcription factor binding sites only the AP-1 binding, not NF-κB binding site, site was necessary for miR-155 induction (Yin et al., 2008b). These studies together suggest that although NF-κB activation is necessary for miR-155 induction, it may only be indirectly involved perhaps through the transcription of intermediate molecules. Here we report that NF-κB-dependent de novo synthesis of c-Jun and c-Fos is necessary for miR-155 induction by innate immune stimuli in monocytes.
Our finding that miR-155-inducing factors could be transferred though sterile filtered media presented two possibilities: the first possibility was that a cytokine such as TNFα, IFNβ, IFNγ, or even IL-1β was inducing miR-155 through autocrine/paracrine signaling (O’Connell et al., 2007; Pottier et al., 2009). We neutralized TNFα and IFNβ, and found that blocking either cytokine alone did not impair miR-155 induction. Monocytes/macrophages are not considered major sources of IFNγ, though we have also used neutralizing antibodies against IFNγ and found no effect on miR-155 induction (data not shown). In our earlier study we found that miR-155 was induced independently of caspase-1 (Cremer et al., 2009), so IL-1β would not be implicated either (Henry et al., 2007). The second possibility was that there are soluble factors originating from the bacterium that lead to miR-155 induction. This possibility is supported by the data in Figure 4E. We believe that it is unlikely that LPS is mediating the induction of miR-155 since Francisella LPS is a poor TLR4 agonist (Gunn and Ernst, 2007) and we previously found the response to be TLR2-dependent (Cremer et al., 2009). On the other hand, Francisella is known to express multiple lipoproteins that activate TLR1/2 or TLR2/6 (Thakran et al., 2008) and thus these are more likely to be the soluble bacterial factors that induce miR-155 expression. Identification of the bacterial factors is a focus for future investigation.
Much work has gone into identifying targets of miR-155. Of the targets identified and verified to date, the most well studied is the inositol phosphatase SHIP. We and others have demonstrated that miR-155 induction leads to SHIP downregulation and results in enhanced activation of the PI3K/Akt pathway. Activation of the PI3K/Akt pathway is beneficial in the context of bacterial infections, as it promotes host-protective inflammatory cytokine production. Attesting to the importance of miR-155 in host-protection against bacterial infection, we have found that virulent strains of F. tularensis (Cremer et al., 2009) and M. tuberculosis (Rajaram et al., 2011) do not support the induction of miR-155 in infected cells. It is unclear as yet whether the attenuated miR-155 induction in human monocytes/macrophages infected with these virulent strains represents active suppression of induction by the bacteria or an inability of the host to sense the virulent bacteria to mount a miR-155 response.
In summary we have uncovered new details on the molecular mechanisms of miR-155 induction by innate immune stimuli. We report that c-Fos and c-Jun are induced during infection in an NF-κB-dependent manner, and are necessary to form the AP-1 complex that is required for miR-155 transcription. Thus NF-κB is indirectly required for miR-155 induction. This study highlights the complexity in the regulation of this immunoregulatory miR.
Experimental Procedures
Cells and Reagents
RAW 264.7 macrophages were obtained from ATCC and cultured in RPMI-1640 (Gibco-BRL, Rockville, MD) supplemented with 5% heat-inactivated fetal bovine serum (FBS; HyClone, Logan, UT) and L-glutamine (Invitrogen, Carlsbad, CA) as previously described (Parsa et al., 2006). The BAY-11-7085 IKK inhibitor was a generous gift from Dr. Denis Guttridge (The Ohio State University). U0126 ERK inhibitor was obtained from Calbiochem (DMSO vehicle control (0.2%) was obtained from Sigma-Aldrich (St. Louis, MO)). Cycloheximide was obtained from Sigma-Aldrich (St. Louis, MO) and dissolved in sterile endotoxin-free water. Brefeldin A dissolved in DMSO was obtained from BioLegend (San Diego, CA) and used at the concentrations recommended by the manufacturer. Brefeldin A was tested for antimicrobial activity against F.n. and we found no effect on CFUs at the concentration used in the experiments with up to 20 h of exposure (data not shown). Likewise no significant effect of Brefeldin A was observed on host cell viability. Recombinant human IFNβ was purchased from PBL Interferon Source (Piscataway, NJ). Passive lysis buffer and luciferase assay reagent was obtained from Promega (Madison, WI). 0.22 μm sterile filtering units were obtained from Millipore (Bedford, MA). R848 (Resiquimod, TLR7/8 ligand) was purchased from Alexis Biochemicals; LPS from E. coli strain 0127:B8 was obtained from Sigma.
Antibodies
Antibodies against phospho-Stat1, c-Fos, and c-Jun were purchased from Cell Signaling (Beverly, Massachusetts). The antibody against actin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-human TNFα neutralizing antibody was obtained from R&D systems (Minneapolis, MN). Anti-human IFNβ neutralizing antibody was obtained from PBL Interferon Source (Piscataway, NJ).
Peripheral blood monocyte isolation
Human PBM were isolated as previously described (Butchar et al., 2008) using centrifugation through a Ficoll gradient followed by CD14-positive selection by Magnet-Assisted Cell Sorting (MACS, Miltenyi Biotec, Auburn, CA).
Bacterial Infections
Francisella novicida U112 (JSG1819) was generously provided by Dr. John Gunn (OSU), and grown on Chocolate II agar plates overnight (Becton, Dickinson and Company, Sparks, MD) at 37°C. Bacteria was suspended in RPMI-1640 culture media without antibiotic and quantified by a spectrometer at 600 nm wave length. Burkholderia cenocepacia K56-2 isolate was grown in L.B. broth (Sigma, St. Louis, MO) for 12–14 h to post-logarithmic phase. Prior to infection, cultures were centrifuged, washed, and re-suspended in macrophage culture media. Lyophilized Mycobacterium smegmatis was obtained from ATCC (#700084), reconstituted and used as single suspension.
Real-time QRT-PCR
Cells were lysed in TRIzol® reagent (Invitrogen, Carlsbad, CA) and RNA isolation was completed according to the manufacturer’s instructions. Reverse transcription was done with 10–100 ng of total RNA. Mature miR-155 expression was assayed with TaqMan® Universal PCR Master Mix, No AmpEraseH UNG (Applied Biosystems, Branchburg, NJ) by qRT-PCR and normalized to RNU44 internal control (Applied Biosystems, Foster City, CA) as previously described (Cremer et al., 2009). Data are expressed as relative expression = 2−(CTmiR-155−CTRNU44) or relative copy number (RCN). RCN for human TNFα mRNA was calculated as previously described and primed for cDNA synthesis with 0.8 nM random hexamer (Applied Biosystems, Foster City, CA; Butchar et al., 2008).
Western Blot Analysis
Cells were lysed in TN1 buffer (50 mM Tris pH 8.0, 10 mM EDTA, 10 mM Na4P2O7, 10 mM NaF, 1% Triton-X 100, 125 mM NaCl, 3 mM Na3VO4, 10 μg/ml each aprotinin and leupeptin). Proteins were separated by SDS/PAGE, transferred to nitrocellulose membranes, and then probed with antibody of interest. Detections were performed using HRP-conjugated secondary antibodies followed by development with enhanced chemiluminescence western blotting substrate (Pierce, Rockford, IL) as previously described (Parsa et al., 2006).
Transfections
12 × 106 RAW 264.7 cells were transfected by electroporation using Amaxa solution V with program U-14 as previously described (Parsa et al., 2006). 2 μg of AP-1 luciferase reporter was used for each transfection (Stratagene, La Jolla, CA).
siRNA
Commercially available siRNA against c-Fos was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). RAW 264.7 cells were transfected with 4.0 μM target or control siRNA via electroporation. After transfection cells were centrifuged and re-suspended in fresh media to remove excess transfection solution and siRNA. Effective knockdown of protein expression was verified by Western blotting for c-Fos.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. S. Akira (Osaka University) for the MyD88-deficient mice, and Dr. Mikhail Gavrilin (The Ohio State University) for providing TNFα PCR primers and Dr. Miguel Valvano (University of Western Ontario) for kindly providing the B. cenocepacia K56-2 isolate. This work was supported by the NIH/NIAID Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (RCE) Program. The authors wish to acknowledge membership within and support from the Region V “Great Lakes” RCE (NIH award 1-U54-AI-057153). This work was also sponsored by NIH/NIAID award # 1-T32-AI-065411, a NRSA training grant administered by the Center for Microbial Interface Biology (CMIB), at The Ohio State University, supporting Thomas J. Cremer, Jonathan P. Butchar is supported by NIH K12 CA133250.
References
Anest, V., Cogswell, P. C., and Baldwin, A. S. Jr. (2004). IkappaB kinase alpha and p65/RelA contribute to optimal epidermal growth factor-induced c-fos gene expression independent of IkappaBalpha degradation. J. Biol. Chem. 279, 31183–31189.
Bushati, N., and Cohen, S. M. (2008). MicroRNAs in neurodegeneration. Curr. Opin. Neurobiol. 18, 292–296.
Butchar, J. P., Cremer, T. J., Clay, C. D., Gavrilin, M. A., Wewers, M. D., Marsh, C. B., Schlesinger, L. S., and Tridandapani, S. (2008). Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS ONE 3, e2924. doi:10.1371/journal.pone.0002924
Cole, L. E., Shirey, K. A., Barry, E., Santiago, A., Rallabhandi, P., Elkins, K. L., Puche, A. C., Michalek, S. M., and Vogel, S. N. (2007). Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect. Immun. 75, 4127–4137.
Costinean, S., Zanesi, N., Pekarsky, Y., Tili, E., Volinia, S., Heerema, N., and Croce, C. M. (2006). Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 103, 7024–7029.
Cremer, T. J., Ravneberg, D. H., Clay, C. D., Piper-Hunter, M. G., Marsh, C. B., Elton, T. S., Gunn, J. S., Amer, A., Kanneganti, T. D., Schlesinger, L. S., Butchar, J. P., and Tridandapani, S. (2009). MiR-155 induction by F. novicida but not the virulent F. tularensis results in SHIP down-regulation and enhanced pro-inflammatory cytokine response. PLoS ONE. 4, Fe8508. doi:10.1371/journal.pone.0008508
Dai, Y., Diao, Z., Sun, H., Li, R., Qiu, Z., and Hu, Y. (2011). MicroRNA-155 is involved in the remodelling of human-trophoblast-derived HTR-8/SVneo cells induced by lipopolysaccharides. Hum. Reprod. 26, 1882–1891.
Eis, P. S., Tam, W., Sun, L., Chadburn, A., Li, Z. M., Gomez, F., Lund, E., and Dahlberg, J. E. (2005). Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. U.S.A. 102, 3627–3632.
Ellis, J., Oyston, P. C., Green, M., and Titball, R. W. (2002). Tularemia. Clin. Microbiol. Rev. 15, 631–646.
Gatto, G., Rossi, A., Rossi, D., Kroening, S., Bonatti, S., and Mallardo, M. (2008). Epstein-Barr virus latent membrane protein 1 trans-activates miR-155 transcription through the NF-kappaB pathway. Nucleic Acids Res. 36, 6608–6619.
Gunn, J. S., and Ernst, R. K. (2007). The structure and function of Francisella lipopolysaccharide. Ann. N. Y. Acad. Sci. 1105, 202–218.
Hambleton, J., Weinstein, S. L., Lem, L., and Defranco, A. L. (1996). Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. U.S.A. 93, 2774–2778.
Hao, S., and Baltimore, D. (2009). The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 10, 281–288.
Henke, J. I., Goergen, D., Zheng, J., Song, Y., Schuttler, C. G., Fehr, C., Junemann, C., and Niepmann, M. (2008). MicroRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 27, 3300–3310.
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J., and Monack, D. M. (2007). Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994.
Hou, J., Wang, P., Lin, L., Liu, X., Ma, F., An, H., Wang, Z., and Cao, X. (2009). MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 183, 2150–2158.
Izar, B., Mannala, G. K., Mraheil, M. A., Chakraborty, T., and Hain, T. (2012). MicroRNA response to Listeria monocytogenes infection in epithelial cells. Int. J. Mol. Sci. 13, 1173–1185.
Jiang, X., Tsitsiou, E., Herrick, S. E., and Lindsay, M. A. (2010). MicroRNAs and the regulation of fibrosis. FEBS J. 277, 2015–2021.
Karin, M., Liu, Z., and Zandi, E. (1997). AP-1 function and regulation. Curr. Opin. Cell Biol. 9, 240–246.
Kieffer, T. L., Cowley, S., Nano, F. E., and Elkins, K. L. (2003). Francisella novicida LPS has greater immunobiological activity in mice than F. tularensis LPS, and contributes to F. novicida murine pathogenesis. Microbes Infect. 5, 397–403.
Kohlhaas, S., Garden, O. A., Scudamore, C., Turner, M., Okkenhaug, K., and Vigorito, E. (2009). Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J. Immunol. 182, 2578–2582.
Li, H., Nookala, S., Bina, X. R., Bina, J. E., and Re, F. (2006). Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. J. Leukoc. Biol. 80, 766–773.
O’Connell, R. M., Kahn, D., Gibson, W. S., Round, J. L., Scholz, R. L., Chaudhuri, A. A., Kahn, M. E., Rao, D. S., and Baltimore, D. (2010). MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 33, 607–619.
O’Connell, R. M., Taganov, K. D., Boldin, M. P., Cheng, G., and Baltimore, D. (2007). MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U.S.A. 104, 1604–1609.
Oyston, P. C., Sjostedt, A., and Titball, R. W. (2004). Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2, 967–978.
Parsa, K. V., Ganesan, L. P., Rajaram, M. V., Gavrilin, M. A., Balagopal, A., Mohapatra, N. P., Wewers, M. D., Schlesinger, L. S., Gunn, J. S., and Tridandapani, S. (2006). Macrophage pro-inflammatory response to Francisella novicida infection is regulated by SHIP. PLoS Pathog. 2, e71. doi:10.1371/journal.ppat.0020071
Pottier, N., Maurin, T., Chevalier, B., Puissegur, M. P., Lebrigand, K., Robbe-Sermesant, K., Bertero, T., Lino Cardenas, C. L., Courcot, E., Rios, G., Fourre, S., Lo-Guidice, J. M., Marcet, B., Cardinaud, B., Barbry, P., and Mari, B. (2009). Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: implication in epithelial-mesenchymal interactions. PLoS ONE 4, e6718. doi:10.1371/journal.pone.0006718
Rajaram, M. V., Butchar, J. P., Parsa, K. V., Cremer, T. J., Amer, A., Schlesinger, L. S., and Tridandapani, S. (2009). Akt and SHIP modulate Francisella escape from the phagosome and induction of the Fas-mediated death pathway. PLoS ONE 4, e7919. doi:10.1371/journal.pone.0007919
Rajaram, M. V., Ganesan, L. P., Parsa, K. V., Butchar, J. P., Gunn, J. S., and Tridandapani, S. (2006). Akt/Protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J. Immunol. 177, 6317–6324.
Rajaram, M. V., Ni, B., Morris, J. D., Brooks, M. N., Carlson, T. K., Bakthavachalu, B., Schoenberg, D. R., Torrelles, J. B., and Schlesinger, L. S. (2011). Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by regulating macrophage MAPK-activated protein kinase 2 (MK2) and microRNA miR-125b. Proc. Natl. Acad. Sci. U.S.A 108, 17408–17413.
Rodriguez, A., Vigorito, E., Clare, S., Warren, M. V., Couttet, P., Soond, D. R., van Dongen, S., Grocock, R. J., Das, P. P., Miska, E. A., Vetrie, D., Okkenhaug, K., Enright, A. J., Dougan, G., Turner, M., and Bradley, A. (2007). Requirement of bic/microRNA-155 for normal immune function. Science 316, 608–611.
Santic, M., Al Khodor, S., and Abu, K. Y. (2010). Cell biology and molecular ecology of Francisella tularensis. Cell. Microbiol. 12, 129–139.
Taganov, K. D., Boldin, M. P., Chang, K. J., and Baltimore, D. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 12481–12486.
Telepnev, M., Golovliov, I., Grundstrom, T., Tarnvik, A., and Sjostedt, A. (2003). Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-alpha and IL-1 from murine macrophages. Cell. Microbiol. 5, 41–51.
Thai, T. H., Calado, D. P., Casola, S., Ansel, K. M., Xiao, C., Xue, Y., Murphy, A., Frendewey, D., Valenzuela, D., Kutok, J. L., Schmidt-Supprian, M., Rajewsky, N., Yancopoulos, G., Rao, A., and Rajewsky, K. (2007). Regulation of the germinal center response by microRNA-155. Science 316, 604–608.
Thakran, S., Li, H., Lavine, C. L., Miller, M. A., Bina, J. E., Bina, X. R., and Re, F. (2008). Identification of Francisella tularensis lipoproteins that stimulate the toll-like receptor (TLR) 2/TLR1 heterodimer. J. Biol. Chem. 283, 3751–3760.
Tili, E., Michaille, J. J., Cimino, A., Costinean, S., Dumitru, C. D., Adair, B., Fabbri, M., Alder, H., Liu, C. G., Calin, G. A., and Croce, C. M. (2007). Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 179, 5082–5089.
Toshchakov, V., Jones, B. W., Perera, P. Y., Thomas, K., Cody, M. J., Zhang, S., Williams, B. R., Major, J., Hamilton, T. A., Fenton, M. J., and Vogel, S. N. (2002). TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat. Immunol. 3, 392–398.
van Rooij, E., Marshall, W. S., and Olson, E. N. (2008). Toward microRNA-based therapeutics for heart disease: the sense in antisense. Circ. Res. 103, 919–928.
Vigorito, E., Perks, K. L., Abreu-Goodger, C., Bunting, S., Xiang, Z., Kohlhaas, S., Das, P. P., Miska, E. A., Rodriguez, A., Bradley, A., Smith, K. G., Rada, C., Enright, A. J., Toellner, K. M., MacLennan, I. C., and Turner, M. (2007). MicroRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 27, 847–859.
Xiao, B., Liu, Z., Li, B. S., Tang, B., Li, W., Guo, G., Shi, Y., Wang, F., Wu, Y., Tong, W. D., Guo, H., Mao, X. H., and Zou, Q. M. (2009). Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J. Infect. Dis. 200, 916–925.
Yin, Q., McBride, J., Fewell, C., Lacey, M., Wang, X., Lin, Z., Cameron, J., and Flemington, E. K. (2008a). MicroRNA-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. J. Virol. 82, 5295–5306.
Yin, Q., Wang, X., McBride, J., Fewell, C., and Flemington, E. (2008b). B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. J. Biol. Chem. 283, 2654–2662.
Keywords: microRNA, microbial pathogenesis, miR-155
Citation: Cremer TJ, Fatehchand K, Shah P, Gillette D, Patel H, Marsh RL, Besecker BY, Rajaram MVS, Cormet-Boyaka E, Kanneganti T-D, Schlesinger LS, Butchar JP and Tridandapani S (2012) MiR-155 induction by microbes/microbial ligands requires NF-κB-dependent de novo protein synthesis. Front. Cell. Inf. Microbio. 2:73. doi: 10.3389/fcimb.2012.00073
Received: 10 April 2012; Accepted: 04 May 2012;
Published online: 01 June 2012.
Edited by:
Anders Sjostedt, Umeå University, SwedenReviewed by:
Mobarak Abu Mraheil, Institut for Medical Microbiology, GermanyKithiganahalli Balaji, Indian Institute of Science, India
Copyright: © 2012 Cremer, Fatehchand, Shah, Gillette, Patel, Marsh, Besecker, Rajaram, Cormet-Boyaka, Kanneganti, Schlesinger, Butchar and Tridandapani. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jonathan P. Butchar and Susheela Tridandapani, Department of Internal Medicine, The Ohio State University, 415 DHLRI, 473 W 12th Avenue, Columbus, OH 43210, USA. e-mail: butchar.2@osu.edu; tridandapani.2@osu.edu