Deep sequencing of Porphyromonas gingivalis and comparative transcriptome analysis of a LuxS mutant
- 1 Center for Oral Health and Systemic Disease, School of Dentistry, University of Louisville, Louisville, KY, USA
- 2 Department of Chemical Engineering, University of Washington, Seattle, WA, USA
- 3 eScience Institute, University of Washington, Seattle, WA, USA
Porphyromonas gingivalis is a major etiological agent in chronic and aggressive forms of periodontal disease. The organism is an asaccharolytic anaerobe and is a constituent of mixed species biofilms in a variety of microenvironments in the oral cavity. P. gingivalis expresses a range of virulence factors over which it exerts tight control. High-throughput sequencing technologies provide the opportunity to relate functional genomics to basic biology. In this study we report qualitative and quantitative RNA-Seq analysis of the transcriptome of P. gingivalis. We have also applied RNA-Seq to the transcriptome of a ΔluxS mutant of P. gingivalis deficient in AI-2-mediated bacterial communication. The transcriptome analysis confirmed the expression of all predicted ORFs for strain ATCC 33277, including 854 hypothetical proteins, and allowed the identification of hitherto unknown transcriptional units. Twelve non-coding RNAs were identified, including 11 small RNAs and one cobalamin riboswitch. Fifty-seven genes were differentially regulated in the LuxS mutant. Addition of exogenous synthetic 4,5-dihydroxy-2,3-pentanedione (DPD, AI-2 precursor) to the ΔluxS mutant culture complemented expression of a subset of genes, indicating that LuxS is involved in both AI-2 signaling and non-signaling dependent systems in P. gingivalis. This work provides an important dataset for future study of P. gingivalis pathophysiology and further defines the LuxS regulon in this oral pathogen.
Introduction
Porphyromonas gingivalis, a Gram-negative anaerobe, is a major pathogen in periodontal disease, one of the most common bacterial infections of humans in developed countries (Sabbah et al., 2010). Moreover, emerging evidence indicates an association between periodontal infections and serious systemic conditions such as coronary artery disease and preterm delivery of low birth weight infants (Bahekar et al., 2007; Humphrey et al., 2008; Katz et al., 2009). The pathogenicity of P. gingivalis is multifactorial and is associated with adhesins such as fimbriae that mediate attachment to surfaces and invasion of epithelial cells; proteases such as the gingipains that degrade the structural components of the periodontium and immune effector molecules; and toxic metabolites that can induce apoptotic and necrotic cell death (Lamont and Jenkinson, 1998).
A major ecological niche of P. gingivalis is the subgingival crevice or periodontal pocket (Lamont and Jenkinson, 1998), and this region experiences fluctuation in oxygen levels, temperature, pH, and nutrient availability. P. gingivalis can sense and respond at the transcriptional level to these environmental cues (Diaz et al., 2006; Okano et al., 2006; Duran-Pinedo et al., 2007; Meuric et al., 2008; Johnson et al., 2011), although the mechanistic basis of gene regulation has not been fully defined.
Porphyromonas gingivalis can accumulate into complex multispecies consortia with other organisms (Kuboniwa and Lamont, 2010) resulting in the differential expression of a large percentage of its genome and proteome (Kuboniwa et al., 2009; Redanz et al., 2011). P. gingivalis and its community partner S. gordonii participate in inter-species communication based on the soluble mediator AI-2 (Chung et al., 2001; McNab et al., 2003), produced by the action of the AI-2 synthase, LuxS (Schauder et al., 2001). LuxS deficient mutants of P. gingivalis and S. gordonii co-adhere normally but do not accrete into microcolonies, indicating that AI-2 is required for P. gingivalis-S. gordonii community development (McNab et al., 2003). In P. gingivalis, LuxS regulates the expression of a number of genes involved in the uptake of hemin and inorganic iron, including hmuR, fetB, feoB1, and ftn, which are negatively regulated, and tlr and kgp which are positively regulated (James et al., 2006). Expression of the luxS gene is controlled by a phospho-dependent signaling pathway that converges on the transcriptional regulator CdhR (Maeda et al., 2008). Moreover, AI-2 also participates in a positive feedback loop that suppresses CdhR expression (Chawla et al., 2010). A microarray analysis of P. gingivalis has shown that LuxS is involved in the control of resistance to stress (Yuan et al., 2005); however, little else is known regarding the broader LuxS regulon.
High-throughput sequencing of cDNAs (deep sequencing) is a powerful technique for quantitative transcriptional profiling. As deep sequencing can detect and quantify RNA present in low levels, the technique is more sensitive than microarrays. A number of bacteria have been analyzed at the transcriptional level by deep sequencing approaches (Oliver et al., 2009; Perkins et al., 2009; Yoder-Himes et al., 2009; Beck et al., 2011; Isabella and Clark, 2011) and the methodology has provided novel insights into pathogenic regulons, as well as identifying small non-coding RNAs. To analyze the P. gingivalis transcriptome and elucidate the P. gingivalis LuxS/AI-2 regulatory circuit in more detail, we employed transcriptome Sequencing by Oligonucleotide Ligation and Detection (SOLiD) deep sequencing. We applied RNA-Seq in a semi-targeted fashion as an alternative to a quantitative microarray experiment. We quantitated differences in gene expression in a luxS mutant, identified novel regions under control of this system and showed that loss of LuxS affects both AI-2 dependent signaling and non-signal dependent systems in P. gingivalis. In addition, we acquired more qualitative information about the transcriptome that would not have been possible using a targeted array approach. We have identified many putative novel small non-coding RNAs (sRNAs), and we have validated the expression of previously hypothetical genes.
Results and Discussion
Sequencing the P. gingivalis Transcriptome
In order to characterize the P. gingivalis transcriptome by SOLiD based RNA-Seq, RNA was prepared from three biological replicate cultures of ATCC 33277 and its ΔluxS mutant. We previously showed that AI-2 synthesis in P. gingivalis peaks in early exponential phase (James et al., 2006), therefore cultures were grown to early exponential phase (OD600 0.5). Reads from the RNA-Seq data were mapped to the annotated P. gingivalis ATCC 33277 genome (NCBI GI:188593544), and gene expression was quantified as reads per kilobase of coding sequence per million reads sequenced (RPKM). Expression data for each replicate were plotted against each other (Figure 1). The replicates were in good agreement with each other, with correlations ranging from 0.8 to 0.95. The number of 50 bp reads in the samples ranged from 44 to 76 million. On average, BWA software (see Materials and Methods) was able to assign 43.5% of the color space reads to the P. gingivalis chromosome. The majority of mapped reads were attributed to ribosomal RNA with the mean across conditions of 80%.
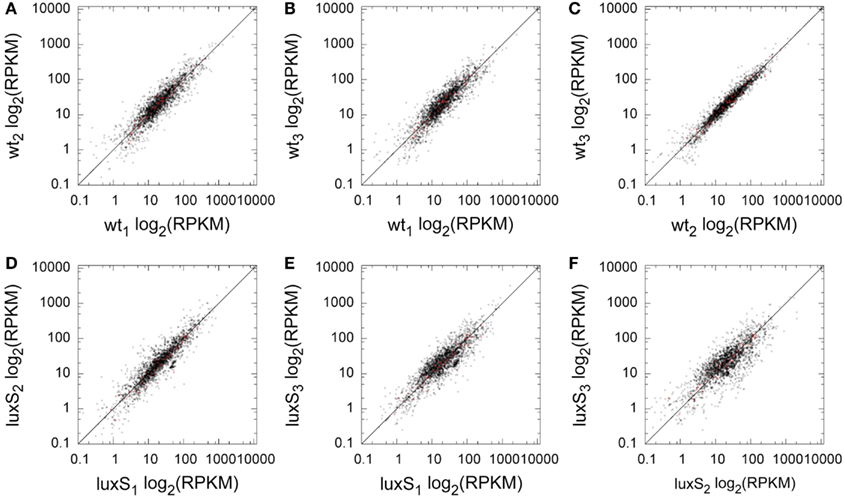
Figure 1. Replicate comparison of per-locus RPKM for coding sequences. For a pair of replicates, each locus has one point with the log2 RPKM in one replicate plotted against the log2 RPKM of a second. The diagonal line shows identity. (A) WT1 vs. WT2, (B) WT1 vs. WT3, (C) WT2 vs. WT3, (D) ΔluxS1 vs. ΔluxS2, (E) ΔluxS 1 vs. ΔluxS 3, (F) ΔluxS2 vs. ΔluxS3. Red colored data points indicate ORFs that demonstrated statistically significant relative abundance differences between WT and ΔluxS.
The average per base coverage is shown in Table S1 in Supplementary Material. The coverage of protein-coding regions for each sample was strongly correlated to rRNA removal efficacy and the size of the read pool. In each sample, the transcriptome penetration was such that all non-hypothetical ORFs were detected. When the replicates for each condition were pooled, every predicted ORF (Naito et al., 2008) was detected. The entire comparative gene expression profiles for WT and luxS mutant are shown in Figure S1 in Supplementary Material. Transcript abundance for WT samples sorted by RPKM ratio is shown in Table S2 in Supplementary Material. Genes for three hypothetical proteins, PGN_0215, PGN_2047, and PGN_0483 had exceptionally high reads compared to other genes. Interestingly, these genes are located in front of 16S ribosomal RNA, PGN_r0001, PGN_r0010, and PGN_r0004, respectively, and may be co-transcribed, suggesting that these gene products might be important for ribosome function. Another 16S ribosomal RNA gene, PGN_r0009 has no such hypothetical gene, indicating a specific role for these hypothetical proteins. Predictably, genes for outer membrane proteins, ribosome proteins, and cysteine proteinases had high reads. Genes encoding the major (fimA) and minor (mfa1) fimbriae also had high numbers of reads mapped.
Hypothetical Proteins
We identified 854 hypothetical genes, which were expressed with at least average read counts of 10 in either the WT or ΔluxS mutant samples (Table S3 in Supplementary Material). In both strains, three genes, PGN_2047, PGN_0215, and PGN_0438, which are fully conserved at the amino acid level, had large numbers of reads mapped. The corresponding predicted proteins are annotated as cytoplasmic proteins with unknown function. Moreover, the nucleotide sequences 3′ of the 43rd nt are identical to the sequence spanning the region 5′ upstream to the first 32 nucleotides of PGN_r0009 16S rRNA. Hypotheticals PGN_0217, PGN_0440, PGN_2049, and PGN_1398, also fully conserved at the amino acid level, were highly expressed in both strains. These results suggest that several of the relatively smaller proteins might have physiologically important functions as yet to be determined.
Identification of Small (s)RNAs
Small RNAs (sRNA) are widespread in bacteria and the significance of sRNAs in gene regulation is increasingly recognized (Storz et al., 2011). Several potential sRNAs in P. gingivalis strain W83 have been identified by transcriptome analysis but the 5′ and 3′ ends have not been determined experimentally (Hovik et al., 2012). Analysis of the SOLiD data (see Materials and Methods) revealed over 2000 intergenic transcripts that will include sRNAs (Table S4 in Supplementary Material). To identify sRNAs and define their 5′ and 3′, we employed a modified rapid amplification of cDNA ends (RACE) technique. Firstly we validated the modified RACE technique in P. gingivalis by detection of Bacteroidales-1, which has been identified by bioinformatic approaches as an RNA structural motif (rfam #; RF01693) conserved in Bacteroidales, including P. gingivalis (Weinberg et al., 2010). This RNA motif is predicted to be 170 bases long and is coded on the reverse strand in an intergenic region (IGR) located between PGN_0141 and PGN_0142. Our deep sequencing results confirmed the expression of this region at average reading of 999 and 3115 in the WT and ΔluxS strains, respectively (Table S4 in Supplementary Material, ID158). Self-ligated, circularized total RNAs from WT were reverse-transcribed to cDNA, two sets of primers in the same IGR region were then used for nested PCR and the fragments were sequenced. This modified RACE technique successfully detected a 173 base Bacteroidales-1 sRNA on the negative strand from 146754 to 146582. Next we focused on IGRs that showed reading counts higher than 1000 in the WT, did not appear as UTR of orthologs in the P. gingivalis strain W831, and were not adjacent to rRNA genes. Thirteen IGRs were examined by RACE and 11 sRNAs were identified with lengths between 50 and 783 bases in 10 IGRs (Table 1), none of which are listed in rfam database. Two stable fragments were identified that started from the same position in IGR0247 and ended at 268 and 218 bases, suggesting sRNA processing or read-through. The fragments in IGR0960 and IGR1662 located as part of the 3′-UTR but could be processed to the stable sRNA fragment since these two mRNA have unusually long 3′-UTR regions reading to 300 and 700 bases beyond the translational stop, respectively. The forward strand in IGR0960 also encodes tRNA-Arg-4. tRNA-Lys-1 in IGR1524 has been predicted to be 73 and 76 bases long in the rfam database web site and the Oralgen database web site, respectively, however a 221 base fragment including the 76 bases segment was detected by RACE.
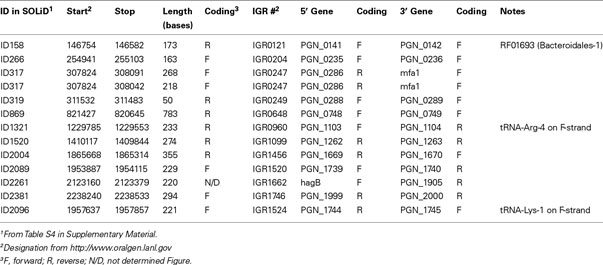
Table 1. List of sRNAs identified by RACE.
Cobalamin Riboswitch
Riboswitches are regulatory elements that modulate expression of the downstream gene or operon (Breaker, 2011). We identified a potential cobalamin-type riboswitch sequence cbl-1 (BLAST e-value of 9.714e−21) that was located between PGN_0703 and PGN_0704 and is predicted to be transcribed in the same direction as PGN_0704. PGN_0704 encodes a BtuB homolog, an outer membrane cobalamin receptor protein for which the riboswitch mechanism has been well characterized in other organisms (Nahvi et al., 2004). Since riboswitches are long 5′ non-coding regulatory regions of the downstream coding region, we examined co-transcription of cbl-1 with PGN_0704, by reverse transcription PCR (RT-PCR) using specific primer sets binding to the riboswitch region and the downstream protein-coding region. RT-PCR showed cbl-1 was co-transcribed with downstream coding regions (Figure 2B). Hereafter PGN_0704 is designated as btuB, and the transcriptional start site for btuB was determined by 5′-RACE. The potential 5′-end position from three independent cbl-1-btuB 5′-RACE clones was found at 430 nt upstream from ATG translational start site of btuB (Figure 2A).
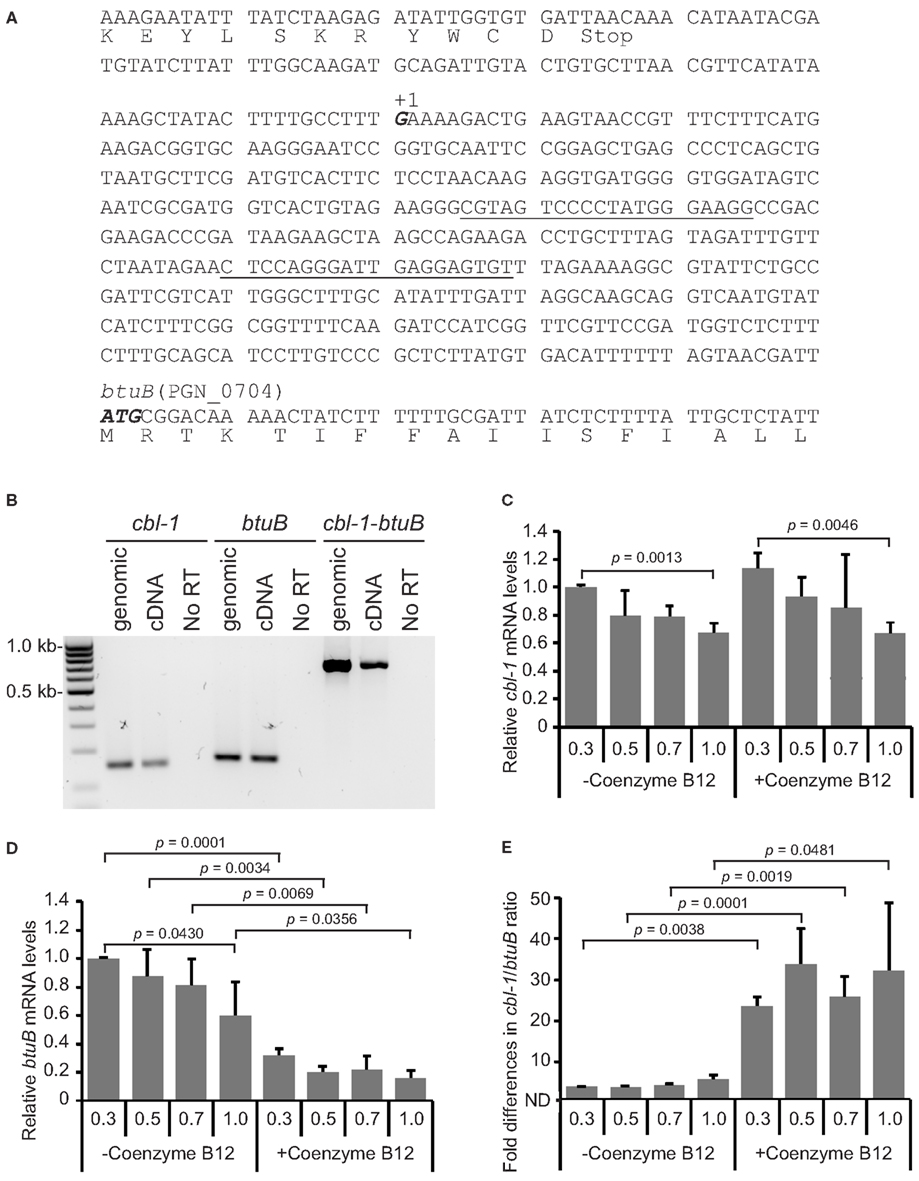
Figure 2. Transcriptional organization and regulation of the cbl-1-btuB (PGN_0704) region in the P. gingivalis chromosome. (A) Based on 5′-RACE, the transcriptional start site for cbl-1-btuB mRNA was mapped to a G (in bold italic and labeled +1) 430 bp upstream of the ATG start codon of btuB. Primers in the cbl-1 region used for RT-PCR in (B) and qRT-PCR in (C–E) are underlined. (B) RT-PCR of genomic DNA or cDNA with primers amplifying cbl-1, btuB or cbl-1, and btuB. The presence of an amplicon with cDNA demonstrates co-transcription of cbl-1 and btuB. The No RT lanes are controls without reverse transcriptase. (C) cbl-1 levels were not affected by 100 μM Coenzyme B12. (D) btuB transcripts were significantly reduced by culture with 100 μM Coenzyme B12. (E) Ratio of cbl-1 to btuB mRNA copy number. Results in (C–E) are means with standard deviation of three biological replicates (n = 9).
The structure of riboswitches can be modulated upon binding to a chemical, usually one of metabolites of the pathway encoded by the downstream genes. The cobalamin transporter BtuB is part of the vitamin B12 synthetic pathway, and in E. coli and Salmonella one of the metabolites of this pathway, Coenzyme B12, binds to the riboswitch region of btuB mRNA (Ravnum and Andersson, 1997; Nahvi et al., 2004). Subsequent structural changes lead to susceptibility to cleavage causing transcriptional arrest, and/or to inhibition of binding to the ribosome. Hence, we examined the stability of cbl-1-btuB at the transcriptional and the translational levels in P. gingivalis. To assess the effects of Coenzyme B12, cells were grown in the presence or absence of 100 μM Coenzyme B12. qRT-PCR using the same primer sets as for RT-PCR was carried out to show the relative amount of cbl-1 riboswitch transcripts compared to those of the btuB coding region (Figures 2C–E). qRT-PCR for the cbl-1 region showed the highest levels were at early log phase with a gradual decrease to almost 60% during culture to OD600 1 (Figure 2C). This signature as well as copy numbers at each sampling point were not significantly changed by the addition of Coenzyme B12. btuB transcripts levels were the highest at early log phase and then similarly decreased in the absence of Coenzyme B12 (Figure 2D). However, culture in the presence of Coenzyme B12 resulted in a significant decrease in the level of btuB mRNA (Figure 2D). The ratios of cbl-1 to btuB were similar in the absence of Coenzyme B12, but significantly higher in the presence of Coenzyme B12 through the growth phase (Figure 2E). These results indicate that under Coenzyme B12 limitation, transcription of the cbl-1-btuB region is most active at early log phase (OD600 0.3) followed by a gradual transcriptional stalling over the growth curve. In contrast, in the presence of Coenzyme B12, although promoter activity of cbl-1-btuB regulon is unchanged, transcription of the btuB coding region stalls more frequently upon Coenzyme B12 binding to cbl-1 riboswitch region.
To verify btuB expression at the translational level, BtuB protein was C-terminally tagged with FLAG. Western blotting of whole cell lysates with anti-FLAG monoclonal antibody demonstrated reduced BtuB protein levels in the presence of Coenzyme B12 (Figure 3A). Levels of FLAG-tagged BtuB decreased to almost undetectable at OD 1.0. Western blotting with antibodies to the Mfa1 fimbrial protein of P. gingivalis was performed as a loading control (data not shown). FLAG/Mfa1 ratios (Figure 3B) confirmed the inhibition of BtuB expression in the presence of Coenzyme B12, and the decrease in BtuB as the cells progress through the growth curve. These results corroborate the concept that cbl-1 upstream of btuB acts as a conventional cobalamin-type riboswitch in P. gingivalis.
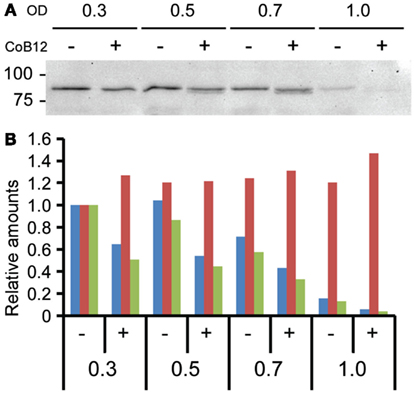
Figure 3. BtuB regulation by the cbl-1 riboswitch. (A) Western blot of lysates of P. gingivalis expressing BtuB::FLAGx3 protein, probed with anti-FLAG antibody. OD: optical density at 600 nm; CoB12: culture with 100 μM Coenzyme B12. Molecular mass markers are shown in kDa. (B) Ratio (green bars) of BtuB::FLAGx3 protein levels to Mfa loading control using Image lab software (Bio-Rad) from western blot results with anti-FLAG (blue bars) and anti-Mfa (red bars) antibodies. For normalization, protein levels at OD = 0.3 were taken as 1.0.
Effect of Lux S Mutation on the P. gingivalis Transcriptome
LuxS AI-2 synthase is responsible for the production of the AI-2 family of signaling molecules, and the enzyme is a component of the activated methyl cycle (Schauder et al., 2001); however, the LuxS regulon in P. gingivalis has not been fully defined. Comparative transcriptome analysis of WT and ΔluxS strains identified 57 ORFs as differentially expressed with a p-value cutoff of 0.05 (Figure S2 in Supplementary Material). The differentially regulated ORFs are depicted by red dots in Figure 1 from which it is clear that they are well reproduced by the replicates for each condition with inter-replicate correlations better than all ORFs, ranging from 0.86 to 0.98. Of these 57, 17 were annotated as hypothetical proteins, and 15 of these were flagged as possessing signal peptides by JGI’s annotation pipeline (Markowitz et al., 2008), with 10 as transmembrane domain containing proteins.
Nearly half of the differentially abundant ORFs identified were found in clusters of two or more neighbor or near-neighbor ORFs in the genome that were similarly differentially abundant. The largest such cluster spanned six ORFs from PGN_0553 to PGN_0558, encompassing the hmu hemin uptake operon, which was up-regulated in the ΔluxS mutant strain. This is consistent with earlier work showing that transcription of hmuR (PGN_0557) is elevated in the same ΔluxS mutant strain as compared to WT (James et al., 2006). Also up-regulated in the ΔluxS mutant were the potential operons spanning PGN_1791 to PGN_1790, and PGN_1472 to PGN_1468. The former region encodes a flavodoxin FldA and a hypothetical protein, respectively. The later region comprises genes involved in diverse functions such as tRNA-guanine transglycosylase, probable YjgP/YjgQ family permease, ssrA RNA binding protein, along with dpp and lipoyl synthase.
In the P. gingivalis chromosome, luxS (PGN_1474) is encoded on the negative strand, preceded and co-transcribed with pfs which encodes an enzyme that synthesizes the substrate for LuxS. mRNA for pfs was differentially abundant, over threefold higher in the mutant. This result suggests the existence of a regulatory feedback mechanism that senses the degree of production of AI-2 and acts to maintain optimal levels.
Down-regulated genes contained insertion sequences: ISPg4 (one), and hypothetical proteins (10). In addition, PGN_0481 and PGN_0482, encoding a hypothetical protein and an immunoreactive protein respectively, and PGN_1403 to PGN_1401, involved in arginine and proline metabolic pathways, were down-regulated.
No systematic bias in COG (Tatusov et al., 2000), pfam (Finn et al., 2010), or KEGG (Kanehisa and Goto, 2000) classifications was observed for the differentially abundant ORFs. We mapped all of the detected ORFs onto the KEGG pathway maps. The depictions of these pathways can be found in the Supplementary Material (Figures S3A–C in Supplementary Material) with genes shown in red and green (up and down in the ΔluxS mutant relative to WT, respectively) and the statistical significance as enzyme background (dark to light, increasing). The streptomycin biosynthesis pathway (KEGG pathway pgn00521, Figure S3A in Supplementary Material) had several enzyme transcripts that were more abundant in the ΔluxS mutant, although not all were statistically significant. One product of this pathway is L-rhamnose (pgn00520), a saccharide that has been shown to be important for virulence in some oral bacteria (Henderson et al., 2009). P. gingivalis has been reported as streptomycin-resistant (Loos et al., 1990). However, since P. gingivalis does not have key enzymes downstream from dTDP-L-rhamnose in the pathway, the cell does not produce streptomycin. Deletion of dTDP-L-rhamnose biosynthesis genes in the rml operon (PGN_0546 to PGN_0549) is lethal in P. gingivalis (Shibata et al., 1999). dTDP-L-rhamnose is a component of lipopolysaccharide (LPS) in P. gingivalis, and thus another role of LuxS may be to regulate LPS biogenesis. In the lipoic pathway (pgn00785, Figure S3B in Supplementary Material), the transcript for LipA (Lipoic acid synthase, PGN_1468) which is important for biosynthesis of cofactors, prosthetic groups, and carriers including lipoate (Kondo et al., 2010), was observed to be more abundant in the ΔluxS mutant. However, most of the KEGG pathway reconstructions had limited numbers of changed transcripts and exhibited mixed abundance change trends, e.g., pgn00550, the peptidoglycan synthesis pathway (Figure S3C in Supplementary Material). The lack of clear trends may be a consequence, at least in part, of the dual function of LuxS in signaling and metabolism (see below).
Verification of Differentially Expressed Genes and Complementation by DPD
To corroborate the sequencing results, expression of a subset of genes was determined by qRT-PCR analysis (Figure 4). RNA samples were prepared from WT and ΔluxS cultures grown in the same conditions as those for the deep sequencing and converted to cDNA for qRT-PCR. Although the magnitude of fold differences was different between the two techniques, the up- or down-regulated profiles for the genes tested were concordant.
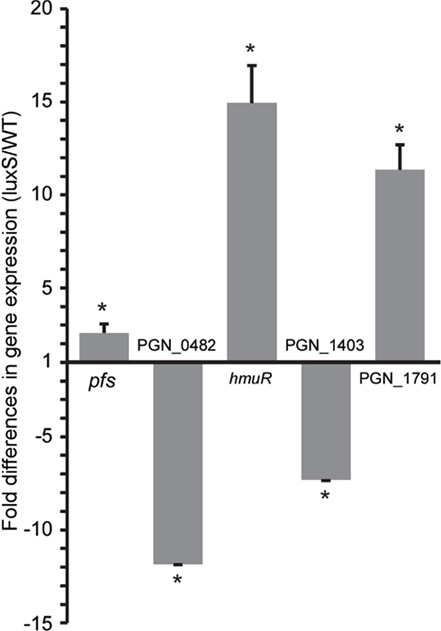
Figure 4. Corroboration of differential gene expression by qRT-PCR. mRNA from WT and ΔluxS strains was examined by qRT-PCR. 16S rRNA was used for sample normalization. Asterisk denotes a significant difference at p < 0.0001 (unpaired t-test) in levels of mRNA in isogenic ΔluxS mutant strain in comparison with WT. Data are shown as means with standard deviations of three biological replicates (n = 9).
In order to distinguish between differential regulation due to a defect in AI-2 signaling or disruption of the methyl cycle, exogenous 4,5-dihydroxy-2,3-pentanedione (DPD, AI-2 precursor) was introduced into the mutant culture, and complementation of the phenotype examined (Figure 5). WT and luxS mutant strains at OD600 = 0.3 were cultured anaerobically in the presence or absence of 4 μM DPD and total RNAs were purified for qRT-PCR. Expression levels of PGN_0482 and PGN_1403 in the luxS mutant were not restored by exogenous DPD, suggesting that regulation of these genes is independent of AI-2 signaling (Figures 5A,B). In contrast, expression levels of PGN_1791 and hmuR in the luxS mutant were significantly (p = 0.0001 and p = 0.0012, respectively) reduced by culture with DPD for 60 min (Figures 5C,D). These results indicate that LuxS has both an AI-2 signaling function, and a metabolic function through the activated methyl cycle, with independent regulons. Similar AI-2 dependent and independent functions for LuxS have also been shown in Streptococcus suis (Cao et al., 2011) and the potential dual role of LuxS may partially explain why no common theme has emerged for the larger LuxS regulon across bacterial species, and indeed different bacteria may utilize LuxS for different functions (Fong et al., 2003; Zhu et al., 2003; Vendeville et al., 2005; Doherty et al., 2006; Lebeer et al., 2007; Siller et al., 2008; Learman et al., 2009). In P. gingivalis, a mutation in the luxS gene affects hemin and iron acquisition, protease activity, and also impacts stress response pathways (Burgess et al., 2002; Yuan et al., 2005; James et al., 2006). Moreover, hemin levels in P. gingivalis can affect virulence (Marsh et al., 1994; Genco, 1995; Genco et al., 1995) and resistance to oxidative stress (Diaz and Rogers, 2004), and the HmuR protein is required for maximal community formation by P. gingivalis with F. nucleatum and S. gordonii (Kuboniwa et al., 2009). Hence AI-2 signaling has the potential to impact a number of important pathogenic properties of P. gingivalis.
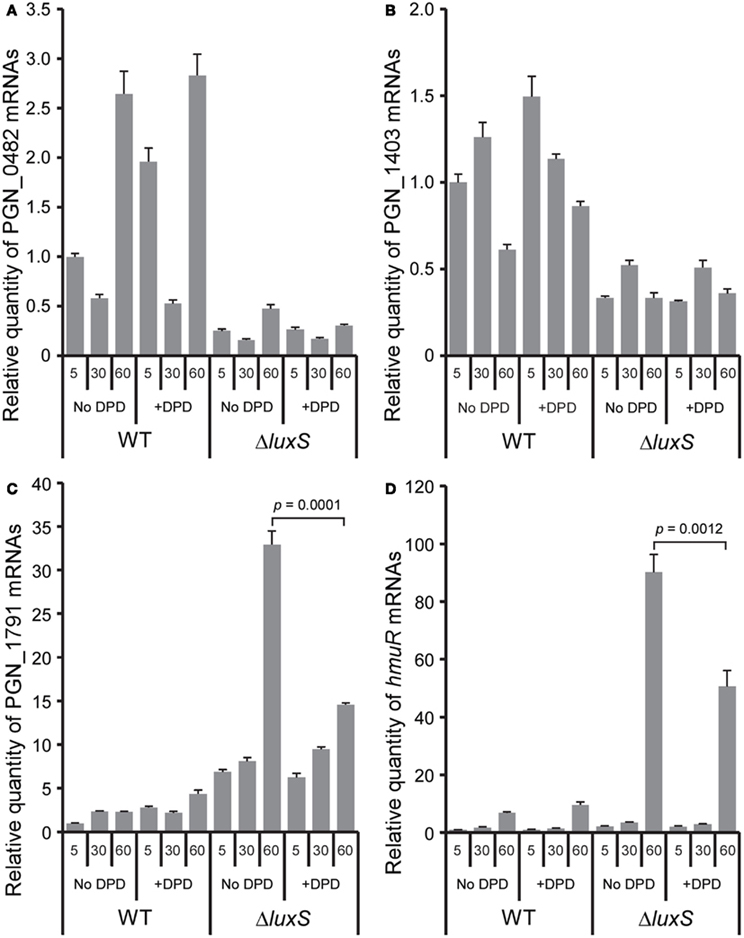
Figure 5. Exogenous DPD complementation of the ΔluxS phenotype. Levels of differentially expressed genes PGN_0482 (A), PGN_1403 (B), PGN_1791 (C), and hmuR(D), in the ΔluxS mutant strain were examined by qRT-PCR following incubation with 4 μM of synthetic DPD for the indicated times. Results are representative of three independent assays. p-values were calculated by unpaired t-test. Error bars represent standard deviations (n = 3).
The non-signal effects of LuxS mutation may be related to disruption of the activated methyl cycle. As previously described (James et al., 2006), the ΔluxS mutant was generated by insertion of PermF::ermF fragment into the luxS coding region and it is unlikely that the gene disruption would have a positive polar effect (i.e., up-regulation) on neighboring gene transcription, especially on the region upstream of the insertion site. Given that the ΔluxS strain lacks the ability to catalyze the reaction to convert the LuxS substrate, SRH, to homocysteine and DPD, we predict that SRH would accumulate in the cell. Since Pfs is required for the conversion of S-adenosyl homocysteine, SAH, to SRH, this also strengthens the case for increased intracellular SRH. Elevated levels of SRH within the cell could be beneficial if there were a means by which SRH could be spontaneously converted to homocysteine and DPD, in the absence of the active LuxS enzyme. In this manner elevated amounts of SRH might partially compensate for the defect in LuxS by providing a minimum amount of homocysteine to run the methyl cycle. This could explain why cell-free media from a P. gingivalis W83 luxS mutant still possesses the ability to induce low levels of bioluminescence (Yuan et al., 2005). Some bacterial species with a mutation in luxS show growth defects, whereas the P. gingivalis luxS mutant strain does not exhibit a similar growth defect. This also supports the notion that the methyl cycle in the mutant strain only suffers a minimal defect in the absence of functional LuxS. To recycle SAH, there exists another pathway to convert SAH to homocysteine using SAH-hydrolase (SahH; Sun et al., 2004). In this pathway, SAH is converted to adenosine and homocysteine without generation of DPD. While several closely related bacteria belonging to the bacteroidia class have been found to possess SahH, P. gingivalis does not harbor this enzyme. It seems that the Pfs-LuxS pathway is the only method of SAH detoxification in P. gingivalis.
Conclusion
The deep sequencing data provided insights into global regulation in P. gingivalis. A small percentage of the total transcriptome was regulated by LuxS, nonetheless the sequencing and analyses were sufficiently sensitive for detection of these differentially abundant transcripts. As the total data set of comparisons is orders of magnitude greater, this demonstrates the utility of RNA-Seq for the study of systems, like this one, where very subtle changes in transcript abundance on a global scale can have biologically important effects on pathogenic potential or specific regulatory or metabolic pathways. Furthermore, the results validated the involvement of LuxS in regulation of the hmu hemin uptake operon described previously, giving additional weight to the newly recognized components of the LuxS regulon. Several novel sRNAs were identified in P. gingivalis which are likely to be important components of gene regulatory pathways.
Materials and Methods
Bacterial Strains and RNA Preparation for Sequencing
Wild-type (WT) P. gingivalis ATCC 33277 and isogenic ΔluxS (James et al., 2006) were used in this study. Bacteria were cultured in trypticase soy broth (TSB) supplemented with yeast extract (1 mg ml–1), hemin (5 μg ml–1), and menadione (1 μg ml–1), anaerobically, at 37°C. Overnight cultures were diluted in TSB to OD600 0.25 and grown to OD600 0.5. Three biological replicates were prepared for each strain. RNA was isolated by Trizol according to the manufacturer’s instructions (Invitrogen). RNA was treated with Turbo-DNA free (Ambion) at 37°C for 1 h, and then purified by an RNeasy mini kit (Qiagen). 16S and 23S rRNAs were removed by a MICROB Express Bacterial mRNA Enrichment Kit according to the manufacturer’s instructions (Ambion). Double-stranded cDNAs were synthesized by a SuperScript Double-stranded cDNA synthesis kit according to the manufacturer’s instructions (Invitrogen).
A fragment DNA library was prepared according to the SOLiD v3 plus procedure (Applied Biosystems). Briefly, cDNA was sheared to 100–110 bp fragments using a Covaris S2 sonication system. The fragmented DNA was subsequently blunted using end-polishing enzymes, and short oligonucleotide adaptors P1 and P2 were ligated to the ends of the end-repaired purified DNA. The oligonucleotide adaptors provide priming sequence for both amplification and sequencing of sample-library DNA fragments. Adaptor-ligated, purified, and size selected 150–200 bp DNA fragments were nick translated and amplified by library PCR primers 1 and 2 using Platinum PCR amplification mix. The sample-library was quantitated by a NanoDrop spectrophotometer. The quality of the libraries and fragment distribution was verified using an Agilent Bioanalyzer. The appropriate amount of dsDNA library was immobilized onto SOLiD P1 DNA beads. The bead-bound library was then clonally amplified by emPCR according to the manufacturer’s protocol.
SOLiD Sequencing and Analysis
After amplification, beads containing clonally amplified template DNA were P2-enriched and extended with a bead linker by terminal transferase. The quantity of the beads was determined using a NanoDrop ND1000 spectrophotometer. Approximately 96 M beads of each enriched library were deposited on slides and sequenced using “sequencing by ligation” chemistry and 50 bp protocol on a SOLiD v3 plus sequencer (Applied Biosystems, CA, USA) at the Interdisciplinary Centre of Biotechnology, University of Florida. Results were obtained as good and best beads as color space FASTA files. Each library was sequenced using 1/4th region of the slide. Figure S4 in Supplementary Material shows an example of the raw reads for all replicates, prior to normalization, for the luxS transcript (PGN_1474).
RNA-Seq Alignment and Coverage
The 50 base unpaired SOLiD short reads were aligned in color space to the P. gingivalis genome scaffold using BWA version 0.5.8 using default parameters (Li and Durbin, 2009) This process began by combining the separate color space read and quality files from the sequencer into a single color space FASTQ file with the “solid2fastq.pl” script distributed with BWA. Next, the P. gingivalis scaffold was indexed by BWA, and the alignments were generated. The SAMtools software was used to generate a “pileup” file from the BWA output (Li et al., 2009). Finally, the pileup data were loaded in a MySQL database and the reads mapped per ORF. Statistics were generated by a standard SQL “SUM over GROUP BY” operation.
Differential abundance analyses were computed using R version 2.11.1 (R Development Core Team, 2010). In order to normalize the samples prior to differential analysis, we applied “quantile normalization,” to assure the reads mapped per ORF distribution had the same empirical distribution across all replicates and experiments (Bolstad et al., 2003). The quantile normalization was performed with the “normalizeBetweenArrays” function of LIMMA (Smyth et al., 2003). In addition, we calculated RPKMs from the normalized reads mapped per ORF as follows: the quantile normalized number of reads mapped was divided by the size of each gene in kb and the count of the millions of reads mapped in the quantile normalized sample (Mortazavi et al., 2008).
To evaluate the significance of differential abundance between experiments we used a t-test with the Welch approximation to account for unequal variances. The p-values derived from the t-statistics were corrected for multiple hypothesis testing using the R package “fdrtool” (Strimmer, 2008). This package calculates Storey’s q-values (Storey, 2003) and Efron’s local false discovery rate (lfdr; Efron et al., 2001) from a set of p-values.
The sequence data have been deposited in the GEO repository2, accession numbers GSM844728–GSM844733.
Quantitative Real-Time PCR
Total RNA was isolated from three independent cultures at OD600 0.5. RNA was converted to cDNA with an iScript cDNA synthesis kit (Bio-Rad). qRT-PCR was performed by StepOne plus by the ΔΔCt method using 16S rRNA as an internal control. Three experimental replicates were analyzed for each biological sample. Primers used are listed in Table S5 in Supplementary Material. Data analysis was performed using both DataAssist v3.0 and StepOne software v2.2.2 from Applied Biosystems.
ΔluxS complementation by synthetic DPD
Overnight cultures of WT and ΔluxS strains were diluted in TSB and the cells were grown to OD600 = 0.3 to avoid accumulation of endogenous AI-2. The cells were washed once in PBS and resuspended in TSB supplemented with 4 μM synthetic DPD (OMM Scientific). The cells were further cultured anaerobically and samples collected after 5, 30, and 60 min. Total RNAs were purified for qRT-PCR.
sRNA Identification
Reads mapped per base “pileup” created with SAMtools from the BWA alignments was used to create a list of all possible intergenic non-coding RNAs as follows. A loop over the base pair positions in the genome was used to scan for continuous stretches of sequence with the running average number of reads mapped greater than a minimum determined by the size of each dataset. In addition, the stretches had to be within 500 bp upstream of a coding region and 100 bp downstream. All loci within 1 kb of each potential ncRNA were flagged for further manual investigation. The potential ncRNAs were assigned unique identifiers based on their position in the genome. For each ncRNA stretch, the RPKM was computed as abundance, and log ratios between conditions were calculated in the same manner as for the ORFs.
sRNAs showing RPKM reading greater than 1000 were chosen for further analysis. Modified RACE for simultaneous identification of 5′- and 3′-end was performed (Britton et al., 2007). Total RNAs were purified from the cells grown in the same condition as for SOLiD analysis. Five μg of total RNAs were treated with 20 U of Tobacco Acid Pyrophosphatase (TAP; Epicenture Biotechnologies) in 1xTAP buffer at 37°C for 1 h. Two μg of TAP-treated RNA were self-ligated by 40 U of RNA ligase I (New England Biolabs) at 17°C overnight. Reverse transcription was performed by using either random primer or IGR-specific forward or reverse primer with the High Capacity cDNA reverse transcription kit (Life Technologies). Nested PCR was carried out by using two sets of PCR primers (Table S5 in Supplementary Material) with GoTaq Green master mix (Promega). Fragments specifically amplified from TAP-treated RNA were TA-cloned into the TOPO-pCR2.1 vector for sequencing (Life technologies).
5′ RACE
Total RNA was isolated using Trizol from WT culture grown to OD600 0.5 followed by RNeasy mini kit including DNase I treatment to remove genomic DNA. The transcriptional start site was determined by 5′-RACE using the FirstChoice RLM-RACE kit (Ambion). Primers used for nested PCR are listed in Table S5 in Supplementary Material. The fragments were ligated into pCR2.1 TOPO vector (Invitrogen) and the ligated DNA was transformed into E. coli Top10 (Invitrogen). The inserts were confirmed by colony PCR using M13 primers and sequenced by the University of Louisville sequencing facility.
Transcriptional and Translational Analysis of btu B
C-terminally in-frame FLAG-tagged btuB (PGN_0704) was constructed in P. gingivalis. For efficient exposure of the FLAG tag sequence from the BtuB protein structure, a linker sequence (GAGAGA) was introduced in between BtuB and three tandem repeats of FLAG sequences (FLAGx3). FLAGx3-tag and ermF alleles were amplified using genomic DNA from a laboratory stock strain as a template to make GAGAGA::FLAGx3::ermF. 700 bases of upstream and downstream of the 3′ end of btuB coding region were amplified using P. gingivalis genomic DNA. The resulting three fragments were fused by PCR and electroporated into electrocompetent cells. Transformants were selected for ermF insertion on blood agar plates containing 5 μg/ml erythromycin and 150 μg/ml gentamicin. The insertion was confirmed by both PCR and sequencing of the recombined region.
To examine the regulation of BtuB, the P. gingivalis strain harboring BtuB::FLAGx3 was inoculated at OD600 0.2 and incubated anaerobically in the presence or absence of 100 μM of Coenzyme B12 (Sigma-Aldrich). Total RNA samples and whole cell lysates were prepared at OD600 0.3, 0.5, 0.7, and 1.0. qRT-PCR was performed to examine the transcriptional level of both the 5′ leader region and the coding region of btuB. For quantifying translational levels of BtuB, lysates from equal cell numbers were Western blotted and probed with anti-FLAG monoclonal antibody (Sigma-Aldrich) or anti-Mfa antibody (Park et al., 2005) followed by probing with HRP conjugated secondary antibody (Cell signaling). Signal was illuminated by the Pierce ECL kit (Thermo) and the bands were quantified by Image lab software on a ChemiDoc™ XRS + system (Bio-Rad).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Erik L. Hendrickson for help with the pathway analysis, and Fred Taub for database programming. This work was facilitated through the use of advanced computational, storage, and networking infrastructure provided by the Hyak supercomputer system, supported in part by the University of Washington eScience Institute. The support of the NIDCR through DE14605 (Donald R. Demuth), DE12505 (Richard J. Lamont), and DE14372 (Murray Hackett) is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Cellular_and_Infection_Microbiology/10.3389/fcimb.2012.00079/abstract
Table S1. RNA-Seq coverage statistics by RPKM.
Table S2. Transcript abundance in WT samples sorted by RPKM.
Table S3. Hypothetical proteins expressed in either WT or ΔluxS strain.
Table S4. Intergenic transcripts sorted by WT reads (large to small).
Table S5. Primers used in this study.
Figure S1. RNA-Seq results of all P. gingivalis genes. Relative gene expression levels of three replicates each for the WT and ΔluxS mutant strains summarized in log2 ratio with p-value. Yellow bars extending to the left or right show down-regulation or up-regulation, respectively, of genes in ΔluxS compared to WT. When statistically significant, the bars for up-regulation or down-regulation of Δlux relative to WT are colored red or green, respectively.
Figure S2. Wild-type vs. ΔluxS by RPKM method. RPKMs of genes in the ΔluxS mutant were compared to those of WT. Fifty-seven ORFs were differentially expressed with a p-value threshold of 0.05. Red and green show up and down-regulation in the ΔluxS strain relative to WT, respectively. Statistical significance is indicated from dark to light with increasing significance.
Figure S3. Pathway analysis for differentially regulated genes in ΔluxS strain. Differentially expressed genes were mapped to the KEGG pathway database, (A) Streptomycin biosynthesis, (B) Lipoic acid biosynthesis, and (C) Peptidoglycan biosynthesis. Red and green denote up and down-regulation in the ΔluxS mutant relative to WT, respectively. Statistically significant relative abundance changes are shown from dark to light with increasing significance.
Figure S4. Un-normalized log2 per base coverage of the luxS (PGN_1474) in RNA-Seq. In gray are the wild-type samples and in green the ΔluxS strain. The deleted region is clearly shown as the uncovered region in the ΔluxS strain. The base numbering is relative to the 3′ end of the ORF. Of note is the consistency of the sequencing depth profile between the three replicates for each sample.
Footnotes
References
Bahekar, A. A., Singh, S., Saha, S., Molnar, J., and Arora, R. (2007). The prevalence and incidence of coronary heart disease is significantly increased in periodontitis: a meta-analysis. Am. Heart J. 154, 830–837.
Beck, D. A., Hendrickson, E. L., Vorobev, A., Wang, T., Lim, S., Kalyuzhnaya, M. G., Lidstrom, M. E., Hackett, M., and Chistoserdova, L. (2011). An integrated proteomics/transcriptomics approach points to oxygen as the main electron sink for methanol metabolism in Methylotenera mobilis. J. Bacteriol. 193, 4758–4765.
Bolstad, B. M., Irizarry, R. A., Astrand, M., and Speed, T. P. (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193.
Britton, R. A., Wen, T., Schaefer, L., Pellegrini, O., Uicker, W. C., Mathy, N., Tobin, C., Daou, R., Szyk, J., and Condon, C. (2007). Maturation of the 5’ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol. Microbiol. 63, 127–138.
Burgess, N. A., Kirke, D. F., Williams, P., Winzer, K., Hardie, K. R., Meyers, N. L., Aduse-Opoku, J., Curtis, M. A., and Camara, M. (2002). LuxS-dependent quorum sensing in Porphyromonas gingivalis modulates protease and haemagglutinin activities but is not essential for virulence. Microbiology 148, 763–772.
Cao, M., Feng, Y., Wang, C., Zheng, F., Li, M., Liao, H., Mao, Y., Pan, X., Wang, J., Hu, D., Hu, F., and Tang, J. (2011). Functional definition of LuxS, an autoinducer-2 (AI-2) synthase and its role in full virulence of Streptococcus suis serotype 2. J. Microbiol. 49, 1000–1011.
Chawla, A., Hirano, T., Bainbridge, B. W., Demuth, D. R., Xie, H., and Lamont, R. J. (2010). Community signalling between Streptococcus gordonii and Porphyromonas gingivalis is controlled by the transcriptional regulator CdhR. Mol. Microbiol. 78, 1510–1522.
Chung, W. O., Park, Y., Lamont, R. J., McNab, R., Barbieri, B., and Demuth, D. R. (2001). Signaling system in Porphyromonas gingivalis based on a LuxS protein. J. Bacteriol. 183, 3903–3909.
Diaz, P. I., and Rogers, A. H. (2004). The effect of oxygen on the growth and physiology of Porphyromonas gingivalis. Oral Microbiol. Immunol. 19, 88–94.
Diaz, P. I., Slakeski, N., Reynolds, E. C., Morona, R., Rogers, A. H., and Kolenbrander, P. E. (2006). Role of oxyR in the oral anaerobe Porphyromonas gingivalis. J. Bacteriol. 188, 2454–2462.
Doherty, N., Holden, M. T., Qazi, S. N., Williams, P., and Winzer, K. (2006). Functional analysis of luxS in Staphylococcus aureus reveals a role in metabolism but not quorum sensing. J. Bacteriol. 188, 2885–2897.
Duran-Pinedo, A. E., Nishikawa, K., and Duncan, M. J. (2007). The RprY response regulator of Porphyromonas gingivalis. Mol. Microbiol. 64, 1061–1074.
Efron, B., Tibshirani, R., Storey, J. D., and Tusher, V. (2001). Empirical bayes analysis of a microarray experiment. J. Am. Stat. Assoc. 96, 1151–1160.
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., Gavin, O. L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L., Eddy, S. R., and Bateman, A. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222.
Fong, K. P., Gao, L., and Demuth, D. R. (2003). luxS and arcB control aerobic growth of Actinobacillus actinomycetemcomitans under iron limitation. Infect. Immun. 71, 298–308.
Genco, C. A. (1995). Regulation of hemin and iron transport in Porphyromonas gingivalis. Adv. Dent. Res. 9, 41–47.
Genco, C. A., Simpson, W., Forng, R. Y., Egal, M., and Odusanya, B. M. (1995). Characterization of a Tn4351-generated hemin uptake mutant of Porphyromonas gingivalis: evidence for the coordinate regulation of virulence factors by hemin. Infect. Immun. 63, 2459–2466.
Henderson, B., Curtis, M., Seymour, R., and Donos, N. (2009). Periodontal Medicine and Systems Biology. Oxford: John Wiley & Sons-Blackwell.
Hovik, H., Yu, W. H., Olsen, I., and Chen, T. (2012). Comprehensive transcriptome analysis of the periodontopathogenic bacterium Porphyromonas gingivalis W83. J. Bacteriol. 194, 100–114.
Humphrey, L. L., Fu, R., Buckley, D. I., Freeman, M., and Helfand, M. (2008). Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J. Gen. Int. Med. 23, 2079–2086.
Isabella, V. M., and Clark, V. L. (2011). Deep sequencing-based analysis of the anaerobic stimulon in Neisseria gonorrhoeae. BMC Genomics 12, 51. doi:10.1186/1471-2164-12-51
James, C. E., Hasegawa, Y., Park, Y., Yeung, V., Tribble, G. D., Kuboniwa, M., Demuth, D. R., and Lamont, R. J. (2006). LuxS involvement in the regulation of genes coding for hemin and iron acquisition systems in Porphyromonas gingivalis. Infect. Immun. 74, 3834–3844.
Johnson, N. A., McKenzie, R. M., and Fletcher, H. M. (2011). The bcp gene in the bcp-recA-vimA-vimE-vimF operon is important in oxidative stress resistance in Porphyromonas gingivalis W83. Mol. Oral Microbiol. 26, 62–77.
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30.
Katz, J., Chegini, N., Shiverick, K. T., and Lamont, R. J. (2009). Localization of P. gingivalis in preterm delivery placenta. J. Dent. Res. 88, 575–578.
Kondo, Y., Ohara, N., Sato, K., Yoshimura, M., Yukitake, H., Naito, M., Fujiwara, T., and Nakayama, K. (2010). Tetratricopeptide repeat protein-associated proteins contribute to the virulence of Porphyromonas gingivalis. Infect. Immun. 78, 2846–2856.
Kuboniwa, M., Hendrickson, E. L., Xia, Q., Wang, T., Xie, H., Hackett, M., and Lamont, R. J. (2009). Proteomics of Porphyromonas gingivalis within a model oral microbial community. BMC Microbiol. 9, 98. doi:10.1186/1471-2180-9-98
Lamont, R. J., and Jenkinson, H. F. (1998). Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62, 1244–1263.
Learman, D. R., Yi, H., Brown, S. D., Martin, S. L., Geesey, G. G., Stevens, A. M., and Hochella, M. F. Jr. (2009). Involvement of Shewanella oneidensis MR-1 LuxS in biofilm development and sulfur metabolism. Appl. Environ. Microbiol. 75, 1301–1307.
Lebeer, S., De Keersmaecker, S. C., Verhoeven, T. L., Fadda, A. A., Marchal, K., and Vanderleyden, J. (2007). Functional analysis of luxS in the probiotic strain Lactobacillus rhamnosus GG reveals a central metabolic role important for growth and biofilm formation. J. Bacteriol. 189, 860–871.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., and Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079.
Loos, B. G., Mayrand, D., Genco, R. J., and Dickinson, D. P. (1990). Genetic heterogeneity of Porphyromonas (Bacteroides) gingivalis by genomic DNA fingerprinting. J. Dent. Res. 69, 1488–1493.
Maeda, K., Tribble, G. D., Tucker, C. M., Anaya, C., Shizukuishi, S., Lewis, J. P., Demuth, D. R., and Lamont, R. J. (2008). A Porphyromonas gingivalis tyrosine phosphatase is a multifunctional regulator of virulence attributes. Mol. Microbiol. 69, 1153–1164.
Markowitz, V. M., Szeto, E., Palaniappan, K., Grechkin, Y., Chu, K., Chen, I. M., Dubchak, I., Anderson, I., Lykidis, A., Mavromatis, K., Ivanova, N. N., and Kyrpides, N. C. (2008). The integrated microbial genomes (IMG) system in 2007: data content and analysis tool extensions. Nucleic Acids Res. 36, D528–D533.
Marsh, P. D., McDermid, A. S., McKee, A. S., and Baskerville, A. (1994). The effect of growth rate and haemin on the virulence and proteolytic activity of Porphyromonas gingivalis W50. Microbiol. 140, 861–865.
McNab, R., Ford, S. K., El-Sabaeny, A., Barbieri, B., Cook, G. S., and Lamont, R. J. (2003). LuxS-based signaling in Streptococcus gordonii: autoinducer 2 controls carbohydrate metabolism and biofilm formation with Porphyromonas gingivalis. J. Bacteriol. 185, 274–284.
Meuric, V., Gracieux, P., Tamanai-Shacoori, Z., Perez-Chaparro, J., and Bonnaure-Mallet, M. (2008). Expression patterns of genes induced by oxidative stress in Porphyromonas gingivalis. Oral Microbiol. Immunol. 23, 308–314.
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628.
Nahvi, A., Barrick, J. E., and Breaker, R. R. (2004). Coenzyme B12 riboswitches are widespread genetic control elements in prokaryotes. Nucleic Acids Res. 32, 143–150.
Naito, M., Hirakawa, H., Yamashita, A., Ohara, N., Shoji, M., Yukitake, H., Nakayama, K., Toh, H., Yoshimura, F., Kuhara, S., Hattori, M., and Hayashi, T. (2008). Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res. 15, 215–225.
Okano, S., Shibata, Y., Shiroza, T., and Abiko, Y. (2006). Proteomics-based analysis of a counter-oxidative stress system in Porphyromonas gingivalis. Proteomics 6, 251–258.
Oliver, H. F., Orsi, R. H., Ponnala, L., Keich, U., Wang, W., Sun, Q., Cartinhour, S. W., Filiatrault, M. J., Wiedmann, M., and Boor, K. J. (2009). Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs. BMC Genomics 10, 641. doi:10.1186/1471-2164-10-641
Park, Y., Simionato, M. R., Sekiya, K., Murakami, Y., James, D., Chen, W., Hackett, M., Yoshimura, F., Demuth, D. R., and Lamont, R. J. (2005). Short fimbriae of Porphyromonas gingivalis and their role in coadhesion with Streptococcus gordonii. Infect. Immun. 73, 3983–3989.
Perkins, T. T., Kingsley, R. A., Fookes, M. C., Gardner, P. P., James, K. D., Yu, L., Assefa, S. A., He, M., Croucher, N. J., Pickard, D. J., Maskell, D. J., Parkhill, J., Choudhary, J., Thomson, N. R., and Dougan, G. (2009). A strand-specific RNA-Seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi. PLoS Genet. 5, e1000569. doi:10.1371/journal.pgen.1000569
Ravnum, S., and Andersson, D. I. (1997). Vitamin B12 repression of the btuB gene in Salmonella typhimurium is mediated via a translational control which requires leader and coding sequences. Mol. Microbiol. 23, 35–42.
R Development Core Team. (2010). R: A Language and Environment for Statistical Computing, Version 2.11.1. Vienna: R Foundation for Statistical Computing.
Redanz, S., Standar, K., Podbielski, A., and Kreikemeyer, B. (2011). A five-species transcriptome array for oral mixed-biofilm studies. PLoS ONE 6, e27827. doi:10.1371/journal.pone.0027827
Sabbah, W., Sheiham, A., and Bernabe, E. (2010). Income inequality and periodontal diseases in rich countries: an ecological cross-sectional study. Int. Dent. J. 60, 370–374.
Schauder, S., Shokat, K., Surette, M. G., and Bassler, B. L. (2001). The LuxS family of bacterial autoinducers: biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol. 41, 463–476.
Shibata, Y., Yamashita, Y., Nakano, Y., and Koga, T. (1999). Isolation and characterization of the rml gene homologs from Porphyromonas gingivalis. Oral Microbiol. Immunol. 14, 339–347.
Siller, M., Janapatla, R. P., Pirzada, Z. A., Hassler, C., Zinkl, D., and Charpentier, E. (2008). Functional analysis of the group A streptococcal luxS/AI-2 system in metabolism, adaptation to stress and interaction with host cells. BMC Microbiol. 8, 188. doi:10.1186/1471-2180-8-188
Smyth, G. K., Yang, Y. H., and Speed, T. (2003). Statistical issues in cDNA microarray data analysis. Methods Mol. Biol. 224, 111–136.
Storey, J. D. (2003). The positive false discovery rate: a Bayesian interpretation and the q-value. Ann. Stat. 31, 2013–2035.
Storz, G., Vogel, J., and Wassarman, K. M. (2011). Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43, 880–891.
Strimmer, K. (2008). fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462.
Sun, J., Daniel, R., Wagner-Dobler, I., and Zeng, A. P. (2004). Is autoinducer-2 a universal signal for interspecies communication: a comparative genomic and phylogenetic analysis of the synthesis and signal transduction pathways. BMC Evol. Biol. 4, 36. doi:10.1186/1471-2148-4-36
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36.
Vendeville, A., Winzer, K., Heurlier, K., Tang, C. M., and Hardie, K. R. (2005). Making ‘sense’ of metabolism: autoinducer-2, LuxS and pathogenic bacteria. Nat. Rev. Microbiol. 3, 383–396.
Weinberg, Z., Wang, J. X., Bogue, J., Yang, J., Corbino, K., Moy, R. H., and Breaker, R. R. (2010). Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol. 11, R31.
Yoder-Himes, D. R., Chain, P. S., Zhu, Y., Wurtzel, O., Rubin, E. M., Tiedje, J. M., and Sorek, R. (2009). Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc. Natl. Acad. Sci. U.S.A. 106, 3976–3981.
Yuan, L., Hillman, J. D., and Progulske-Fox, A. (2005). Microarray analysis of quorum-sensing-regulated genes in Porphyromonas gingivalis. Infect. Immun. 73, 4146–4154.
Keywords: LuxS, ncRNA, periodontal pathogen
Citation: Hirano T, Beck DAC, Demuth DR, Hackett M and Lamont RJ (2012) Deep sequencing of Porphyromonas gingivalis and comparative transcriptome analysis of a LuxS mutant. Front. Cell. Inf. Microbio. 2:79. doi: 10.3389/fcimb.2012.00079
Received: 10 April 2012; Accepted: 21 May 2012;
Published online: 12 June 2012.
Edited by:
Francois Vandenesch, Immunity Infection Vaccination, FranceReviewed by:
Tom Geissmann, Institut National de la Santé et de la Recherche MédicaleGerhart Wagner, Uppsala University, Sweden
Copyright: © 2012 Hirano, Beck, Demuth, Hackett and Lamont. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Richard J. Lamont, School of Dentistry, University of Louisville, 501 S Preston Street, Louisville, KY 40292, USA. e-mail: rich.lamont@louisville.edu
†Takanori Hirano and David A. C. Beck have contributed equally to this work.