Central sympathetic activation and arrhythmogenesis during acute myocardial infarction: modulating effects of endothelin-B receptors
 Theofilos M. Kolettis1*
Theofilos M. Kolettis1*  Marianthi Kontonika1
Marianthi Kontonika1
 Eleonora Barka1
Eleonora Barka1
 Evangelos P. Daskalopoulos1
Evangelos P. Daskalopoulos1
 Giannis G. Baltogiannis1
Giannis G. Baltogiannis1
 Christos Tourmousoglou1
Christos Tourmousoglou1
 Apostolos Papalois1,2
Apostolos Papalois1,2  Zenon S. Kyriakides1
Zenon S. Kyriakides1
- 1Cardiovascular Research Institute, Ioannina and Athens, Greece
- 2Experimental Research Center ELPEN, Athens, Greece
Sympathetic activation during acute myocardial infarction (MI) is an important arrhythmogenic mechanism, but the role of central autonomic inputs and their modulating factors remain unclear. Using the in vivo rat-model, we examined the effects of clonidine, a centrally acting sympatholytic agent, in the presence or absence of myocardial endothelin-B (ETB) receptors. We studied wild-type (n = 20) and ETB-deficient rats (n = 20) after permanent coronary ligation, with or without pretreatment with clonidine. Cardiac rhythm was continuously recorded for 24 h by implantable telemetry devices, coupled by the assessment of autonomic and heart failure indices. Sympathetic activation and arrhythmogenesis were more prominent in ETB-deficient rats during the early phase post-ligation. Clonidine improved these outcomes throughout the observation period in ETB-deficient rats, but only during the delayed phase in wild-type rats. However, this benefit was counterbalanced by atrioventricular conduction abnormalities and by higher incidence of heart failure, the latter particularly evident in ETB-deficient rats. Myocardial ETB-receptors attenuate the arrhythmogenic effects of central sympathetic activation during acute MI. ETB-receptor deficiency potentiates the sympatholytic effects of clonidine and aggravates heart failure. The interaction between endothelin and sympathetic responses during myocardial ischemia/infarction and its impact on arrhythmogenesis and left ventricular dysfunction merits further investigation.
Introduction
Sudden cardiac death (SCD) accounts for 13% of total mortality from natural causes in the general population and has emerged as an important health-related problem. In most cases, SCD is caused by ventricular tachyarrhythmias, i.e., ventricular tachycardia (VT) and ventricular fibrillation (VF), triggered by acute myocardial infarction (MI) (1). Driven by its high-societal impact, in-depth understanding of the pathophysiology of ischemia-induced VT/VF has attracted multifaceted research efforts, with a view toward decreasing SCD rates.
The role of sympathetic activation in the pathogenesis of VT/VF during acute MI has been known for decades (2); myocardial ischemia leads to local norepinephrine release from sympathetic nerve terminals, primarily via non-exocytotic mechanisms, following an immediate exocytotic phase (3). The elicited positive inotropic response serves the maintenance of cardiac output, but local catecholamine surge alters ventricular electrophysiological properties and creates an arrhythmogenic milieu (4). These mechanisms may predominate during the early period post-MI, corresponding clinically to the critical pre-hospital stage, during which VT/VF carries a dismal prognosis (1).
Recently, accumulated evidence indicates endothelin-1 (ET-1), a ubiquitous 21-aminoacid peptide, as an important moderator of sympathetic activation (5) and arrhythmogenesis (6) during acute-MI. Both (ETA and ETB) ET-receptors, located in sympathetic nerve varicosities in the ventricular myocardium (7), exert significant, albeit opposing effects; ETA-receptors inhibit norepinephrine re-uptake, whereas ETB-receptors attenuate exocytotic norepinephrine release (8). These actions gain pathophysiological significance in the MI-setting, during which ETA-receptor stimulation enhances norepinephrine release, but the role of ETB-receptors remains unclear (9). Experimental studies have shown increased sympathetic activation and arrhythmogenesis in the absence of functional ETB-receptors in the ischemic ventricular myocardium (7, 10); however, most data originate from ex vivo, isolated (hence, denervated) beating hearts, thereby hindering the deduction of firm conclusions on the role of ETB-receptors, in the presence of intact autonomic innervation (11).
In addition to myocardial norepinephrine release from intrinsic nerve terminals, central sympathetic activation occurs after ischemia, mediated by locally produced metabolites that stimulate afferent myocardial nerve fibers; in turn, efferent autonomic discharges from the brain stem modulate left ventricular (LV) function and electrophysiology (12). Earlier studies in anesthetized dogs demonstrated increased cardiac sympathetic nerve activity during MI that was prevented by clonidine, a centrally acting α2-adrenergic-receptor agonist (13). Efferent inputs to the heart are arrhythmogenic, as shown by continuous recordings of the left stellate ganglion in conscious dogs, revealing the precedence of VT/VF by enhanced sympathetic nerve activity (14), and by effective clinical management of intractable VT/VF by surgical or thoracoscopic denervation (15). Despite this knowledge, the relative impact of central sympathetic activation on VT/VF along with the course of acute-MI, and the potential modulating effects of ETB-receptors in the ventricular myocardium are not well defined.
Here, we examined arrhythmogenesis, as well as indices of sympathetic activation and LV failure, in the in vivo acute-MI rat-model; we compared these outcomes in groups with or without pretreatment with clonidine, in the presence or absence of myocardial ETB-receptors.
Materials and Methods
Animal Study Population and Ethics
The animal study population consisted of 40 rats, 20–24 weeks of age, weighing 225–300 g. The animals were housed in plexiglas-cages in groups of two, with free access to standard rodent pellet-diet and water. The laboratory conditions were kept optimal, in terms of temperature (20–22°C), humidity (~70%), and light/dark cycles (12/12 h). The study protocol adheres to the guiding principles of the declaration of Helsinki (on animal research) and to European legislation (European Union directive for the protection of animals used for scientific purposes 609/1986, revised in 2010/63/EU); all procedures were approved by the institutional ethics’ committee and by the regulatory state authorities.
The role of ETB-receptors in the ventricular myocardium was examined by a “subtraction model,” using a previously characterized (16) Wistar–Imamichi rat strain (n = 20), kindly provided by Professor M. Yanagisawa (Southwestern Medical Center, Dallas, TX, USA and University of Tsukuba, Tsukuba, Japan). These animals carry a (naturally occurring) deletion in the gene encoding for the ETB-receptor and die prematurely of intestinal obstruction. This phenotype is rescued by directed ETB transgene-expression, leading to normal development of enteric nervous system and brain function, and provides a valuable tool in the study of ETB-receptors in the cardiovascular system (17).
Study Protocol
Cardiac rhythm was recorded continuously for 24 h post-MI, with the use of miniature electrocardiography (ECG) telemetry transmitters; these devices permit long-term assessment in conscious animals, without the confounding effects of anesthesia (18). The survival duration was accurately determined from the stored ECG-recordings; mortality was further classified as tachyarrhythmic (i.e., ventricular asystole, immediately preceded by VT/VF) or bradyarrhythmic [i.e., gradual increase in sinus heart rate (HR), followed by an abrupt onset of complete atrioventricular block and asystole], the latter indicative of death due to heart failure (18, 19).
Transmitter Implantation
The telemetry transmitters (Dataquest, Data Sciences International, DSI, Transoma Medical, Arden Hills, MN, USA) were implanted in the abdominal cavity, as described previously (20); both leads were tunneled under the skin and secured at the right axillary and left inguinal areas, respectively. The animals were housed in individual cages, placed on a receiver that continuously captured the ECG-signal, processed by a software program (A.R.T. 2.2, DSI).
Clonidine Administration
Central sympathetic activation was examined with the use of clonidine, a commonly applied pharmacological model; after an initial peripheral α1-stimulation, the central action of clonidine prevails, resulting in inhibition of sympathetic preganglionic neurons and decreased sympathetic drive (21). As previously (22), clonidine was given intraperitoneally (0.5 mg/kg), 1 h prior to the experiments.
Induction of Myocardial Infarction
After tracheal intubation, the rats were mechanically ventilated (rodent apparatus model 7025, Ugo Basile, Comerio, Italy), and anesthesia was maintained with a mixture of oxygen and 2.5% sevoflurane. MI was generated by permanent ligation at the middle segment of the left coronary artery, guided by the anatomic landmarks provided by the pulmonary cone, the left atrium, and the ventricular apex (23). MI was validated by inspection of a pale, akinetic area, and by ST-segment elevation in a six-lead ECG (QRS-Card digital PC-ECG, Pulse Biomedical Inc., PBI, Norristown, PA, USA, amplified by Cardiology Suite, version 4.05, PBI). The incision was closed in three layers and pneumothorax was evacuated. Spontaneous respiration and consciousness resumed within ~2 min after discontinuation of anesthesia.
Arrhythmia Analysis
The analysis of ECG-recordings adhered to the guidelines provided by the (recently updated) Lambeth conventions for determination of experimental arrhythmias (24). The tracings were manually scrolled and the number of tachy- and bradyarrhythmic events were recorded; the count of premature ventricular contractions, couplets, or triplets was omitted, based on their uncertain significance. As distinction between VT (four or more consecutive premature ventricular contractions) and VF (indistinguishable QRS deflections) may be occasionally difficult (19, 20), we report them collectively as VT/VF; the duration of each episode was measured, aided by the time-scale provided by the software. Similarly, we recorded transient atrioventricular conduction abnormalities, not associated with bradyarrhythmic death. To account for differences in mortality, VT/VF duration during the entire observation period was normalized for survival (18).
Arrhythmia Time-Intervals
The incidence of VT/VF is reported separately for phase I (i.e., during the first hour post-ligation, corresponding to ischemia and the onset of myocardial necrosis), and phase II (i.e., from the 2nd until the 24th hour post-ligation, corresponding to evolving MI, until the completion of necrosis). Such separation presents the inherent limitation of producing temporal dissimilarities in the characteristics within an animal-population in cases of excessive early-phase mortality, but it is useful for its translational value in the clinical setting, and also provides information on the underlying arrhythmogenic mechanisms (25, 26).
ECG-Indices of Sympathetic Activation
Mean values of HR, calculated from sinus beats, are given separately for phases I and II. Furthermore, heart rate variability (HRV), derived by fast Fourier transformation, is reported as the ratio of low-frequency (LF, 0.195–0.605 Hz) to high-frequency (HF, 0.605–2.5 Hz) bands (27). This index correlates with catecholamine measurements (28), and permits the continuous assessment of autonomic balance in conscious animals (29). For HRV-analysis, we used the Kubios HRV-software (version 2.1, Biosignal Analysis and Medical Imaging Group, Department of Applied Physics, University of Eastern Finland, Kuopio, Finland) (30).
Activity Measurement
Acute LV failure was assessed by voluntary motor activity, recorded by the analysis program (A.R.T. 2.2, DSI). The software records strength-variations in the telemetry-signal, in relation to the location of the animal; changes in signal-amplitude are depicted as counts, the number of which depends on total animal activity. This variable correlates with the functional status, and has been used as a surrogate marker for heart failure (27, 31, 32).
Statistical Analysis
Values are presented as mean ± SEM. Kaplan–Meier survival curves were constructed and differences in mortality were assessed with Gehan’s Wilcoxon test. Categorical variables were compared with Fisher’s exact (two-tailed) test. Continuous variables were compared with analysis of variance, whereas their changes over time were compared with analysis of variance for repeated measures, both followed by post hoc Newman–Keuls multi-stage test. Statistical significance was defined at an alpha level of 0.05.
Results
Mortality
As seen in Kaplan–Meier survival curves (Figure 1), mortality was higher (p = 0.028) in ETB-deficient than in wild-type rats. Clonidine had no overall effect, but a shift in the mode of death was observed, from tachyarrhythmic (83% of mortality in ETB-deficient and 33% in wild-type rats) to bradyarrhythmic (100% of mortality in both groups); this difference reached statistical significance (p = 0.0152) in ETB-deficient rats.
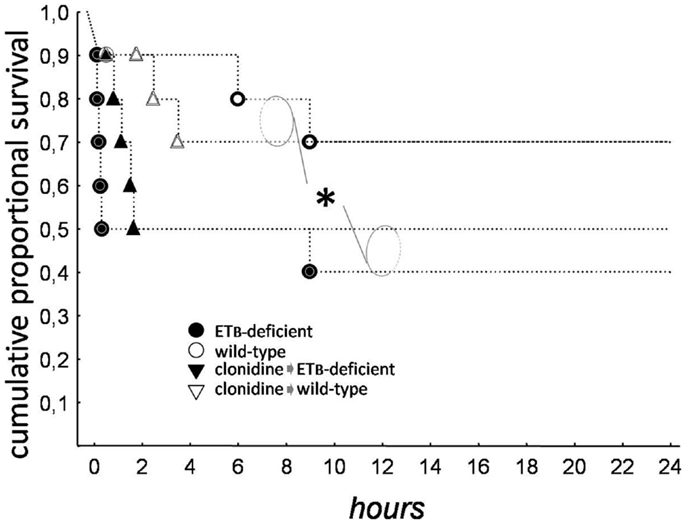
Figure 1. Kaplan–Meier survival curves. Total mortality was higher in ETB-deficient (asterisk), than in wild-type rats.
Sympathetic Activation
During phase I, HR and LF/HF ratio were higher in ETB-deficient than in wild-type rats (Figure 2). Compared to the respective untreated groups, both variables were lower after clonidine at baseline and during phase I, without differences between groups. During phase II, HR and LF/HF ratio were lower in ETB-deficient than in wild-type rats. After clonidine, LF/HF ratio was similar, although HR tended to be higher (p = 0.064) in ETB-deficient rats.
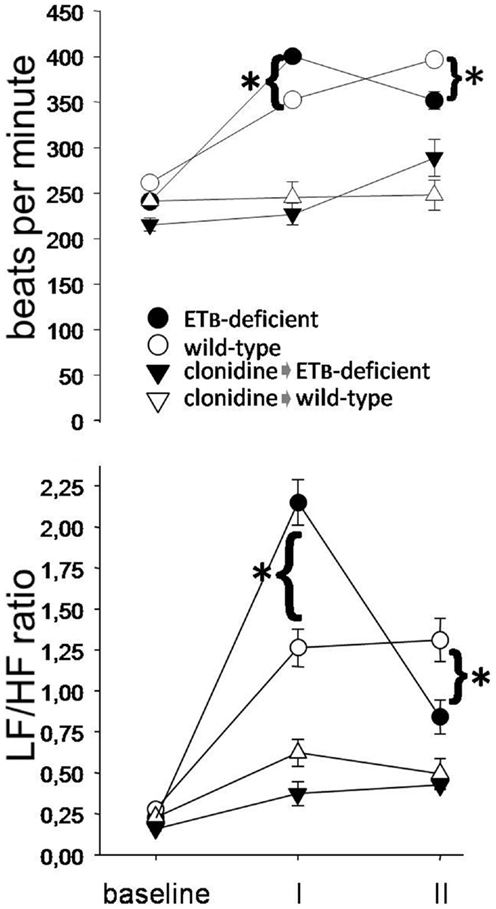
Figure 2. Sympathetic activation. Heart rate (upper panel) and low (LF) to high (HF) frequency ratio (lower panel) were higher in ETB-deficient rats during phase I (asterisks). Note their marked decrease after clonidine.
Ventricular Arrhythmias and A–V Block
During phase I, the number of VT/VF episodes was comparable in the two groups, but their mean duration was longer in ETB-deficient rats, resulting in longer (p = 0.00013) total VT/VF duration. Compared to the respective untreated group, VT/VF duration did not change significantly in clonidine-treated wild-type rats, although variable responses were observed. In contrast, VT/VF duration decreased in ETB-deficient rats (p = 0.000159) after clonidine, resulting from (marginally) fewer (p = 0.053) and (mainly) shorter (p = 0.000641) episodes (Figure 3). Of note, transient second degree atrioventricular block was seen after clonidine in both groups, namely, 1.0 ± 1.0 (of 8.7 ± 8.7 s duration) and 3.7 ± 2.4 (18.5 ± 13.5 s) episodes in ETB-deficient and wild-type rats, respectively; these differences (between groups) did not reach statistical significance.
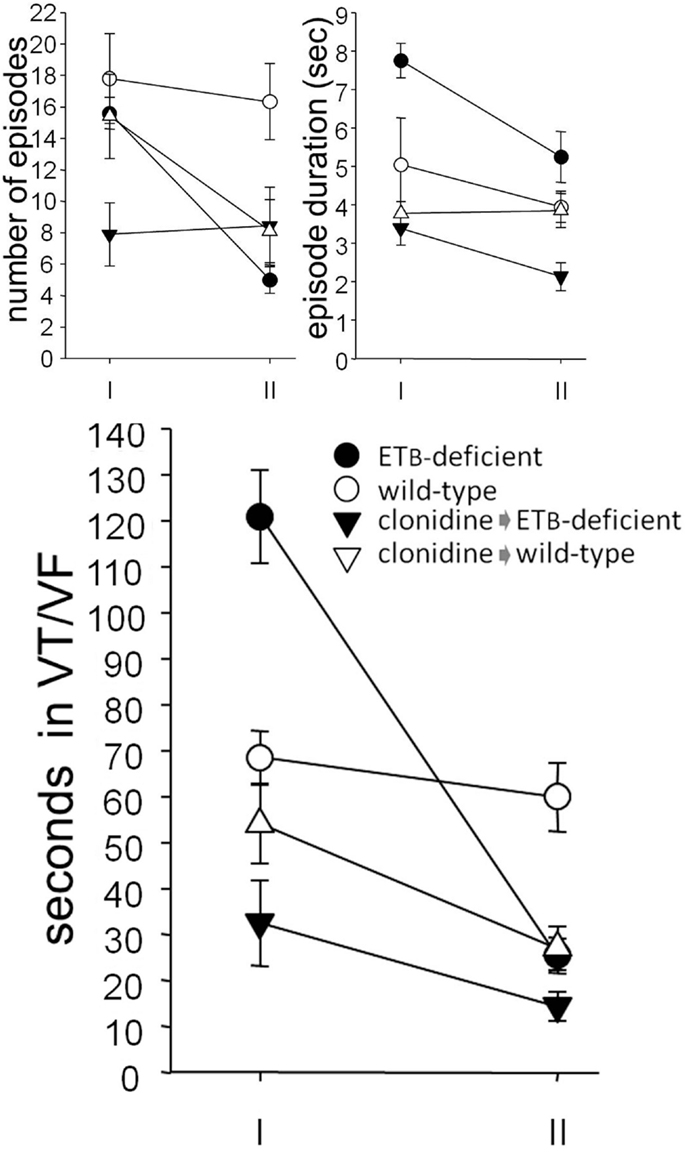
Figure 3. Ventricular arrhythmogenesis. Number and mean duration of ventricular tachycardia (VT) and fibrillation (VF) episodes (upper panel). Note, the decreased total duration (lower panel) in clonidine-treated ETB-deficient rats during phase I, and in clonidine-treated wild-type rats during phase II.
Compared to wild-type rats, less (p = 0.0031) VT/VF episodes occurred in ETB-deficient rats during phase II, resulting in shorter (p = 0.0010) total duration. After clonidine pretreatment, VT/VF duration was comparable in the two groups, due to decreased values (p = 0.0014) in wild-type rats. As in phase I, a mean of 0.7 ± 0.4 (of 1.8 ± 1.1 s duration) and 2.4 ± 1.3 episodes (10.5 ± 9.0 s) of second degree atrioventricular block were seen in ETB-deficient and wild-type rats, respectively; again, these differences (between groups) did not reach statistical significance.
When normalized for survival, total VT/VF duration (during both phases) was markedly higher in untreated ETB-deficient rats (414.6 ± 162.8 s), compared to the remaining three groups [i.e., untreated wild-type rats (26.1 ± 18.9 s, p = 0.0020), clonidine-treated ETB-deficient rats (6.3 ± 2.7 s, p = 0.0063) and clonidine-treated wild-type rats (7.8 ± 2.5 s, p = 0.0035)]. Representative examples of VT/VF and atrioventricular block are shown in Figure 4.
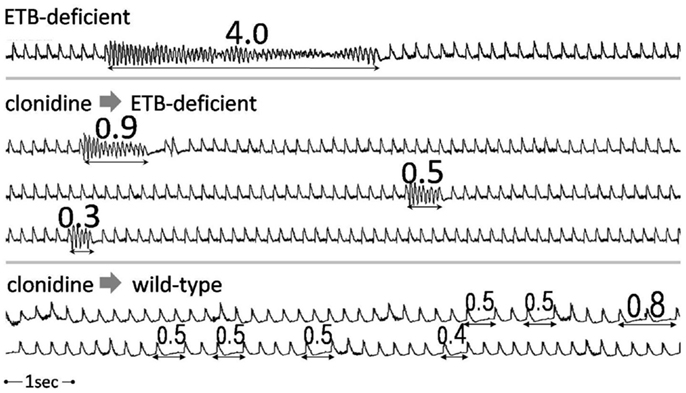
Figure 4. Examples of tachy- and bradyarrhythmias. Examples of ventricular arrhythmias (continuous strips) from an untreated (upper panel) and a clonidine-treated (middle panel) ETB-deficient rat. Frequent episodes of transient atrioventricular conduction abnormalities in a clonidine-treated wild-type rat (lower panel). Numbers indicate duration in seconds.
Activity
Activity-counts were lower in ETB-deficient (1307 ± 472) than in wild-type rats (2296 ± 524) during the entire observation period post-MI, albeit of marginal statistical significance (p = 0.088, Figure 5). Clonidine did not change activity-counts in ETB-deficient rats during phase I, but decreased them (p = 0.036) in wild-type rats. Compared to the respective untreated groups, lower values were found after clonidine in both groups during phase II, but differences reached statistical significance (p = 0.0054) only in ETB-deficient rats.
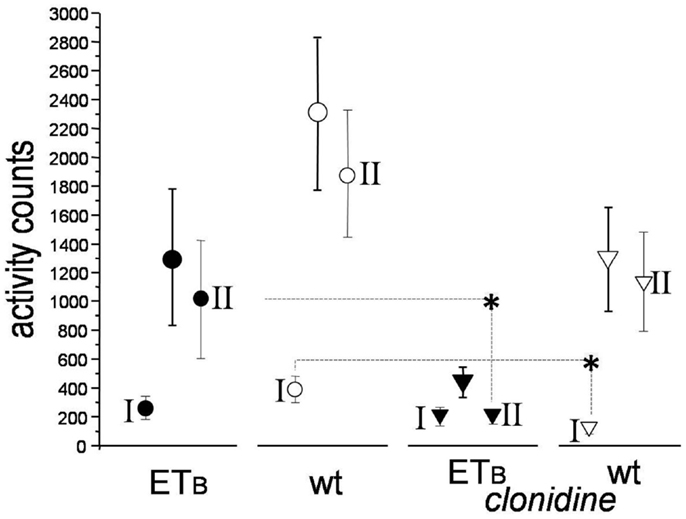
Figure 5. Activity-counts. Compared to the respective untreated groups, activity-counts were lower in clonidine-treated wild-type rats during phase I and in clonidine-treated ETB-deficient rats during phase II (asterisks).
Discussion
Sympathetic activation is an essential mechanism responsible for arrhythmogenesis during acute-MI, but the contribution of central autonomic inputs relative to local norepinephrine release remains under investigation. We examined the effects of (the centrally acting sympatholytic agent) clonidine on rhythm disturbances in the in vivo MI-rat-model, focusing on the modulating actions of myocardial ETB-receptors.
Bradycardic Effects of Clonidine
Clonidine-pretreatment decreased HR and deteriorated atrioventricular conduction in both rat-groups throughout the observation period. Our findings are similar to those seen after intrathecal clonidine (from the spinal cord) in a canine-model of ischemia (superimposed on healed-MI and heart failure); this study (33) reported 32% reduction in HR, 22% increase in PQ interval, and 122% increase in Wenckebach cycle-length. The bradycardic effects of clonidine on the sinus and atrioventricular nodes are thought to be secondary to autonomic modulation, rather than a direct effect on cardiac tissues (33).
Central Sympathetic Activation and Arrhythmogenesis in Wild-Type Rats
The effects of clonidine in wild-type rats differed between the two (post-ligation) phases. Phase I was characterized by variable responses, but clonidine had no overall effect on VT/VF duration at this stage. Thus, the involvement of central sympathetic activation on early-phase arrhythmogenesis post-MI seems to be minor. Conversely, clonidine-pretreatment decreased (by ~50%) the number and total duration of VT/VF episodes during phase II.
Using neurographic recordings from postganglionic cardiac efferent nerves in conscious dogs, Zhou et al. (14) found augmented nerve activity immediately preceding most VT/VF episodes, underscoring its role in triggering arrhythmic events. Additional arrhythmogenic action appears to be exerted via enhanced dispersion of refractoriness, demonstrated by ventricular electrograms following left stellate ganglion stimulation in anesthetized dogs, although marked variation in the individual responses was present (34, 35). Further corroborating these observations, attenuated cardiac sympathetic activity and VT/VF were reported in dogs [in the aforementioned study by Issa et al. (33)] after intrathecal clonidine. Nonetheless, the temporal pattern of arrhythmogenesis, caused by central sympathetic inputs, along with the course of acute-MI cannot be defined from these studies (14, 33–35).
In a single experiment in the ovine model, Jardine et al. (36) reported increased nerve activity 30 min after coronary occlusion, leading to VF. This topic was subsequently systematically addressed by the same researchers (37) in a group of 11 sheep; enhanced cardiac sympathetic nerve frequency was evident (a mean of) 1 h after coronary occlusion, but again, variable degree of activation was noted (37). Examined together, the findings of the present work in the context of previous studies (14, 33–37), indicate that (in the presence of ETB-receptors in the ventricular myocardium) cardiac sympathetic nerve activity participates only to a minor extent in the mechanisms underlying early-phase VT/VF, although variable responses occur; in contrast, central sympathetic activation contributes to delayed-phase arrhythmogenesis. In addition to autonomic responses, this temporal pattern can be attributed to differences in the substrate, created by ischemia versus evolving MI (1, 25). Furthermore, LV-afterload reduction [often observed as a delayed effect after clonidine administration (38)] may have contributed to this pattern, by decreasing wall-stress and, thereby, stretch-induced VT/VF (39).
The contribution of central sympathetic activation to delayed-phase arrhythmogenesis, indicated by our results, may appear contradictory to previous experiments in Langendorff-perfused rat-hearts, in which catecholamines failed to restore phase II VT/VF (40); further pointing toward this direction, relatively limited contribution of the adrenal medulla on arrhythmogenesis was recently shown in the adrenalectomized in vivo rat-MI-model (41). However, it should be pointed out that neurally released and circulating norepinephrine activate adrenergic-receptors at different myocardial areas, thereby producing dissimilar electrophysiologic milieu and arrhythmogenic actions (42). We feel that this important point needs further investigation in future studies.
Central Sympathetic Activation and Arrhythmogenesis in ETB-Deficient Rats
Compared to wild-type rats, the course of sympathetic activity and arrhythmogenesis differed in our ETB-deficient group, strongly suggesting modulating effects of ETB-receptors. As in previous work from our laboratory (28, 41), we found higher incidence of VT/VF in ETB-deficient rats during phase I, along with a corresponding increase in mortality. This outcome concurs also with observations in pharmacological studies, in which dual-(ETA and ETB)-blockade (43) mitigated the protective effects of ETA-receptor-blockade during this time-frame (44). Thus, functioning myocardial ETB-receptors ameliorate arrhythmogenesis during the early post-MI phase; decreased norepinephrine overflow, mediated by attenuated exocytotic release at this stage, has been proposed as a likely explanation (45).
Contrasting phase I, phase II was characterized by lower arrhythmogenesis in untreated ETB-deficient rats, in accordance with previous results (28). The explanation for this finding is difficult, but catecholamine-depletion, secondary to excessive sympathetic activation during phase I, has been put forward as a possible mechanism (41).
In the present work, we found lower incidence of VT/VF in clonidine-treated ETB-deficient rats during phase I; this resulted mainly from shorter mean episode duration, in line with the well-described sympathetic effects on the maintenance of VT/VF over the ischemic-border (34). Thus, in contrast to wild-type rats, central sympathetic inputs appear to exert an important role on early-phase arrhythmogenesis in ETB-deficient rats. This observation can be hardly explained solely by the absence of protective effects exerted by myocardial ETB-receptors, raising the possibility of enhanced central sympathetic activation in this rat strain. This notion is reinforced by cellular studies in the brain of these rats, demonstrating elevated endothelin-converting-enzyme and extracellular ET-1 levels at basal conditions (46).
The ET-system is known to be abundantly present in the central nervous system (47), where it regulates several processes, including brain stem function (48); thus, enhanced central sympathetic activation may have contributed to arrhythmogenesis in our ETB-deficient group. This hypothesis is supported by higher HR and LF/HF ratio, observed in untreated ETB-deficient animals during phase I, as these indices reflect the autonomic balance on the sinus node, irrespective of myocardial catecholamine concentrations. Further studies are needed on this intriguing topic, focusing on the complex interaction between ET-1 and central sympathetic activation.
LV Dysfunction and ETB-Receptors
We observed a trend toward lower activity-counts (used as a surrogate marker of acute LV failure) in untreated ETB-deficient rats, when compared to untreated wild-type rats. The higher incidence of heart failure, implied by our findings, is in agreement with experiments in Langendorff-perfused rat-hearts in the setting of global (10) or regional (49) ischemia, demonstrating prominent LV dysfunction, caused by genetic deficiency or pharmacological blockade of myocardial ETB-receptors. The (likely) increased incidence of acute LV failure after clonidine in our experiments (offsetting the reduction in arrhythmogenesis) adds important information on this issue; although evident in both rat-groups, this effect was more pronounced in ETB-deficient rats, as suggested by a shift toward bradyarrhythmic mortality, by markedly decreased activity-counts, and by higher HR during phase II.
The pathophysiologic link between ETB-receptors and LV dysfunction (in the setting of myocardial ischemia) is poorly understood, although increased metabolic demand caused by excessive sympathetic activation has been proposed (10). Nonetheless, the (likely) higher incidence of heart failure after central sympatholytic action (in our in vivo experiments) point toward additional mechanisms that may be operative. Pharmacological interaction between clonidine and cardiovascular ETB-receptors is also possible, based on previous reports in rats (38), demonstrating augmented sympatholytic effects of clonidine after ETB-receptor-blockade (and the reverse after ETB-receptor-stimulation). This topic requires thorough investigation in future work, using more specific assessment of LV function and heart failure, including methods of highlighting potential underlying mechanisms.
Strengths and Limitations
The topic examined in the present work is of high clinical significance, fueled by accumulated evidence linking central sympathetic activation with acute-MI and SCD (50). We investigated the modulating effects of ET-1 on this process, driven by experimental data (7–10) and by clinical findings, demonstrating a twofold increase in plasma ET-1 levels in patients with acute coronary syndromes triggered by emotional stress (51). Focusing on the role of ETB-receptors, we used a “subtraction” rat-model that circumvents certain caveats associated with pharmacological ET-receptor-blockade (8, 45). Lastly, our data, derived from conscious animals, were not confounded by the effects of anesthesia on sympathetic activation or arrhythmogenesis. Despite these merits, four limitations should be acknowledged: first, implantable transmitters, utilized in our experiments, enable thorough evaluation in conscious animals, but HRV-indices reflect the net autonomic balance on the sinus node and cannot assess sympathetic and vagal discharges separately. Second, we assessed the incidence of heart failure based on bradyarrhythmic death rates, HR, and activity-counts. However, (particularly the latter two of) these end-points are non-specific and should be viewed only as suggestive of acute heart failure, as they may have been confounded by known actions of clonidine, despite the fact that these were likely exerted equally to both animal-groups. Specifically, increased HR may have ensued from reflex tachycardia secondary to hypotension, and low activity counts may be attributed to the sedative effects of clonidine (52). Thus, more solid evidence of depressed LV function should be sought in future studies. Third, we did not explore the electrophysiological mechanisms underlying VT/VF, triggered or sustained by central autonomic inputs. Lastly, as the pretreatment-protocol was tailored for pathophysiological investigation, implications on treatment cannot be immediately drawn.
Conclusion
Myocardial ETB-receptors modulate autonomic responses during acute-MI. Their genetic deficiency potentiates the arrhythmogenic effects of central sympathetic activation, leading to increased arrhythmic-mortality. Clonidine induces potent sympatholytic effects and lowers the incidence of VT/VF, but possibly aggravates heart failure, particularly in the absence of ETB-receptors. Further research is needed on the central and myocardial interaction between ET-1 and sympathetic activation in the setting of ischemia/infarction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Maria-Eleni Baibaki, Eleftheria Karampela, Nikolaos Psychalakis, Kalliopi Tsarea, Maria Karamperi, and Antonios Karaiskos, for their valuable help during the experiments. The meticulous animal-care by Stergios Gerakis and Evripidis Gerakis is gratefully acknowledged. Eleni Goga, M.Sc., efficiently coordinated this project. We are indebted to Experimental Research Center ELPEN for providing research scholarships to MK and EB. Funding: This study was funded, in part, by a grant from the Hellenic Society of Cardiology (2013-HCS-grants).
References
1. Kolettis TM. Coronary artery disease and ventricular tachyarrhythmia: pathophysiology and treatment. Curr Opin Pharmacol (2013) 13(2):210–7. doi: 10.1016/j.coph.2013.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Schömig A, Haass M, Richardt G. Catecholamine release and arrhythmias in acute myocardial ischaemia. Eur Heart J (1991) 12(Suppl F):38–47. doi:10.1093/eurheartj/12.suppl_F.38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Schömig A. Adrenergic mechanisms in myocardial infarction: cardiac and systemic catecholamine release. J Cardiovasc Pharmacol (1988) 12(Suppl 1):S1–7. doi:10.1097/00005344-198806121-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Curtis MJ. Characterisation, utilisation and clinical relevance of isolated perfused heart models of ischaemia-induced ventricular fibrillation. Cardiovasc Res (1998) 39(1):194–215. doi:10.1016/S0008-6363(98)00083-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Tawa M, Yamamoto S, Ohkita M, Matsumura Y. Endothelin-1 and norepinephrine overflow from cardiac sympathetic nerve endings in myocardial ischemia. Cardiol Res Pract (2012) 2012:789071. doi:10.1155/2012/789071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Kolettis TM, Barton M, Langleben D, Matsumura Y. Endothelin in coronary artery disease and myocardial infarction. Cardiol Rev (2013) 21(5):249–56. doi:10.1097/CRD.0b013e318283f65a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Isaka M, Kudo A, Imamura M, Kawakami H, Yasuda K. Endothelin receptors, localized in sympathetic nerve terminals of the heart, modulate norepinephrine release and reperfusion arrhythmias. Basic Res Cardiol (2007) 102(2):154–62. doi:10.1007/s00395-006-0623-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Backs J, Bresch E, Lutz M, Kristen AV, Haass M. Endothelin-1 inhibits the neuronal norepinephrine transporter in hearts of male rats. Cardiovasc Res (2005) 67(2):283–90. doi:10.1016/j.cardiores.2005.03.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Tawa M, Fukumoto T, Ohkita M, Matsumura Y. Role of endogenous endothelin-1 in post-ischemic cardiac dysfunction and norepinephrine overflow in rat hearts. Eur J Pharmacol (2008) 591(1–3):182–8. doi:10.1016/j.ejphar.2008.06.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Yamamoto S, Matsumoto N, Kanazawa M, Fujita M, Takaoka M, Gariepy CE, et al. Different contributions of endothelin-A and endothelin-B receptors in postischemic cardiac dysfunction and norepinephrine overflow in rat hearts. Circulation (2005) 111(3):302–9. doi:10.1161/01.CIR.0000153351.86708.F7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Kolettis TM. Ventricular tachyarrhythmias during acute myocardial infarction: the role of endothelin-1. Life Sci (2014) 118(2):136–40. doi:10.1016/j.lfs.2014.01.060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Malliani A, Montano N. Emerging excitatory role of cardiovascular sympathetic afferents in pathophysiological conditions. Hypertension (2002) 39(1):63–8. doi:10.1161/hy0102.099200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Heusch G, Schipke J, Thamer V. Clonidine prevents the sympathetic initiation and aggravation of poststenotic myocardial ischemia. J Cardiovasc Pharmacol (1985) 7(6):1176–82. doi:10.1097/00005344-198511000-00026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Zhou S, Jung BC, Tan AY, Trang VQ, Gholmieh G, Han SW, et al. Spontaneous stellate ganglion nerve activity and ventricular arrhythmia in a canine model of sudden death. Heart Rhythm (2008) 5(1):131–9. doi:10.1016/j.hrthm.2007.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Ajijola OA, Wisco JJ, Lambert HW, Mahajan A, Stark E, Fishbein MC, et al. Extracardiac neural remodeling in humans with cardiomyopathy. Circ Arrhythm Electrophysiol (2012) 5(5):1010–116. doi:10.1161/CIRCEP.112.972836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Gariepy CE, Ohuchi T, Williams SC, Richardson JA, Yanagisawa M. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. J Clin Invest (2000) 105:925–33. doi:10.1172/JCI8609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Riechers CC, Knabe W, Siren AL, Gariepy CE, Yanagisawa M, Ehrenreich H. Endothelin B receptor deficient transgenic rescue rats: a rescue phenomenon in the brain. Neuroscience (2004) 124(4):719–23. doi:10.1016/j.neuroscience.2003.10.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Opitz CF, Mitchell GF, Pfeffer MA, Pfeffer JM. Arrhythmias and death after coronary artery occlusion in the rat. Continuous telemetric ECG monitoring in conscious, untethered rats. Circulation (1995) 92(2):253–61. doi:10.1161/01.CIR.92.2.253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Agelaki MG, Pantos C, Korantzopoulos P, Tsalikakis DG, Baltogiannis GG, Fotopoulos A, et al. Comparative antiarrhythmic efficacy of amiodarone and dronedarone during acute myocardial infarction in rats. Eur J Pharmacol (2007) 564(1–3):150–7. doi:10.1016/j.ejphar.2007.02.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Kolettis TM, Agelaki MG, Baltogiannis GG, Vlahos AP, Mourouzis I, Fotopoulos A, et al. Comparative effects of acute vs. chronic oral amiodarone treatment during acute myocardial infarction in rats. Europace (2007) 9(11):1099–104. doi:10.1093/europace/eum196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Guyenet PG, Cabot JB. Inhibition of sympathetic preganglionic neurons by catecholamines and clonidine: mediation by an alpha-adrenergic receptor. J Neurosci (1981) 1(8):908–17.
22. Yelken B, Dorman T, Erkasap S, Dundar E, Tanriverdi B. Clonidine pretreatment inhibits stress-induced gastric ulcer in rats. Anesth Analg (1999) 89(1):159–62. doi:10.1213/00000539-199907000-00028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Elaiopoulos DA, Tsalikakis DG, Agelaki MG, Baltogiannis GG, Mitsi AC, Fotiadis DI, et al. Growth hormone decreases phase II ventricular tachyarrhythmias during acute myocardial infarction in rats. Clin Sci (Lond) (2007) 112(7):385–91. doi:10.1042/CS20060193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Curtis MJ, Hancox JC, Farkas A, Wainwright CL, Stables CL, Saint DA, et al. The Lambeth conventions (II): guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacol Ther (2013) 139(2):213–48. doi:10.1016/j.pharmthera.2013.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Di Diego JM, Antzelevitch C. Ischemic ventricular arrhythmias: experimental models and their clinical relevance. Heart Rhythm (2011) 8(12):1963–8. doi:10.1016/j.hrthm.2011.06.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Clements-Jewery H, Hearse DJ, Curtis MJ. Phase 2 ventricular arrhythmias in acute myocardial infarction: a neglected target for therapeutic antiarrhythmic drug development and for safety pharmacology evaluation. Br J Pharmacol (2005) 145(5):551–64. doi:10.1038/sj.bjp.0706231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Kolettis TM, Kontonika M, Valenti MC, Vilaeti AD, Baltogiannis GG, Papalois A, et al. Arrhythmogenesis after acute myocardial necrosis with and without preceding ischemia in rats. J Basic Clin Physiol Pharmacol (2014) 25(2):143–53. doi:10.1515/jbcpp-2013-0117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Oikonomidis DL, Tsalikakis DG, Baltogiannis GG, Tzallas AT, Xourgia X, Agelaki MG, et al. Endothelin-B receptors and ventricular arrhythmogenesis in the rat model of acute myocardial infarction. Basic Res Cardiol (2010) 105(2):235–45. doi:10.1007/s00395-009-0066-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kruger C, Kalenka A, Haunstetter A, Schweizer M, Maier C, Ruhle U, et al. Baroreflex sensitivity and heart rate variability in conscious rats with myocardial infarction. Am J Physiol (1997) 273:H2240–7.
30. Niskanen JP, Tarvainen MP, Ranta-Aho PO, Karjalainen PA. Software for advanced HRV analysis. Comput Methods Programs Biomed (2004) 76(1):73–81. doi:10.1016/j.cmpb.2004.03.004
31. Howarth FC, Jacobson M, Shafiullah M, Adeghate E. Effects of insulin treatment on heart rhythm, body temperature and physical activity in streptozotocin-induced diabetic rat. Clin Exp Pharmacol Physiol (2006) 33(4):327–31. doi:10.1111/j.1440-1681.2006.04370.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Fukushima S, Coppen SR, Lee J, Yamahara K, Felkin LE, Terracciano CM, et al. Choice of cell-delivery route for skeletal myoblast transplantation for treating post-infarction chronic heart failure in rat. PLoS One (2008) 3(8):e3071. doi:10.1371/journal.pone.0003071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Issa ZF, Ujhelyi MR, Hildebrand KR, Zhou X, Rosenberger J, Groh WJ, et al. Intrathecal clonidine reduces the incidence of ischemia-provoked ventricular arrhythmias in a canine postinfarction heart failure model. Heart Rhythm (2005) 2(10):1122–7. doi:10.1016/j.hrthm.2005.06.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Opthof T, Coronel R, Vermeulen JT, Verberne HJ, van Capelle FJ, Janse MJ. Dispersion of refractoriness in normal and ischaemic canine ventricle: effects of sympathetic stimulation. Cardiovasc Res (1993) 27(11):1954–60. doi:10.1093/cvr/27.11.1954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Opthof T, Misier AR, Coronel R, Vermeulen JT, Verberne HJ, Frank RG, et al. Dispersion of refractoriness in canine ventricular myocardium. Effects of sympathetic stimulation. Circ Res (1991) 68(5):1204–15.
36. Jardine DL, Charles CJ, Forrester MD, Whitehead M, Nicholls MG. A neural mechanism for sudden death after myocardial infarction. Clin Auton Res (2003) 13(5):339–41. doi:10.1007/s10286-003-0109-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Jardine DL, Charles CJ, Ashton RK, Bennett SI, Whitehead M, Frampton CM, et al. Increased cardiac sympathetic nerve activity following acute myocardial infarction in a sheep model. J Physiol (2005) 565(Pt 1):325–33. doi:10.1113/jphysiol.2004.082198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Lavhale MS, Briyal S, Parikh N, Gulati A. Endothelin modulates the cardiovascular effects of clonidine in the rat. Pharmacol Res (2010) 62(6):489–99. doi:10.1016/j.phrs.2010.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Evans SJ, Dalton GR, Levi AJ. Experimental studies on myocardial stretch and ventricular arrhythmia in hypertrophied and non-hypertrophied hearts. J Cardiovasc Risk (2000) 7(3):163–75.
40. Clements-Jewery H, Hearse DJ, Curtis MJ. Independent contribution of catecholamines to arrhythmogenesis during evolving infarction in the isolated rat heart. Br J Pharmacol (2002) 135(3):807–15. doi:10.1038/sj.bjp.0704509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Kolettis TM, Oikonomidis DL, Baibaki ME, Barka E, Kontonika M, Tsalikakis DG, et al. Endothelin B-receptors and sympathetic activation: impact on ventricular arrhythmogenesis during acute myocardial infarction. Life Sci (2014) 18(2):281–7. doi:10.1016/j.lfs.2014.01.069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Warner MR, Kroeker TS, Zipes DP. Sympathetic stimulation and norepinephrine infusion modulate extracellular potassium concentration during acute myocardial ischemia. Circ Res (1992) 71(5):1078–87. doi:10.1161/01.RES.71.5.1078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Kolettis TM, Baltogiannis GG, Tsalikakis DG, Tzallas AT, Agelaki MG, Fotopoulos A, et al. Effects of dual endothelin receptor blockade on sympathetic activation and arrhythmogenesis during acute myocardial infarction in rats. Eur J Pharmacol (2008) 580(1–2):241–9. doi:10.1016/j.ejphar.2007.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Baltogiannis GG, Tsalikakis DG, Mitsi AC, Hatzistergos KE, Elaiopoulos D, Fotiadis DI, et al. Endothelin receptor-A blockade decreases ventricular arrhythmias after myocardial infarction in rats. Cardiovasc Res (2005) 67(4):647–54. doi:10.1016/j.cardiores.2005.04.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Lehmann LH, Stanmore DA, Backs J. The role of endothelin-1 in the sympathetic nervous system in the heart. Life Sci (2014) 118(2):165–72. doi:10.1016/j.lfs.2014.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Ehrenreich H, Oldenburg J, Hasselblatt M, Herms J, Dembowski C, Loffler BM, et al. Endothelin B receptor-deficient rats as a subtraction model to study the cerebral endothelin system. Neuroscience (1999) 91(3):1067–75. doi:10.1016/S0306-4522(98)00663-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Jones CR, Hiley CR, Pelton JT, Mohr M. Autoradiographic visualization of the binding sites for [125I]endothelin in rat and human brain. Neurosci Lett (1989) 97(3):276–9. doi:10.1016/0304-3940(89)90610-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Dashwood MR, Loesch A. Endothelin-1 as a neuropeptide: neurotransmitter or neurovascular effects? J Cell Commun Signal (2010) 4(1):51–62. doi:10.1007/s12079-009-0073-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Bibli SI, Toli EV, Vilaeti AD, Varnavas VC, Baltogiannis GG, Papalois A, et al. Endothelin-B receptors and left ventricular dysfunction after regional versus global ischaemia-reperfusion in rat hearts. Cardiol Res Pract (2012) 2012:986813. doi:10.1155/2012/986813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Lampert R. Emotion and sudden cardiac death. Expert Rev Cardiovasc Ther (2009) 7(7):723–5. doi:10.1586/erc.09.75
51. Wilbert-Lampen U, Nickel T, Leistner D, Guthlin D, Matis T, Volker C, et al. Modified serum profiles of inflammatory and vasoconstrictive factors in patients with emotional stress-induced acute coronary syndrome during World Cup Soccer 2006. J Am Coll Cardiol (2010) 55(7):637–42. doi:10.1016/j.jacc.2009.07.073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: acute myocardial infarction, sympathetic activation, ventricular arrhythmias, endothelin receptors
Citation: Kolettis TM, Kontonika M, Barka E, Daskalopoulos EP, Baltogiannis GG, Tourmousoglou C, Papalois A and Kyriakides ZS (2015) Central sympathetic activation and arrhythmogenesis during acute myocardial infarction: modulating effects of endothelin-B receptors. Front. Cardiovasc. Med. 2:6. doi: 10.3389/fcvm.2015.00006
Received: 14 January 2015; Paper pending published: 02 February 2015;
Accepted: 10 February 2015; Published online: 23 February 2015.
Edited by:
Mario Daniel Gonzalez, Penn State University, USAReviewed by:
Javier Eduardo Banchs, Baylor Scott & White Health, USATalal Moukabary, Carondelet Heart and Vascular Institute Physicians, USA
Copyright: © 2015 Kolettis, Kontonika, Barka, Daskalopoulos, Baltogiannis, Tourmousoglou, Papalois and Kyriakides. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theofilos M. Kolettis, Department of Cardiology, University of Ioannina, 1 Stavrou Niarxou Avenue, Ioannina 45500, Greece e-mail: theofilos.m.kolettis@gmail.com