 Alice Liguori Antonella Puglianiello Daniela Germani Annalisa Deodati Emanuela Peschiaroli Stefano Cianfarani*
Alice Liguori Antonella Puglianiello Daniela Germani Annalisa Deodati Emanuela Peschiaroli Stefano Cianfarani*
- Molecular Endocrinology Unit, D.P.U.O. Bambino Gesù Children’s Hospital, ‘Rina Balducci’ Center of Pediatric Endocrinology, Tor Vergata University, Rome, Italy
Epidemiologic studies have demonstrated an association between intrauterine growth retardation and a greater risk of chronic disease, including coronary heart disease, hypertension, stroke, and type 2 diabetes in adulthood. An adverse intrauterine environment may affect both growth and development of the organism, permanently programming endocrine and metabolic functions. One of the mechanisms of programming is the epigenetic modification of gene promoters involved in the control of key metabolic pathways. The aim of this review is to provide an overview of the experimental evidence showing the effects of early exposure to suboptimal environment on epigenome. The knowledge of the epigenetic markers of programming may allow the identification of susceptible individuals and the design of targeted prevention strategies.
Intrauterine Programming: Causes and Consequences
Early life events may play a critical role in determining the susceptibility to chronic diseases (Gluckman et al., 2005). Epidemiological evidence suggests a close relationship between the exposure to a suboptimal in utero environment, whose consequence is intrauterine growth retardation (IUGR), and the development of insulin resistance, type 2 diabetes, hypertension, hyperlipidemia, and cardiovascular disease in adult life (Barker et al., 1989; Robinson et al., 1992; Ravelli et al., 1998).
Lucas (1991) introduced the concept of programming, based on the observation that early diet influenced brain development and growth in preterm babies. This concept was subsequently expanded to include the long-term effects of in utero programming induced by maternal cues on metabolic and endocrine functions of the fetus. The programming process occurs during “critical periods” of embryo–fetal life characterized by high cell proliferation rate in the developing tissues and may involve structural and functional changes in genes, cells, tissues, and even whole organs. This ability of the organism to change structure and function in response to environmental signals is named “developmental plasticity” (Gluckman and Hanson, 2004). Such plasticity permits a range of phenotypes to develop from a single genotype and is finalized to allow the organism to match its environment (Gluckman et al., 2009).
“Developmental plasticity” acts through epigenetic changes in gene transcription, alterations in tissue differentiation, and changes in homeostatic processes (Gluckman and Hanson, 2004). Epigenetic changes are established in early life and modulate gene expression during development, thus mediating the adaptation of the organism to the environment (Gluckman and Hanson, 2004; Gluckman et al., 2008). However, when environmental conditions change a mismatch may occur, rendering the organism less adapt to cope to the new environmental conditions (such as postnatal overfeeding), eventually leading to disease (Gluckman et al., 2009).
Epigenetic mechanisms are commonly associated to gene silencing, genomic imprinting and transcriptional regulation of tissue-specific genes during cellular differentiation (Schübeler et al., 2000). The epigenetic control of gene expression is based on modulation of chromatin structure and accessibility to transcription factors (Figure 1). This type of control is achieved by multiple mechanisms such as methylation–demethylation of cytidine–guanosine (CpG) sequences in the promoter regions, acetylation–deacetylation of lysine residues of core histones in the nucleosome and presence of microRNA molecules which bind to complementary sequences in the 3′ end of mRNA and reduce the rate of protein synthesis (Goldberg et al., 2007). CpG-rich regions of DNA reside in 60% of promoters utilized by human RNA polymerase II and are often found in association with housekeeping and tissue-specific genes. Transcriptionally active chromatin is characterized by unmethylation of CpG sequences, which permits an open structure of the chromatin, thus allowing access to transcription factors. The same effect is given by acetylation of lysine residues of histones, which decreases their binding to DNA (Wolffe and Matzke, 1999).
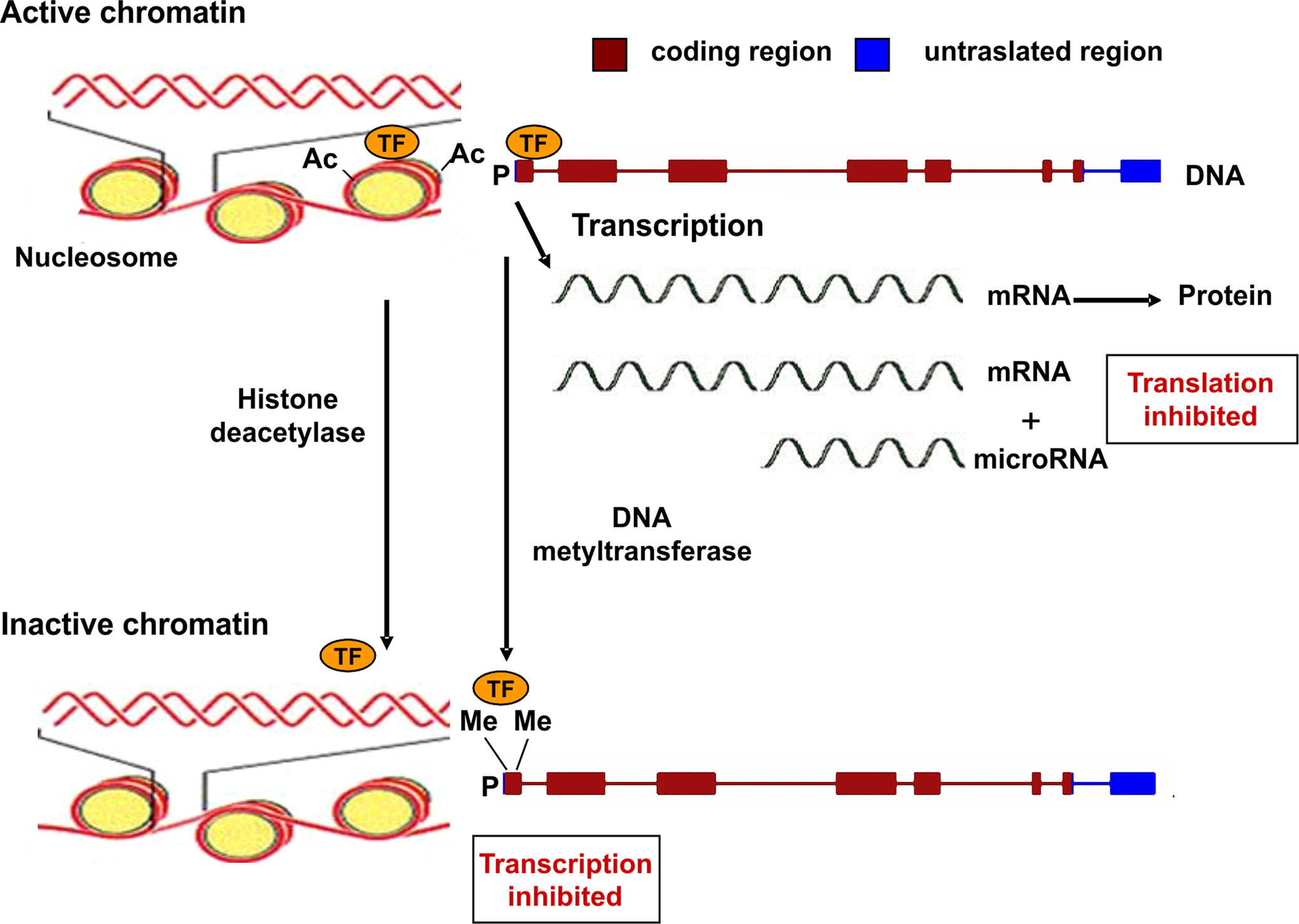
Figure 1. Epigenetic mechanisms regulating gene expression. Transcriptionally active chromatin is characterized by the presence of acetyl groups (Ac) on specific lysine residues of histones in the nucleosome, which decreases their binding to DNA, eventually leading to an open chromatin structure that permits access to transcription factors (TF). In addition, demethylation of cytidine–guanosine (CpG) sequences in the promoter region (P) of actively transcribed genes allows for the binding of transcription factors (TF). Transcriptionally inactive chromatin is characterized by histone deacetylation, promoter CpG methylation (as indicated by the presence of methyl groups, Me), and decreased binding of transcription factors. A further level of epigenetic control is provided by microRNA molecules (19–22 nucleotides in length) which bind to mRNA thus reducing the rate of translation.
Major epigenetic “programming,” involving the removal of epigenetic marks in the nucleus, followed by establishment of a different set of marks occurs physiologically upon fertilization when many gametic marks are erased and replaced with embryonic marks important for early embryonic development and toti- or pluripotency. Major programming also takes place in primordial germ cells in which parental imprints are erased and totipotency is restored (Morgan et al., 2005).
One of the mechanisms that trigger programming and, hence, epigenetic changes, is intrauterine malnutrition (Cianfarani et al., 1999), which can be induced by several causes affecting the placental transfer of nutrients from mother to fetus (Fowden et al., 2006). To explain the relationship between prenatal undernourishment and postnatal risk of metabolic disease, Hales and Barker proposed the “thrifty phenotype” hypothesis (Hales and Barker, 1992). According to the “thrifty phenotype” model, the growing fetus exposed to nutritional deprivation adopts at least two strategies to aid survival. First, it diverts nutrients to the brain to preserve brain growth at the expense of body growth and the development of other organs such as pancreas, liver, and muscle. Second, metabolic programming occurs in a manner that is beneficial to survival under conditions of poor postnatal nutrition. However, if the organism is born into conditions of adequate or overnutrition, then this may conflict with the earlier programming and insulin resistance, and, later on, type 2 diabetes, may result (Geremia and Cianfarani, 2004).
Experimental Evidence of Epigenetic Programming
Several animal models have been created to reproduce a poor uterine environment leading to fetal undernourishment and, consequently, developmental programming. Most animal models of IUGR are based on uteroplacental insufficiency (Simmons et al., 2001), which limits the supply of substrates to the fetus, or suboptimal maternal nutrition (Armitage et al., 2004).
Altered intrauterine milieu associated with uteroplacental (placental) insufficiency affects DNA methylation and histone H3 acetylation. In liver, increased levels of S-adenosylhomocysteine together with DNA hypomethylation and histone hyperacetylation of histone H3 on lysine 9 (H3K9), lysine 14 (H3K14), and lysine 18 (H3K18) are present at birth (MacLennan et al., 2004). These features persist up to day 21 of postnatal life, suggesting a permanent change in hepatic gene expression. Fu et al. (2004) showed that the hyperacetylation on histone H3 in the liver of IUGR rats occurs in association with decreased nuclear protein levels of histone deacetylase 1 (HDAC1) and HDAC activity. These site-specific changes in histone H3 acetylation alter the histone association with the promoter regions of PPAR-gamma coactivator (PGC-1) and carnitine–palmitoyl-transferase I (CPTI), two genes that we have previously demonstrated to be persistently altered in the IUGR rat. PGC-1 expression is increased whereas CPTI expression is reduced in IUGR rats who will develop diabetes (Lane et al., 2001, 2002). PGC-1 is a transcriptional coactivator that mediates hepatic glucose production by controlling mRNA levels of key gluconeogenic enzymes, such as glucose-6-phosphatase, phosphoenolpyruvate carboxykinase, and fructose-1,6-bisphosphatase (Yoon et al., 2001). CPTI is a part of the carnitine shuttle and is considered to be a rate-limiting transporter in mitochondrial fatty acid β-oxidation (McGarry and Brown, 1997). Altered mRNA levels of these genes characterize the IUGR liver at birth, and these changes persist postnatally. Finally, these epigenetic modifications may be gender specific as at day 21, the neonatal pattern of H3 hyperacetylation persist only in male IUGR rats (Fu et al., 2004). The major limitations of these studies are the lack of information on the effects of the epigenetic changes on gene expression and the short postnatal follow-up of the study animals.
A key developmental gene whose epigenetic modulation has been studied to explain the intrauterine metabolic programming predisposing to type 2 diabetes is Pdx1 (pancreatic and duodenal homeobox 1). Pdx1 is a transcription factor that regulates pancreas development and β-cell differentiation. Reductions in Pdx1 expression in human and animal models have been shown to cause type 2 diabetes, β-cell dysfunction, and impaired islet compensation in the presence of insulin resistance (Holland et al., 2005; Stoffers et al., 1997). Uteroplacental insufficiency causes multiple epigenetic changes of Pdx1 involving histone modifications, DNA methylation, and chromatin remodeling in rats (Park et al., 2008). In this animal model, β-cell mass is normal at birth, whilst Pdx1 expression is reduced. In adult animals, β-cell mass is markedly decreased and Pdx1 expression is nearly absent. The epigenetic mechanisms underlying these events are characterized by the progressive histone H3 and H4 deacetylation, lysine 4 on histone H3 (H3K4) demethylation, and lysine 9 on histone H3 (H3K9) methylation in Pdx1 proximal promoter. All these changes lead to a silenced chromatin, with decreased USF-1 (a key transcription factor) binding and increased recruitment of histone deacetylase 1 (HDAC1) and its corepressor Sin3A. During the neonatal period, these epigenetic changes and the reduction in Pdx1 expression could be reversed by HDAC1 inhibition (islets cultured in the presence of trichostatin). However, as H3K9me2 accumulates, DNMT3A (a DNA methyltransferase) is recruited to the promoter and initiates de novo DNA methylation, which locks in the silenced state the pancreas, eventually resulting in diabetes (Park et al., 2008).
Exendin-4 (Ex-4), a pancreatic β-cell trophic factor, has been shown to reverse the silencing of Pdx-1. Administration of Ex-4 during the prediabetic neonatal period dramatically prevents the development of diabetes in IUGR rats by restoring expression of Pdx1 to normal levels and normalizing islet β-cell proliferation rate (Stoffers et al., 2003). However, it is not yet known whether Ex-4 may influence Pdx-1 epigenetics.
Raychaudhuri et al. (2008) explored epigenetic mechanisms underlying diminished skeletal muscle GLUT4 mRNA in a rodent model of IUGR obtained by nutrient restriction. GLUT4 is the major insulin-responsive isoform in the family of membrane-spanning glycoproteins with the function of glucose transporters. At 450 days of life the female offspring demonstrated a significant decrease in skeletal muscle total GLUT4 mRNA concentrations. This gender-specific difference may originate from early developmental perturbations in pancreatic β-islet cell insulin synthesis and secretion (Chamson-Reig et al., 2006), young females demonstrating persistent postnatal hypoinsulinemia that regulates skeletal muscle GLUT4 transcription. No significant increase in the methylation of CpG regions within GLUT4 promoter was observed in skeletal muscle of IUGR rats. On the contrary, hypomethylation of most CpG islands was found. In this IUGR model deacetylation and di-methylation of specific amino acid residues in the N-tail of histone 3 were identified. Taken together these findings indicate that epigenetic changes of histone code may inhibit skeletal muscle GLUT4 transcription in adult female IUGR rats.
Thompson et al. (2010) have recently studied DNA methylation of the whole genome in pancreatic islets of IUGR rats at 7 weeks of age. Using the HpaII tiny fragment enrichment by ligation-mediated PCR (HELP) assay, they generated a DNA methylation map at almost 1 million unique sites throughout the rat genome in normal pancreatic islet cells. Male IUGR animals showed a different cytosine methylation pattern in approximately 1400 loci at 7 weeks of age. Epigenetic dysregulation occurred preferentially in conserved intergenic sequences, frequently near genes regulating processes involved in vascularization, β-cell proliferation, insulin secretion, and cell death. Intergenic sequences may represent important regulatory sites influencing local gene expression. This epigenomic dysregulation precedes the development of diabetes and probably represents a link between intrauterine growth restriction and development of type 2 diabetes in adulthood. Candidate dysregulated loci were investigated with quantitative assay of cytosine methylation and gene expression was tested by RT-PCR. GTP cyclohydrolase 1 (CGH-1) gene (a gene with a role in the endothelial dysfunction and in β-cell development) showed a three-fold reduction in mRNA expression, associated with hypermethylation at a conserved intergenic site. Hypomethylation of fibroblast growth factor receptor 1 (FGFR1) gene (whose signaling is modulated by β-cell microenvironment) was associated with increased mRNA expression. Proprotein convertase subtilisin/ketin type 5 (PCSK5) gene (a gene that impairs β-cell activity and regulates β-cell adhesion to extracellular matrix) showed significantly reduced mRNA expression associated with hypermethylation. These findings strongly suggest that early epigenetic modifications in pancreatic islets may mediate the long-term metabolic consequences of exposure to suboptimal intrauterine environment (Thompson et al., 2010).
The epigenetic changes affecting glucose metabolism are reported in Table 1.
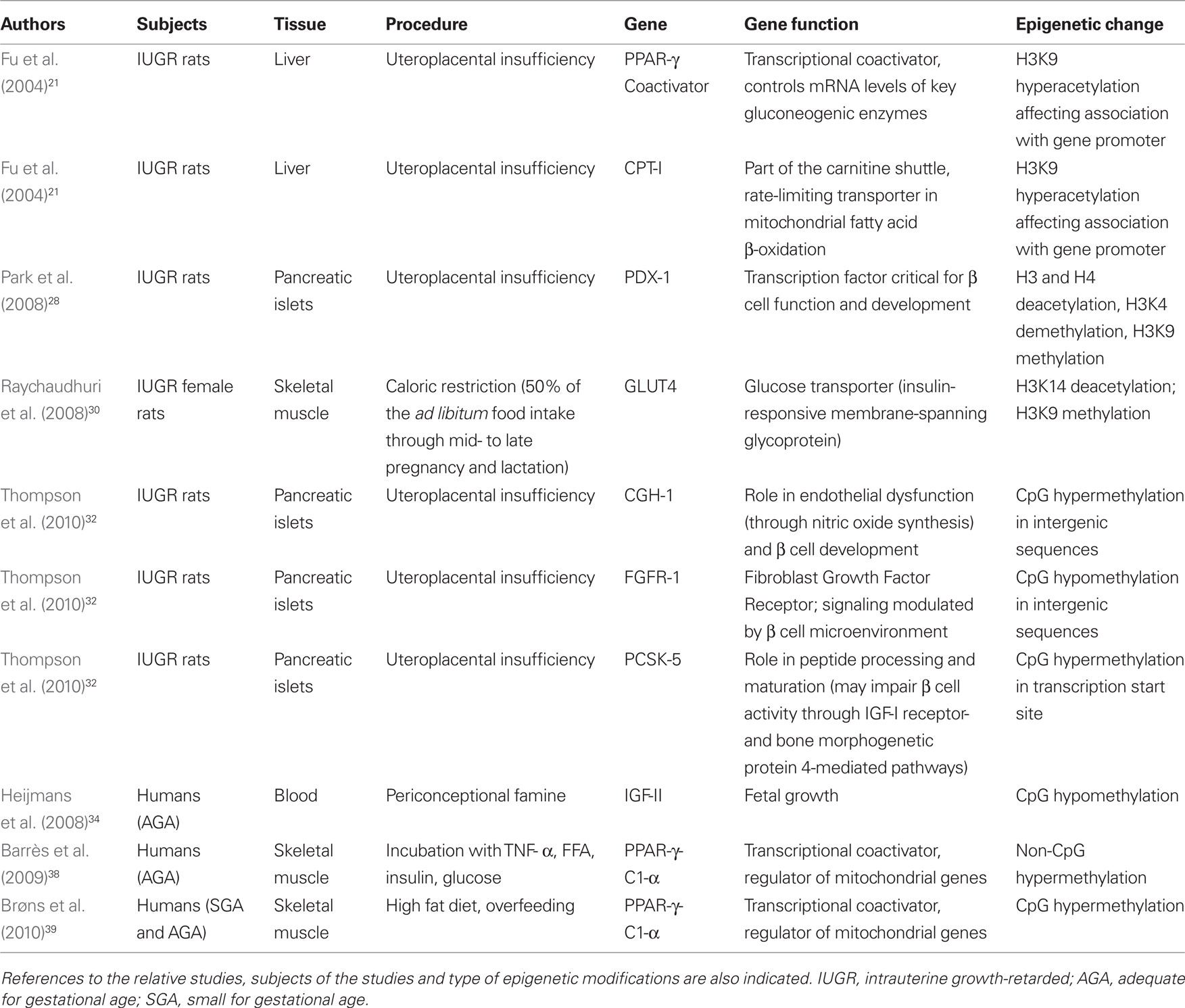
Table 1. The different genes whose function is linked to the development of type 2 diabetes (and its complications).
Clinical Evidence of Epigenetic Programming
In humans, evidence that epigenetic changes predispose the organism to type 2 diabetes stems from the studies on the individuals who were prenatally exposed to famine during the Dutch Hunger Winter in 1944–45. This period of famine was the consequence of a German-imposed food embargo in the western part of The Netherlands toward the end of World War II during the winter of 1944–45. As official food rations were documented, the reported average daily rations were 667 kcal and there was little variation in the percentage of calories from proteins (≈12%, of which 4% of animal origin), fat (19%), and carbohydrates (69%). It was demonstrated that individuals exposed to famine prenatally were at higher risk of developing cardiovascular and metabolic diseases in adulthood (Lumey et al., 2007).
Heijmans et al. (2008) selected 60 individuals whose mothers were exposed to famine during the periconceptional period and 62 individuals exposed late in gestation for at least 10 weeks. They studied the epigenetic status of the IGF-II gene. IGF-II gene is maternally imprinted and strongly epigenetically regulated (hypomethylation leads to bi-allelic expression). Among the 60 individuals exposed to famine periconceptionally, all cytosine–guanine (CpG) sites of the IGF-II gene but one were significantly less methylated (in comparison with the same-sex siblings). Among the 62 individuals exposed to famine late in gestation, no difference in IGF-II methylation was found between the exposed individuals and their unexposed siblings. They concluded that periconceptional exposure to famine is associated with lower methylation of the IGF-II gene 6 decades later (Heijmans et al., 2008). The reduced methylation of IGF-II may represent the consequence of intrauterine exposure to deficient methyl donors supply, such as the aminoacid methionine, although additional contribution of other stressors such as cold and emotional stress cannot be ruled out. Consistent with the potential role of methyl donors in determining IGF-II gene methylation status is the recent observation that periconceptional folic acid use of the mother is related to an increased methylation of the IGF-II gene of the offspring (Steegers-Theunissen et al., 2009). However, we point out that remains to be determined whether the changes in IGF-II gene methylation are associated with changes in gene expression.
The transcriptional coactivator peroxisome proliferator activated receptor γ coactivator-1 α (protein PGC-1α; gene PPAR-γ-C1-α) is an important factor regulating the expression of genes for oxidative phosphorylation and ATP production in target tissues through coactivation of nuclear receptors (Lin et al., 2005). PPAR-γ-C1-α shows an epigenetically regulated decrease of expression in muscle and pancreatic islets from patients with type 2 diabetes. Insulin secretion in pancreatic islets is dependent upon mitochondrial function and this transcriptional coactivator is a master regulator of mitochondrial genes: this protein can interact with and regulate the activities of cAMP response element binding protein and nuclear respiratory factors. It provides a direct link between external physiological stimuli and the regulation of mitochondrial biogenesis. The epigenetic modulation results in increased DNA methylation on PPAR-γ-C1-α gene promoter (Ling et al., 2008).
The highest proportion of cytosine methylation within PPAR-γ-C1-α is found within non-CpG nucleotides. As alterations in the extracellular milieu, including hyperglycemia, hyperinsulinemia, elevated free fatty acids (FFA), and elevated cytokines can cause peripheral insulin resistance, Barrès et al. (2009) incubated human skeletal muscle from normal glucose-tolerant subjects with tumor necrosis factor-α (TNF-α), FFA, insulin and glucose. In these experimental conditions non-CpG methylation was acutely increased in human myotubes by exposure to TNF-α or FFA. Selective silencing of the DNA methyltransferase 3B (DNMT3B) prevented palmitate-induced non-CpG methylation of PPAR-γ-C1-α and decreased mitochondrial DNA and PGC-1α mRNA. These data suggest that DNMT3B is linked to the acute fatty acid-induced non-CpG methylation of PPAR-γ-C1-α promoter.
DNA methylation and gene expression of PPAR-γ-C1-α in human muscle is influenced by high-fat diet in a birth weight dependent manner (Brøns et al., 2010). Brøns et al. studied 26 young, healthy, lean men with low birth weight (LBW, <10th percentile) and 20 subjects with normal birth weight (NBW, 50–90th percentile). All subjects were born at term, and the groups were matched according to age and body mass index (BMI). Subjects were examined twice with a hyperinsulinemic–euglycemic clamp after intake of a 3-days control diet including 30% fat and after a 5-days high-fat diet containing 50% extra calories and 60% fat. When challenged with high-fat diet, LBW subjects developed peripheral insulin resistance and reduced PPAR-γ-C1-α and OXPHOS (the cluster of genes from both the nuclear and mitochondrial genomes controlling the mitochondrial oxidative phosphorylation) gene expression in the muscle. PPAR-γ-C1-α methylation was significantly higher in LBW subjects during the control diet. When exposed to overfeeding, methylation of PPAR-γ-C1-α increased in the NBW group only. LBW subjects showed similar PPAR-γ-C1-α methylation during the two diets. When shifted to the control diet after overfeeding, reversibility of PPAR-γ-C1-α methylation was observed only in NBW. Epigenetic alterations observed in LBW subjects at increased risk of developing type 2 diabetes could reflect DNA methylation remnants established during fetal life, possibly affecting tissue development in organs relevant to type 2 diabetes pathophysiology during phases of active cell divisions. This study shows that the same PPAR-γ-C1-α CpG sites reported to be more highly methylated in pancreatic β cells from type 2 diabetes subjects are methylated to a higher extent in young and lean LBW compared with NBW subjects when studied during an isocaloric control diet (Brøns et al., 2010). The increased methylation of PPAR-γ-C1-α observed after overfeeding in NBW only may reflect a more widespread genome-wide response, probably part of a normal physiological response involved in the day-to-day regulation of mechanisms influenced by the diet.
These epigenetic changes are reported in Table 1.
Transgenerational Transmission of Epigenetic Changes
The majority of known environmentally induced epigenetic effects will involve direct exposures and action on somatic tissues. However, experimental evidence suggests that programming induced by suboptimal in utero environment can be transmitted to the following generations (Jablonka and Raz, 2009). The daughters of women exposed to nutrient restriction and environmental stress during pregnancy as a result of the Dutch Hunger Winter, showed a decreased birth weight and an increased risk of insulin resistance, and their daughters were born with a lower birth weight (Stein and Lumey, 2000; Painter et al., 2005). In rats, feeding a protein-restricted diet to the F0 generation during pregnancy resulted in alterations of glucose and insulin metabolism in male and female F2 offspring (Zambrano et al., 2005). The adverse effects on glucose homeostasis of feeding a protein-restricted diet during pregnancy in the F0 generation have been found in the offspring up to F3 generation (Benyshek et al., 2006). The administration of dexamethasone to dams in late pregnancy induced an increased expression of the glucocorticoid receptor (GR) and its target gene phosphoenolpyruvate carboxykinase (PEPCK) in the liver of the F1 and F2 offspring (Drake et al., 2005).
Hepatic peroxisome proliferated activated receptor α (PPARα) and GR promoter methylation has been reported to be significantly reduced in rats prenatally exposed to protein-restricted diet in both F1 and F2 generation (Burdge et al., 2007).
The mechanisms underlying the intergenerational transfer of epigenetic traits are not clear. Intrinsic epigenetic transgenerational processes will require the involvement of the germline to allow the transmission of an epigenetic abnormality between multiple generations. It is known that postfertilization deletion of epigenetic marks is incomplete for imprinted genes and, probably, some non-imprinted genes (Chong and Whitelaw, 2004).
Conclusions
The process by which early cues at critical stages of development lead to permanent changes in tissue structure and function is known as intrauterine programming and is finalized to allow the organism to match its environment. Programming induces epigenetic changes that may permanently affect the expression of specific genes in selected target tissues. Epigenetic changes permit a range of phenotypes to develop from a single genotype, and may be transmitted across generations (Figure 2). However, when the organism is exposed to a different environment, the epigenetic pattern induced in utero may become detrimental and may predispose the individual to the development of chronic diseases. Therefore, the identification of the cues and the target genes involved in programming would permit early interventions to prevent severe adult illnesses such as type 2 diabetes and cardiovascular disease. Epidemiological and experimental evidence shows the importance of the periconceptional period, when maternal suboptimal nutrition can have long-lasting effects on the offspring, and suggests the potential importance of designing specific nutritional supplementation programs during the pre-pregnancy period. In addition, the identification of the epigenetic markers associated with increased risk of metabolic disease may allow to identify the individuals at metabolic risk and to propose a prevention strategy based on a healthy life-style as well as nutritional and pharmacological interventions.
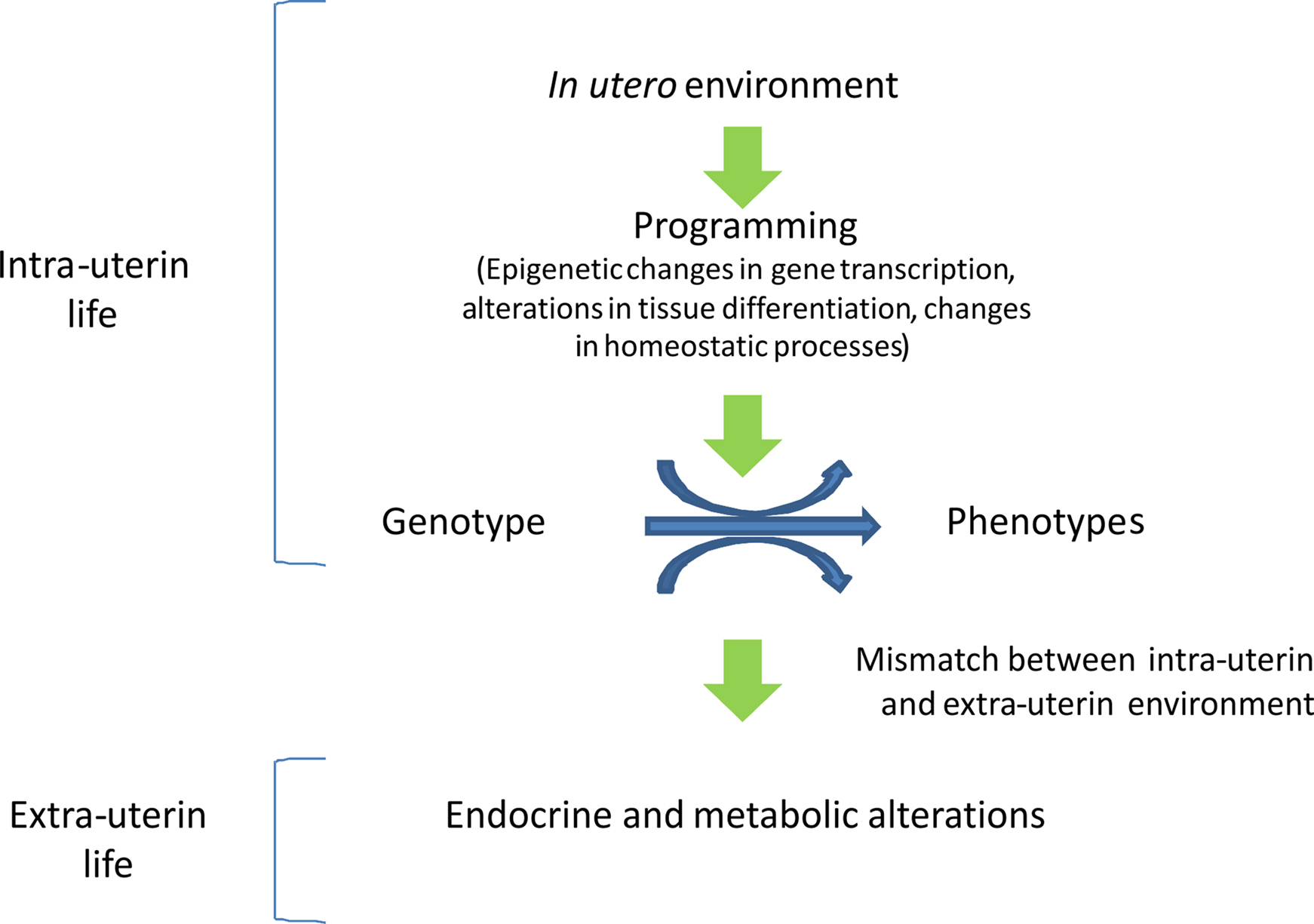
Figure 2. In utero epigenetic programming. Epigenetic programming permits a range of phenotypes to develop from a single genotype and is finalized to allow the organism to match its environment. When environmental conditions change a mismatch may occur, rendering the organism less adapt to cope to the new environment, eventually leading to disease.
Conflict of Interest Statement
A. Liguori, A. Puglianiello, D. Germani, A. Deodati, and E. Peschiaroli have nothing to declare. S. Cianfarani received lecture fees from Ipsen, Eli Lilly, Novo Nordisk, and Pfizer, and consulting fees from Ipsen, Eli Lilly, and Pfizer.
References
Armitage, J. A., Khan, I. Y., Taylor, P. D., Nathanielsz, P. W., and Poston, L. (2004). Developmental programming of the metabolic syndrome by maternal nutritional imbalance: how strong is the evidence from experimental models in mammals? J. Physiol. 561 (Pt 2), 355–377.
Barker, D. J., Winter, P. D., Osmond, C., Margetts, B., and Simmonds, S. J. (1989). Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580.
Barrès, R., Osler, M. E., Yan, J., Rune, A., Fritz, T., Caidahl, K., Krook, A. K., and Zierath, J. R. (2009). Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 10, 189–198.
Benyshek, D. C., Johnston, C. S., and Martin, J. F. (2006). Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. Diabetologia 49, 1117–1119.
Brøns, C., Jacobsen, S., Nilsson, E., Rönn, T., Jensen, C. B., Storgaard, H., Poulsen, P., Groop, L., Ling, C., Astrup, A., and Vaag, A. (2010). Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J. Clin. Endocrinol. Metab. 95, 3048–3056.
Burdge, G. C., Slater-Jefferies, J., Torrens, C., Phillips, E. S., Hanson, M. A., and Lillycrop, K. A. (2007). Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br. J. Nutr. 97, 435–439.
Chamson-Reig, A., Thyssen, S. M., Arany, E., and Hill, D. J. (2006). Altered pancreatic morphology in the offspring of pregnant rats given reduced dietary protein is time and gender specific. J. Endocrinol. 191, 83–92.
Chong, S., and Whitelaw, E. (2004). Epigenetic germline inheritance. Curr. Opin. Genet. Dev. 14, 692–696.
Cianfarani, S., Germani, D., and Branca, F. (1999). Low birthweight and adult insulin resistance: the “catch-up growth” hypothesis. Arch. Dis. Child Fetal Neonatal. Ed. 81, F71–F73.
Drake, A. J., Walker, B. R., and Seckl, J. R. (2005). Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R34–R38.
Fowden, A. L., Giussani, D. A., and Forhead, A. J. (2006). Intrauterine programming of physiological systems: causes and consequences. Physiology (Bethesda) 21, 29–37.
Fu, Q., McKnight, R. A., Yu, X., Wang, L., Callaway, C. W., and Lane, R. H. (2004). Uteroplacental insufficiency induces site-specific changes in histone H3 covalent modifications and affects DNA-histone H3 positioning in day 0 IUGR rat liver. Physiol. Genomics 20, 108–116.
Geremia, C., and Cianfarani, S. (2004). Insulin sensitivity in children born small for gestational age (SGA). Rev. Diabet. Stud. 1, 58–65.
Gluckman, P. D., and Hanson, M. A. (2004). Living with the past: evolution, development, and patterns of disease. Science 305, 1733–1736.
Gluckman, P. D., and Hanson, M. A. (2004). The developmental origins of the metabolic syndrome. Trends Endocrinol. Metab. 15, 183–187.
Gluckman, P. D., Hanson, M. A., Bateson, P., Beedle, A. S., Law, C. M., Bhutta, Z. A., Anokhin, K. V., Bougnères, P., Chandak, G. R., Dasgupta, P., Smith, G. D., Ellison, P. T., Forrester, T. E., Gilbert, S. F., Jablonka, E., Kaplan, H.,Prentice, A. M., Simpson, S. J., Uauy, R., and West-Eberhard, M. J. (2009). Towards a new developmental synthesis: adaptive developmental plasticity and human disease. Lancet 373, 1654–1657.
Gluckman, P. D., Hanson, M. A., Cooper, C., and Thornburg, K. L. (2008). Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 359, 61–73.
Gluckman, P. D., Hanson, M. A., and Pinal, C. (2005). The developmental origins of adult disease. Matern. Child Nutr. 1, 130–141.
Goldberg, A. D., Allis, C. D., and Bernstein, E. (2007). Epigenetics: a landscape takes shape. Cell 128, 635–638.
Hales, C. N., and Barker, D. J. (1992). Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35, 595–601.
Heijmans, B. T., Tobi, E. W., Stein, A. D., Putter, H., Blauw, G. J., Susser, E. S., Slagboom, P. E., and Lumey, L. H. (2008). Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 17046–17049.
Holland, A. M., Góñez, L. J., Naselli, G., Macdonald, R. J., and Harrison, L. C. (2005). Conditional expression demonstrates the role of the homeodomain transcription factor Pdx1 in maintenance and regeneration of beta-cells in the adult pancreas. Diabetes 54, 2586–2595.
Jablonka, E, and Raz, G. (2009). Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 84, 131–176.
Lane, R. H., Kelley, D. E., Gruetzmacher, E. M., and Devaskar, S. U. (2001). Uteroplacental insufficiency alters hepatic fatty acid-metabolizing enzymes in juvenile and adult rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R183–R190.
Lane, R. H., MacLennan, N. K., Hsu, J. L., Janke, S. M., and Pham, T. D. (2002). Increased hepatic peroxisome proliferator-activated receptor-gamma coactivator-1 gene expression in a rat model of intrauterine growth retardation and subsequent insulin resistance. Endocrinology 143, 2486–2490.
Lin, J., Handschin, C., and Spiegelman, B. M. (2005). Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370.
Ling, C., Del Guerra, S., Lupi, R., Rönn, T., Granhall, C., Luthman, H., Masiello, P., Marchetti, P., Groop, L., and Del Prato, S., (2008). Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 51, 615–622.
Lucas, A. (1991). Programming by early nutrition in man. Ciba Found. Symp. 156, 38–50; discussion 50–55.
Lumey, L. H., Stein, A. D., Kahn, H. S.,van der Pal-de Bruin, K. M., Blauw, G. J., Zybert, P. A., and Susser, E. S. (2007). Cohort profile: the Dutch Hunger Winter families study. Int. J. Epidemiol. 36, 1196–1204.
MacLennan, N. K., James, S. J., Melnyk, S., Piroozi, A., Jernigan, S., Hsu, J. L., Janke, S. M., Pham, T. D., and Lane, R. H. (2004). Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiol. Genomics 18, 43–50.
McGarry, J. D., and Brown, N. F. (1997). The mitochondrial carnitine palmitoyltransferase system – From concept to molecular analysis. Eur. J. Biochem. 244, 1–14.
Morgan, H. D., Santos, F., Green, K., Dean, W., and Reik, W. (2005). Epigenetic reprogramming in mammals. Hum. Mol. Genet. 14, R47–R58.
Painter, R. C., Roseboom, T. J., and Bleker, O. P. (2005). Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod. Toxicol. 20, 345–352.
Park, J. H., Stoffers, D. A., Nicholls, R. D., and Simmons, R. A. (2008). Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Invest. 118, 2316–2324.
Ravelli, A. C., van der Meulen, J. H., Michels, R. P., Osmond, C., Barker, D. J., Hales, C. N., and Bleker, O. P. (1998). Glucose tolerance in adults after prenatal exposure to famine. Lancet 351, 173–177.
Raychaudhuri, N., Raychaudhuri, S., Thamotharan, M., and Devaskar, S. U. (2008). Histone code modifications repress glucose transporter 4 expression in the intrauterine growth-restricted offspring. J. Biol. Chem. 283, 13611–13626.
Robinson, S., Walton, R. J., Clark, P. M., Barker, D. J., Hales, C. N., and Osmond, C. (1992). The relation of fetal growth to plasma glucose in young men. Diabetologia 35, 444–446.
Schübeler, D., Lorincz, M. C., Cimbora, D. M., Telling, A., Feng, Y. Q., Bouhassira, E. E., and Groudine, M. (2000). Genomic targeting of methylated DNA: influence of methylation on transcription, replication, chromatin structure, and histone acetylation. Mol. Cell Biol. 20, 9103–9112.
Simmons, R. A., Templeton, L. J., and Gertz, S. J. (2001). Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50, 2279–2286.
Steegers-Theunissen, R. P., Obermann-Borst, S. A., Kremer, D., Lindemans, J., Siebel, C., Steegers, E. A., Slagboom, P. E., and Heijmans, B. T. (2009). Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS One 4:e7845. doi: 10.1371/journal.pone.0007845.
Stein, A. D., and Lumey, L. H. (2000). The relationship between maternal and offspring birth weights after maternal prenatal famine exposure: the Dutch Famine Birth Cohort Study. Hum. Biol. 72, 641–654.
Stoffers, D. A., Desai, B. M., DeLeon, D. D., and Simmons, R. A. (2003). Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes 52, 734–740.
Stoffers, D. A., Zinkin, N. T., Stanojevic, V., Clarke, W. L., and Habener, J. F. (1997). Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 15, 106–110.
Thompson, R. F., Fazzari, M. J., Niu, H., Barzilai, N., Simmons, R. A., and Greally, J. M. (2010). Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J. Biol. Chem. 285, 15111–15118.
Wolffe, A. P., and Matzke, M. A. (1999). Epigenetics: regulation through repression. Science 286, 481–486.
Yoon, J. C., Puigserver, P., Chen, G., Donovan, J., Wu, Z., Rhee, J., Adelmant, G., Stafford, J., Kahn, C. R., Granner, D. K., Newgard, C. B., and Spiegelman, B. M. (2001). Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–128.
Zambrano, E., Martínez-Samayoa, P. M., Bautista, C. J., Deás, M., Guillén, L., Rodríguez-González, G. L., Guzmán, C., Larrea, F., and Nathanielsz, P. W. (2005). Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. J. Physiol. 566, 225–236.
Keywords: epigenetics, intrauterine growth retardation, diabetes
Citation: Liguori A, Puglianiello A, Germani D, Deodati A, Peschiaroli E and Cianfarani S (2010) Epigenetic changes predisposing to type 2 diabetes in intrauterine growth retardation. Front. Endocrin. 1:5. doi: 10.3389/fendo.2010.00005
Received: 18 August 2010;
Paper pending published: 13 September 2010;
Accepted: 08 November 2010;
Published online: 22 November 2010.
Edited by:
Raimo Voutilainen, University of Kuopio, FinlandReviewed by:
Wieland Kiess, University of Leipzig, GermanyRebecca Simmons, University of Pennsylvania, USA
Copyright: © 2010 Liguori, Puglianiello, Germani, Deodati, Peschiaroli and Cianfarani. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Stefano Cianfarani, Department of Public Health and Cell Biology, Tor Vergata University, Room E-178, Via Montpellier 1, 00133 Rome, Italy. e-mail: stefano.cianfarani@uniroma2.it