
- Metabolic Research Labs, Institute of Metabolic Science, Addenbrooke’s Hospital, University of Cambridge, Cambridge, UK
Few would dispute that the current obesity epidemic has been driven by lifestyle and environmental changes. However, it is clear that individuals respond differently to these “obesigenic” changes and this variation in response has a strong genetic element. Genome-wide association studies have revealed that single nucleotide polymorphisms in Fat mass and obesity-associated transcript (FTO) are robustly associated with body mass index and obesity. Although the effect of these risk alleles are modest, with heterozygous and homozygous carriers weighing approximately 1.5 and 3 kg more respectively, there are an estimated one billion homozygous carriers in the world, spanning multiple different ethnicities and populations. Yet despite its broad impact, the biological function of FTO, particularly its role in controlling energy balance, remains unknown. Although the study of severe Mendelian obesity has been invaluable in illuminating critical pathways controlling food intake, the major burden of disease is carried by those of us with “common obesity,” which to date has resisted yielding meaningful biological insights. FTO has at last given us a handle on a huge, worldwide, common problem. In this review, we focus on the available genetic and in vivo evidence to date that implicates FTO in the control of energy balance.
Introduction
Obesity is a severe problem facing most developed nations and many other emerging economies (Zimmet et al., 2001). The World Health Organization estimates there will be more than 2 billion overweight and 700 million obese adults in the world by 2015, and by 2050, 90% of today’s children will be overweight or obese. This is a huge drain on the healthcare systems of affected countries not because of obesity itself per se, but because of the associated increased risk to several unpleasant co-morbidities, including type 2 diabetes mellitus (T2D), hypertension, cardiovascular diseases, and certain forms of cancer (Calle et al., 1999; Katzmarzyk et al., 2003). In addition, other consequences of being obese often include social stigmatization and discrimination.
It is clear that lifestyle and environmental changes that have occurred over the past 20 years, including a more convenient supply of high calorie food and decreased physical activity, underlie the etiology of this phenomenon. However, it is also clear that genetic factors have a powerful role to play, with different individuals responding in different ways to this changing environment. In fact, genetic influence plays an important role in individual body mass index (BMI) differences, with twin and adoption studies estimating the heritability of fat mass to be between 40 and 60% (Raman, 2002). Thus, obesity is determined by interactions between individual genetics and environmental factors (Hill and Peters, 1998).
Over the past 15 years, studies of Mendelian inherited syndromes of obesity in humans, together with the use of monogenic mouse models have been invaluable in illuminating critical pathways controlling food intake and body weight. The most well characterized to date is the central leptin–melanocortin pathway, where disruption of most of its components by monogenic defects, either in humans or mice, results in severe early onset obesity (Vaisse et al., 1998; Yeo et al., 1998; Stutzmann et al., 2007). In humans however, these monogenic causes of severe obesity are extremely rare. In contrast to the Mendelian obesity syndromes, common obesity is likely to have a “polygenic” etiology, with multiple variations each having a subtle effect. It is therefore unsurprising that conventional candidate gene analysis either failed in reliably detecting novel variants or validating these minor gene defects.
It was only within the past few years that the required technology to interrogate hundreds of thousands (now more than a million) single nucleotide polymorphisms (SNPs) in parallel was developed. These technologies, which allow for genome-wide association studies (GWAS), coupled with unprecedented international collaborations resulting in the collection of increasing large study cohorts, have become extremely powerful for detecting common genetic variants associated with complex disorders like obesity. Thus in many ways, the rapidly evolving environment coupled with the emergence of new technologies have served to magnify previously subtle genetic susceptibility.
Discovery of FTO from GWAS
In 2007, a T2D GWAS identified multiple SNPs in the first intron of a gene called Fat mass and obesity-related transcript (FTO) associated with disease. However the association between these FTO SNPs and T2D disappeared after adjustment for BMI, suggesting that these SNPs were actually associated with BMI, with increased weight being a risk factor for T2D. Confirmation of this association with BMI was replicated in 13 independent cohorts including a total of 38,759 participants (Frayling et al., 2007). Subsequently and in close succession, a number of other independent studies encompassing adults and children from several European populations(Dina et al., 2007; Scuteri et al., 2007; Peeters et al., 2008; Attaoua et al., 2009; Gonzalez-Sanchez et al., 2009; Jonsson et al., 2009), as well as populations of Asian (Cha et al., 2008; Chang et al., 2008; Hotta et al., 2008; Tan et al., 2008), and African ancestries (Grant et al., 2008; Hennig et al., 2009; Adeyemo et al., 2010; Bollepalli et al., 2010; Keebler et al., 2010), reported associations of FTO intron 1 SNPs with obesity-related traits including increased hip circumference, waist to hip ratio and body weight. Thus, FTO became the first of the post-GWAS obesity genes. In this review, we examine the current status of human genetic and rodent model data and how these have begun to inform us of the complex biology of FTO and its involvement in the control of energy balance.
Carriers of FTO Variants Weigh 3 Kg Heavier
The FTO SNP rs9939609 has the strongest known effect on increased BMI, particularly within European populations. Frayling et al. (2007) reported that individuals homozygous for the A “risk” allele weigh approximately 3 kg more and have a 1.67-fold increased risk of developing obesity, while heterozygous carriers display an intermediate weight gain of 1.5 kg. The association between rs9939609 and increased BMI/obesity was replicated and confirmed in both adults and children in multiple ethnic groups (Al-Attar et al., 2008; Andreasen et al., 2008; Karasawa et al., 2010). However the allelic frequency of rs9939609 is different across ethnic groups, for example close to 60% of most European populations carrying one or more copy of the A “risk” allele as compared to 17% in some Asian populations (Song et al., 2008). Despite the variation in frequency, the modest effect size of 3 kg remains consistent regardless of ethnicity. With an estimated 1 billion homozygous carriers in the world, FTO is clearly influencing in some way the body weight of a significant proportion of the world’s population (Leibel, 2008).
As regulation of body weight is of course a balance between energy intake and energy expenditure, many of the subsequent studies have examined the influence of FTO variants on measures of appetite, food intake, or energy expenditure (Hakanen et al., 2009; Haupt et al., 2009). Subjects homozygous for the A risk allele of rs9939609 eat significantly more (Speakman et al., 2008; Wardle et al., 2009), have reduced satiety (Wardle et al., 2008; den Hoed et al., 2009; Tanofsky-Kraff et al., 2009), prefer higher caloric food and have a higher fat mass (Cecil et al., 2008; Timpson et al., 2008) than subjects homozygous for the T allele. The association of FTO SNPs with energy intake are seen even in small sample sizes (n = 150), which lack the required power to identify the association with small increases in BMI (Speakman et al., 2008). Thus, based on the weight of evidence to date, FTO risk alleles are unequivocally associated with increased food intake and appetitive behavior. In contrast, the association with the other half of the energy balance equation is less clear. Certainly, FTO SNPs are not associated with reduced energy expenditure (Speakman et al., 2008; Goossens et al., 2009; Haupt et al., 2009) or physical activity (Jonsson et al., 2009). However, there is some evidence showing an association with increased energy expenditure, as well as increased physical activity levels (Jonsson and Franks, 2009).
Association of FTO SNPs with other metabolic traits including higher plasma levels of C-reactive protein (CRP; Fisher et al., 2009; Sun et al., 2010) and an increased degree of insulin resistance (Jacobsson et al., 2008), independent of their association with BMI have also been reported. However the results are not consistent, with other researchers finding that these associations disappear after correction for BMI (Legry et al., 2009).
FTO or RPGRIP1L?
Powerful though it has been, it is easy to forget that GWAS is a gene agnostic approach. SNPs reaching the appropriate statistical threshold for a given phenotype or disease can appear anywhere in the genome, within, near or far away from any coding sequence. The current assumption that the closest coding region, which is sometimes hundreds of kilobases away, is the likely candidate is perhaps a reasonable first guess, but not necessarily true (Cantor et al., 2010). In this instance, the “FTO” risk alleles are actually located in the first intron of the FTO gene. However, they are also very close to the transcriptional start site of RPGRIP1L (human ortholog of mouse Ftm), which is adjacent to and coded for on the opposite DNA strand to FTO (Stratigopoulos et al., 2008), as shown in Figure 1. So why, in this situation, was FTO invoked, instead of RPGRIP1L?
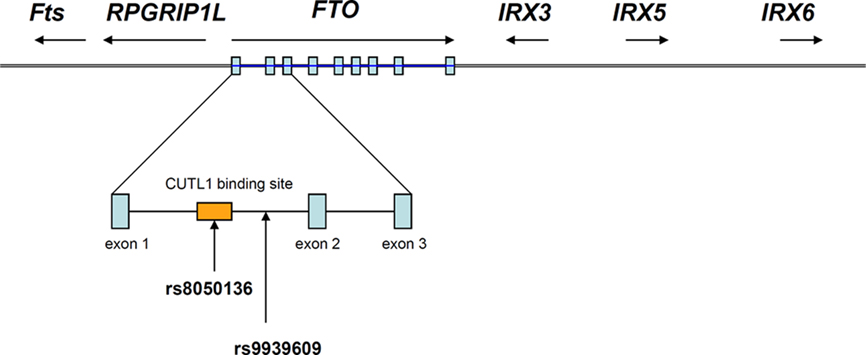
Figure 1. Genomic organization of FTO and its neighboring genes (not drawn to scale). The FTO gene contains nine exons which are depicted in blue rectangles and the most replicated FTO SNP rs9939609 is found in intron 1 of the gene. CUTL1 binding site overlaps with the FTO SNP rs8050136 (Stratigopoulos et al., 2011).
Rpgrip1l is previously known to localize in the primary cilia and centrosomes of ciliated cells (Arts et al., 2007). The protein binds to nephrocystin-4 and is involved in mechanisms including programmed cell death, craniofacial development, patterning of the limbs, and formation of the left–right axis (Tokue et al., 2009). Crucially, defects in human RPGRIP1L exist and cause two rare genetic disorders; Joubert syndrome type 7 (JBTS) and Meckel syndrome type 5 (MKS5). JBTS is caused by homozygous loss-of-function missense mutations in RPGRIP1L, and presents clinically with a peculiar cerebellar and brainstem malformation known as the “molar tooth sign” (MTS) and renal failure (Brancati et al., 2008; Tokue et al., 2009). MKS5 is a far more severe variant of the disease which results from non-sense or frameshift mutations in RPGRIP1L, thereby resulting in a complete loss-of-function of the protein (Delous et al., 2007). The patients do not present with any obvious obesity-related phenotypes, with the caveat that any potential “lean” phenotype is difficult to ascertain in a healthy individual, let alone someone who is severely ill. Deletion of the mouse ortholog Rpgrip1l (Ftm) recapitulates the cerebral, renal, and hepatic defects seen in the patients. Therefore it is less likely that Rpgrip1l, in of itself, is related to the BMI phenotype.
However, there is evidence for possible co-regulatory mechanisms between FTO and RPGRIP1L. Stratigopoulos et al. (2008) report an overlapping regulatory region within intron 1 of FTO that contains at least two putative transcription factor binding sites (CUTL1), one of which overlaps with another obesity-associated SNP (rs8050136). The group reported a reduced expression level of both FTO and RPGRIP1L when the CUTL1 site is knocked-down human fibroblasts, however the mechanism of this phenomenon remains to be determined (Stratigopoulos et al., 2008, 2011). They speculate that the association between FTO SNPs and body weight regulation is mediated through changing the expression of both FTO and RPGRIP1L.
What is FTO?
Fto was originally identified in 1999 as one of six contiguous genes in a naturally occurring 1.6 Mb deletion on chromosome 8 in a mouse model known as fused toes (Ft). Homozygous Ft mutants are embryonic lethal and display neural tube defects, whereas heterozygous mice have fused toes, and are not obese. In addition to Fto, the other five genes are Irx3, Irx5, Irx6, Fts, and the aforementioned Rpgrip1 (van der Hoeven et al., 1994; Peters et al., 1999, 2002). The genomic region adjacent to FTO together with the relative location of these other genes are shown in Figure 1. Irx3, Irx5, and Irx6 belong to the Iroquois-class of homeodomain proteins, which are transcriptional factors involved in SHH-dependent neural patterning (Briscoe et al., 2000). Fts encodes a Hook-interacting protein of unknown function but is predicted by sequence similarity to be involved in vesicle transport (Okazaki et al., 2003). None of these other genes have been implicated in any obese phenotype.
FTO is a large gene of nine exons spanning more than 400 kb on chromosome 16 in humans. All the SNPs identified so far are located in the first and largest intron of the gene, a region where the sequence is strongly conserved across species (Loos et al., 2008). Evolutionarily, FTO is found only in vertebrates and marine algae, and is absent from all invertebrates, fungi, green plants, or bacteria, suggesting that the gene was present 450 million years ago and has undergone horizontal gene transfer (Robbens et al., 2008).
FTO is widely expressed in fetal and adult tissues in humans, mice, and rats, with the highest expression in the brain (Vaisse et al., 1998; Frayling et al., 2007; Stutzmann et al., 2007). Within the brain, Fto expression is relatively high in a number of hypothalamic nuclei: arcuate (ARC), paraventricular (PVN), dorsomedial (DMN), and ventromedial (VMN) nuclei, where control of energy homeostasis is centered (Gerken et al., 2007; Fredriksson et al., 2008). Soon after the first report of FTO, we and others used a bioinformatics approach to show that human FTO has the highest sequence similarity with the E. coli DNA repair protein AlkB and its mammalian homologues ABH2 and 3, which all belong to the family of Fe2+ and 2-oxoglutarate (2-OG)-dependent dioxygenases (Gerken et al., 2007). Members of the family are involved in various cellular processes including DNA repair, fatty acid metabolism and post-translational modifications. Gerken et al. (2007) showed that in vitro, FTO catalyzes the demethylation of 3-meT in single-stranded DNA in the presence of Fe2+ and 2-OG, with concomitant production of succinate, formaldehyde, and carbon dioxide. A subsequent study reported FTO has a slight preference for 3-meU in single-stranded RNA, than for 3-meT, suggesting that FTO has a substrate preference for methylated RNA over DNA (Jia et al., 2008). These in vitro studies suggest that FTO’s physiological role could be linked with nucleic acid repair or modification. In addition, FTO is present in the cellular nucleus (Gerken et al., 2007), which supports its functional involvement in nucleic acid demethylation. However we still do not know the endogenous substrates of FTO and how this demethylation activity of FTO is linked to human obesity.
Recently, the X-ray crystallography structure of FTO provided a basis for the study of structure–function relationships. The protein is composed of two domains: an N-terminal domain carrying a catalytic core and a C-terminal domain of unknown structural homology as shown in Figure 2 (Han et al., 2010). In the catalytic domain are five obligate amino acid residues found in all members of this enzyme superfamily; two residues, a histidine (H) and an aspartic acid (D), required for binding Fe(II); and three residues, an H and two arginines (R) separated by six amino acids, required for 2-OG binding. Structural based sequence alignment showed that FTO has an extra region, referred to as Loop 1, which is highly conserved among FTOs from different species, but is absent from AlkB and ABH proteins. The crystal structure reveals that Loop 1 sterically hinders the unmethylated strand of dsDNA/RNA from gaining access to the substrate-binding site, explaining why FTO has a substrate preference for single-stranded nucleic acid (Han et al., 2010).
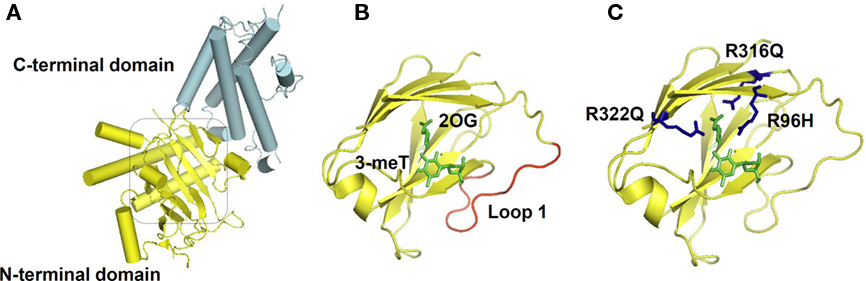
Figure 2. FTO Crystal Structure. (A) Overall structure of FTO protein. The C-terminal with an unknown structural homology, and the N-terminal domain that contains the catalytic core, are colored in cyan and yellow respectively. (B) FTO’s catalytic site is shown, with its bound substrates: 3-meT and 2-oxoglutarate (2-OG) highlighted in green. The extra loop 1 from FTO that physically hinder the entrance of dsDNA/RNA is in red. (C) Residues that cause a compete loss-of-function of FTO: R316 and R322 that are required for 2-OG binding, and R96 that occurs in the substrate recognition lid, are shown in blue. The molecular graphics were generated using PyMOL (Version 1.1r1, Schrödinger, LLC).
Human FTO Deficiency
There are several examples in metabolic disease where common variants close to a particular gene are associated with alterations in risk of common phenotypes such as fat mass or risk of obesity, while rare loss or gain of function mutations in the same gene are associated with a more severe version of the same metabolic phenotype, e.g., MCR4 (Vaisse et al., 1998; Yeo et al., 1998; Geller et al., 2004; Stutzmann et al., 2007; Chambers et al., 2008; Loos et al., 2008), POMC (Krude et al., 1998; Challis et al., 2002), BDNF (Gray et al., 2006; Han et al., 2008; Thorleifsson et al., 2009), and PCSK1 (Jackson et al., 1997; Benzinou et al., 2008). When information is available from both types of variants, clarity of mechanistic understanding is greatly enhanced. Can the same thing be said for FTO?
In 2009, we in collaboration with others, reported a large consanguineous family of Palestinian Arab origin with nine members affected by a previously unknown polymalformation syndrome, all of whom were homozygous for an arginine to glutamine change at position 316 (R316Q) in FTO. Unfortunately for the affected individuals, because R316 is one of the obligate residues required for binding of 2-OG, the R316Q mutation completely abolishes its demethylase activity. The syndromic characteristics observed in the affected individuals include postnatal growth retardation, microcephaly, severe functional brain deficits, facial dysmorphism, cardiac abnormalities, and cleft palate. Tragically, the severity of the phenotype was such that all affected children died within the first 30 months of life. Although the heterozygous carriers within this family did not have their clinical phenotype specifically studied, no overt obesity-related phenotypes were reported (Boissel et al., 2009).
In another study, we screened the coding regions of FTO in 1400 lean and morbidly obese individuals to determine if there was an increased burden of non-synonymous variants in either cohort. In short, the answer was no. However, we did identify two novel loss-of-function FTO mutations; R96H, which occurs in the substrate recognition lid and R322Q, the second obligate arginine required for 2-OG binding, both of which were found in heterozygous form and resulted in complete loss of catalytic activity. Unfortunately, these two mutations did nothing to clarify the role of FTO in energy balance, as the mutations were each identified twice in both lean and obese individuals (Meyre et al., 2010). It is a puzzle that while SNPs in intron 1 of FTO are unequivocally associated with obesity in multiple populations, it appears that loss of one functional copy of FTO in humans is compatible with being either lean or obese.
There is an oddity about the distribution of the mutations throughout FTO, which may yet prove relevant. Although non-synonymous variants elsewhere in the molecule are found equally in obese and lean subjects, eight such variants found in the carboxy-terminus were detected in obese subjects and only two in lean (Meyre et al., 2010). It remains to be seen if this clue transpires into anything of functional significance. What is clear is FTO seems to have a critical, but as yet undetermined role, in the development of several major organ systems, including the central nervous and cardiovascular systems.
At this point, it is worth noting that to date, we do not know if or how the FTO risk alleles are influencing the FTO protein. Considering the intronic location of all the FTO obesity-related SNPs (spanning across ∼40 kb), they are unlikely to cause functional mutations. Instead, the SNPs are more likely to be playing a transcriptional regulatory role, either to up- or down-regulate FTO expression. In addition, it is also possible that the SNPs could be involved in post-transcriptional modifications, which could result with various FTO splice variants and isoforms of different functions.
Lessons from FTO Rodent Models of FTO
Mouse models of Fto deficiency, although far from being straightforward, have been more helpful in illuminating a role for FTO energy balance. Phenotypic characteristics of the five recent Fto rodent models are summarized in Table 1.
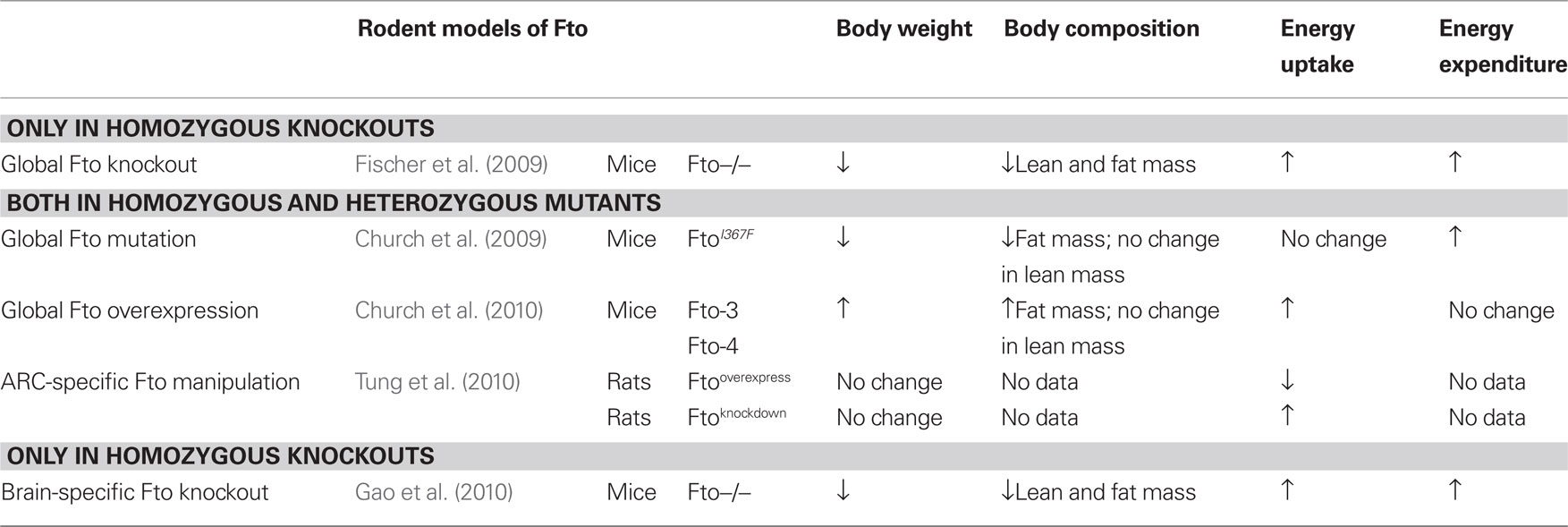
Table 1. Summary of phenotypes from the five different Fto rodent models.
Global FTO Knockout
Mice homozygous for a targeted deletion in Fto display a complex phenotype (Fischer et al., 2009). They are postnatally growth retarded with decreased fat and lean body mass, and although are born with a normal body weight and at the expected Mendelian ratio, display 50% lethality by the time of weaning. That they are born normally implies that implantation and embryonic development are not grossly affected. The cause of the postnatal lethality is still unknown, however it appears that a complete ablation of Fto in mice causes retarded postnatal development, consistent with FTO deficiency in humans, supporting the hypothesis that Fto is involved in normal body development. They also suffer from a curious and as yet unexplained reduction in ambulatory movement. A detailed study of the organs from these Fto−/− mice has yet to be reported and could prove informative.
In support of the human data showing that FTO risk SNPs strongly correlate with increased food intake, Fto−/− mice appear to display hyperphagia and increased energy expenditure when corrected for lean body mass. The authors suggested that the increase in energy expenditure occurs in the presence of a higher sympathetic activity, which is demonstrated by a higher circulating concentration of adrenaline in the Fto−/− mice.
Resonant with the studies exploring the link between the risk alleles and metabolic rate in humans, the observation that Fto null mice have increased energy expenditure has not been without controversy. In fact, it has ignited a broader debate within the metabolic field about the methodologies underlying the general measurement of energy expenditure in rodents.
Since the hypothalamus is important for control of energy balance, and it is a region where Fto is highly expressed, Fischer et al. (2009) then went on to examine whether the hyperphagic phenotype of Fto−/− mice is caused by hypothalamic functional defects. However neither abnormal hypothalamic development nor changes in expression levels of feeding-related neuropeptides in the arcuate nucleus of the hypothalamus (ARC) of adult Fto−/− mice were found. The most crucial piece of evidence in this report supporting a role for Fto in energy balance is that the Fto+/− mice, who are not growth retarded, appear to be resistant to high-fat diet (HFD) induced obesity, when compared with their wild-type littermates.
As discussed above, since Fto lies in close proximity to Rpgrip1l, the authors checked for Rpgrip1l mRNA expression levels in the Fto−/− and Fto+/− mice and found no changes, arguing against the co-regulation of both genes playing a role in body weight regulation.
ENU Induced FTO Mutation
Another mouse model that harbors a global Fto mutation I367F subsequently followed (Church et al., 2009). The group reported that an N-ethyl-N-nitrosourea (ENU) mutagenesis induced point mutation in Fto (I367F) resulted in a partial loss-of-function, a 60% drop in catalytic activity, when compared to wild-type control. Both FtoI367F homozygous and heterozygous male mice had reduced body weight compared to their wild-type littermates from 12 weeks onward, and presented with a reduction of fat mass, but not lean mass. Given the fact that no difference in food intake and physical activity was found between these Fto mutant male mice and their control littermates, the authors suggested that the reduction of their body weight is a consequence of an increase in resting metabolic rate. FtoI367F mice also have a higher level of glucagon compared to wild-type controls, suggesting they may have an increased sympathetic activity. Thus, FtoI367F mice resemble Fto−/− mice in many areas, except with a milder phenotype and the absence of developmental problems.
Mice Overexpressing FTO
More recently, Church et al. (2010)have generated a mouse model that carries one or two additional copies of Fto (Fto-3 for three additional copies; one plus the two endogenous copies and Fto-4 for two additional copies). Mice overexpressing Fto showed a dramatic increase in food intake resulting in a marked increase in body weight and fat mass when they were fed either chow or a HFD. However despite the significant increase in fat mass, lean mass was not found to be changed in mice overexpressing Fto (with the exception of the female Fto-4 mice), leading the authors to suggest that overexpression of Fto has a positive influence on fat-to-lean tissue mass ratio in these mice. There was no significant change in either energy expenditure or physical activity.
Although the increase in weight with the overexpression of Fto seems consistent with Fto deficiency resulting in a “lean” phenotype, the increase in food intake seen in these mice is not. The authors go on to look at circulating leptin concentrations, and find that, curiously, at the age of 8 weeks, both Fto-3 and Fto-4 mice had reduced leptin levels after a 16-h overnight fast compared to wild-type controls. It is fascinating that this phenomenon occurs, keeping in mind that these are obese mice, whose normal response would be to have increased circulating leptin, reflecting their increased fat mass. The authors comment that the hyperphagic phenotype is possibly due to an Fto dependent reduction of leptin, although no mechanism is invoked. However at the age of 20 weeks, circulating leptin levels returned to levels commensurate with the increased fat mass resulting from the hyperphagia seen in the Fto overexpressing mice. The observation seems to have contradicted their regression analysis that showed a positive correlation between leptin level and fat mass content.
FTO in the Hypothalamus Modulates Food Intake
As discussed above, FTO is found ubiquitously in many tissues in human, mice, and rats, but is highest expressed in the brain, especially in feeding-related nuclei in the hypothalamus where control of energy homeostasis is centered (Gerken et al., 2007; Fredriksson et al., 2008). Given the fact the Fto is highly expressed in a multiple feeding-related nuclei in the hypothalamus, it was interesting to ask whether Fto mRNA levels are nutritionally regulated. Indeed, expression of Fto in the ARC is bi-directionally regulated as a function of nutritional status; decreasing following a 48-h fast (Fredriksson et al., 2008) and increasing after 10 weeks of exposure to a HFD (Tung et al., 2010). Since fasting, a strong stimulus to eat, reduces Fto mRNA levels in the ARC, if Fto was having a direct action on the control of energy intake one would predict that reducing its expression would recapitulate the fasting state, thereby resulting in an increase in food intake. This does indeed seem to be the case. Tung et al. (2010) knocked-down and overexpressed Fto specifically in the ARC in rats, using adeno-associated virus (AAV) vectors coupled with stereotactic injections. When FTO is overexpressed by 2.5-fold in the ARC, it results in a 14% reduction in average daily food intake. Conversely, knocking down Fto expression by 40% increases food intake by 16% (Tung et al., 2010). Unlike genetic mouse models, the observed phenotypic effects are transient. However it has proved advantageous for studying acute changes in eating behavior of the “mutant” rats, and this spatial and temporal specific approach allows us to observe only direct action of Fto, because possible influences of Fto on early development are bypassed.
Brain-Specific FTO KO
Central nervous system (CNS) specific Fto deleted mice have now been generated by Gao et al. (2010). Surprisingly, these brain-specific Fto deficient mice recapitulate the phenotype of the whole-body knockouts, although this is yet to be exhaustively examined. This suggests that much of Fto’s function, including its link to the regulation of energy homeostasis (and in keeping with the observations by Tung et al. (2010), is mediated in the brain.
In the past 3 years, 23 other genetic loci have been identified to be associated with increased BMI from independent GWAS in adults. Most of the presumed candidate in these loci, as we have discussed here with FTO, are also highly expressed or are known to act in the CNS, emphasizing, as in rare monogenic forms of obesity, the role of the CNS in predisposition to obesity (Willer et al., 2009). Perhaps there is some truth to the statement that “obesity is all in your head” after all!
Conclusion
FTO is the first and the most robust gene identified to be associated with increased BMI in humans in the post-GWAS era. Yet despite its broad impact, FTO’s biological function, particularly the molecular mechanisms underlying its role in controlling energy balance, remains unknown. The major burden of disease is carried by those of us with “common obesity,” which to date has resisted yielding meaningful biological insights. FTO has at last given us a handle on a huge, worldwide, common problem and demands exploration. Understanding the biology of FTO could uncover novel pathways and mechanisms controlling energy balance, revealing new therapeutic targets in our battle against obesity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the UK Medical Research Council Centre for Obesity and Related Metabolic Disorders (MRC-CORD) and the EU FP7-HEALTH-2009-241592 EurOCHIP.
References
Adeyemo, A., Chen, G., Zhou, J., Shriner, D., Doumatey, A., Huang, H., and Rotimi, C. (2010). FTO genetic variation and association with obesity in West Africans and African Americans. Diabetes 59, 1549–1554.
Al-Attar, S. A., Pollex, R. L., Ban, M. R., Young, T. K., Bjerregaard, P., Anand, S. S., Yusuf, S., Zinman, B., Harris, S. B., Hanley, A. J., Connelly, P. W., Huff, M. W., and Hegele, R. A. (2008). Association between the FTO rs9939609 polymorphism and the metabolic syndrome in a non-Caucasian multi-ethnic sample. Cardiovasc. Diabetol. 7, 5.
Andreasen, C. H., Stender-Petersen, K. L., Mogensen, M. S., Torekov, S. S., Wegner, L., Andersen, G., Nielsen, A. L., Albrechtsen, A., Borch-Johnsen, K., Rasmussen, S. S., Clausen, J. O., Sandbaek, A., Lauritzen, T., Hansen, L., Jørgensen, T., Pedersen, O., and Hansen, T. (2008). Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes 57, 95–101.
Arts, H. H., Doherty, D., van Beersum, S. E., Parisi, M. A., Letteboer, S. J., Gorden, N. T., Peters, T. A., Märker, T., Voesenek, K., Kartono, A., Ozyurek, H., Farin, F. M., Kroes, H. Y., Wolfrum, U., Brunner, H. G., Cremers, F. P., Glass, I. A., Knoers, N. V., and Roepman, R. (2007). Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 39, 882–888.
Attaoua, R., Ait El Mkadem, S., Lautier, C., Kaouache, S., Renard, E., Brun, J. F., Fedou, C., Gris, J. C., Bringer, J., and Grigorescu, F. (2009). Association of the FTO gene with obesity and the metabolic syndrome is independent of the IRS-2 gene in the female population of Southern France. Diabetes Metab. 35, 476–483.
Benzinou, M., Creemers, J. W., Choquet, H., Lobbens, S., Dina, C., Durand, E., Guerardel, A., Boutin, P., Jouret, B., Heude, B., Balkau, B., Tichet, J., Marre, M., Potoczna, N., Horber, F., Le Stunff, C., Czernichow, S., Sandbaek, A., Lauritzen, T., Borch-Johnsen, K., Andersen, G., Kiess, W., Körner, A., Kovacs, P., Jacobson, P., Carlsson, L. M., Walley, A. J., Jørgensen, T., Hansen, T., Pedersen, O., Meyre, D., and Froguel, P. (2008). Common nonsynonymous variants in PCSK1 confer risk of obesity. Nat. Genet. 40, 943–945.
Boissel, S., Reish, O., Proulx, K., Kawagoe-Takaki, H., Sedgwick, B., Yeo, G. S., Meyre, D., Golzio, C., Molinari, F., Kadhom, N., Etchevers, H. C., Saudek, V., Farooqi, I. S., Froguel, P., Lindahl, T., O’Rahilly, S., Munnich, A., and Colleaux, L. (2009). Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am. J. Hum. Genet. 85, 106–111.
Bollepalli, S., Dolan, L. M., Deka, R., and Martin, L. J. (2010). Association of FTO gene variants with adiposity in African-American adolescents. Obesity (Silver Spring) 18, 1959–1963.
Brancati, F., Travaglini, L., Zablocka, D., Boltshauser, E., Accorsi, P., Montagna, G., Silhavy, J. L., Barrano, G., Bertini, E., Emma, F., Rigoli, L; International JSRD Study Group, Dallapiccola, B., Gleeson, J. G., and Valente, E. M. (2008). RPGRIP1L mutations are mainly associated with the cerebello-renal phenotype of Joubert syndrome-related disorders. Clin. Genet. 74, 164–170.
Briscoe, J., Pierani, A., Jessell, T. M., and Ericson, J. (2000). A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101, 435–445.
Calle, E. E., Thun, M. J., Petrelli, J. M., Rodriguez, C., and Heath, C. W. Jr. (1999). Body-mass index and mortality in a prospective cohort of U.S. adults. N. Engl. J. Med. 341, 1097–1105.
Cantor, R. M., Lange, K., and Sinsheimer, J. S. (2010). Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am. J. Hum. Genet. 86, 6–22.
Cecil, J. E., Tavendale, R., Watt, P., Hetherington, M. M., and Palmer, C. N. (2008). An obesity-associated FTO gene variant and increased energy intake in children. N. Engl. J. Med. 359, 2558–2566.
Cha, S. W., Choi, S. M., Kim, K. S., Park, B. L., Kim, J. R., Kim, J. Y., and Shin, H. D. (2008). Replication of genetic effects of FTO polymorphisms on BMI in a Korean population. Obesity (Silver Spring) 16, 2187–2189.
Challis, B. G., Pritchard, L. E., Creemers, J. W., Delplanque, J., Keogh, J. M., Luan, J., Wareham, N. J., Yeo, G. S., Bhattacharyya, S., Froguel, P., White, A., Farooqi, I. S., and O’Rahilly, S. (2002). A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum. Mol. Genet. 11, 1997–2004.
Chambers, J. C., Elliott, P., Zabaneh, D., Zhang, W., Li, Y., Froguel, P., Balding, D., Scott, J., and Kooner, J. S. (2008). Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat. Genet. 40, 716–718.
Chang, Y. C., Liu, P. H., Lee, W. J., Chang, T. J., Jiang, Y. D., Li, H. Y., Kuo, S. S., Lee, K. C., and Chuang, L. M. (2008). Common variation in the fat mass and obesity-associated (FTO) gene confers risk of obesity and modulates BMI in the Chinese population. Diabetes 57, 2245–2252.
Church, C., Lee, S., Bagg, E. A., McTaggart, J. S., Deacon, R., Gerken, T., Lee, A., Moir, L., Mecinović, J., Quwailid, M. M., Schofield, C. J., Ashcroft, F. M., and Cox, R. D. (2009). A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet. 5, e1000599. doi: 10.1371/journal.pgen.1000599
Church, C., Moir, L., McMurray, F., Girard, C., Banks, G. T., Teboul, L., Wells, S., Brüning, J. C., Nolan, P. M., Ashcroft, F. M., and Cox, R. D. (2010). Overexpression of Fto leads to increased food intake and results in obesity. Nat. Genet. 42, 1086–1092.
Delous, M., Baala, L., Salomon, R., Laclef, C., Vierkotten, J., Tory, K., Golzio, C., Lacoste, T., Besse, L., Ozilou, C., Moutkine, I., Hellman, N. E., Anselme, I., Silbermann, F., Vesque, C., Gerhardt, C., Rattenberry, E., Wolf, M. T., Gubler, M. C., Martinovic, J., Encha-Razavi, F., Boddaert, N., Gonzales, M., Macher, M. A., Nivet, H., Champion, G., Berthélémé, J. P., Niaudet, P., McDonald, F., Hildebrandt, F., Johnson, C. A., Vekemans, M., Antignac, C., Rüther, U., Schneider-Maunoury, S., Attié-Bitach, T., and Saunier, S. (2007). The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 39, 875–881.
den Hoed, M., Westerterp-Plantenga, M. S., Bouwman, F. G., Mariman, E. C., and Westerterp, K. R. (2009). Postprandial responses in hunger and satiety are associated with the rs9939609 single nucleotide polymorphism in FTO. Am. J. Clin. Nutr. 90, 1426–1432.
Dina, C., Meyre, D., Gallina, S., Durand, E., Körner, A., Jacobson, P., Carlsson, L. M., Kiess, W., Vatin, V., Lecoeur, C., Delplanque, J., Vaillant, E., Pattou, F., Ruiz, J., Weill, J., Levy-Marchal, C., Horber, F., Potoczna, N., Hercberg, S., Le Stunff, C., Bougnères, P., Kovacs, P., Marre, M., Balkau, B., Cauchi, S., Chèvre, J. C., and Froguel, P. (2007). Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 39, 724–726.
Fischer, J., Koch, L., Emmerling, C., Vierkotten, J., Peters, T., Brüning, J. C., and Rüther, U. (2009). Inactivation of the Fto gene protects from obesity. Nature 458, 894–898.
Fisher, E., Schulze, M. B., Stefan, N., Häring, H. U., Döring, F., Joost, H. G., Al-Hasani, H., Boeing, H., and Pischon, T. (2009). Association of the FTO rs9939609 single nucleotide polymorphism with C-reactive protein levels. Obesity (Silver Spring) 17, 330–334.
Frayling, T. M., Timpson, N. J., Weedon, M. N., Zeggini, E., Freathy, R. M., Lindgren, C. M., Perry, J. R., Elliott, K. S., Lango, H., Rayner, N. W., Shields, B., Harries, L. W., Barrett, J. C., Ellard, S., Groves, C. J., Knight, B., Patch, A. M., Ness, A. R., Ebrahim, S., Lawlor, D. A., Ring, S. M., Ben-Shlomo, Y., Jarvelin, M. R., Sovio, U., Bennett, A. J., Melzer, D., Ferrucci, L., Loos, R. J., Barroso, I., Wareham, N. J., Karpe, F., Owen, K. R., Cardon, L. R., Walker, M., Hitman, G. A., Palmer, C. N., Doney, A. S., Morris, A. D., Smith, G. D., Hattersley, A. T., and McCarthy, M. I. (2007). A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894.
Fredriksson, R., Hägglund, M., Olszewski, P. K., Stephansson, O., Jacobsson, J. A., Olszewska, A. M., Levine, A. S., Lindblom, J., and Schiöth, H. B. (2008). The obesity gene, FTO, is of ancient origin, up-regulated during food deprivation and expressed in neurons of feeding-related nuclei of the brain. Endocrinology 149, 2062–2071.
Gao, X., Shin, Y. H., Li, M., Wang, F., Tong, Q., and Zhang, P. (2010). The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS ONE 5, e14005. doi: 10.1371/journal.pone.0014005
Geller, F., Reichwald, K., Dempfle, A., Illig, T., Vollmert, C., Herpertz, S., Siffert, W., Platzer, M., Hess, C., Gudermann, T., Biebermann, H., Wichmann, H. E., Schäfer, H., Hinney, A., and Hebebrand, J. (2004). Melanocortin-4 receptor gene variant I103 is negatively associated with obesity. Am. J. Hum. Genet. 74, 572–581.
Gerken, T., Girard, C. A., Tung, Y. C., Webby, C. J., Saudek, V., Hewitson, K. S., Yeo, G. S., McDonough, M. A., Cunliffe, S., McNeill, L. A., Galvanovskis, J., Rorsman, P., Robins, P., Prieur, X., Coll, A. P., Ma, M., Jovanovic, Z., Farooqi, I. S., Sedgwick, B., Barroso, I., Lindahl, T., Ponting, C. P., Ashcroft, F. M., O’Rahilly, S., and Schofield, C. J. (2007). The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318, 1469–1472.
González-Sánchez, J. L., Zabena, C., Martínez-Larrad, M. T., Martínez-Calatrava, M. J., Pérez-Barba, M., and Serrano-Ríos, M. (2009). Variant rs9939609 in the FTO gene is associated with obesity in an adult population from Spain. Clin. Endocrinol. (Oxf) 70, 390–393.
Goossens, G. H., Petersen, L., Blaak, E. E., Hul, G., Arner, P., Astrup, A., Froguel, P., Patel, K., Pedersen, O., Polak, J., Oppert, J. M., Martinez, J. A., Sørensen, T. I., and Saris WH; NUGENOB, Consortium. (2009). Several obesity- and nutrient-related gene polymorphisms but not FTO and UCP variants modulate postabsorptive resting energy expenditure and fat-induced thermogenesis in obese individuals: the NUGENOB study. Int. J. Obes. (Lond.) 33, 669–679.
Grant, S. F., Li, M., Bradfield, J. P., Kim, C. E., Annaiah, K., Santa, E., Glessner, J. T., Casalunovo, T., Frackelton, E. C., Otieno, F. G., Shaner, J. L., Smith, R. M., Imielinski, M., Eckert, A. W., Chiavacci, R. M., Berkowitz, R. I., and Hakonarson, H. (2008). Association analysis of the FTO gene with obesity in children of Caucasian and African ancestry reveals a common tagging SNP. PLoS ONE 3, e1746. doi: 10.1371/journal.pone.0001746
Gray, J., Yeo, G. S., Cox, J. J., Morton, J., Adlam, A. L., Keogh, J. M., Yanovski, J. A., El Gharbawy, A., Han, J. C., Tung, Y. C., Hodges, J. R., Raymond, F. L., O’Rahilly, S., and Farooqi, I. S. (2006). Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 55, 3366–3371.
Hakanen, M., Raitakari, O. T., Lehtimäki, T., Peltonen, N., Pahkala, K., Sillanmäki, L., Lagström, H., Viikari, J., Simell, O., and Rönnemaa, T. (2009). FTO genotype is associated with body mass index after the age of seven years but not with energy intake or leisure-time physical activity. J. Clin. Endocrinol. Metab. 94, 1281–1287.
Han, J. C., Liu, Q. R., Jones, M., Levinn, R. L., Menzie, C. M., Jefferson-George, K. S., Adler-Wailes, D. C., Sanford, E. L., Lacbawan, F. L., Uhl, G. R., Rennert, O. M., and Yanovski, J. A. (2008). Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 359, 918–927.
Han, Z., Niu, T., Chang, J., Lei, X., Zhao, M., Wang, Q., Cheng, W., Wang, J., Feng, Y., and Chai, J. (2010). Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature 464, 1205–1209.
Haupt, A., Thamer, C., Staiger, H., Tschritter, O., Kirchhoff, K., Machicao, F., Häring, H. U., Stefan, N., and Fritsche, A. (2009). Variation in the FTO gene influences food intake but not energy expenditure. Exp. Clin. Endocrinol. Diabetes 117, 194–197.
Hennig, B. J., Fulford, A. J., Sirugo, G., Rayco-Solon, P., Hattersley, A. T., Frayling, T. M., and Prentice, A. M. (2009). FTO gene variation and measures of body mass in an African population. BMC Med. Genet. 10, 21. doi: 10.1186/1471-2350-10-21
Hill, J. O., and Peters, J. C. (1998). Environmental contributions to the obesity epidemic. Science 280, 1371–1374.
Hotta, K., Nakata, Y., Matsuo, T., Kamohara, S., Kotani, K., Komatsu, R., Itoh, N., Mineo, I., Wada, J., Masuzaki, H., Yoneda, M., Nakajima, A., Miyazaki, S., Tokunaga, K., Kawamoto, M., Funahashi, T., Hamaguchi, K., Yamada, K., Hanafusa, T., Oikawa, S., Yoshimatsu, H., Nakao, K., Sakata, T., Matsuzawa, Y., Tanaka, K., Kamatani, N., and Nakamura, Y. (2008). Variations in the FTO gene are associated with severe obesity in the Japanese. J. Hum. Genet. 53, 546–553.
Jackson, R. S., Creemers, J. W., Ohagi, S., Raffin-Sanson, M. L., Sanders, L., Montague, C. T., Hutton, J. C., and O’Rahilly, S. (1997). Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 16, 303–306.
Jacobsson, J. A., Klovins, J., Kapa, I., Danielsson, P., Svensson, V., Ridderstråle, M., Gyllensten, U., Marcus, C., Fredriksson, R., and Schiöth, H. B. (2008). Novel genetic variant in FTO influences insulin levels and insulin resistance in severely obese children and adolescents. Int. J. Obes. (Lond.) 32, 1730–1735.
Jia, G., Yang, C. G., Yang, S., Jian, X., Yi, C., Zhou, Z., and He, C. (2008). Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 582, 3313–3319.
Jonsson, A., and Franks, P. W. (2009). Obesity, FTO gene variant, and energy intake in children. N. Engl. J. Med. 360, 1571–1572; author reply 1572.
Jonsson, A., Renström, F., Lyssenko, V., Brito, E. C., Isomaa, B., Berglund, G., Nilsson, P. M., Groop, L., and Franks, P. W. (2009). Assessing the effect of interaction between an FTO variant (rs9939609) and physical activity on obesity in 15,925 Swedish and 2,511 Finnish adults. Diabetologia 52, 1334–1338.
Karasawa, S., Daimon, M., Sasaki, S., Toriyama, S., Oizumi, T., Susa, S., Kameda, W., Wada, K., Muramatsu, M., Fukao, A., Kubota, I., Kawata, S., Kayama, T., and Kato, T. (2010). Association of the common fat mass and obesity associated (FTO) gene polymorphism with obesity in a Japanese population. Endocr. J. 57, 293–301.
Katzmarzyk, P. T., Janssen, I., and Ardern, C. I. (2003). Physical inactivity, excess adiposity and premature mortality. Obes. Rev. 4, 257–290.
Keebler, M. E., Deo, R. C., Surti, A., Konieczkowski, D., Guiducci, C., Burtt, N., Buxbaum, S. G., Sarpong, D. F., Steffes, M. W., Wilson, J. G., Taylor, H. A., and Kathiresan, S. (2010). Fine-mapping in African Americans of 8 recently discovered genetic loci for plasma lipids: the Jackson Heart Study. Circ. Cardiovasc. Genet. 3, 358–364.
Krude, H., Biebermann, H., Luck, W., Horn, R., Brabant, G., and Grüters, A. (1998). Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157.
Legry, V., Cottel, D., Ferrières, J., Arveiler, D., Andrieux, N., Bingham, A., Wagner, A., Ruidavets, J. B., Ducimetière, P., Amouyel, P., and Meirhaeghe, A. (2009). Effect of an FTO polymorphism on fat mass, obesity, and type 2 diabetes mellitus in the French MONICA Study. Metab. Clin. Exp. 58, 971–975.
Leibel, R. L. (2008). Energy in, energy out, and the effects of obesity-related genes. N. Engl. J. Med. 359, 2603–2604.
Loos, R. J., Lindgren, C. M., Li, S., Wheeler, E., Zhao, J. H., Prokopenko, I., Inouye, M., Freathy, R. M., Attwood, A. P., Beckmann, J. S., Berndt, S. I.; Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, Jacobs, K. B., Chanock, S. J., Hayes, R. B., Bergmann, S., Bennett, A. J., Bingham, S. A., Bochud, M., Brown, M., Cauchi, S., Connell, J. M., Cooper, C., Smith, G. D., Day, I., Dina, C., De, S., Dermitzakis, E. T., Doney, A. S., Elliott, K. S., Elliott, P., Evans, D. M., Sadaf Farooqi, I., Froguel, P., Ghori, J., Groves, C. J., Gwilliam, R, Hadley, D., Hall, A. S., Hattersley, A. T., Hebebrand, J., Heid, I. M.; KORA, Lamina, C., Gieger, C., Illig, T., Meitinger, T., Wichmann, H. E., Herrera, B., Hinney, A., Hunt, S. E., Jarvelin, M. R., Johnson, T., Jolley, J. D., Karpe, F., Keniry, A., Khaw, K. T., Luben, R. N., Mangino, M., Marchini, J., McArdle, W. L., McGinnis, R., Meyre, D., Munroe, P. B., Morris, A. D., Ness, A. R., Neville, M. J., Nica, A. C., Ong, K. K., O’Rahilly, S., Owen, K. R., Palmer, C. N., Papadakis, K., Potter, S., Pouta, A., Qi L; Nurses’ Health Study, Randall, J. C., Rayner, N. W., Ring, S. M., Sandhu, M. S., Scherag, A., Sims, M. A., Song, K., Soranzo, N., Speliotes, E. K.; Diabetes Genetics Initiative, Syddall, H. E., Teichmann, S. A., Timpson, N. J., Tobias, J. H., Uda M; SardiNIA Study, Vogel, C. I., Wallace, C., Waterworth, D. M., Weedon, M. N.; Wellcome Trust Case Control Consortium, Willer, C. J.; FUSION, Wraight, Yuan, X., Zeggini, E., Hirschhorn, J. N., Strachan, D. P., Ouwehand, W. H., Caulfield, M. J., Samani, N. J., Frayling, T. M., Vollenweider, P., Waeber, G., Mooser, V., Deloukas, P., McCarthy, M. I., Wareham, N. J., Barroso, I., Jacobs, K. B., Chanock, S. J., Hayes, R. B., Lamina, C., Gieger, C., Illig, T., Meitinger, T., Wichmann, H. E., Kraft, P., Hankinson, S. E., Hunter, D. J., Hu, F. B., Lyon, H. N., Voight, B. F., Ridderstrale, M., Groop, L., Scheet, P., Sanna, S., Abecasis, G. R., Albai, G., Nagaraja, R., Schlessinger, D., Jackson, A. U., Tuomilehto, J., Collins, F. S., Boehnke, M., and Mohlke, K. L. (2008). Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet. 40, 768–775.
Meyre, D., Proulx, K., Kawagoe-Takaki, H., Vatin, V., Gutiérrez-Aguilar, R., Lyon, D., Ma, M., Choquet, H., Horber, F., Van Hul, W., Van Gaal, L., Balkau, B., Visvikis-Siest, S., Pattou, F., Farooqi, I. S., Saudek, V., O’Rahilly, S., Froguel, P., Sedgwick, B., and Yeo, G. S. (2010). Prevalence of loss-of-function FTO mutations in lean and obese individuals. Diabetes 59, 311–318.
Okazaki, N., Kikuno, R., Ohara, R., Inamoto, S., Aizawa, H., Yuasa, S., Nakajima, D., Nagase, T., Ohara, O., and Koga, H. (2003). Prediction of the coding sequences of mouse homologues of KIAA gene: II. The complete nucleotide sequences of 400 mouse KIAA-homologous cDNAs identified by screening of terminal sequences of cDNA clones randomly sampled from size-fractionated libraries. DNA Res. 10, 35–48.
Peeters, A., Beckers, S., Verrijken, A., Roevens, P., Peeters, P., Van Gaal, L., and Van Hul, W. (2008). Variants in the FTO gene are associated with common obesity in the Belgian population. Mol. Genet. Metab. 93, 481–484.
Peters, T., Ausmeier, K., Dildrop, R., and Ruther, U. (2002). The mouse Fused toes (Ft) mutation is the result of a 1.6-Mb deletion including the entire Iroquois B gene cluster. Mamm. Genome 13, 186–188.
Peters, T., Ausmeier, K., and Ruther, U. (1999). Cloning of Fatso (Fto), a novel gene deleted by the Fused toes (Ft) mouse mutation. Mamm. Genome 10, 983–986.
Robbens, S., Rouzé, P., Cock, J. M., Spring, J., Worden, A. Z., and Van de Peer, Y. (2008). The FTO gene, implicated in human obesity, is found only in vertebrates and marine algae. J. Mol. Evol. 66, 80–84.
Scuteri, A., Sanna, S., Chen, W. M., Uda, M., Albai, G., Strait, J., Najjar, S., Nagaraja, R., Orrú, M., Usala, G., Dei, M., Lai, S., Maschio, A., Busonero, F., Mulas, A., Ehret, G. B., Fink, A. A., Weder, A. B., Cooper, R. S., Galan, P., Chakravarti, A., Schlessinger, D., Cao, A., Lakatta, E., and Abecasis, G. R. (2007). Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 3, e115. doi: 10.1371/journal.pgen.0030115
Song, Y., You, N. C., Hsu, Y. H., Howard, B. V., Langer, R. D., Manson, J. E., Nathan, L., Niu, T., F Tinker, L., and Liu, S. (2008). FTO polymorphisms are associated with obesity but not diabetes risk in postmenopausal women. Obesity (Silver Spring) 16, 2472–2480.
Speakman, J. R., Rance, K. A., and Johnstone, A. M. (2008). Polymorphisms of the FTO gene are associated with variation in energy intake, but not energy expenditure. Obesity (Silver Spring) 16, 1961–1965.
Stratigopoulos, G., Padilla, S. L., LeDuc, C. A., Watson, E., Hattersley, A. T., McCarthy, M. I., Zeltser, L. M., Chung, W. K., and Leibel, R. L. (2008). Regulation of Fto/Ftm gene expression in mice and humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1185–R1196.
Stratigopoulos, G., Leduc, C. A., Cremona, M. L., Chung, W. K., and Leibel, R. L. (2011). Cut-like homeobox 1 (CUX1) regulates expression of the fat mass and obesity-associated and retinitis pigmentosa GTPase regulator-interacting protein-1-like (RPGRIP1L) genes and coordinates leptin receptor signaling. J. Biol. Chem. 286, 2155–2170.
Stutzmann, F., Vatin, V., Cauchi, S., Morandi, A., Jouret, B., Landt, O., Tounian, P., Levy-Marchal, C., Buzzetti, R., Pinelli, L., Balkau, B., Horber, F., Bougnères, P., Froguel, P., and Meyre, D. (2007). Non-synonymous polymorphisms in melanocortin-4 receptor protect against obesity: the two facets of a Janus obesity gene. Hum. Mol. Genet. 16, 1837–1844.
Sun, Y., Sun, J., Wang, X., You, W., and Yang, M. (2010). Variants in the fat mass and obesity associated (FTO) gene are associated with obesity and C-reactive protein levels in Chinese Han populations. Clin. Invest. Med. 33, E405–E412.
Tan, J. T., Dorajoo, R., Seielstad, M., Sim, X. L., Ong, R. T., Chia, K. S., Wong, T. Y., Saw, S. M., Chew, S. K., Aung, T., and Tai, E. S. (2008). FTO variants are associated with obesity in the Chinese and Malay populations in Singapore. Diabetes 57, 2851–2857.
Tanofsky-Kraff, M., Han, J. C., Anandalingam, K., Shomaker, L. B., Columbo, K. M., Wolkoff, L. E., Kozlosky, M., Elliott, C., Ranzenhofer, L. M., Roza, C. A., Yanovski, S. Z., and Yanovski, J. A. (2009). The FTO gene rs9939609 obesity-risk allele and loss of control over eating. Am. J. Clin. Nutr. 90, 1483–1488.
Thorleifsson, G., Walters, G. B., Gudbjartsson, D. F., Steinthorsdottir, V., Sulem, P., Helgadottir, A., Styrkarsdottir, U., Gretarsdottir, S., Thorlacius, S., Jonsdottir, I., Jonsdottir, T., Olafsdottir, E. J., Olafsdottir, G. H., Jonsson, T., Jonsson, F., Borch-Johnsen, K., Hansen, T., Andersen, G., Jorgensen, T., Lauritzen, T., Aben, K. K., Verbeek, A. L., Roeleveld, N., Kampman, E., Yanek, L. R., Becker, L. C., Tryggvadottir, L., Rafnar, T., Becker, D. M., Gulcher, J., Kiemeney, L. A., Pedersen, O., Kong, A., Thorsteinsdottir, U., and Stefansson, K. (2009). Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 41, 18–24.
Timpson, N. J., Emmett, P. M., Frayling, T. M., Rogers, I., Hattersley, A. T., McCarthy, M. I., and Davey Smith, G. (2008). The fat mass- and obesity-associated locus and dietary intake in children. Am. J. Clin. Nutr. 88, 971–978.
Tokue, S., Sasaki, M., and Nakahata, N. (2009). Thromboxane A2-induced signal transduction is negatively regulated by KIAA1005 that directly interacts with thromboxane A2 receptor. Prostaglandins Other Lipid Mediat. 89, 8–15.
Tung, Y. C., Ayuso, E., Shan, X., Bosch, F., O’Rahilly, S., Coll, A. P., and Yeo, G. S. (2010). Hypothalamic-specific manipulation of Fto, the ortholog of the human obesity gene FTO, affects food intake in rats. PLoS ONE 5, e8771. doi: 10.1371/journal.pone.0008771
Vaisse, C., Clement, K., Guy-Grand, B., and Froguel, P. A. (1998). frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 20, 113–114.
van der Hoeven, F., Schimmang, T., Volkmann, A., Mattei, M. G., Kyewski, B., and Rüther, U. (1994). Programmed cell death is affected in the novel mouse mutant Fused toes (Ft). Development 120, 2601–2607.
Wardle, J., Llewellyn, C., Sanderson, S., and Plomin, R. (2008). Obesity associated genetic variation in FTO is associated with diminished satiety. J. Clin. Endocrinol. Metab. 93, 3640–3643.
Wardle, J., Llewellyn, C., Sanderson, S., and Plomin, R. (2009). The FTO gene and measured food intake in children. Int. J. Obes. (Lond.) 33, 42–45.
Willer, C. J., Speliotes, E. K., Loos, R. J., Li, S., Lindgren, C. M., Heid, I. M., Berndt, S. I., Elliott, A. L., Jackson, A. U., Lamina, C., Lettre, G., Lim, N., Lyon, H. N., McCarroll, S. A., Papadakis, K., Qi, L., Randall, J. C., Roccasecca, R. M., Sanna, S., Scheet, P., Weedon, M. N., Wheeler, E., Zhao, J. H., Jacobs, L. C., Prokopenko, I., Soranzo, N., Tanaka, T., Timpson, N. J., Almgren, P., Bennett, A., Bergman, R. N., Bingham, S. A., Bonnycastle, L. L., Brown, M., Burtt, N. P., Chines, P., Coin, L., Collins, F. S., Connell, J. M., Cooper, C., Smith, G. D., Dennison, E. M., Deodhar, P., Elliott, P., Erdos, M. R., Estrada, K., Evans, D. M., Gianniny, L., Gieger, C., Gillson, C. J., Guiducci, C., Hackett, R., Hadley, D., Hall, A. S., Havulinna, A. S., Hebebrand, J., Hofman, A., Isomaa, B., Jacobs, K. B., Johnson, T., Jousilahti, P., Jovanovic, Z., Khaw, K. T., Kraft, P., Kuokkanen, M., Kuusisto, J., Laitinen, J., Lakatta, E. G., Luan, J., Luben, R. N., Mangino, M., McArdle, W. L., Meitinger, T., Mulas, A., Munroe, P. B., Narisu, N., Ness, A. R., Northstone, K., O’Rahilly, S., Purmann, C., Rees, M. G., Ridderstråle, M., Ring, S. M., Rivadeneira, F., Ruokonen, A., Sandhu, M. S., Saramies, J., Scott, L. J., Scuteri, A., Silander, K., Sims, M. A., Song, K., Stephens, J., Stevens, S., Stringham, H. M., Tung, Y. C., Valle, T. T., Van Duijn, C. M., Vimaleswaran, K. S., Vollenweider, P., Waeber, G., Wallace, C., Watanabe, R. M., Waterworth, D. M., Watkins N; Wellcome Trust Case Control Consortium, Witteman, J. C., Zeggini, E., Zhai, G., Zillikens, M. C., Altshuler, D., Caulfield, M. J., Chanock, S. J., Farooqi, I. S., Ferrucci, L., Guralnik, J. M., Hattersley, A. T., Hu, F. B., Jarvelin, M. R., Laakso, M., Mooser, V., Ong, K. K., Ouwehand, W. H., Salomaa, V., Samani, N. J., Spector, T. D., Tuomi, T., Tuomilehto, J., Uda, M., Uitterlinden, A. G., Wareham, N. J., Deloukas, P., Frayling, T. M., Groop, L. C., Hayes, R. B., Hunter, D. J., Mohlke, K. L., Peltonen, L., Schlessinger, D., Strachan, D. P., Wichmann, H. E., McCarthy, M. I., Boehnke, M., Barroso, I., Abecasis, G. R., Hirschhorn, J. N.; Genetic Investigation of ANthropometric Traits Consortium. (2009). Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 41, 25–34.
Yeo, G. S., Farooqi, I. S., Aminian, S., Halsall, D. J., Stanhope, R. G., and O’Rahilly, S. (1998). A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat. Genet. 20, 111–112.
Keywords: FTO, obesity, food intake, hypothalamus, GWAS, gene
Citation: Cheung M-KM and Yeo GSH (2011) FTO biology and obesity: why do a billion of us weigh 3 kg more? Front. Endocrin. 2:4. doi: 10.3389/fendo.2011.00004
Received: 28 December 2010;
Paper pending published: 18 January 2011;
Accepted: 07 February 2011;
Published online: 22 February 2011.
Edited by:
Paul Thomas Pfluger, University of Cincinnati, USAReviewed by:
Miguel Lopez, University of Santiago de Compostela, SpainDarlene Evans Berryman, Ohio University, USA
Copyright: © 2011 Cheung and Yeo. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Giles S. H. Yeo, Metabolic Research Labs, Level 4, Institute of Metabolic Science, Box 289, Addenbrooke’s Hospital, University of Cambridge, Cambridge CB2 0QQ, UK.e-mail: gshy2@cam.ac.uk