 Cédric Dessimoz1 Patrick Browaeys2 Philippe Maeder2 Benoît Lhermitte3 Nelly Pitteloud4 Shahan Momjian5 François P. Pralong4*
Cédric Dessimoz1 Patrick Browaeys2 Philippe Maeder2 Benoît Lhermitte3 Nelly Pitteloud4 Shahan Momjian5 François P. Pralong4*- 1 Centre for Infectious Diseases and Epidemiology, Central Institute of the Valais Hospitals, Sion, Switzerland
- 2 Department of Radiology, Lausanne University Hospital, Lausanne, Switzerland
- 3 Department of Histopathology, Lausanne University Hospital, Lausanne, Switzerland
- 4 Service of Endocrinology, Diabetology and Metabolism, Lausanne University Hospital, Lausanne, Switzerland
- 5 Service of Neurosurgery, Hôpitaux Universitaires de Genève, Geneva, Switzerland
Combined prolactin (PRL) and growth hormone (GH) secretion by a single pituitary tumor can occur in approximately 5% of cases. However, in all previously reported patients, combined secretion of both hormones was present at the time of diagnosis. Here we describe a patient initially diagnosed with a pure prolactin-secreting microadenoma, who experienced the progressive apparition of symptomatic autonomous GH secretion while on intermittent long term dopamine agonist therapy. She was operated on, and immunohistochemical analysis of tumor tissue confirmed the diagnosis of pituitary adenoma with uniform co-staining of all cells for both GH and PRL. This patient represents the first documented occurrence of asynchronous development of combined GH and PRL secretion in a pituitary adenoma. Although pathogenic mechanisms implicated remain largely speculative, it emphasizes the need for long term hormonal follow up of patients harboring prolactinomas.
Introduction
Approximately 10% of all clinically overt intracranial neoplasms arise from the pituitary gland and at autopsy, if histological examination of unselected autopsy material is to be believed, adenomas are present in as many as one in five cases (Levy and Lightman, 1993). Prolactin-secreting adenomas are the most common of them, representing about 40% of adenomas (Mindermann and Wilson, 1994). Prevalence has classically been estimated at 6–10 per 100,000 inhabitants, but more recent data suggest it could be as high as 62 per 100,000 inhabitants (Daly et al., 2006). Early diagnosis of mixed adenomas is rendered difficult by the fact that they generally present with signs or symptoms that can be related to overproduction of a single hormone. Clinical diagnosis of combined anterior pituitary hormone excess is thus a rare instance, and these adenomas are mostly identified after neurosurgery, at the time of pathological diagnosis.
The most frequent combination of pituitary hormone excess associates growth hormone (GH) and prolactin (PRL); the combination of GH, PRL, and the alpha subunit (α-SU) of the glycoprotein hormones is also possible, whereas other hormone combinations within a single tumor are extremely rare (Kovacs et al., 1998; Ma et al., 2002). Ultrastructural and immune electron microscopic data allow to classify bi-hormonal or pluri-hormonal adenomas in three separate classes: monomorphous, bimorphous, and plurimorphous tumors. Monomorphous adenomas consist of a single cell type with synthesis of two or more hormones in the same cell. Bimorphous and plurimorphous adenomas are composed of two or more cell types, and each hormone is produced by a different cell population. However, overlaps may exist, and some tumors can consist in a mix of cells, some of them producing only one hormone and others producing two or more hormones. In some adenomas, one cell type can predominate; in others, various areas contain groups of similar cells, or a gradual transformation seems to exist between the cell types. Despite these overlaps, the classification of tumors into monomorphous, bimorphous, and plurimorphous types for diagnostic purposes retains its value and remains a useful practical tool (Kovacs et al., 1998). In this paper, we describe a patient who presented initially with a pure PRL secreting pituitary adenoma, and who secondarily developed co-secretion of GH with time.
Case Report
Here we present the case of female patient who delivered her first child in July 1996 at the age of 31 years, after spontaneous conception and an uneventful pregnancy. She did not breast feed her baby, and was seen again in May 1997, where she complained of persistent secondary amenorrhea and intermittent galactorrhea of 10 months’ duration. No specific investigations were performed at the time, and substitutive treatment with Estradiol and Norgestrel (Cyclacur®) was initiated by her treating gynecologist. This resulted in regular menstruations, but the treatment was stopped by the patient in September 1997 because of a desire of pregnancy, allowing to confirm the persistence of secondary amenorrhea.
She was otherwise in good general health, had no particular complaint and was on no medication, only reporting having lost 10 kg after her first pregnancy. On physical exam, she was a healthy appearing young woman weighing 51 kg for 162 cm, the only positive finding being the presence of bilateral galactorrhea on stimulation. A pregnancy test was negative, and investigation of anterior pituitary function disclosed the following baseline hormone levels (November 1997): LH 1.6 U/l (N: 2–12), FSH 4.8 U/l (N: 4–12), oestradiol 0.2 nmol/l (N: 0.14–0.34); PRL 42 μg/l (N: 3.4–24.1); TSH 0.79 mU/l (N: 0.2–3.5), free T4 15 pmol/l (N: 8–22). GH and IGF-1 levels were reportedly within normal limits at the time. The patient refused to undergo a pituitary MRI because of claustrophobia, and a head CT scan disclosed the presence of a microadenoma (7 × 4 mm) in the left lobe of the pituitary gland.
The diagnosis of microprolactinoma was retained, based on the good correlation between the relatively small size of the adenoma and the moderate, albeit symptomatic, elevation of prolactin levels. Thus, dopamine agonist therapy with Quinagolide (Norprolac® 75 mg/day) was initiated in January 1998. A few weeks later, the patient became pregnant, treatment was stopped, and an uneventful pregnancy resulted in the delivery of a normal second child in October 1998. The patient was able to breast feed her baby. At the follow up visit in March 1999, the patient was still amenorrheic and she had persistent bilateral galactorrhea on stimulation. Her baseline prolactin level was 66 μg/l (N: 2–19), and IGF-1 was normal at 342 μg/l (N: 98–442) in July 1999. A repeat head CT scan confirmed the presence of a stable microadenoma. She then refused to take any medication, and also refused to take oral contraception.
Over the course of the following years, her baseline prolactin levels increased slowly but steadily, reaching 92 μg/l (N: 2–19) in 2002. Because of the risk of osteoporosis carried by long term amenorrhea, the patient then agreed upon taking dopamine agonist therapy and was started on oral Cabergoline (Dostinex® 0.5 mg/week) in June 2002. As anticipated, her serum prolactin levels declined, galactorrhea disappeared and by October 2002, regular ovulatory cycles had resumed. However, she experienced a minor depression on cabergoline that required transient treatment (August 2002–June 2003) with the selective serotonin re-uptake inhibitor nefazodone (Nefadar®). In May 2004, recurrence of amenorrhea under treatment motivated a repeat head CT scan which disclosed a small decrease in size of the adenoma. At the time, serum PRL levels were stable at 57 μg/l and notably did not exhibit any significant change despite the transient treatment with a selective serotonin re-uptake inhibitor that had occurred a few months before. Cabergoline was increased to 0.75 mg/week, with return of the menses. The first MRI was obtained in June 2005, showing an oval hypointense (T1) nodule in the left lobe of the pituitary gland, measuring 5 mm by 3 mm (i.e., smaller than on all previous imaging studies). In December of that year, the patient decided to stop taking cabergoline. She became amenorrheic again shortly thereafter, with Prolactin levels rising to 110 μg/l in August 2006, prompting re-introduction of the treatment. IGF-1 levels at that time were evaluated in a novel assay because of a change in the methodology used at our hospital and amounted to 356 μg/l (N: 106–276, see Figure 1).
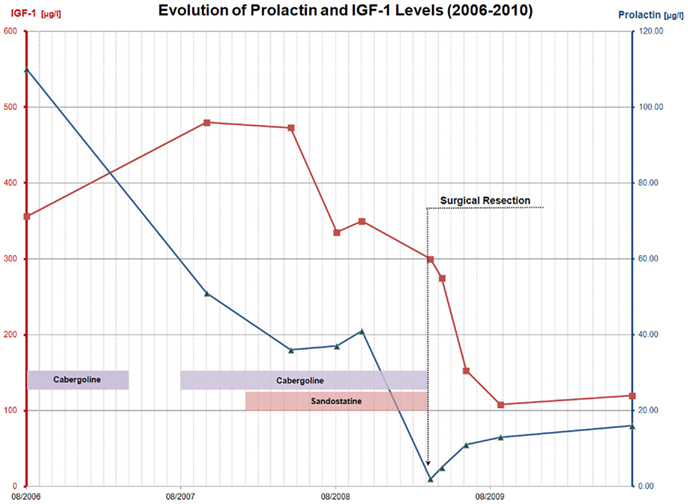
Figure 1. Summary of the evolution of prolactin and IGF-1 levels over time, and with respect to the various treatments administered between August 2006 and July 2010. This graph illustrates the sudden rise in IGF-1 levels (squares) between 2006 and 2007, as well as the good response to long-acting octreotide therapy and the resolution of both IGF-1 and prolactin (triangles) hypersecretion after trans-sphenoidal surgery.
Once again, therapy was stopped by the patient in April 2007, an interruption that was followed by another episode of amenorrhea accompanied by intermittent galactorrhea. At her next yearly follow up visit in November 2007, she mentioned spontaneously an increase in size of her fingers. On specific questioning, she also reported disseminated arthralgias, and symptoms suggestive of carpal tunnel syndrome. Baseline hormone work up at the time was as follows: PRL was stable at 51 μg/l (N: 4–29), but IGF-1 was significantly higher than a few months before, at 480 μg/l (N: 98–261). A repeat pituitary MRI was therefore obtained in November 2007 (see Figure 2A) disclosing the existence of a mixed solid and cystic nodule in the left lobe of the anterior pituitary gland, measuring 6 by 10 mm. This nodule was larger than on previous imaging studies, and was located in contact with the left cavernous sinus without sign of invasion. This MRI exam also showed a second lesion localized at the root of the pituitary stalk, measuring 3 by 3 mm, corresponding to a small cyst. A GH suppression test with oral glucose was then performed, confirming autonomous GH secretion (Table 1).
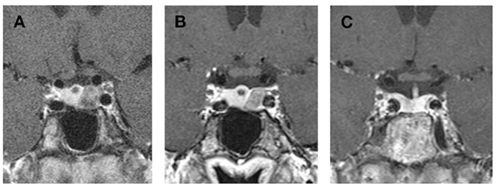
Figure 2. Evolution of the adenoma on long-acting octreotide therapy (A,B) and result of trans-sphenoidal surgery [(C), i.e., 17 months after surgery]. (A) MRI November 2007 (Coronal T1 after gadolinium injection): oval hypointense nodule in the left lobe of pituitary gland, measuring 10 mm on 6 mm. No invasion of the cavernous sinus. Small cyst at the root of the pituitary stalk. (B) MRI March 2009 (Coronal T1 after gadolinium injection): in comparison to November 2007, small decrease (under long-acting octreotide therapy) of left microadenoma, still in contact with the left cavernous sinus (without any sign of invasion). (C) MRI August 2010 (Coronal T1 after gadolinium injection): the adenoma has been removed leaving only minor scar tissue in the left anterior pituitary lobe. The small cyst has gradually disappeared.
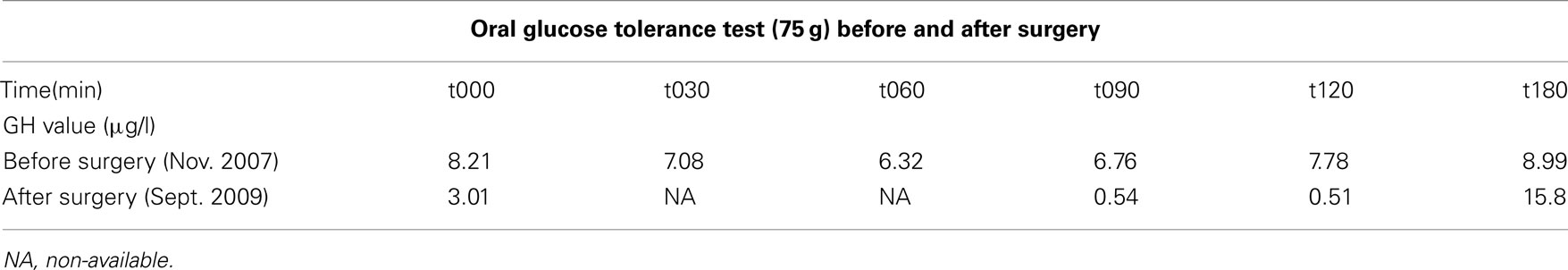
Table 1. Results of the oral glucose tolerance tests (=oGTT) performed before and after successful trans-sphenoidal surgery, demonstrating the lack of inhibition of GH before surgery, and the inhibition of GH levels to a nadir of 0.51 ng/mL followed by physiological rebound at 180 min after surgery.
Because of the apparition of GH hypersecretion, long-acting octreotide therapy (Sandostatin LAR® 10 mg a month, increased to 20 mg after 3 months) was started in January 2008, with good resolution of symptoms. IGF-1 levels, which were still elevated at 473 μg/l in April 2008, decreased within a few months to reach a nadir of 335 μg/l (N: 98–261) in August 2008. At that time, IGF-BP3 levels were within normal limits at 5.2 mg/l (N: 3.3–6–6) and prolactin was stable at 37 μg/l (N: 4–29) on oral Cabergoline (Dostinex® 0.5 mg/week). A repeat pituitary MRI after 6 months (not shown) disclosed no change. The dose of long-acting octreotide therapy was increased to 30 mg a month, without complete normalization of IGF-1 values (350 μg/l, N: 98–261) in October 2008. A repeat pituitary MRI in March 2009 showed a small decrease of the microadenoma, which was still in contact with the left cavernous sinus. The cystic lesion at the root of pituitary stalk was stable (see Figure 2B).
Resection of this microadenoma was performed by trans-phenoidal approach in March 2009 (i.e., after 13 months of long-acting octreotide treatment). Preoperative IGF-1 and IGF-BP3 levels were 275 μg/l (N: 98–261) and 3.8 mg/l (N: 3.3–6–6), respectively. Prolactin was normal at 5 μg/l (N: 4–29). Peri-operative course was uneventful. Pathological exam of tumor tissue confirmed the diagnosis of pituitary adenoma with monomorphous staining for GH and PRL at immunohistochemistry (see Figure 3). There was no expression of LH, FSH, TSH, ACTH, or HCG by the tumor.
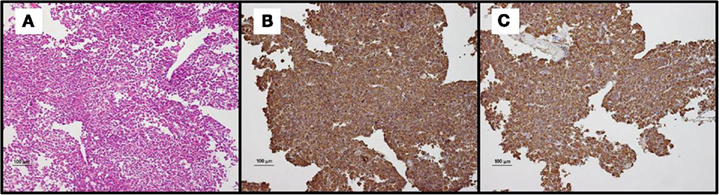
Figure 3. Histopathological exam of resected tissue. (A) Hematoxylin-eosin staining (Magnification ×10): pituitary adenoma composed of round to polygonal cells with abundant eosinophilic or granular cytoplasm. The cells are arranged in solid nests or trabeculae. (B) Immunohistochemical analysis (Magnification ×10) with an anti-GH antibody (polyclonal), demonstrating monomorphous expression of GH in the majority of cells. (C) Immunohistochemical analysis (Magnification ×10) with an anti-PRL antibody (polyclonal), demonstrating monomorphous expression of PRL in the majority of cells.
All therapy was withheld and at the 3-months follow up visit, IGF-1 and IGF-BP3 levels were normal at 153 μg/l (N: 98–261) and 4.8 mg/l (N: 3.3–6–6) respectively. Prolactin was also normal at 11 μg/l (N: 4–29), as was the remaining of anterior pituitary function. Biological remission of the disease was confirmed at 6 months, and performance of a GH suppression test with oral glucose showed a nadir GH level of 0.51 μg/l after 120 min. The control MRI realized in 2010 showed no residual tumor and the small cyst had also disappeared (see Figure 2C). Blood hormone levels were as follows (July 2010): PRL 16 μg/l (N: 4–29), IGF-1 120 μg/l (N: 98–261), and IGF-BP3 3.8 mg/l (N: 3.3–6–6).
Discussion
Combined prolactin and GH secretion has been reported in 5% of all pituitary tumors, an co-secretion is usually diagnosed simultaneously (Kasantikul and Shuangshoti, 1990). Mixed prolactin and GH secreting pituitary adenomas are relatively common because somatotrophs and lactotrophs share the common somato-mammotroph progenitor lineage. Conversely, the occurrence of a prolactinoma evolving into clinically and biochemically active acromegaly seems to be a rare phenomenon. In a recent report, Lania et al. (2010) describe secondary apparition of GH hypersecretion. However, in contrast to the patient described here, the primary tumor in their report was an aggressive prolactinoma, suggesting a different pathophysiological mechanism from the start. In this context, older papers describing the occurrence of combined elevations of GH and prolactin do not allow to differentiate between concomitant or sequential development of the individual hypersecretion syndromes (Tournaire et al., 1985; Goldman and Klinges, 1989; Pagesy et al., 1991). Moreover, it should also be reminded that elevated prolactin levels in the context of a somatotroph adenoma can also result from desinhibition of lactotroph cells by pituitary stalk compression.
In contrast to these previous reports, the patient discussed here is remarkable in the sense that symptoms of GH excess appeared progressively, several years after the diagnosis of microprolactinoma. Very importantly, clinical symptoms were well correlated with the biological findings. Indeed, an elevation of IGF-1 levels was not observed before 2006, and the first spontaneous complaint suggestive of GH excess was recorded in 2007. The fact that once documented, these elevated IGF-1 levels continued to increase steadily on further testing, together with the good correlation with clinical symptoms, argues very much in favor of secondary apparition of GH hypersecretion and against an artifact related to the change in the IGF-1 assay that occurred in 2005. This is further confirmed by the clearly abnormal results of the oral glucose suppression test, confirming the abnormal GH secretion.
Interestingly, this change in phenotype from a pure prolactin-secreting adenoma into a mixed somato-lactotroph adenoma was accompanied by significant growth of the known pituitary tumor, after several years of stability in size. This unfortunate evolution, in parallel with the change in the secretory profile of the tumor, could therefore be interpreted as a sign of de-differentiation. Indeed, hormone-specific anterior pituicytes are embryologically derived from a pluripotent precursor. The process of anterior pituitary cell development and differentiation arises as a consequence of concerted spatio-temporal expression or repression of a series of transcription factors to produce fully differentiated cells of the various lineages (Melmed, 2003). In this differentiation process, prolactin-expressing cells are just one step downstream from cells expressing both GH and prolactin.
At that point of the clinical evolution, and regardless of the pathogenic mechanisms implicated, the indication for surgical resection of the adenoma was retained. Given the close vicinity of the cavernous sinus, pre-treatment with a long-acting somatostatin analog was initiated in order to increase the chance of curative trans-sphenoidal surgery (Ferone et al., 2000). This option proved successful since medical treatment induced a significant reduction of the tumor size, ultimately allowing radiologically complete resection of all tumor tissue. Immunohistochemical exam of the removed tumor disclosed a pituitary adenoma with uniform co-staining for both GH and PRL, confirming the hypothesis of an evolution into a mixed somato-mammotroph pituitary adenoma.
The cytogenesis of pituitary adenomas that consist of two different cell populations is not known and remains to be elucidated (Kovacs et al., 1998), although different hypotheses have been proposed. Most, if not all, of these adenomas are monoclonal, as demonstrated by X-inactivation studies (Herman et al., 1990; Ma et al., 2002). Moreover, at the end of the eighties, an activating mutation of the α-SU of G proteins was identified in somatotroph cells of up to 40% of sporadic GH-secreting pituitary adenomas in Caucasians (Landis et al., 1990; Levy and Lightman, 1993). The resulting oncogene, gsp, was thought to induce tumorigenesis by virtue of persistent activation of adenylyl cyclase, with subsequent GH hypersecretion (Landis et al., 1989; Eugster and Pescovitz, 1999). This report constitutes the demonstration that specific molecular abnormalities may form the basis of pituitary adenoma formation and hormone hypersecretion, at least in some specific cases.
Alternatively, it may be hypothesized that such mixed pituitary adenomas are not monoclonal, and that the insult that causes the neoplastic transformation may affect two different cell types. It is also conceivable that some pituitary tumors originate in an uncommitted stem cell which, because of unknown factors, can differentiate into two separate cell types. Such multidirectional differentiation could explain the development of some pluri-hormonal tumors (Kovacs et al., 1998).
Another hypothesis would be that one cell type can “transdifferentiate” to another cell type as a result of subsequent mutations during tumor progression. Such phenomenon involves reversible transformation of one cell type to another by phenotypic switches, without cell division (Senovilla et al., 2004). It takes place in a few pituitary cell types under physiological conditions. This concept of transdifferentiation was for example introduced to explain the existence of “somato-mammotrope” cells, a cell type that stores and secretes both GH and PRL which is generated by the conversion of somatotropes into mammotrophs during situations demanding large amounts of PRL such as lactation (Frawley and Boockfor, 1991). Transdifferentiation has also been shown in the pituitaries of rats made hypothyroid by chemical thyroidectomy (Horvath et al., 1990), and reversible phenotypic switching of GH and PRL gene expression has long been reported in experimental rat pituitary tumor cells (Ivarie and Morris, 1983; Melmed, 2003).
Thus, pathogenic mechanisms of pituitary adenoma formation remain poorly understood, especially in sporadic occurrences such as in the patient discussed here. It should also be noted that this change in the phenotype of the tumor was observed while the patient was on long term dopamine agonist treatment, but also that therapy had been discontinued spontaneously by the patient on several occasions over the years. One could therefore speculate that poor adherence to dopamine agonist therapy may have played a role in this rather unusual evolution, by allowing emergence of a less differentiated adenomatous cell lineage.
In conclusion, we believe that this patient represents the first definitive documented evidence of the secondary apparition of GH autonomous secretion within a known microprolactinoma. This phenotypic change of the adenoma was accompanied by a change in its growth potential, suggesting some degree of de-differentiation. Complements of investigations by immunohistochemistry, DNA analyses, and clonality of tumor specimens would have been interesting to obtain, in order to understand what was responsible for this change. Pathogenic mechanisms implicated in this rather unusual evolution for a microprolactinoma remain largely unclear, and the potential role played by poor therapeutic adherence is only speculative. However and regardless of the pathogenic mechanisms implicated, the clinical course of this unusual patient emphasizes the need for long term hormonal follow up of all pituitary adenomas.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Daly, A. F., Rixhon, M., Adam, C., Dempegioti, A., Tichomirowa, M. A., and Beckers, A. (2006). High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J. Clin. Endocrinol. Metab. 91, 4769–4775.
Ferone, D., Colao, A., van der Lely, A. J., and Lamberts, S. W. (2000). Pharmacotherapy or surgery as primary treatment for acromegaly? Drugs Aging 17, 81–92.
Frawley, L. S., and Boockfor, F. R. (1991). Mammosomatotropes: presence and functions in normal and neoplastic pituitary tissue. Endocr. Rev. 12, 337–355.
Goldman, M. H., and Klinges, K. (1989). Acromegaly as the amenorrhea-galactorrhea syndrome. N. J. Med. 86, 887–888.
Herman, V., Fagin, J., Gonsky, R., Kovacs, K., and Melmed, S. (1990). Clonal origin of pituitary adenomas. J. Clin. Endocrinol. Metab. 71, 1427–1433.
Horvath, E., Lloyd, R. V., and Kovacs, K. (1990). Propylthiouracyl-induced hypothyroidism results in reversible transdifferentiation of somatotrophs into thyroidectomy cells. A morphologic study of the rat pituitary including immunoelectron microscopy. Lab. Invest. 63, 511–520.
Ivarie, R., and Morris, J. (1983). Phenotypic switching in GH3 rat pituitary tumor cells: linked expression of growth hormone and another hormonally responsive protein. DNA 2, 113–120.
Kasantikul, V., and Shuangshoti, S. (1990). Pituitary adenomas: immunohistochemical: study of 90 cases. J. Med. Assoc. Thai. 73, 514–521.
Kovacs, K., Horvath, E., Stefaneanu, L., Bilbao, J., Singer, W., Muller, P. J., Thapar, K., and Stone, E. (1998). Pituitary adenoma producing growth hormone and adrenocorticotropin: a histological, immunocytochemical, electron microscopic, and in situ hybridization study. Case report. J. Neurosurg. 88, 1111–1115.
Landis, C. A., Harsh, G., Lyons, J., Davis, R. L., McCormick, F., and Bourne, H. R. (1990). Clinical characteristics of acromegalic patients whose pituitary tumors contain mutant Gs protein. J. Clin. Endocrinol. Metab. 71, 1416–1420.
Landis, C. A., Masters, S. B., Spada, A., Pace, A. M., Bourne, H. R., and Vallar, L. (1989). GTPase inhibitng mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumors. Nature 340, 692–696.
Lania, A. G., Ferrero, S., Pivonello, R., Mantovani, G., Peverelli, E., Di Sarno, A., Beck-Peccoz, P., Spada, A., and Colao, A. (2010). Evolution of an aggressive prolactinoma into a growth hormone secreting pituitary tumor coincident with GNAS gene mutation. J. Clin. Endocrinol. Metab. 95, 13–17.
Levy, A., and Lightman, S. L. (1993). The pathogenesis of pituitary adenomas. Clin. Endocrinol. (Oxf.) 38, 559–570.
Ma, W., Ikeda, H., and Yoshimoto, T. (2002). Clinicopathologic study of 123 cases of prolactin-secreting pituitary adenomas with special reference to multihormone production and clonality of the adenomas. Cancer 95, 258–266.
Melmed, S. (2003). Mechanisms for pituitary tumorigenesis: the plastic pituitary. J. Clin. Invest. 112, 1603–1618.
Mindermann, T., and Wilson, C. B. (1994). Age-related and gender-related occurrence of pituitary adenomas. Clin. Endocrinol. (Oxf.) 41, 359–364.
Pagesy, P., Li, J. Y., Kujas, M., Peillon, F., Delalande, O., Visot, A., and Derome, P. (1991). Apparently silent somatotroph adenomas. Pathol. Res. Pract. 187, 950–956
Senovilla, L., Núñez, L., de Campos, J. M., de Luis, D. A., Romero, E., Sánchez, A., García-Sancho, J., and Villalobos, C. (2004). Multifonctional cells in human pituitary adenomas: implications for paradoxical secretion and tumorigenesis. J. Clin. Endocrinol. Metab. 89, 4545–4552.
Keywords: microprolactinoma, growth hormone, IGF-1, galactorrhea, amenorrhea, somatostatin analog
Citation: Dessimoz C, Browaeys P, Maeder P, Lhermitte B, Pitteloud N, Momjian S and Pralong FP (2012) Transformation of a microprolactinoma into a mixed growth hormone and prolactin-secreting pituitary adenoma. Front. Endocrin. 2:116. doi: 10.3389/fendo.2011.00116
Received: 09 September 2011;
Accepted: 27 December 2011;
Published online: 12 January 2012.
Edited by:
Paolo Beck-Peccoz, University of Milan, ItalyCopyright: © 2012 Dessimoz, Browaeys, Maeder, Lhermitte, Pitteloud, Momjian and Pralong. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: François P. Pralong, Service of Endocrinology, Lausanne University Hospital, BH 10-563, Bugnon 21, CH-1011 Lausanne, Switzerland. e-mail: francois.pralong@chuv.ch