
- Department of Physiology, Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen Medical Centre, Nijmegen, Netherlands
The succinate receptor (also known as GPR91) is a G protein-coupled receptor that is closely related to the family of P2Y purinoreceptors. It is expressed in a variety of tissues, including blood cells, adipose tissue, the liver, retina, and kidney. In these tissues, this receptor and its ligand succinate have recently emerged as novel mediators in local stress situations, including ischemia, hypoxia, toxicity, and hyperglycemia. Amongst others, the succinate receptor is involved in recruitment of immune cells to transplanted tissues. Moreover, it was shown to play a key role in the development of diabetic retinopathy. However, most prominently, the role of locally increased succinate levels and succinate receptor activation in the kidney, stimulating the systemic and local renin–angiotensin system, starts to unfold: the succinate receptor is a key mediator in the development of hypertension and possibly fibrosis in diabetes mellitus and metabolic syndrome. This makes the succinate receptor a promising drug target to counteract or prevent cardiovascular and fibrotic defects in these expanding disorders. Recent development of SUCNR1-specific antagonists opens novel possibilities for research in models for these disorders and may eventually provide novel opportunities for the treatment of patients.
Introduction
The sequencing of the human genome lead to the cloning, de-orphanization, and characterization of many novel G protein-coupled receptors. In 2001, a cluster of six GPCRs was identified on chromosome 3q24–3q25, including the four orphan receptors GPR86, GPR87, H963, and GPR91 (Wittenberger et al., 2001). Of these receptors, the latter shares high sequence homology with the family of P2Y purinoreceptors. Thus, GPR91 was expected to be activated by purinergic compounds. Surprisingly, however, He et al. (2004) showed that this GPCR was specifically activated by succinate, a citric acid cycle intermediate. Therefore, the gene encoding GPR91 was re-named succinate receptor 1 (SUCNR1). At the same time, a second and structurally related receptor, GPR99, was shown to be activated by another citric acid cycle intermediate α-ketoglutarate (or oxoglutarate). As such, this receptor was re-named oxoglutarate receptor 1 (OXGR1). However, 7 years after its de-orphanization, the (patho)physiological function of this receptor remains unknown.
Over the last years, the involvement of SUCNR1 in multiple (patho)physiological processes and its potential as a drug target has become increasingly evident. The tissue distribution of SUCNR1 revealed high expression in the kidney, but also in other tissues (He et al., 2004; Regard et al., 2008). In this review, we will discuss ligand specificity of agonists and recently developed antagonists on this receptor, its downstream signaling pathways and the current knowledge on the role of this receptor in various tissues in (patho)physiology.
SUCNR1 Ligands and Receptor Structure
According to most database entries, the human SUCNR1 (e.g., Acc. No. EAW78789) encodes a protein of 334 amino acids. However, within this sequence, a methionine residue is present at position 5 (Figure 1). Analysis of the Kozak sequence of this area point to this second methionine being the actual start site, resulting in a 330 amino acid receptor (Wittenberger et al., 2001). The SUCNR1 sequence has a high degree of homology between human, rat (Acc. No. NP_001001518), and mouse (Acc. No. NP_115776), with exception of the C-terminus, which is 12 amino acids shorter in rodents (Figure 1). Whether and how this affects receptor signaling has not been studied in detail, but succinate shows slightly reduced potency on the rat and mouse receptor, compared to the human SUCNR1 (He et al., 2004).
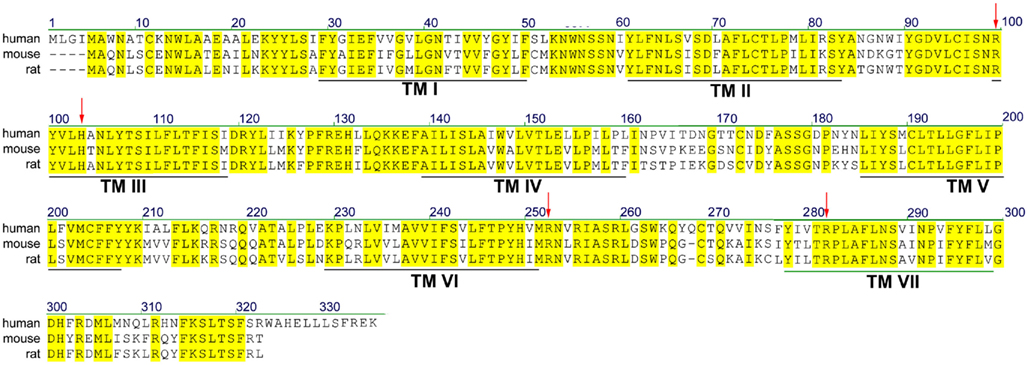
Figure 1. Sequence alignment of human, rat, and mouse SUCNR1. Yellow residues are conserved between species. Predicted transmembrane (TM) domains I–VII are indicated by black lines. Residues important for succinate binding (Arg99, His103, Arg252, and Arg281) are indicated with red arrows.
At present, our knowledge of the regulation of SUCNR1 on the molecular and cellular level is still limited. It is clear that SUCNR1 is a plasma membrane receptor (He et al., 2004; Robben et al., 2009), where it acts as a sensor for extracellular succinate. As for all GPCRs, SUCNR1 has an extracellularly located N-terminus, which contains a consensus sequence for N-linked glycosylation at Asn4. A second consensus sequence for N-linked glycosylation is contained within extracellular loop 2 at position 164. Although we demonstrated that SUCNR1 is subject to complex N-glycosylation (Robben et al., 2009), the precise role of this posttranslational modification remains to be determined.
In order to prevent prolonged signaling, most GPCRs are subject to desensitization followed by sequestration, followed by recycling or degradation (Kohout and Lefkowitz, 2003). Indeed, in HEK293 cells, SUCNR1 was shown to internalize into endosomes/lysosomes upon stimulation with succinate (He et al., 2004). In contrast, in polarized renal Madin–Darby Canine Kidney (MDCK) cells, we found that succinate-induced temporal desensitization of the receptor, but that this did not involve significant internalization of the receptor from the plasma membrane. In line with this, resensitization of the receptor occurred within 15 min upon washout of succinate (Robben et al., 2009).
Succinate was identified as a ligand for the then orphan GPCR GPR91 using an ion-exchange size-exclusion and reversed-phase, fast performance liquid chromatography, combined with mass spectrometry. EC50 values for receptor activation were in the 20–50 μM range, depending on the assay used. It was unequivocally shown that succinate was the endogenous ligand for this receptor by testing 800 pharmacologically active compounds and known GPCR ligands, as well as 200 carboxylic acids and compounds structurally related to succinate, including the citric acid cycle intermediates α-ketoglutarate, citrate, isocitrate, malate, and oxaloacetate, and multiple purinergic compounds. Only maleate and methylmalonate were able to activate SUCNR1, albeit with a 5- to 10-fold lower potency compared to succinate (He et al., 2004). The selectivity of SUCNR1 for succinate is mediated by four positively charged key residues in the binding cavity enclosed by the receptor’s 7 transmembrane (TM) domains. Individual mutation of Arg99 (3.29 according to the Ballesteros–Weinstein numbering scheme), His103 (3.33), Arg252 (6.55), and Arg281 (7.39; Figure 1; arrowheads) abolished receptor activation (He et al., 2004). Since these residues are also present in OXGR1, they are likely involved in the binding of the electrostatic binding to the dicarboxylate headgroups of these compounds.
Succinate Accumulation and Release
As part of the citric acid – or Krebs – cycle in the mitochondrial matrix, succinate is formed from succinyl-CoA by succinyl-CoA synthetase and is subsequently converted by succinate dehydrogenase to generate fumarate (Figures 2A,B). As may be expected for a component of the citric acid cycle, succinate is normally present in the mitochondria, however, it can be released to the extracellular space due to local energy metabolism disturbances (Krebs, 1970). Plasma succinate concentrations measured by liquid chromatography tandem mass spectrometry in rodents vary from 6 to 20 μM, whereas in human succinate levels have been detected from 2 to 3 μM (Sadagopan et al., 2007) and 2 to 20 μM (Kushnir et al., 2001) in serum and plasma, respectively. Moreover, the succinate concentration in urine of mice is around 20–30 μM under physiological conditions (Toma et al., 2008). Using an aequorin luminescence assay, He et al. (2004) determined that the half-maximal response concentration for succinate-induced activation of human and mouse SUCNR1 was 56 ± 8 and 28 ± 5 μM, respectively, indicating that under normal physiological conditions, plasma, and urinary succinate levels are too low for receptor activation. However, since values under normal physiological conditions are only approximately twofold lower than the level required for a half-maximal response, only a slight elevation of the plasma or urinary succinate concentration may be needed to fully active SUCNR1.
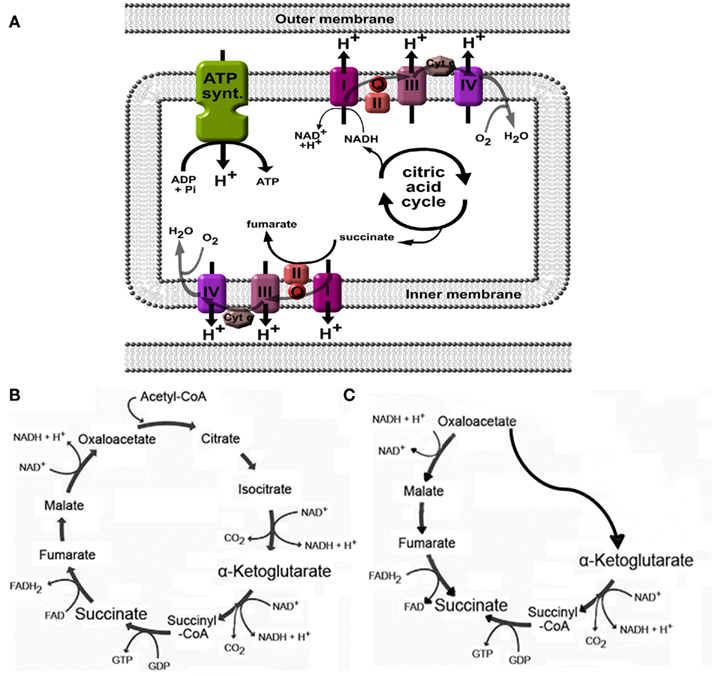
Figure 2. Generation of succinate in mitochondria. (A) Succinate is an intermediate in the citric acid cycle, and is converted by succinate dehydrogenase (also called complex II) to fumarate. When high H+ gradients over the inner membrane are present, or when the oxygen pressure is low, complex I, II, III, and IV will be inhibited, leading to accumulation of succinate. (B) Overview of the citric acid cycle under normal physiological conditions. (C) Cellular stress such as hypoxia may affect normal functioning of the citric acid cycle and induce a part of the cycle to run in reverse, ultimately leading to increased succinate production. See text for more details and references.
Changes in the energy balance may affect the production and release of succinate. As discussed in more detail below, this was demonstrated in animal models for diabetes mellitus (Toma et al., 2008), metabolic disease (Sadagopan et al., 2007), and liver damage (Correa et al., 2007). In hyperglycemia, as with diabetes mellitus and metabolic syndrome, high activity of the Krebs cycle due to high substrate availability may increase the H+ gradient over the mitochondrial membrane (Figure 2A), leading to inhibition of enzymatic steps mediated by complexes within the electron transport chain, including succinate dehydrogenase (Complex II; Figure 2A; Brownlee, 2005). This may then lead to intracellular accumulation, and eventually, release of succinate into the blood stream. As the succinate dehydrogenase complex is involved in the electron transport chain over the mitochondrial membrane, its activity indirectly depends on the availability of oxygen. As such, in situations when pO2 is low, succinate will accumulate due to low activity of succinate dehydrogenase or other enzymes in the electron transport chain that affect its activity (Weinberg et al., 2000; Jassem and Heaton, 2004; Brownlee, 2005).
Increased production of succinate in low oxygen states may also be caused by alteration of the flux in the normal citric acid cycle (Figure 2B) to an alternative, partially inverted citric acid cycle (Figure 2C) as demonstrated by Weinberg et al. (2000). In addition, succinate formation may occur non-enzymatically (Fedotcheva et al., 2006). Indeed, low oxygen states, such as with ischemia (Hems and Brosnan, 1970), or exercise (Hochachka and Dressendorfer, 1976; Lewis et al., 2010), have been shown to increase circulating succinate levels. Using the one-legged knee extension exercise model, Gibala et al. (1998) demonstrated that succinate intramuscular concentrations increased more than threefold after maximal dynamic knee extensor exercise. Similar results were obtained in plasma from subjects at the peak of diagnostic treadmill or bicycle ergometry cardiopulmonary exercise testing. The effect of low oxygen states on increased succinate levels was also obvious in rats anesthetized with 100% CO2 instead of oxygenized isoflurane, succinate levels increased from 7 to 40 μM in left ventricle, and even 173 μM when (low oxygen) blood was collected from the vena cava (Sadagopan et al., 2007).
Since under normal physiological pH, succinate is a charged molecule, it is unable to efficiently diffuse through membranes. Thus, following its accumulation within the mitochondrial matrix, succinate requires specific transporters or exchangers to facilitate its transport from within the mitochondria to the cytosol, and subsequently across the plasma membrane to the extracellular environment. The inner mitochondrial membrane contains a number of dicarboxylate transporters and exchangers, of which the succinate–fumarate/malate transporter SLC25A10 (Oswald et al., 2007) is the most likely candidate to facilitate transport of accumulated succinate. Transport of succinate across the outer mitochondrial membrane likely occurs through porins, which are large channels that are permeable to most molecules under 1.5 kDa. Candidate proteins for succinate transport across the plasma membrane will likely be tissue or cell type-specific, as no ubiquitously expressed plasma membrane dicarboxylate transporters exist. Likely candidates, however, are members of the organic anion transporter families, multidrug resistance proteins, and sodium–dicarboxylate exchangers (Markovich and Murer, 2004; Pajor, 2006). However, no data is currently available regarding their role in succinate accumulation in oxidative or metabolic stress. In the future, the use of specific antagonists or knockout mice for these cotransporters may reveal pathways involved in pathological states due to succinate accumulation.
Tissue Distribution and (Patho)Physiological Roles of SUCNR1
Initially, Q-RT-PCR showed SUCNR1 expression in kidney, liver, and spleen (He et al., 2004). A later study confirmed expression in kidney and liver, and in addition demonstrated expression in white adipose tissue (Regard et al., 2008). Subsequently, several studies described the presence and function of SUCNR1 in specific cell types of these tissues, and aimed to elucidate its role, as described in more detail below. Although its function remains to be established in most tissues, it is clear that this receptor is a detector of disturbances in the body’s energy balance.
When the body is a hypoglycemic state, hormones such as glucagon trigger adipocytes in white adipose tissue to degrade triglycerides into free fatty acids for energy production. In SUCNR1-positive adipocytes, succinate inhibits lipolysis (Regard et al., 2008) and may thus prevent the release of fatty acids from adipocyte for energy consumption. As increased succinate levels are found in rodent models for diabetes mellitus and metabolic syndrome (Sadagopan et al., 2007; Toma et al., 2008), high succinate levels may prevent lipolysis in states when fuel molecules such as glucose and free fatty acids are abundant.
The liver is crucial for regulating the body’s metabolism by storing fuel molecules such as glycogen, and plays a major role in lipid and amino acid conversion and synthesis. Besides, it plays a major role in the degradation of toxic compounds. Likely, this organ is subject to multiple and various stress factors, often caused by an unhealthy lifestyle. In the liver, the SUCNR1 is exclusively expressed in quiescent hepatic stellate cells (HSC). Application of ischemia in a perfused liver model increased succinate levels of the perfusate 14-fold to approximately 1 mM (Correa et al., 2007). Moreover, HSC cells treated with succinate showed increased levels of myofibroblastic markers compared to activated control cells, indicating that succinate independently enhances HSC activation. However, upon activation of HSC, the expression of SUCNR1 decreased rapidly (Correa et al., 2007; De Minicis et al., 2007), suggesting that SUCNR1 serves as an early detector of hepatic stress or damage. Together, this suggests that SUCNR1 signaling plays an enhancing role in HSC activation to restore damaged tissue in the ischemic liver, but may thereby also contribute to the formation of fibrosis.
Although no SUCNR1 mRNA was detected in the heart by RT-PCR experiments (He et al., 2004; Regard et al., 2008), a recent study by Aguiar et al. (2010) demonstrated the presence of SUCNR1 mRNA and protein in freshly isolated preparations of ventricular cardiomyocytes, where it is localized in the sarcolemmal membrane and the T-tubules. In these cardiomyocytes, succinate administration may affect cell contraction. Importantly, prolonged incubation of cardiomyocytes with high concentrations (10 mM) succinate-induced apoptosis (Aguiar et al., 2010). As such, SUCNR1 may regulate apoptosis in the heart in states of ischemia and hypoxia.
Although also not initially identified in tissue panels (He et al., 2004; Regard et al., 2008), it has now become evident that the SUCNR1 is also expressed in hematopoietic precursor cells and multiple types of blood and immune cells (Macaulay et al., 2007; Hakak et al., 2009). When administered to platelets, succinate potentiates platelet activation and aggregation in a dose-dependent manner, increasing the maximum aggregation compared to controls without succinate (Macaulay et al., 2007; Hogberg et al., 2011). This suggests a role for succinate in atherothrombosis, in which succinate levels may be increased due to local hypoxia. In hematopoietic progenitor cells, the activated SUCNR1 induces cell proliferation and protects erythroleukemic cells from serum starvation-induced apoptosis. Together, this explains how administration of succinate in a mouse model of chemotherapy-induced myelosuppression leads to increased levels of hemoglobin, platelets, and neutrophils (Hakak et al., 2009), and thus, succinate administration may be beneficial for patients recovering from chemotherapy.
Rubic et al. (2008) detected SUCNR1 mRNA in immature dendritic cells (DCs), but not in monocytes, T-, or B-cells, suggesting that SUCNR1 expression is induced when monocytes develop into immature DCs. In immature DCs, succinate stimulates cell migration in a concentration-dependent manner, and thus mediates chemotaxis. Moreover, SUCNR1 and toll-like receptors act in synergy to potentiate the production of the inflammatory cytokines tumor necrosis factor α (TNFα) and interleukin-1β. Upon activation, immature DCs will mature to mature antigen-presenting DCs, which will subsequently activate T-cells. Succinate treatment of the DCs promoted the interferon-γ production of activated CD4+ T-cells. Furthermore, and underscoring the fact that the above observations are SUCNR1-mediated, it was shown that mice challenged with tetanus toxin accumulate higher levels of mature DCs in their lymph nodes compared to SUCNR1−/− mice. It was subsequently shown that grafts from SUCNR1−/− mice show improved outcome in skin transplantation graft rejection experiments (Rubic et al., 2008). As such, interfering with SUCNR1 signaling by, e.g., specific receptor antagonists or preventing succinate accumulation, may be beneficial for patients receiving organ transplantation.
In the retina, SUCNR1 is predominantly expressed in the cell bodies of the retinal ganglion cell (RGC) layer (Sapieha et al., 2008). To study the role of SUCNR1 in the developing retina, SUCNR1 siRNA was injected into the eye of newborn rat pups, which decreased the vascularization of the retina at day 4 post-partum compared to controls. In line with this, injection of succinate resulted in increased vessel numbers in the retina, clearly demonstrating a positive role for SUCNR1 in retinal vascularization. The SUCNR1-regulated increase in vessel growth occurred through the production and release of pro-angiogenic hormones. Together, these data revealed that SUCNR1 expression in RGCs are essential for proper vascularization of the eye. In this respect, it has been suggested that increased succinate levels may cause the high rate of neovascularization in diabetes mellitus or retinal ischemia, leading to retinopathy (Rubic et al., 2008). If so, inhibitors of SUCNR1 may provide a potential treatment. Recently, also the expression of SUCNR1 in the apical membrane of retinal pigment epithelium (RPE) was demonstrated. Since RPE expresses VEGF and excess iron levels promote angiogenesis, the role of SUCNR1 in iron-dependent angiogenesis was evaluated in Hfe−/− mice, a model of hereditary hemochromatosis that results in increased iron absorption and accumulation (Gnana-Prakasam et al., 2011). Indeed, SUCNR1 expression in total retina and RPE from Hfe−/− mice is higher compared with WT animals. This observation was also demonstrated in human RPE cell line ARPE19, primary RPE cells subjected to CMV US2 infection or ferric ammonium administration, cellular models that mimic Hfe deficiency. Using SUCNR1-specific shRNA, the succinate-induced expression of VEGF could be could be reduced in RPE cells, confirming a role of SUCNR1 in the regulation of vascular adaptations in the retina.
In the kidney, the SUCNR1 localizes to the renal vascular lumen, in particular the afferent arteriole and the glomerular vasculature. Moreover, SUCNR1 is expressed in the luminal membrane of multiple segments of the renal tubules: the cortical thick ascending limb (cTAL) of Henle’s loop, including the macula densa (MD), and the cortical and medullary collecting duct (CD; Figure 3A; Toma et al., 2008; Robben et al., 2009; Vargas et al., 2009). Recent work by the Peti-Peterdi group demonstrated that the SUCNR1 mediates the release of renin from the JGA via SUCNR1 located in the vascular luminal membrane (Toma et al., 2008) and in the apical membrane of MD cells (Robben et al., 2009; Vargas et al., 2009). Elegant microperfusion studies combined with live imaging of isolated glomeruli showed that perfusion with a succinate-containing buffer induced renin release from the granular cells of the JGA and rapidly induced vasodilation of the afferent arteriole. Together, this suggests that SUCNR1 plays a pivotal role in development of glomerular hyperfiltration and activation of the renal renin–angiotensin system. This clearly explained how infusion of succinate in mice, as initially performed by He et al. (2004) resulted hypertension. The SUCNR1-mediated release of renin from the JGA is mediated by nitric oxide and prostaglandin E2 that will subsequently transactivate EP2 and/or EP4 receptors on the granular cells (Figures 3A,B; Toma et al., 2008). Moreover, it was shown that SUCNR1 activation in the luminal membrane of the MD cells triggers renin release from the JGA via a similar mechanism, although in this case SUCNR1 serves as a sensor for succinate in the pro-urine rather than in the blood (Vargas et al., 2009). As SUCNR1 in the renal tubules senses (pro)urinary succinate levels, measurement of succinate in excreted urine may provide an easy, non-invasive way to determine SUCNR1 activity in the kidney compared to circulating succinate levels.
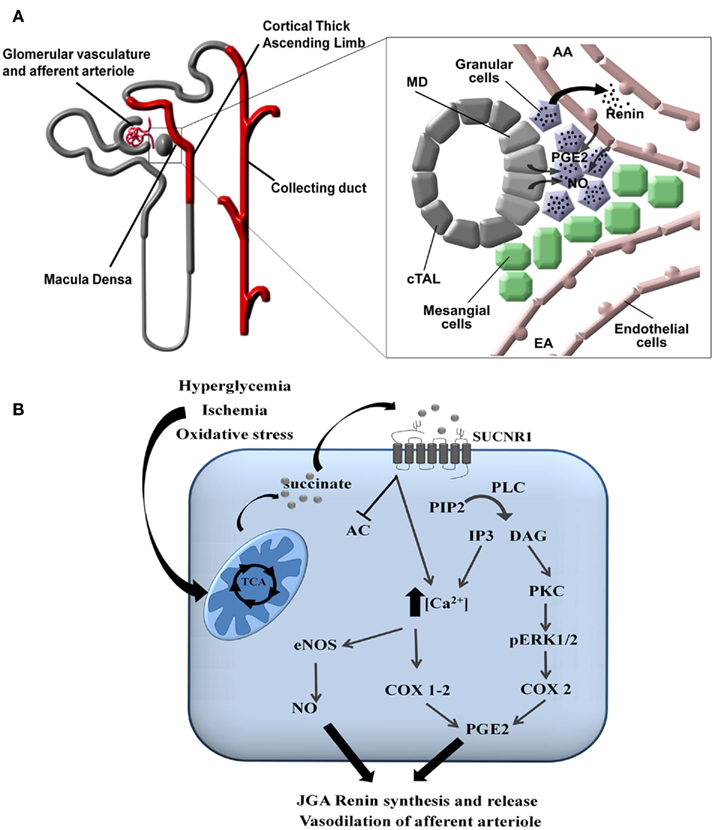
Figure 3. Signaling of SUCNR1 in the kidney. (A) Signaling in the juxtaglomerular apparatus. SUCNR1 is expressed in several parts of the nephron (indicated in red), where it contributes to renin release from the JGA (see inset on the right). Succinate receptor activation on macula densa (MD) cells or on endothelial cells of the afferent arteriole (AA) induce the release of prostaglandin E2 (PGE2) and nitric oxide (NO), which trigger the release of renin form the granular cells of the juxtaglomerular apparatus (JGA). Additional abbreviations: EA, efferent arteriole; cTAL, cortical thick ascending limb. (B) Intracellular signaling. In tubular cells, diabetes mellitus, ischemia/reperfusion injury, or oxidatives stress affect the functioning of the mitochondria, resulting in the release of succinate into the cytoplasm and eventually to the extracellular environment. There, it can bind to SUCNR1 on the cell surface. This triggers a signaling cascade that eventually leads to the secretion of nitric oxide (NO) and prostaglandin E2 (PGE2). These signaling molecules will subsequently promote the release of renin from the JGA as described under (A). Abbreviations: PIP2, Phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; PKC, protein kinase C; pERK1/2, phosphorylated extracellularly regulated kinase 1/2; COX, cyclo-oxogenase, eNOS, endothelial nitric oxide synthase.
Analogous to the development of hypertension upon administration of succinate to mice (He et al., 2004), plasma levels of succinate were shown to be elevated in several rodent models of hypertension and metabolic disease. Spontaneous hypertensive rats, fatty Zucker fa/fa rats, db/db diabetic mice, and ob/ob mice had succinate levels that were elevated two- to four-fold compared to their respective non-hypertensive or lean controls (Sadagopan et al., 2007). However, in serum of hypertensive or diabetic patients, succinate levels were similar to healthy age-matched controls (Sadagopan et al., 2007). The cause of this discrepancy between rodent models and patients remains to be established. Although the above succinate measurements indicated that SUCNR1 may play a role in diabetes and metabolic syndrome, the relationship between diabetes and development of hypertension was first unquestionably demonstrated using SUCNR1−/− mice. The JGA and whole-kidney renin content of diabetic mice is elevated compared to non-diabetic controls, and renin release is stimulated by perfusion of the afferent arteriole (Toma et al., 2008) or the MD-containing cTAL (Vargas et al., 2009) with a high glucose or succinate buffer. The observed release of renin, combined with the aforementioned dilation of the afferent arteriole resulting in hyperfiltration, are hallmarks of the diabetic kidney.
Nowadays, it is well established that the production and release of renin is no longer restricted to the JGA and individual components of the renin–angiotensin system have been detected throughout the nephron (Rohrwasser et al., 1999) and its role is increasingly being elucidated (Suzaki et al., 2006; Schweda et al., 2007). In the kidney of diabetic mice, activation of SUCNR1 in the CD (Robben et al., 2009) leads to increased phosphorylation of the extracellularly regulated kinases 1 and 2 (ERK1/2), whereas this effect is absent in SUCNR1−/− mice. Sustained tubular ERK1/2 phosphorylation is associated with proliferation of tubular cells and the development of tubulo-interstitial fibrosis (Sakai et al., 2005; Steckelings et al., 2009) and the SUCNR1 has been postulated to be instrumental in the development of fibrosis in diabetic nephropathy and diabetes-induced hypertension (Peti-Peterdi et al., 2008). However, the exact role of the SUCNR1 activation and mechanisms underlying these processes require further investigation.
Receptor Signaling Pathways
Its expression within a large number of tissues and in a multitude of cell types within these tissues suggests a highly specific signaling machinery downstream of SUCNR1 activation. Indeed, this seems to be the case: in kidney (derived) cells, such as HEK293 and MDCK cells, SUCNR1 signals through Gq and Gi mediated pathways that induce intracellular calcium mobilization, increase inositol phosphate accumulation, activate the (ERK1/2; Figure 3B) and inhibit cAMP accumulation in a dose-dependent manner (He et al., 2004). Similar downstream signaling pathways were identified in other tissues or cell types: in SUCNR1-positive adipocytes, succinate inhibits lipolysis in a pertussis toxin-dependent manner (Regard et al., 2008), demonstrating that SUCNR1 signaling inhibits cAMP formation that is induced by lipolytic hormones. Similarly, in hematopoietic progenitor cells, the activated SUCNR1 signals via the Gi/o protein to induce cell proliferation via ERK1/2 (Hakak et al., 2009). Although the identity of its associated G proteins remain to be elucidated, in DCs, SUCNR1 acts in synergy with toll-like receptors via activation ERK1/2 (Rubic et al., 2008).
Interestingly, stimulation of cardiac myocytes with succinate leads to increased PKA activity that subsequently triggers intracellular calcium transients. Moreover, the maximum peak height and frequency of the calcium transients is affected by SUCNR1 activation. The succinate-induced apoptosis observed in cardiac myocytes is most likely orchestrated by a combination of PKA activation and increased intracellular calcium levels (Aguiar et al., 2010). Activation of the PKA activation suggests signaling of SUCNR1 through the Gs/cAMP pathway. Indeed, this pathway has also been shown to be involved in the SUCNR1-mediated activation of platelets (Hogberg et al., 2011). The signaling pathways involved in HSC activation remain to be elucidated. In contrast to adipocytes or renal cells, administration of succinate to HSC failed to induce an intracellular Ca2+ response, nor did it decrease basal or forskolin-induced cAMP levels or increase cAMP levels by itself (Correa et al., 2007).
SUCNR1-Mediated Transactivation Pathways
Besides through intracellular pathways, SUCNR1 is an important initiator of transactivation signals by inducing the release of extracellular factors or hormones. The release of renin from the JGA is mediated in part by the formation of nitric oxide. Moreover, SUCNR1 activation increases intracellular release of arichidonic acid that, through the actions of cyclooxygenase (COX)-2, leads to the production and release of prostaglandin E2 (Robben et al., 2009) that subsequently transactivate EP2 and/or EP4 receptors on the granular cells (Figures 3A,B; Toma et al., 2008; Vargas et al., 2009). SUCNR1-mediated local activation of the prostaglandin system has also been suggested as a potential mechanism responsible for the succinate-induced apoptosis observed in cardiac myocytes (Aguiar et al., 2010), although experiments clearly demonstrating this are currently lacking.
In the eye, SUCNR1 activation induces VEGF and other angiogenic factors (Sapieha et al., 2008). Although the full cellular pathways triggering this release remain unknown, the study by Sapieha et al. revealed revolutionary insights in the role of succinate in this process: for a long time, the production of VEGF has been linked to increased intracellular succinate levels that lead to stabilization of hypoxia inducing factor (HIF)1α (Taylor, 2008). Interestingly, Sapieha et al. (2008) showed that extracellular succinate, through action of the succinate receptor, triggers the increased expression and release of VEGF under hypoxic conditions, independently from HIF1α. This validates critical re-analysis of the role of HIF1α in tissue adaptive responses in cellular or tissue hypoxic conditions, as part of its actions may be regulated by SUCNR1.
Development and Action of SUCNR1 Antagonists
Because of the involvement of SUCNR1 in a multitude of diseases, receptor-specific antagonists are promising drug candidates. Recently, using a systematic structure-activity relationship approach, Bhuniya et al. (2011) were the first to identify selective antagonists for both human and rat SUCNR1. The IC50 for the different compounds ranges from 7 to 180 and 7 to 435 nM for human SUCNR1 and rat SUCNR1, respectively. Of these, compound “4c” (Figure 4) was shown to be the most efficient antagonist in vitro (IC50 = 7 nM). Bolus intraperitoneal injection of 100 mg/kg “4c” in rats resulted in plasma levels of 200–300 nM of this compound, which ameliorated a succinate-induced change of mean arterial pressure up to 76%. Moreover, and of high pharmaceutical and clinical importance, two compounds structurally related to “4c” (“5g” and “7e”), were shown to be active upon oral administration and will thus serve as leads for further drug development. Interestingly, none of these antagonists show any obvious structural relationship to succinate (Figure 4), and their binding sites within the receptor remain to be determined. However, the discovery and characterization of these compounds opens new possibilities to unravel the role of SUCNR1 in many pathological states and will likely establish SCURN1 as a promising drug target with high clinical potential.
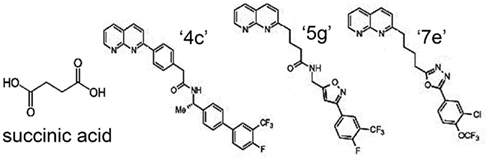
Figure 4. Structure of SUCNR1 ligands. From left to right: succinic acid, compound “4g,” compound “5g,” and compound “7e.” Structures were derived from (Bhuniya et al., 2011).
Future Perspectives
As illustrated above, signaling of the SUCNR1 is involved in various pathologies in multiple organs. These pathologies are particularly linked to local stress factors that affect the energy balance of a tissue, such as ischemia, hypoxia, metabolic syndrome, and diabetes mellitus. Besides, SUCNR1 senses local damage and increases inflammatory responses. Therefore, this receptor is a sensor of local stress situations that affect the cellular metabolism, as reflected by increased formation and release of succinate.
It is clear that SUCNR1 is a major regulator of blood pressure in diabetes mellitus, and may contribute to the development of tubulo-interstitial fibrosis in diabetic nephropathy. Moreover, it has an obvious role in the development of excess retinal vascularization in diabetic retinopathy, activation of the innate immune system. Future challenges lie in elucidating the cellular and molecular mechanisms responsible for these effects, and identifying means, e.g., the development of specific receptor antagonists, to prevent, cure, or ameliorate these pathological effects.
The presence of SUCNR1 on immune cells may also affect pathology of various tissues. Succinate’s role as chemotactic compound via SUCNR1 on immature DCs may induce infiltration of immune cells in transplanted organs or damaged tissues. In, e.g., renal transplantation, ischemia, and hypoxia will likely increase renal succinate formation, similar as observed in ischemic retinopathy. This may, analogous to the skin transplantation effects described above, promote maturation of immature DCs in the kidney. In renal ischemia-reperfusion experiments, which serves as an accurate model for transplantation, DCs are the major source of TNFα produced early in inflammatory responses (Dong et al., 2007). The synergistic effect of SUCNR1 and toll-like receptors (Rubic et al., 2008) may contribute significantly to the release of high levels of TNFα, thus increasing inflammation, renal epithelial apoptosis, and recruitment, binding, and migration of leukocytes. Eventually, this may result in graft injury and rejection (Rogers et al., 2009).
Besides promoting retinal vascularization during development (Sapieha et al., 2008), no clear role for SUCNCR1 in physiology has currently been established. This may in part be due to the relative recent discovery that succinate can act as a signaling molecule, and this realization may incite researchers to investigate the regulatory role of succinate and its receptor further. Alternatively, its role in physiology may be very subtle or redundant, and extracellular succinate may exclusively act as a stress or damage signal, as illustrated by its involvement in the multiple pathologies described above. Although its role in (patho)physiology remains to be investigated in more detail, it is clear that this receptor is a highly promising drug target in a multitude of disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Peter M. T. Deen is a recipient of VICI grant 865.07.002 of the Netherlands Organization for Scientific research (NWO). PMTD is supported by grants from the Dutch Kidney Foundation (C03-2060), NWO (865.07.002), and Coordination Theme 1 (Health) of the European Community’s seventh Framework Program (HEALTH-F2–2007-201590, entitled EUNEFRON). Joris Hubertus Robben is supported by the Dutch Kidney Foundation (KJPB 09.012).
References
Aguiar, C. J., Andrade, V. L., Gomes, E. R., Alves, M. N., Ladeira, M. S., Pinheiro, A. C., Gomes, D. A., Almeida, A. P., Goes, A. M., Resende, R. R., Guatimosim, S., and Leite, M. F. (2010). Succinate modulates Ca(2+) transient and cardiomyocyte viability through PKA-dependent pathway. Cell Calcium 47, 37–46.
Bhuniya, D., Umrani, D., Dave, B., Salunke, D., Kukreja, G., Gundu, J., Naykodi, M., Shaikh, N. S., Shitole, P., Kurhade, S., De, S., Majumdar, S., Reddy, S. B., Tambe, S., Shejul, Y., Chugh, A., Palle, V. P., Mookhtiar, K. A., Cully, D., Vacca, J., Chakravarty, P. K., Nargund, R. P., Wright, S. D., Graziano, M. P., Singh, S. B., Roy, S., and Cai, T. Q. (2011). Discovery of a potent and selective small molecule hGPR91 antagonist. Bioorg. Med. Chem. Lett. 21, 3596–3602.
Brownlee, M. (2005). The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54, 1615–1625.
Correa, P., Kruglow, E., Thompson, M., Leite, M., Dranoff, J., and Nathanson, M. (2007). Succinate is a paracrine signal for liver damage. J. Hepatol. 47, 262–269.
De Minicis, S., Seki, E., Uchinami, H., Kluwe, J., Zhang, Y., Brenner, D. A., and Schwabe, R. F. (2007) Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 132, 1937–1946.
Dong, X., Swaminathan, S., Bachman, L. A., Croatt, A. J., Nath, K. A., and Griffin, M. D. (2007). Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 71, 619–628.
Fedotcheva, N. I., Sokolov, A. P., and Kondrashova, M. N. (2006). Nonezymatic formation of succinate in mitochondria under oxidative stress. Free Radic. Biol. Med. 41, 56–64.
Gibala, M. J., MacLean, D. A., Graham, T. E., and Saltin, B. (1998). Tricarboxylic acid cycle intermediate pool size and estimated cycle flux in human muscle during exercise. Am. J. Physiol. 275, E235–E242.
Gnana-Prakasam, J. P., Ananth, S., Prasad, P. D., Zhang, M., Atherton, S. S., Martin, P. M., Smith, S. B., and Ganapathy, V. (2011). Expression and iron-dependent regulation of succinate receptor GPR91 in retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 52, 3751–3758.
Hakak, Y., Lehmann-Bruinsma, K., Phillips, S., Le, T., Liaw, C., Connolly, D. T., and Behan, D. P. (2009). The role of the GPR91 ligand succinate in hematopoiesis. J. Leukoc. Biol. 85, 837–843.
He, W., Miao, F. J. P., Lin, D. C. H., Schwandner, R. T., Wang, Z., Gao, J., Chen, J. L., Tian, H., and Ling, L. (2004). Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429, 188–193.
Hems, D. A., and Brosnan, J. T. (1970). Effects of ischaemia on content of metabolites in rat liver and kidney in-vivo. Biochem. J. 120, 105.
Hochachka, P. W., and Dressendorfer, R. H. (1976). Succinate accumulation in man during exercise. Eur. J. Appl. Physiol. Occup. Physiol. 35, 235–242.
Hogberg, C., Gidlof, O., Tan, C., Svensson, S., Nilsson-Ohman, J., Erlinge, D., and Olde, B. (2011). Succinate independently stimulates full platelet activation via cAMP and phosphoinositide 3-kinase-beta signaling. J. Thromb. Haemost. 9, 361–372.
Jassem, W., and Heaton, N. D. (2004). The role of mitochondria in ischemia//reperfusion injury in organ transplantation. Kidney Int. 66, 514–517.
Kohout, T. A., and Lefkowitz, R. J. (2003). Regulation of g protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 63, 9–18.
Kushnir, M. M., Komaromy-Hiller, G., Shushan, B., Urry, F. M., and Roberts, W. L. (2001). Analysis of dicarboxylic acids by tandem mass spectrometry. High-throughput quantitative measurement of methylmalonic acid in serum, plasma, and urine. Clin. Chem. 47, 1993–2002.
Lewis, G. D., Farrell, L., Wood, M. J., Martinovic, M., Arany, Z., Rowe, G. C., Souza, A., Cheng, S., McCabe, E. L., Yang, E., Shi, X., Deo, R., Roth, F. P., Asnani, A., Rhee, E. P., Systrom, D. M., Semigran, M. J., Vasan, R. S., Carr, S. A., Wang, T. J., Sabatine, M. S., Clish, C. B., and Gerszten, R. E. (2010). Metabolic signatures of exercise in human plasma. Sci. Transl. Med. 2, 33ra37.
Macaulay, I. C., Tijssen, M. R., Thijssen-Timmer, D. C., Gusnanto, A., Steward, M., Burns, P., Langford, C. F., Ellis, P. D., Dudbridge, F., Zwaginga, J. J., Watkins, N. A., van der Schoot, C. E., and Ouwehand, W. H. (2007). Comparative gene expression profiling of in vitro differentiated megakaryocytes and erythroblasts identifies novel activatory and inhibitory platelet membrane proteins. Blood 109, 3260–3269.
Markovich, D., and Murer, H. (2004). The SLC13 gene family of sodium sulphate/carboxylate cotransporters. Pflugers Arch. 447, 594–602.
Oswald, S., Grube, M., Siegmund, W., and Kroemer, H. K. (2007). Transporter-mediated uptake into cellular compartments. Xenobiotica 37, 1171–1195.
Pajor, A. M. (2006). Molecular properties of the SLC13 family of dicarboxylate and sulfate transporters. Pflugers Arch. 451, 597–605.
Peti-Peterdi, J., Kang, J. J., and Toma, I. (2008). Activation of the renal renin-angiotensin system in diabetes – new concepts. Nephrol. Dial. Transplant. 23, 3047–3049.
Regard, J. B., Sato, I. T., and Coughlin, S. R. (2008). Anatomical profiling of G protein-coupled receptor expression. Cell 135, 561–571.
Robben, J. H., Fenton, R. A., Vargas, S. L., Schweer, H., Peti-Peterdi, J., Deen, P. M., and Milligan, G. (2009). Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 76, 1258–1267.
Rogers, N. M., Matthews, T. J., Kausman, J. Y., Kitching, A. R., and Coates, P. T. (2009). Review article: kidney dendritic cells: their role in homeostasis, inflammation and transplantation. Nephrology (Carlton) 14, 625–635.
Rohrwasser, A., Morgan, T., Dillon, H. F., Zhao, L., Callaway, C. W., Hillas, E., Zhang, S., Cheng, T., Inagami, T., Ward, K., Terreros, D. A., and Lalouel, J. M. (1999). Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension 34, 1265–1274.
Rubic, T., Lametschwandtner, G., Jost, S., Hinteregger, S., Kund, J., Carballido-Perrig, N., Schwarzler, C., Junt, T., Voshol, H., Meingassner, J. G., Mao, X., Werner, G., Rot, A., and Carballido, J. M. (2008). Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 9, 1261–1269.
Sadagopan, N., Li, W., Roberds, S. L., Major, T., Preston, G. M., Yu, Y., and Tones, M. A. (2007). Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am. J. Hypertens. 20, 1209–1215.
Sakai, N., Wada, T., Furuichi, K., Iwata, Y., Yoshimoto, K., Kitagawa, K., Kokubo, S., Kobayashi, M., Hara, A., Yamahana, J., Okumura, T., Takasawa, K., Takeda, S. I., Yoshimura, M., Kida, H., and Yokoyama, H. (2005). Involvement of extracellular signal-regulated kinase and p38 in human diabetic nephropathy. Am. J. Kidney Dis. 45, 54–65.
Sapieha, P., Sirinyan, M., Hamel, D., Zaniolo, K., Joyal, J. S., Cho, J. H., Honore, J. C., Kermorvant-Duchemin, E., Varma, D. R., Tremblay, S., Leduc, M., Rihakova, L., Hardy, P., Klein, W. H., Mu, X., Mamer, O., Lachapelle, P., Di, P. A., Beausejour, C., Andelfinger, G., Mitchell, G., Sennlaub, F., and Chemtob, S. (2008). The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med. 14, 1067–1076.
Schweda, F., Friis, U., Wagner, C., Skott, O., and Kurtz, A. (2007). Renin release. Physiology 22, 310–319.
Steckelings, U. M., Rompe, F., Kaschina, E., and Unger, T. (2009). The evolving story of the RAAS in hypertension, diabetes and CV disease: moving from macrovascular to microvascular targets. Fundam. Clin. Pharmacol. 23, 693–703.
Suzaki, Y., Prieto-Carrasquero, M. C., and Kobori, H. (2006). Intratubular renin-angiotensin system in hypertension. Curr. Hypertens. Rev. 2, 151–157.
Taylor, C. T. (2008). Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 409, 19–26.
Toma, I., Kang, J. J., Sipos, A., Vargas, S., Bansal, E., Hanner, F., Meer, E., and Peti-Peterdi, J. (2008). Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Invest. 118, 2526–2534.
Vargas, S. L., Toma, I., Kang, J. J., Meer, E. J., and Peti-Peterdi, J. (2009). Activation of the succinate receptor GPR91 in macula densa cells causes renin release. J. Am. Soc. Nephrol. 20, 1002–1011.
Weinberg, J. M., Venkatachalam, M. A., Roeser, N. F., and Nissim, I. (2000). Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. U.S.A. 97, 2826–2831.
Keywords: oxidative stress, GPCR, SUCNR1, GPR91, succinate, diabetes mellitus, renal disease, rennin
Citation: Ariza AC, Deen PMT and Robben JH (2012) The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrin. 3:22. doi: 10.3389/fendo.2012.00022
Received: 02 November 2011;
Accepted: 29 January 2012;
Published online: 16 February 2012.
Edited by:
Nicola J. Smith, Victor Chang Cardiac Research Institute, AustraliaReviewed by:
Nicola J. Smith, Victor Chang Cardiac Research Institute, AustraliaChengcheng “Alec” Zhang, UT Southwestern Medical Center, USA
Keyong Du, Tufts Medical Center, USA
Copyright: © 2012 Ariza, Deen and Robben. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Joris Hubertus Robben, Department of Physiology, Nijmegen Centre for Molecular Life Sciences, Radboud University Nijmegen Medical Centre, Nijmegen, Netherlands. e-mail: j.robben@fysiol.umcn.nl