
- 1Division of Endocrinology, Department of Medicine, Indiana University Medical School, Indianapolis, IN, USA
- 2Division of Radiation and Cancer Biology, Department of Radiation Oncology, Stanford University School of Medicine, Stanford, CA, USA
Bone marrow is a highly heterogeneous and vascularized tissue. The various cell types populating the bone marrow extensively communicate with each other, and cell-to-cell cross talk is likely to be essential for proper bone development and homeostasis. In particular, the existence of osteogenesis and angiogenesis coupling has been recently proposed. Despite its high degree of vascularization, a gradient of oxygenation is present in the bone marrow, and the endosteal surface of cortical bone appears to be among the most hypoxic areas in the body. Oxygen (O2) is both an essential metabolic substrate and a regulatory signal that is in charge of a specific genetic program. An important component of this program is the family of transcription factors known as hypoxia-inducible factors (HIFs). In this Perspective, we will summarize our current knowledge about the role of the HIF signaling pathway in controlling bone development and homeostasis, and especially in regulating the crosstalk between osteoblasts, progenitor cells, and bone marrow blood vessels.
Bone Marrow: A Highly Vascularized Tissue with a Gradient of Oxygenation
Skeletal development is dependent on two mechanisms: intramembranous ossification and endochondral replacement (1, 2). Intramembranous ossification is a process in which mesenchymal cells differentiate directly into osteoblasts, is responsible for the formation of the flat bones of the skull. In contrast, endochondral replacement, which accounts for the development of most other bones, is a two-step process: mesenchymal cells give origin to chondrocytes, which build a template known as growth plate; this template is next replaced by bone.
Bone marrow is a highly heterogeneous and highly vascularized tissue. In addition to endothelial cells, at least two other cell types populate the adult bone marrow: hematopoietic cells, which originate from the very well characterized hematopoietic stem cell (HSC), and mesenchymal cells, which include stromal cells, osteoblasts, and adipocytes, and are thought to be derived from a still not fully defined mesenchymal stem cell population (3). Stromal cells are a meshwork of osteoblast precursors and specialized fibroblasts known as reticular or adventitial cells. However, the relationship between these two types of stromal cells, if any, remains to be elucidated (4).
It is noteworthy that HSCs can also give rise to osteoclasts, which are specialized cells that resorb bone (5). Furthermore, it has been hypothesized that hematopoietic and endothelial cells probably share a common mesodermal precursor (6).
The clear dichotomy between hematopoietic-endothelial cells and mesenchymal cells and their distinct embryological origin was already recognized more than 40 years ago by Le Douarin, who wrote in one of her seminal papers “The contribution to bone marrow histogenesis of cells of vascular and blood origin, on one hand, and of the elements of the cartilaginous model, on the other hand, was analyzed. It appeared that osteoblasts, osteocytes, and stromal cells of marrow are derived from the perichondrium. In contrast, the endothelium of the vascular buds and the hemopoietic cells which invade the diaphyseal cartilage during the endochondral ossification process do not belong to the mesenchymal bone primordium but have a fully extrinsic origin” (7).
The various cell types populating the bone marrow extensively communicate with each other, and the cellular cross talk is essential for proper control of bone and bone marrow development and homeostasis. For example, endothelial cells, osteoblasts, and stromal cells are components of niches that have an important role in the maintenance of the HSC pool (8–13), and in the regulation of the number of B-lymphocytes (14–16) and granulocytes (15, 17). Conversely, hematopoietic cells, in particular mast cells and megakaryocytes, are known to produce a large set of cytokines and growth factors that significantly affect bone mass (18, 19).
The capillary bed in the bone marrow is formed by “sinusoidal” capillaries (20–22). Sinusoids are discontinuous capillaries as their endothelial cells have both large fenestrations and wide spaces among them (23). Moreover, their basal membrane is also discontinuous. Their diameter is large (30–40 μm), which considerably reduces the rate of blood flow (24). These unique anatomical and functional features allow for maximum exchange of macromolecules as well as cell movement between the bone marrow and the blood. Sinusoidal capillaries are also found in liver and spleen (23), namely in the other two major organs that at different physiological stages and in pathological conditions contribute to hematopoiesis. Mesenchymal cells known as pericytes intermittently surround the sinusoidal endothelial cells (4), and produce their own basal membrane, which blends with the basal membrane produced by the endothelium.
O2, nutrients, and a cornucopia of hormones and growth factors, which are important for bone and bone marrow development and homeostasis, are brought to bone by blood vessels (25).
In addition, it has been convincingly proven that blood vessels supply osteoblast precursors either with their flow (26) or as components of their walls (4, 27). Along these lines, it has been shown that bone marrow pericytes, which are positive for CD146 contribute to colony forming units in vitro and thus, according to the model proposed by Friedenstein decades ago (28, 29), are likely precursors of osteoblasts and bone marrow adipocytes (4). Moreover, it has been elegantly demonstrated that blood vessels guide the migration of osteoblast precursors from the periosteum to the bone marrow (27). Conversely, fully differentiated periosteal osteoblasts have apparently lost the ability to migrate from the periosteum into the bone marrow (27). Lastly, it has been reported that endothelial cells expressing mutant activin 1 receptors (ACVR1) can trans differentiate into osteoblasts, an event that may be critical in the pathogenesis of a devastating disease such as Fibrodysplasia Ossificans Progressiva (FOP) (30–32).
Taken together, the concept that blood vessels regulate the pool of osteoblasts both in physiological and in pathological states is progressively gaining strong support by the scientific community (33). Whether and how cells of the osteoblast lineage control number and shape of blood vessels in physiological conditions in vivo, is also an important question that needs to be investigated in more depth.
Despite its high degree of vascularization, a gradient of oxygenation is present in the bone marrow, and the endosteal surface of cortical bone is among the most hypoxic areas as revealed by staining with the marker of hypoxia pimonidazole (34–36). The high degree of bone marrow cellularity, the high levels of O2 consumption by leukocytes as well as the sluggish blood flow in the sinusoids are all thought to be all contributing factors to the generation of a gradient of oxygenation within the bone marrow (36, 37).
In the next sections of this Perspective, we will discuss the most recent findings about the role of the hypoxia-inducible factor (HIF) signaling pathway in osteoblast biology and in the osteoblast-dependent control of bone marrow angiogenesis.
The HIF Signaling Pathway
Oxygen is both an essential metabolic substrate in numerous enzymatic reactions, including mitochondrial respiration, and a regulatory signal that controls a specific genetic program. An important component of this program is the transcription factor HIF-1α (2, 38 –43), which is a key-mediator of cellular adaptation to low O2 tension (hypoxia). In normoxia, a class of 2-oxoglutarate-dependent and Fe2+-dependent prolyl-4-hydroxylases (PHDs) hydroxylate specific proline residues of the HIF-1α protein (44, 45). Hydroxylated HIF-1α is then targeted to the proteosome for degradation by the E3 ubiquitin ligase von Hippel Lindau (VHL). Under hypoxic conditions, hydroxylation of HIF-1α diminishes, HIF-1α protein accumulates, translocates from the cytoplasm to the nucleus, dimerizes with its constitutively expressed partner HIF-1β, binds in a sequence dependent manner to hypoxia-responsive elements, recruits transcriptional co-activators, and increases the transcription of an ever growing number of hypoxia-responsive genes (44–49). The protein products of these HIF target genes regulate a variety of biological processes, including angiogenesis, non-oxidative glycolysis, and matrix formation (2). While the mRNA encoding HIF-1α is widely expressed (50), the regulation of HIF-1α is largely post-transcriptional. In tissues where oxygen tension is higher than 5%, HIF-1α protein is barely detectable, but when oxygen tension drops below 5% HIF-1α protein progressively accumulates. Mouse embryos lacking HIF-1α die in utero by E11 (51–53), indicating that HIF-1α is critical for embryonic development.
Two other members of the family, HIF-2α and HIF-3α, have been cloned and characterized. Hypoxia controls stability of HIF-2α in a similar fashion as HIF-1α (54). HIF-2α and HIF-1α have common targets as well as specific ones (54–58). Moreover, they exhibit different tissue expression patterns (59). Lastly, in some genetic backgrounds mice lacking HIF-2α survive postnatally (60), where lack of HIF-1α causes early embryonic lethality. In contrast to HIF-1α and HIF-2α, the biological role of the HIF-3α isoform is still largely unknown, though it has been proposed that this protein could have a dominant negative function, since it lacks transactivation domains (61).
The E3 ubiquitin ligase VHL is expressed in most tissues and cell types, and is critical for HIF-1α degradation. Heterozygous missense mutations of the VHL gene have been identified in the VHL syndrome (62). This syndrome is characterized by a dominantly inherited predisposition to develop pheochromocytoma, clear cell renal carcinoma, and hemangioblastomas in the central nervous system and in the retina (62). As discussed above, VHL has E3 ubiquitin ligase activity, and the HIFs are among its main targets. The importance of VHL for proteolysis of HIF-1α and HIF-2α is highlighted by the finding that cells lacking a functional VHL are unable to degrade these transcription factors, ultimately resulting in an accumulation of HIFs (45, 49, 63, 64). However, VHL has also a variety of biological activities including control of the cell cycle, critical interaction with the cytoskeleton and with the primary cilia, and regulation of matrix proteins that have been reported to be HIF-independent (62).
The HIF Signaling Pathway in Osteoblasts and its Impact on Bone and Blood Vessels
Loss of VHL in fully differentiated osteoblasts, resulting in the stabilization of HIF-1α and HIF-2α, and increased activity of HIF signaling in these cells, leads to a high bone mass phenotype in the bones that are formed through the endochondral replacement process. In contrast, the flat bones of the skull, which develop through an intramembranous program, appear to be largely unaffected (36, 65). Strikingly, this increased accumulation of trabecular bone in the long bones is associated with an augmented surface and volume of blood vessels in the bone marrow cavity (16, 65). Therefore, activation of the HIF signaling pathway in osteoblasts is sufficient to modulate bone marrow angiogenesis.
More recently, these findings were confirmed and expanded in another mouse model lacking VHL in osteoprogenitors. Loss of VHL in osteoprogenitors (OSX-VHL) caused a dramatic increase of trabecular bone mass (16). In particular, OSX-VHL mutant mice exhibited excessive accumulation of trabecular bone in the metaphyseal and diaphyseal regions of the long bones (16). Surrounding the numerous trabecule were abundant stromal cells and strikingly dilated blood vessels (angiectasis) (Figure 1) (16). In agreement with previous findings, the frequency of HSCs was increased in the bone marrow of OSX-VHL mutant mice (16). Although it is still uncertain whether this apparent expansion of the HSC pool is a consequence of either the expanded osteoblastic niche, or the augmented blood vessel surface, or both. Dilated blood vessels and expansion of stromal cells are two histological hallmarks of the hemangioblastoma lesions identified in VHL syndrome (66). Loss of HIF-1α and HIF-2α in VHL-deficient osteoprogenitors fully corrects the high bone mass phenotype, the increased number of HSCs, the angiectasis, and the aberrant expansion of the bone marrow stroma secondary (16).
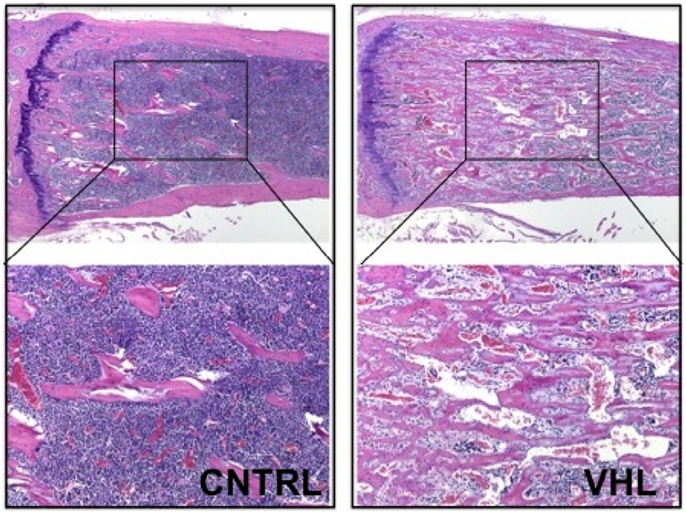
Figure 1. Loss of VHL in cells of the osteoblast lineage increases trabecular bone mass. H and E of histological sections of tibia isolated from 4-month-old control (CNTRL) and OSX-VHL mutant (VHL) mice; bottom panels are blows up of a and b, respectively. For details, see Text.
Taken together, these data demonstrate that augmented HIF activity in osteoprogenitors augments trabecular bone volume, increases the bone marrow stromal population, and expands the HSC pool (16). Moreover, loss of VHL in osteoprogenitor cells demonstrates that stabilization of HIFs in osteoblasts is sufficient to increase number and/or size of blood vessels in the bone marrow. However, it remains unresolved whether osteoblastic HIFs are necessary for regulation of bone marrow angiogenesis in a more physiological setting.
VEGF and EPO: Potential Candidates Mediating the Osteoblasts-Blood Vessels Cross Talk in the Bone Marrow
Numerous proangiogenic factors are transcriptionally regulated by HIFs, and each of them could be responsible for the modulation of blood vessel number and/or size by osteoblasts when the HIF transcriptional pathway is activated in these cells. Among them, vascular endothelial growth factor-A (VEGF) is one of the best-characterized downstream targets of HIFs.
Vascular endothelial growth factor is a glycoprotein that belongs to the dimeric cysteine-knot growth factor super-family (67, 68). The mouse VEGF gene encodes at least three isoforms (VEGF120, VEGF164, and VEGF188) that arise through alternative splicing (69, 70). While VEGF164 and VEGF188 bind the extracellular matrix component heparan sulfate, VEGF120 lacks the binding motif necessary for this interaction (71, 72). VEGF is a principal regulator of blood vessel formation, and it is also a survival factor for HSCs in vivo and in vitro, through an internal autocrine loop mechanism (73). In endochondral bones, VEGF is a survival factor for chondrocytes in vivo, at least in part by regulating angiogenesis in the surrounding soft tissue (74). Moreover, in hypertrophic chondrocytes it controls blood vessel invasion and thus replacement of cartilage by bone in vivo (75). Furthermore, it promotes osteogenesis in vitro (68), and when overexpressed in cells of the osteoblast lineage in vivo it leads to a high bone mass phenotype associated to marrow fibrosis and increased number of blood vessels (76). These effects appear to be secondary, at least in part, to activation of beta catenin in osteoblasts (76). Conversely, mice that lack VEGF in osteoprogenitors display a reduced bone mass phenotype with increased bone marrow fat. Even more intriguing in this mouse model, VEGF regulates the balance between osteoblasts and adipocytes by a novel, intracrine mechanism (77). VEGF is also a positive modulator of osteoclastogenesis as well, both in vivo and in vitro (76, 78, 79).
Vascular endothelial growth factor expression, as expected, is increased in mutant bones lacking VHL in cells of the osteoblast lineage (16, 65). Therefore, it is reasonable to hypothesize that VEGF could be responsible for the increased number and/or size of blood vessels in these mutants (25), although this possibility has still to be experimentally tested.
Notably, mutant mice lacking VHL in cells of the osteoblast lineage develop severe polycythemia, which is due to HIF-2α dependent overproduction of erythropoietin (EPO) by osteoblasts (16). This study provided the first demonstration that cells of the osteoblast lineage are capable of producing and secreting EPO upon activation of the HIF signaling pathway in vivo (16). The HIF-2, rather than the HIF-1, dependence of EPO production is consistent with what has been reported in other experimental models (80, 81). EPO is a hormone that stimulates erythropoiesis through binding and activation of its receptor (EpoR) in erythroid progenitor cells (82 –84). In adults, more than 90% of EPO is produced in the kidney by a subset of peritubular interstitial fibroblasts (84). The key regulatory mechanism of EPO expression is hypoxia through the activation of the HIF pathway (84). Anemia stimulates EPO production by inducing local hypoxia (84). Unlike EPO, EpoR expression is not affected by hypoxia or anemia (83). In the cell membrane, EpoR forms a homodimer that, upon EPO binding, recruits JAK2, which in turns leads to activation of STAT5, PI3 Kinase (PI3K), and Ras-MAPK signaling pathways (83). Both EPO and EpoR are also expressed in a variety of non-hematopoietic tissues, which implies that EPO may have functions that go beyond regulation of erythropoiesis. For example, EpoR has been identified on the surface of endothelial cells (82, 83). Consistent with this finding, the EPO/EpoR complex is thought to be an important regulator of angiogenesis, particularly in the context of malignant tumors (82, 83). Interestingly, in addition to its direct effect on endothelial cells, EPO may control angiogenesis by modulating the release of other angiogenic factors such as VEGF (83).
Taken together, it is tempting to speculate that both EPO and VEGF interact to coordinate bone marrow angiogenesis, when the HIF signaling pathway is activated in osteoblasts (Figure 2).
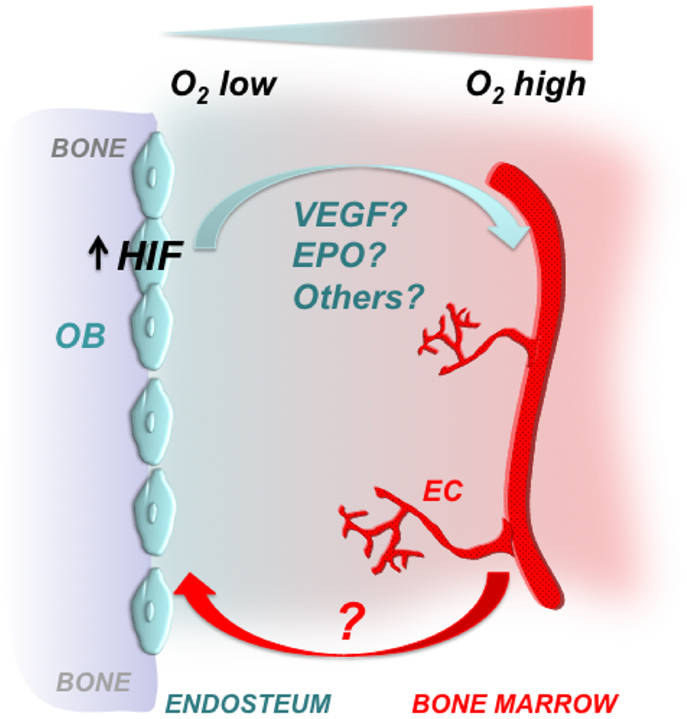
Figure 2. Osteoblastic HIFs control bone marrow angiogenesis. Gradients of oxygenation are present in the bone marrow with osteoblasts at the endosteal surface of bone being among the most hypoxic areas. Osteoblasts express HIFs, which regulate osteoblast activity and angiogenesis. Some of the HIF effects on angiogenesis could be mediated by VEGF, EPO, and/or other proangiogenic factors. It has yet to be defined whether increased angiogenesis is a necessary pre-requisite for the high bone mass phenotype observed upon activation of HIFs in cells of the osteoblast lineage. OB, osteoblast; EC, endothelial cell.
Future Directions
In this Perspective, we have highlighted the critical role of the HIF signaling pathway in regulating bone mass and bone marrow angiogenesis. It will be now important to identify the molecular mechanisms that mediate osteoblasts-blood vessel cross talk, and to define whether increased angiogenesis is a necessary pre-requisite for the high bone mass phenotype observed upon activation of HIFs in cells of the osteoblast lineage (Figure 2). Addressing these two questions may significantly expand our understanding of the osteogenesis–angiogenesis coupling in physiological and pathological states.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Provot S, Schipani E. Molecular mechanisms of endochondral bone development. Biochem Biophys Res Commun (2005) 328:658–65. doi:10.1016/j.bbrc.2004.11.068
2. Maes C, Carmeliet G, Schipani E. Hypoxia-driven pathways in bone development, regeneration and disease. Nat Rev Rheumatol (2012) 8:358–66. doi:10.1038/nrrheum.2012.36
3. Ohishi M, Schipani E. Bone marrow mesenchymal stem cells. J Cell Biochem (2010) 109:277–82. doi:10.1002/jcb.22399
4. Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell (2007) 131:324–36. doi:10.1016/j.cell.2007.08.025
5. Boyle W, Simonet W, Lacey D. Osteoclast differentiation and activation. Nature (2003) 423:337–42. doi:10.1038/nature01658
6. Xiong JW. Molecular and developmental biology of the hemangioblast. Dev Dyn (2008) 237:1218–31. doi:10.1002/dvdy.21542
7. Le Douarin NM, Houssaint E, Jotereau FV, Belo M. Origin of hemopoietic stem cells in embryonic bursa of Fabricius and bone marrow studied through interspecific chimeras. Proc Natl Acad Sci U S A (1975) 72:2701–5. doi:10.1073/pnas.72.7.2701
8. Taichman RS, Emerson SG. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med (1994) 179:1677–82. doi:10.1084/jem.179.5.1677
9. Taichman RS, Reilly MJ, Emerson SG. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood (1996) 87:518–24.
10. Taichman RS, Reilly MJ, Emerson SG. The hematopoietic microenvironment: osteoblasts and the hematopoietic microenvironment. Hematology (2000) 4:421–6.
11. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature (2003) 425:841–6. doi:10.1038/nature02040
12. Méndez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, MacArthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature (2010) 466:829–34. doi:10.1038/nature09262
13. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature (2013) 495:231–5. doi:10.1038/nature11885
14. Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood (2007) 109:3706–12. doi:10.1182/blood-2006-08-041384
15. Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A (2008) 105:16976–81. doi:10.1073/pnas.0802898105
16. Rankin EB, Wu C, Khatri R, Wilson TL, Andersen R, Araldi E, et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell (2012) 149:63–74. doi:10.1016/j.cell.2012.01.051
17. Fulzele K, Krause DS, Panaroni C, Saini V, Barry KJ, Liu X, et al. Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood (2013) 121:930–9. doi:10.1182/blood-2012-06-437160
18. Turner RT, Iwaniec UT, Marley K, Sibonga JD. The role of mast cells in parathyroid bone disease. J Bone Miner Res (2010) 25:1637–49. doi:10.1002/jbmr.49
19. Kacena MA, Shivdasani RA, Wilson K, Xi Y, Troiano N, Nazarian A, et al. Megakaryocyte-osteoblast interaction revealed in mice deficient in transcription factors GATA-1 and NF-E2. J Bone Miner Res (2004) 19:652–60. doi:10.1359/JBMR.0301254
20. Draenert K, Draenert Y. The vascular system of bone marrow. Scan Electron Microsc (1980) 4:113–22.
21. Roche B, David V, Vanden-Bossche A, Peyrin F, Malaval L, Vico L, et al. Structure and quantification of microvascularisation within mouse long bones: what and how should we measure? Bone (2012) 50:390–9. doi:10.1016/j.bone.2011.09.051
22. Kristensen HB, Andersen TL, Marcussen N, Rolighed L, Delaisse JM. Increased presence of capillaries next to remodeling sites in adult human cancellous bone. J Bone Miner Res (2013) 28:574–85. doi:10.1002/jbmr.1760
23. Couvelard A, Scoazec JY, Dauge MC, Bringuier AF, Potet F, Feldmann G. Structural and functional differentiation of sinusoidal endothelial cells during liver organogenesis in humans. Blood (1996) 87:4568–80.
24. Bianco P. Bone and the hematopoietic niche: a tale of two stem cells. Blood (2011) 117:5281–8. doi:10.1182/blood-2011-01-315069
25. Schipani E, Maes C, Carmeliet G, Semenza GL. Regulation of osteogenesis-angiogenesis coupling by HIFs and VEGF. J Bone Miner Res (2009) 24:1347–53. doi:10.1359/jbmr.090602
26. Zvaifler NJ, Marinova-Mutafchieva L, Adams G, Edwards CJ, Moss J, Burger JA, et al. Mesenchymal precursor cells in the blood of normal individuals. Arthritis Res (2000) 2:477–88. doi:10.1186/ar130
27. Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell (2010) 19:329–44. doi:10.1016/j.devcel
28. Friedenstein A, Chailakhjan R, Lalykina K. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet (1970) 3:393–403.
29. Friedenstein A. Osteogenic stem cells in the bone marrow. In: JNM Heersche and JA Kanis editors. Bone and Mineral Research. (Vol. 1), San Diego: Elsevier Science Publisher (1990). p. 243–70.
30. Medici D, Olsen BR. The role of endothelial-mesenchymal transition in heterotopic ossification. J Bone Miner Res (2012) 27:1619–22. doi:10.1002/jbmr.1691
31. Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med (2010) 16:1400–6. doi:10.1038/nm.2252
32. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet (2006) 38:525–7. doi:10.1038/ng1783
33. Bianco P, Sacchetti B, Riminucci M. Osteoprogenitors and the hematopoietic microenvironment. Best Pract Res Clin Haematol (2011) 24:37–47. doi:10.1016/j.beha.2011.01.005
34. Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A (2007) 104:5431–6. doi:10.1073/pnas.0701152104
35. Winkler IG, Barbier V, Wadley R, Zannettino AC, Williams S, and Lévesque JP. Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood (2010) 116:375–85. doi:10.1182/blood-2009-07-233437
36. Rankin EB, Giaccia AJ, Schipani E. A central role for hypoxic signaling in cartilage, bone, and hematopoiesis. Curr Osteoporos Rep (2011) 9:46–52. doi:10.1007/s11914-011-0047-2
37. Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I. Krogh’s model. Biophys J (2001) 81:675–84. doi:10.1016/S0006-3495(01)75733-5
38. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A (1995) 92:5510–4. doi:10.1073/pnas.92.12.5510
39. Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev (1996) 76:839–85.
40. Kaelin WG Jr. How oxygen makes its presence felt. Genes Dev (2002) 16:1441–5. doi:10.1101/gad.1003602
41. Giaccia A, Siim B, Johnson R. HIF-1 as a target for drug development. Nat Rev Drug Discov (2003) 2:803–11. doi:10.1038/nrd1199
42. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3:721–32. doi:10.1038/nrc1187
43. Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther (2004) 3:492–7.
44. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi:10.1126/science.1059796
45. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science (2001) 292:464–8. doi:10.1126/science.1059817
46. Ohh M, Kaelin W. The von Hippel-Lindau tumor suppressor protein: new perspectives. Mol Med Today (1999) 5:257–63. doi:10.1016/S1357-4310(99)01481-1
47. Chan D, Suthphin P, Denko N, Giaccia A. Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1alpha. J Biol Chem (2002) 277:40112–7. doi:10.1074/jbc.M206922200
48. Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr, Pavletich NP. Structure of an HIF-1alpha-pVHL complex: hydroxyproline recognition in signaling. Science (2002) 296:1886–9. doi:10.1126/science.1073440
49. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature (1999) 399:271–5. doi:10.1038/20459
50. Wenger RH, Rolfs A, Spielmann P, Zimmermann DR, Gassmann M. Mouse hypoxia-inducible factor-1alpha is encoded by two different mRNA isoforms: expression from a tissue-specific and a housekeeping-type promoter. Blood (1998) 91:3471–80.
51. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev (1998) 12:149–62. doi:10.1101/gad.12.2.149
52. Ryan HE, Lo J, Johnson RS. HIF 1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J (1998) 17:3005–15. doi:10.1093/emboj/17.11.3005
53. Compernolle V, Brusselmans K, Franco D, Moorman A, Dewerchin M, Collen D, et al. Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1alpha. Cardiovasc Res (2003) 60:569–79. doi:10.1016/j.cardiores.2003.07.003
54. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2011) 12:9–22. doi:10.1038/nrc3183
55. Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev (2008) 9:285–96. doi:10.1038/nrm2354
56. Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol (2003) 23:9361–74. doi:10.1128/MCB.23.24.9361-9374.2003
57. Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol (2005) 25:5675–86. doi:10.1128/MCB.25.13.5675-5686.2005
58. Wang V, Davis DA, Haque M, Huang LE, Yarchoan R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res (2005) 65:3299–306.
59. Wiesener MS, Jürgensen JS, Rosenberger C, Scholze CK, Hörstrup JH, Warnecke C, et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J (2003) 17:271–3.
60. Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet (2003) 35:331–40. doi:10.1038/ng1266
61. Jogi A, Øra I, Nilsson H, Lindeheim A, Makino Y, Poellinger L. Hypoxia alters gene expression in human neuroblastoma cells towards an immature and neural crest-like phenotype. Proc Natl Acad Sci U S A (2002) 99:7021–6. doi:10.1073/pnas.102660199
62. Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer (2008) 8:865–73. doi:10.1038/nrc2502
63. Haase VH. The VHL tumor suppressor in development and disease: functional studies in mice by conditional gene targeting. Semin Cell Dev Biol (2005) 16:564–74. doi:10.1016/j.semcdb.2005.03.006
64. Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF 1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A (2000) 97:10430–5. doi:10.1073/pnas.190332597
65. Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert SR, et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest (2007) 117:1616–26. doi:10.1172/JCI31581
66. Shively SB, Beltaifa S, Gehrs B, Duong H, Smith J, Edwards NA, et al. Protracted haemangioblastic proliferation and differentiation in von Hippel-Lindau disease. J Pathol (2008) 216:514–20. doi:10.1002/path.2435
67. Ferrara N, Gerber H, LeCouter J. The biology of VEGF and its receptors. Nat Med (2003) 9:669–76. doi:10.1038/nm0603-669
68. Zelzer E, Olsen BR. Multiple roles of vascular endothelial growth factor (VEGF) in skeletal development, growth, and repair. Curr Top Dev Biol (2005) 65:169–87. doi:10.1016/S0070-2153(04)65006-X
69. Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocrinol (1991) 5:1806–14. doi:10.1210/mend-5-12-1806
70. Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem (1991) 266:11947–54.
71. Robinson C, Stringer S. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci (2001) 114:853–65.
72. Cohen T, Gitay-Goren H, Sharon R, Shibuya M, Halaban R, Levi BZ. VEGF121, a vascular endothelia growth factor (VEGF) isoform lacking heparin binding ability, requires cell-surface heparan sulfates for efficient binding to the VEGF receptors of human melanoma cells. J Biol Chem (1995) 270:11322–6. doi:10.1074/jbc.270.19.11322
73. Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, et al. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature (2002) 417:954–8. doi:10.1038/nature00821
74. Maes C, Araldi E, Haigh K, Khatri R, Van Looveren R, Giaccia AJ. VEGF-independent cell-autonomous functions of HIF-1alpha regulating oxygen consumption in fetal cartilage are critical for chondrocyte survival. J Bone Miner Res (2012) 27:596–609. doi:10.1002/jbmr.1487
75. Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med (1999) 5:623–8. doi:10.1038/9467
76. Maes C, Goossens S, Bartunkova S, Drogat B, Coenegrachts L, Stockmans I, et al. Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J (2010) 29:424–41. doi:10.1038/emboj.2009.361
77. Liu Y, Berendsen AD, Jia S, Lotinun S, Baron R, Ferrara N, et al. Intracellular VEGF regulates the balance between osteoblast and adipocyte differentiation. J Clin Invest (2012) 122:3101–13. doi:10.1172/JCI61209
78. Niida S, Kondo T, Hiratsuka S, Hayashi S, Amizuka N, Noda T, et al. VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc Natl Acad Sci U S A (2005) 102:14016–21. doi:10.1073/pnas.0503544102
79. Niida S, Kaku M, Amano H, Yoshida H, Kataoka H, Nishikawa S, et al. Vascular endothelial growth factor can substitute for macrophage colony-stimulating factor in the support of osteoclastic bone resorption. J Exp Med (1999) 190:293–8. doi:10.1084/jem.190.2.293
80. Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest (2007) 117:1068–77. doi:10.1172/JCI30117
81. Scortegagna M, Ding K, Zhang Q, Oktay Y, Bennett MJ, Bennett M, et al. HIF-2alpha regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood (2005) 105:3133–40. doi:10.1182/blood-2004-05-1695
82. Jelkmann W. Erythropoietin after a century of research: younger than ever. Eur J Haematol (2007) 78:183–205. doi:10.1111/j.1600-0609.2007.00818.x
83. Cao Y. Erythropoietin in cancer: a dilemma in risk therapy. Trends Endocrinol Metab (2013) 24:190–9. doi:10.1016/j.tem.2012.10.007
Keywords: hypoxia-inducible factor, vascular endothelial growth factor, prolyl hydroxylases, erythropoietin, osteoblasts
Citation: Schipani E, Wu C, Rankin EB and Giaccia AJ (2013) Regulation of bone marrow angiogenesis by osteoblasts during bone development and homeostasis. Front. Endocrinol. 4:85. doi: 10.3389/fendo.2013.00085
Received: 03 May 2013; Accepted: 26 June 2013;
Published online: 10 July 2013.
Edited by:
Rhonda Prisby, University of Delaware, USAReviewed by:
Carmen Huesa, University of Edinburgh, UKMatt Prideaux, University of Missouri-Kansas City, USA
Copyright: © 2013 Schipani, Wu, Rankin and Giaccia. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Ernestina Schipani, Indiana University School of Medicine, 980 West Walnut Street, R3, Room C104, Indianapolis, IN 46202, USA e-mail: eschipan@iu.edu;
Amato J. Giaccia, Division of Radiation and Cancer Biology, Department of Radiation Oncology, Stanford University, CCSR-South, Room 1255, 269 Campus Drive, Stanford, CA 94303-5152, USA e-mail: giaccia@stanford.edu