 Robert Root-Bernstein
Robert Root-Bernstein Abigail Podufaly2
Abigail Podufaly2 Patrick F. Dillon
Patrick F. Dillon- 1Department of Physiology, Michigan State University, East Lansing, MI, USA
- 2College of Osteopathic Medicine, Michigan State University, East Lansing, MI, USA
Rationale: Insulin (INS) resistance associated with hyperestrogenemias occurs in gestational diabetes mellitus, polycystic ovary syndrome, ovarian hyperstimulation syndrome, estrogen therapies, metabolic syndrome, and obesity. The mechanism by which INS and estrogen interact is unknown. We hypothesize that estrogen binds directly to INS and the insulin receptor (IR) producing INS resistance.
Objectives: To determine the binding constants of steroid hormones to INS, the IR, and INS-like peptides derived from the IR; and to investigate the effect of estrogens on the binding of INS to its receptor.
Methods: Ultraviolet spectroscopy, capillary electrophoresis, and NMR demonstrated estrogen binding to INS and its receptor. Horse-radish peroxidase-linked INS was used in an ELISA-like procedure to measure the effect of estradiol on binding of INS to its receptor.
Measurements: Binding constants for estrogens to INS and the IR were determined by concentration-dependent spectral shifts. The effect of estradiol on INS binding to its receptor was determined by shifts in the INS binding curve.
Main Results: Estradiol bound to INS with a Kd of 12 × 10−9 M and to the IR with a Kd of 24 × 10−9 M, while other hormones had significantly less affinity. Twenty-two nanomolars of estradiol shifted the binding curve of INS to its receptor 0.8 log units to the right.
Conclusion: Estradiol concentrations in hyperestrogenemic syndromes may interfere with INS binding to its receptor producing significant INS resistance.
Introduction
Insulin (INS) resistance associated with hyperestrogenemias occurs in gestational diabetes mellitus (GDM), polycystic ovary syndrome (PCOS), ovarian hyperstimulation syndrome (OHSS), estrogen therapies, metabolic syndrome, and obesity. The mechanism by which INS and estrogen interact is unknown, but both in vitro and in vivo studies suggest that estradiol (E2) directly or indirectly inhibits INS binding to, and/or activation of, the insulin receptor (IR).
The concentration of estrogen hormones varies across a biologically extraordinary range of almost three orders of magnitude in the normal human being, ranging between 30 pg/ml (0.06 nM) in normal males and menstruating females and 10 ng/ml (40 nM) in some pregnant women (1–7). At plasma concentrations of E2 above about 1 nM, INS resistance often develops. Some degree of INS resistance occurs in virtually all pregnant women and between 2 and 10% of pregnancies are so severely affected by GDM that they require INS treatment (5–15). Similar rates of INS resistance and diabetes occur as a result of OHSS in which E2 concentrations often exceed 10 nM (16, 17). After parturition, INS sensitivity typically returns rapidly to pre-pregnancy levels (1–10). High-dose estrogen-containing contraceptives, but not low-dose ones, have, in the past, also been associated with INS resistance and hyperinsulinemia (18–24) and in men, elevated E2 (often resulting from aromatase conversion of testosterone to E2) is also an independent risk factor for metabolic syndrome, as it may occur during puberty or following synthetic androgen use in body building (3, 25–27). The development of INS resistance also occurs in women who are treated with cyproterone acetate and ethinyl estradiol for hirsutism and/or acne (28). Administration of E2 or testosterone (which can be converted to E2) in persons undergoing sexual reassignment is another cause of INS resistance (27, 29). And 45–70% of patients suffering from PCOS have varying severities of INS resistance associated with hyperinsulinemia (30–39). In PCOS, concentrations of E2 can exceed 1 ng/ml or 4 nM (36, 37).
All of these clinical observations suggest that INS resistance is somehow related to the concentration of plasma E2.
In vitro experiments have helped to unravel some interactions between E2 and INS. The effects of E2 on INS activity are tri-modal. Cells that are grown in media lacking E2 or which are exposed to hypoestrogenemic conditions develop significant INS resistance as a result of decreased expression of IR and the glucose transporter GLUT4 (40–46). Normal levels of E2 are associated with normal INS and IR function. Cells exposed to hyperestrogenemic conditions develop significant decrease in INS and IR activity. Hypertestosterone syndromes such as PCOS may also contribute indirectly to E2 inhibition of INS and IR since aromatase converts testosterone to E2 resulting in hyperestrogenemia as well (30, 36, 47).
In vivo experiments and clinical studies confirm the in vitro results. Ovariectomized (OVEX) rats respond to E2 levels 100 times those found in normal gestation by decreasing the number of IR, increasing the serum concentration of INS threefold, decreasing phosphorylation of IR substrate-1, and decreasing GLUT4 expression; INS resistance ensues (48–51). Two types of data suggest that these rat studies can be extrapolated to human beings. First, functionally ovariectomized people given E2 as part of sex reassignment treatment also incur INS resistance in direct proportion to the dose of E2 (27–29). Additionally, women undergoing in vitro fertilization often reach supraphysiological levels of E2 >6,000 pg/ml (>10 nM) and develop INS resistance (52–59).
The combination of in vitro and in vivo and clinical studies summarized above strongly suggests that E2 alone is sufficient to induce INS resistance either directly or indirectly. Thus far, all attempts to understand E2-related INS resistance have focused on indirect mechanisms, such as reduced GLUT4 mRNA expression and transport (11, 38), increased INS synthesis and secretion (40, 60), and interactions between IR, IR complex 1 estrogen-receptor α (61). It is not clear whether these phenomena are, however, causes or results of E2-mediated activity.
We propose that a more direct mechanism may contribute to INS resistance in hyperestrogenemias: E2 may bind directly to INS and/or the IR at pathophysiologically high concentrations of E2.
Materials and Methods
Sex Hormones
All hormones, selective estrogen-receptor modulators (SERMS) were obtained from Sigma-Aldrich: 4-andro-stenedione (A9630), cholesterol (C8667), corticosterone (C2505), estradiol (E1024), estriol (E1253), estrone (E1274), hydrocortisone (H3160), 5-Pregnen-3β-ol-20-one (P9129), progesterone (P3972), Raloxifene HCl (R1402), Tamoxifen citrate salt (T9262), and Genistein (92136). We did not test testosterone since our department lacks the approval to obtain this controlled substance.
Peptides
Human recombinant INS (I0259 or I2643), INS A chain, oxidized (I1633) and INS B chain, and oxidized (I6383) and glucagon (G3157) were obtained from Sigma-Aldrich. Humulin R was obtained from Lilly. Peptides from the IR identified as being INS- or glucagon-like peptides (Table 1) were synthesized by the Mass Spectrometry, Synthesis, and Sequencing Facility of the Biochemistry Department of Michigan State University and purified to >98% purity as determined by high pressure liquid chromatography.
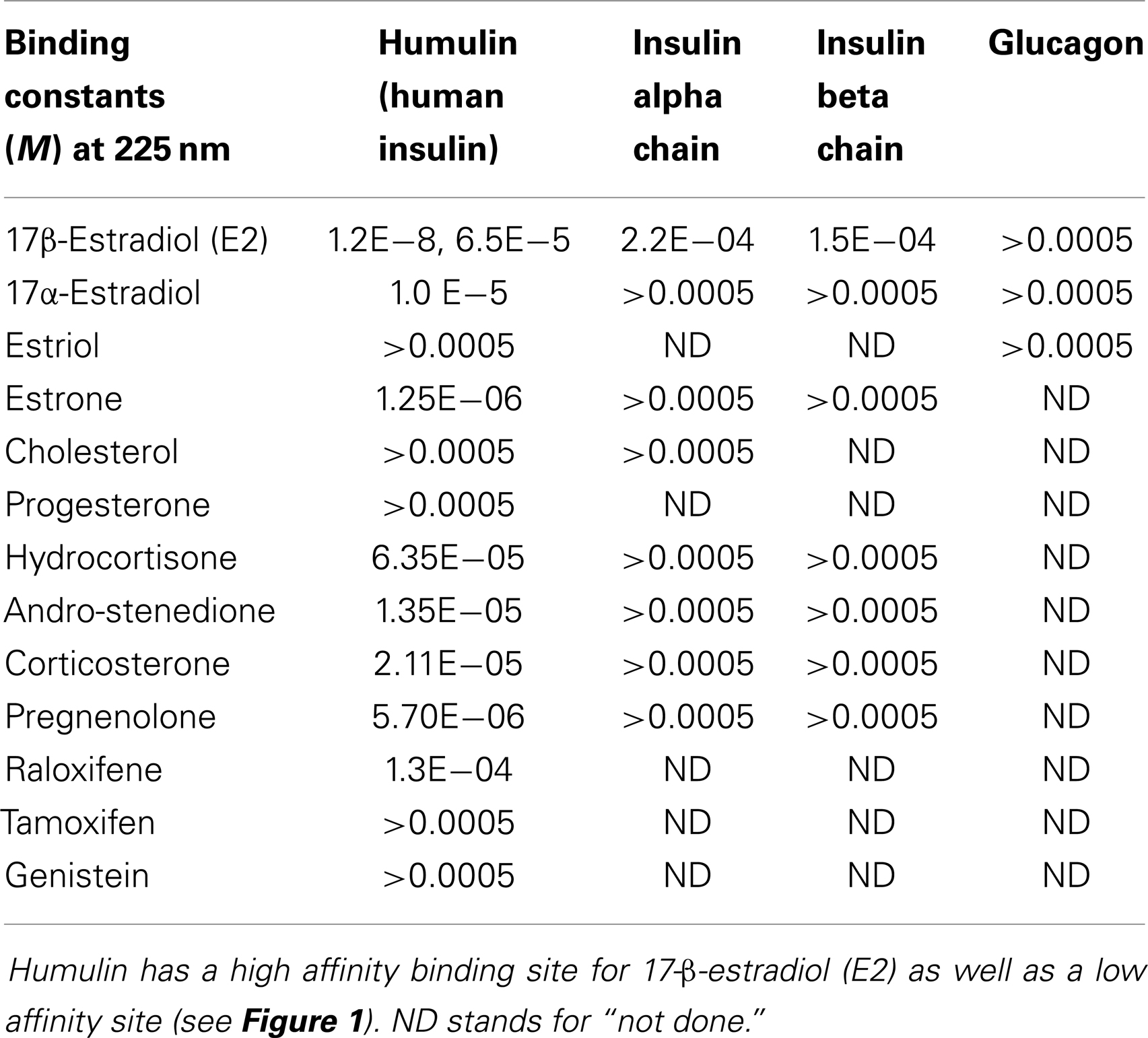
Table 1. Binding constants for various sex hormone binding to recombinant human insulin (Humulin), the oxidized insulin A chain, the oxidized insulin B chain, and glucagon.
Receptors
Rat IR membrane preparation (I-9266) (IR) and human INS-like growth factor receptor (IGFR) (I-4657) were obtained from Sigma-Aldrich. The rat IR was a partially purified preparation in residual membrane and contained 250 units of activity as measured by its ability to catalyze the incorporation of 1 pmol per min of phosphate from γ-32P-ATP into poly(Glu, Tyr), 4:1, at 30°C. The IGFR was recombinant, at least 95% pure by SDS-PAGE, and capable of binding INS-like growth factor. Both receptor preparations, in sum, retained activity.
UV Spectrophotometry
Ultraviolet spectroscopy was used to determine binding constants of sex hormones to INS, INS peptides, IR peptides, IR, and IGFR. This method has been used for decades by other investigators (62–65) as well as by the authors of this paper (55, 56, 66–69).
Insulin receptor peptides were prepared at 100 nM in pH 7.4 phosphate buffer (Fisher Scientific). Humulin and human recombinant INS were prepared at 12.5 and 25 nM in pH 7.4 phosphate buffer. IR (250 units) and INS-like growth factor receptor (50 μg) were each prepared for assay by diluting the manufacturer’s receptor preparation in 1.0 ml pH 7.4 phosphate buffer to about 3.5 × 10−12 M.
Sex hormones and controls were dissolved in 95% ethyl alcohol to 1.0 mM and diluted serially by thirds.
Hormone peptide binding studies were carried out as follows. In two columns of the serially diluted steroid hormone, 100 μL of diluted peptide was added to the wells. One hundred microliters of buffer was added to the other two columns containing serially diluted steroid hormones. As controls, in a fifth column, 100 μL of the diluted peptide was added to 100 μL of 95% ethyl alcohol in two wells. To two additional wells, 100 μL of 95% ethyl alcohol and 100 μL of buffer were added. Plates were incubated at room temperature for 30 min and were then read in a SpectraMax 384Plus from 200 to 290 nm. The differences between the expected absorbance values (determined by adding the individual hormone absorbances and the individual peptide, INS absorbances minus the buffer–ethanol controls) and the observed value of the combinations were then plotted as a function of the concentration of the hormone.
An additional method of ascertaining binding involved serial additions of estrogens to INS, INS peptides, or glucagon (as a control). Peptides were dissolved as above and 200 μL added to a well in a crystal 96-well plate. Two hundred microliters of buffer was placed in an adjacent well in the plate. Five microliters of aliquots of estrogens (1.0 or 0.1 μM in 95% ethanol) or SERMS (100 μM in 95% ethanol) were then added in tandem to the peptide well and the buffer well. An additional buffer well received a 5 μL aliquot of ethanol each time an estrogen or SERM was added to any other well. Additions were made serially. The UV spectrum was obtained after each addition as above at 225 nm. In this way, all wells were diluted with ethanol at the same rate and to the same concentrations at each aliquot. The absorbance value of the buffer well after each aliquot of ethanol was subtracted from each of the other wells (peptides, hormones, or SERMS, and their combinations). The absorbance value of the peptide, glucagon, or INS at each ethanol concentration was determined; the absorbance value of each hormone (or SERM) was determined; then these individual values were added to provide an expected value of their combination in accordance with Beer’s law. The difference between this expected absorbance value after each aliquot addition and that obtained from the actual experimental combination was then determined and this difference was plotted as a function of the estrogen or SERM concentration. All binding experiments were run in duplicate and the results averaged. Binding constants were determined from the inflection point of any S-shaped curves that resulted.
This serial addition method was also used to determine the binding of estradiol to IR and IGFR.
Capillary Electrophoresis
Capillary electropherograms of INS and estrogen were prepared at a variety of voltages. Use of capillary electrophoresis to determine binding between compounds was pioneered by this laboratory (70, 71). Human recombinant INS (0.6 mg/ml; Sigma-Aldrich) and beta estradiol (1.5 mg/ml), and a combination of INS plus beta estradiol at the same concentrations, were prepared in 50 mM pH 7.4 phosphate buffer and allowed to incubate at room temperature for 48 h. The estrogen was in crystalline form and did not dissolve; only the supernatant, avoiding any estrogen crystals, was used in the experiments. Each sample was then diluted in distilled water 1:4 for a buffer concentration of 10 mM. Samples were vacuum injected into an Isco 3950 electropherograph 100 μm inner diameter glass capillary of 49 cm with a distance from injection site to detection window of 27 cm. The carrier buffer was 10 mM sodium phosphate. Samples were driven from injection site to ground using positive voltage. Each sample containing INS alone, estrogen alone, or both, was driven at 10, 15, 20, 25, and 30 kV. Peaks were detected at 200 nm wavelength.
1H Nuclear Magnetic Resonance
Human recombinant INS (0.6 mg/ml; Sigma-Aldrich) and beta estradiol (1.5 mg/ml), and a combination of INS plus beta estradiol at the same concentrations, were prepared in 99.98% D2O (Sigma-Aldrich) and allowed to incubate at room temperature for 48 h. The solutions were at pH 5.5. Neither the E2 nor the INS dissolved completely, so particulates were spun down for a minute in a tabletop centrifuge and only the clear supernatant was used in the experiments. Data were collected on an Agilent Technologies 500 MHz NMR machine in the Max T. Rogers NMR Facility located in the Chemistry Department of Michigan State University. A PROTON pulse sequence (s2pul) was used at 26.0°C. The relaxation delay was 1.000 s; Pulse 45.0°; acquisition time 2.045 s; width 8012.8 Hz; 2352 repetitions. The resulting data were processed to minimize noise and to flatten the baseline.
Enzyme-Linked INS Binding Assay
In order to determine whether estradiol interfered with INS binding to the IR, a modified form of ELISA was used, employing INS conjugated to horse-radish peroxidase (Sigma-Aldrich) in place of an antibody (55, 56). IR was isolated from rat liver (Sigma-Aldrich). A few crystals of estradiol were added to 1.0 ml solutions of INS–HRP and IR solutions (concentrations below) and allowed to bind for 48 h. Only the supernatant of these solutions was utilized for further experimentation (i.e., only the solubilized fraction of E2). For the ELISA, the IR and IGFR (with and without E2) was utilized at 3.5 × 10−12 M and the INS–HRP (with and without E2) was serially diluted by thirds from an initial concentration of 50 μM. The initial concentration of E2 solubilized by the INS–HRP, INSR, and IGFR was, in each case, approximately 200 nM (see below). 100 μL of the IR or IGFR (with and without E2) was plated first and allowed to bind to the Costar EIA/RIA 96-well plate for 1 h with shaking at room temperature. The plate was then triply washed with a 1% Tween 40 solution. 200 μL of 1% polyvinyl alcohol (PVA) was added to each well and served as blocking agent. An additional set of wells were incubated with buffer alone and then the PVA to act as a control for non-specific binding in subsequent steps. The PVA was incubated and washed as described above. Next, 100 μL of the INS–HRP (with and without E2) dilutions was added to each well, incubated, and washed as above. Finally, 100 μL of 2,2′AZINO-bis (3-ethylbenziazoline-6-sulfonic acid) (ABTS single reagent, Chemicon International) was added to all wells, allowed to incubate for 30 min at room temperature, and the absorbance of each well read at 405 nm. All combinations were run in duplicate and the results averaged. Note that, because the E2 was solubilized in INS–HRP, it, too, was serially diluted as the INS–HRP was serially diluted. The concentration of E2 at the mid-point in the resulting binding curves was therefore approximately 22 nM. It is also important to note that the estradiol concentration was not directly controlled in this experiment and had to be determined by capillary electrophoretic (CE) (below). We found that use of defined solutions containing ethanol above 30% destroyed the assay.
To determine the concentration of E2 used in the experiment, a few crystals of estradiol were added to 1.0 ml of buffer containing either IR or INS–HRP and allowing these solutions to incubate at room temperature for at least 48 h. The concentration estradiol was determined using CE (pH 8.4 borate running buffer) and comparing the results to a series of known concentrations of E2 prepared in 95% ethanol. The concentration of E2 in the E2-saturated phosphate buffer in the presence of either INS or IR was found to be approximately 200 nM (ca. 60 ng/ml) or about five times the plasma concentration of E2 observed in third-trimester pregnancy.
Data Analysis
All data from the UV spectroscopy and ELISA experiments were analyzed and plotted using the Microsoft Office Excel spreadsheet program. There are no statistics or error bars because all of the methods used for determining curves and binding constants are differences in absorbance that involved multiple subtractions.
Results
Figure 1 and Table 1 summarize the UV spectroscopy results of recombinant human INS binding to estradiol (E2), estriol, estrone, hydrocortisone, andro-stenedione, corticosterone, and pregnenolone and the SERM raloxifene. Estriol had a binding constant >500 μM, raloxifene 130 μM, while the other five hormones all had binding constants between 1.25 and 63.5 μM. The only hormone or SERM that bound within a physiological range was E2, which had a binding constant of 12 nM. Figure 2 confirms that measurable binding of E2 to INS is readily apparent at 10 nM, but alpha estradiol, a very weak E2 agonist, does not bind significantly to INS even at 1 μM. Binding of E2 to INS requires the cooperation of both the A and B chains of INS, since the binding constants for E2 binding to the individual oxidized chains is only 220 and 150 μM, respectively (Table 1; Figures 3 and 4). The binding of E2 to INS-like peptides derived from IR provides additional information about the specificity of the binding site. E2 does not bind with measurable affinity to glucagon (Table 1).
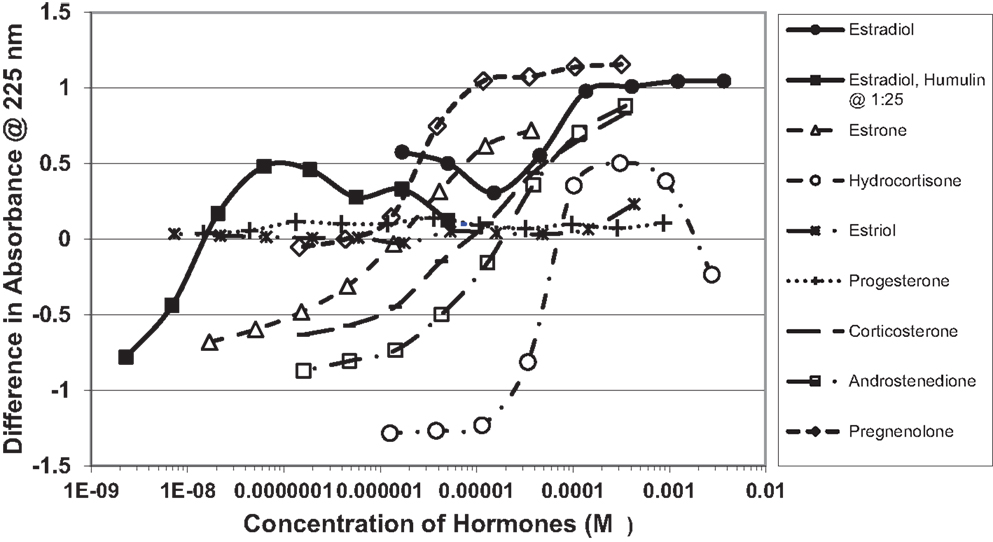
Figure 1. Curves showing concentration-dependent binding of various sex hormones to recombinant human insulin (Humulin). Binding constants (Table 1) were approximated by determining the inflection points of these curves. Note that estradiol has two binding modes, a high affinity one that is apparent at very low concentrations of E2 (Humulin 1/25 dilution) and a lower affinity site that is present at higher concentrations of E2.
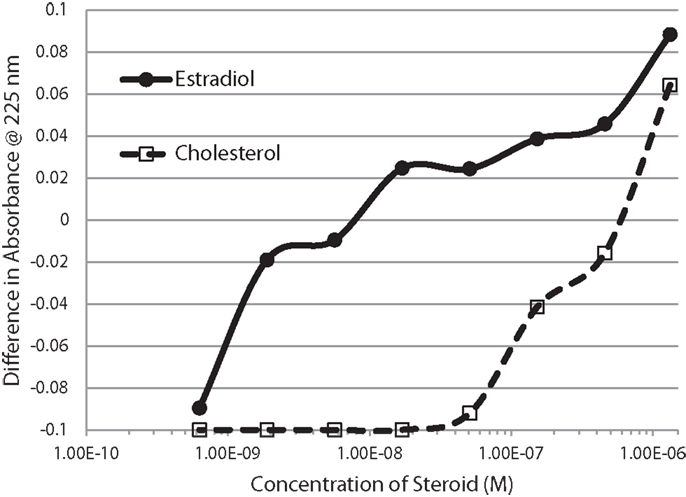
Figure 2. Serial additions of estradiol (E2) also demonstrated concentration-dependent changes in the Humulin spectrum starting at nanomolar concentrations of E2. E2 binding to cholesterol, in contrast, was observable only at micromolar concentrations. These results confirm the high affinity of E2 for insulin shown in Figure 1.
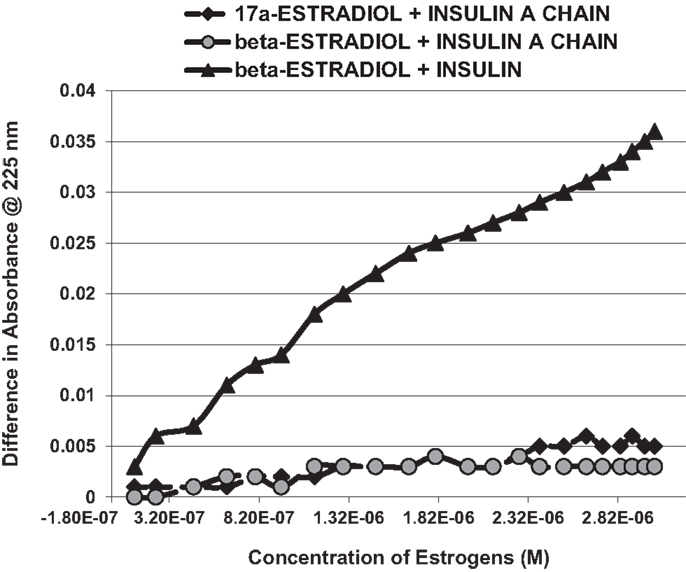
Figure 3. A serial addition experiment like that shown in Figure 2, but starting with higher concentrations of Humulin and estradiols. Even at 1000 times the concentration of E2 used in Figure 2, no binding was apparent to the insulin A chain, nor did 17-α-estradiol, an E2 antagonist, bind significantly to Humulin in this concentration range (Table 1).
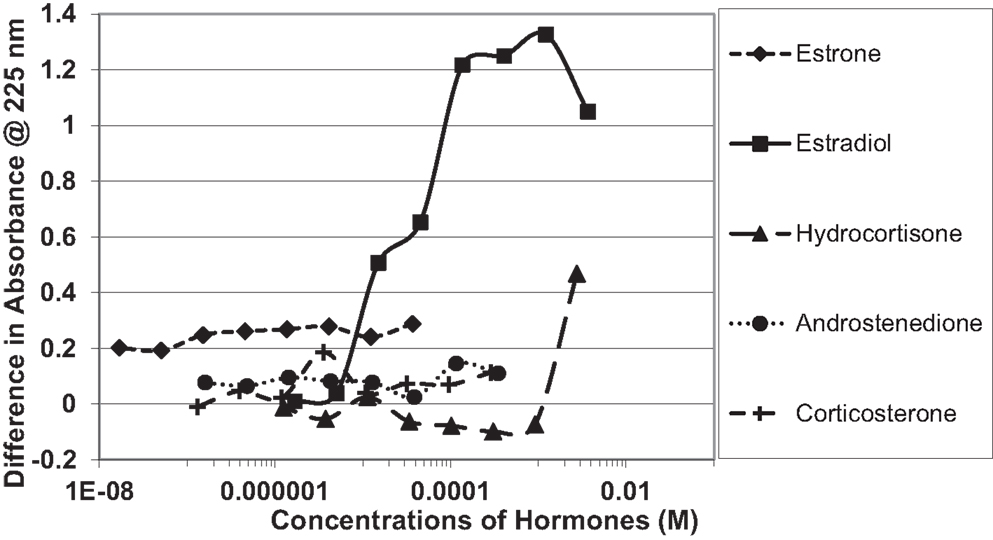
Figure 4. Curves showing concentration-dependent binding of various sex hormones to the oxidized B chain of insulin. Only estradiol (E2) showed measurable binding (see Table 1).
Notably, the binding of E2 to INS is biphasic, displaying high affinity binding (12 nM) and low affinity binding (65 μM) (Table 1; Figures 1 and 2). This biphasic binding suggests that there are at least two binding sites for E2 on INS, but the lower affinity site is unlikely to have any physiological or pharmacological importance since INS and E2 never approach concentrations even in the most extraordinary circumstances that would result in measurable binding to this site.
Binding of E2 to INS was confirmed by CE and 1H nuclear magnetic resonance spectroscopic (NMR) experiments. It is also important to know that the area under a peak representing a chemical species is, in CE, directly proportional to the concentration of the chemical species over a wide range of concentrations. Figure 5 shows the results for 15 and 30 kV experiments. Significantly, more E2 was present in solution when the E2 was mixed with INS than without. Table 2 shows that at each voltage, the peak ratio of estrogen from the estrogen/INS sample compared with the estrogen alone sample was approximately 4:1, indicating that the presence of INS causes a substantial increase in the amount of estrogen in solution. This is possible only if INS binds E2 and is consistent with the other experiments in this paper.
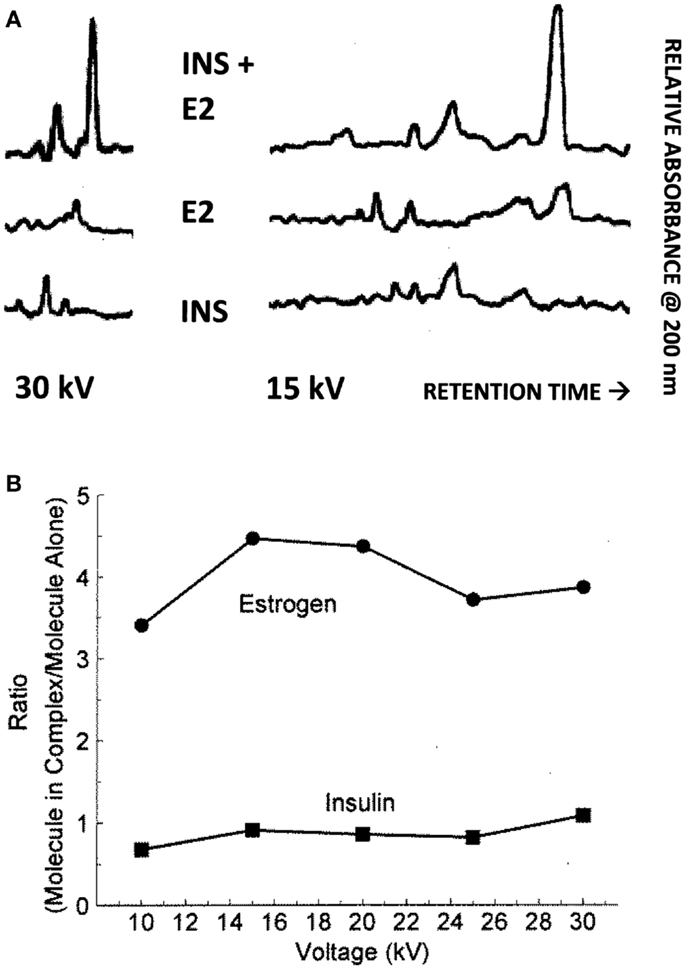
Figure 5. (A) Capillary electropherograms showing the relative absorbance at 225 nm of insulin (INS) at the bottom, estradiol (E2) in the middle, and their combination at the top, in 25 mM pH 7.4 phosphate buffer at 30 and 15 kV. Since the area of a CE peak is proportional to the concentration of the compound present, it is possible to calculate the ratio of the INS and of the E2 present in the combinations as a function of the individual samples as in (B). Note that some peaks are shared in both the E2 and INS electropherograms and represent buffer material. (B) Illustrates the fact that while the presence of E2 has no effect on the amount of INS present in solution, the presence of INS increases the amount of E2 that is solubilized by a factor of about 4. The mean ± SD of the five EI/E ratios is 3.96 ± 0.45.
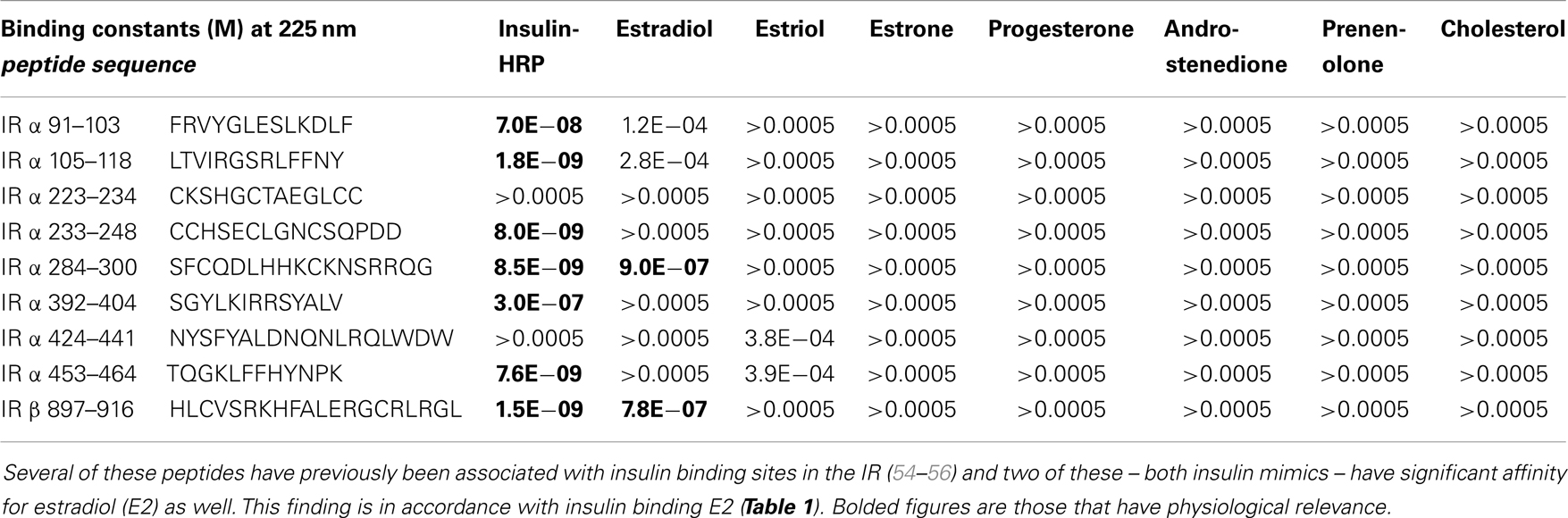
Table 2. Binding constants for various sex hormones and insulin to peptides derived from the human insulin receptor (IR).
The results of the NMR experiments, illustrated in Figures 6A,B, also indicate that E2 binds to INS. Significant changes in the spectrum occur in both the aromatic region of the spectrum, especially around 9.7 and 7.5 ppm, and also in the region of the spectrum around 2 ppm. Most strikingly, a number of peaks in both regions become so broadened or suppressed as to disappear. All such peaks are associated in both INS and E2 with aromatic hydrogens strongly suggesting that binding of E2 to INS is mediated through interactions with the histidine, phenylalanine, and/or tyrosine residues of the peptide, probably involving charge transfer complexing.
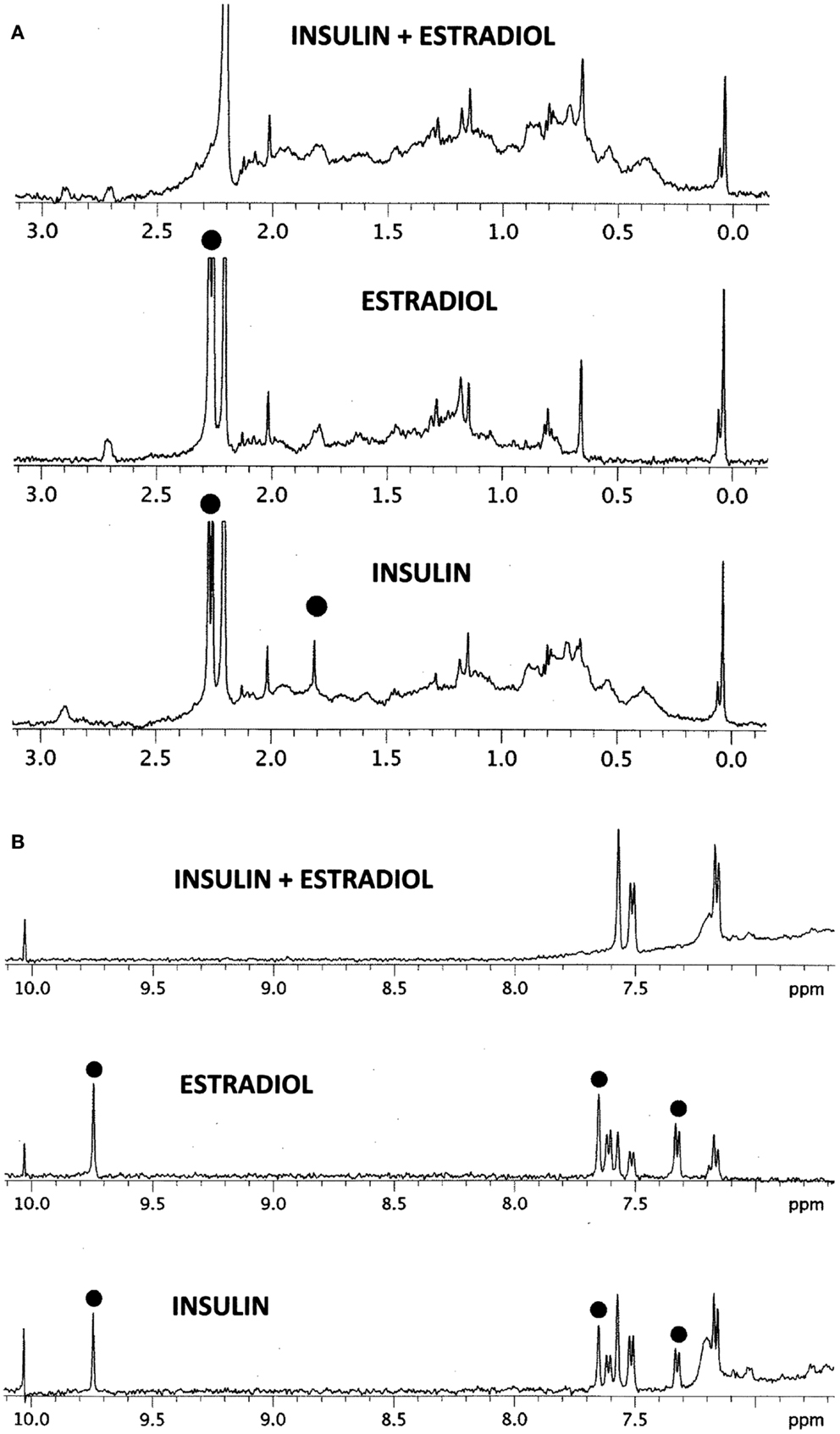
Figure 6. 1H NMR spectra in D2O of insulin (INS) are shown at the bottom, estradiol (E2) in the middle, and their combination (INS + E2) at the top. (A) Spectral region from 0 to 3 ppm. While the various peaks of INS and E2 ranges between 0.2 and 1.5 ppm and 2.5 and 3.0 ppm add to create the INS–E2 spectrum (top), significant changes are also evident. The INS peak at 1.8 ppm (dot) broadens out in the INS–E2 combination, while the INS and E2 peaks at 2.3 ppm (dots) shift to 2.2 ppm. (B) Spectral region from 7 to 10 ppm. Again, some of the INS and E2 peaks are clearly additive to give the INS + E2 spectrum on top, e.g., those at 7.2 and 7.6 ppm. Other peaks in this region show major changes. The INS and E2 peaks (dots) at 9.8 ppm disappear in the combination while those at 7.35 and 7.65 ppm in both INS and E2 (dots) are quenched or shift to positions at 7.2 and 7.6 ppm. These spectral changes are consistent with E2 binding to INS involving mainly aromatic residues on both molecules.
UV spectroscopy revealed that E2 binds to the rat IR membrane preparations with an affinity very similar to that of E2 for INS. The binding of E2 to IR was biphasic with a high affinity binding having a Kd of about 2.4 × 10−8 and a lower affinity binding with a Kd above 10−6 (Figure 7, top). E2 also bound to human recombinant IGFR, again in a biphasic manner, with high affinity binding of about 1.0 × 10−7 and a lower affinity binding with a Kd above 10−5 (Figure 7, bottom). The biphasic binding of E2 to IR and IGFR, like that of E2 to INS itself, suggests the presence of at least two binding sites for E2 on these receptors. As with INS, the low affinity E2-binding sites are extremely unlikely to play any physiological or pharmacological role.
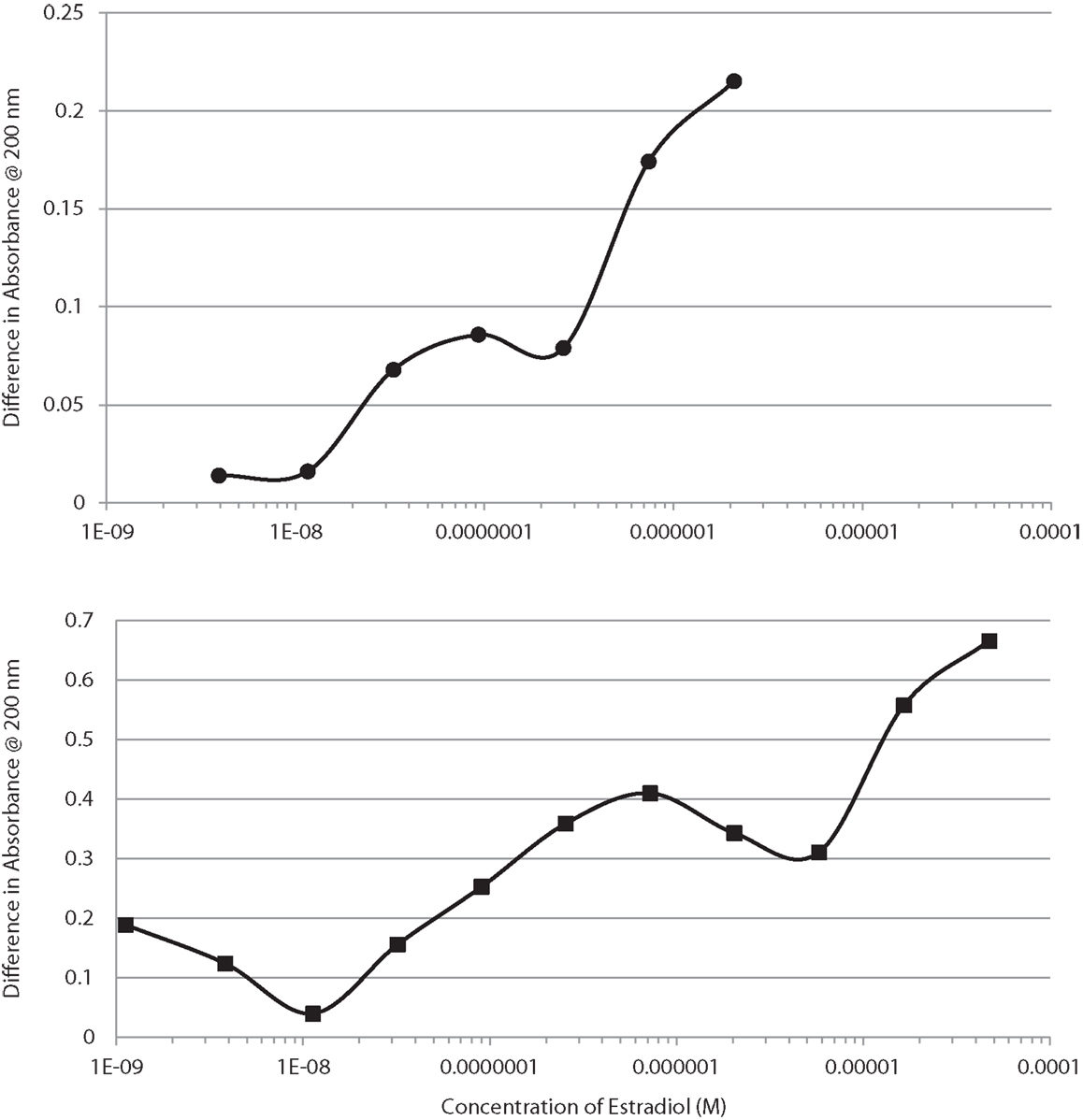
Figure 7. Binding curves showing concentration-dependent effects of increasing estradiol (E2) on the UV spectrum of the insulin receptor (IR) at the top, and on the insulin-like growth factor receptor (IGFR) at the bottom. Both curves are bimodal, showing high affinity and low affinity (probably non-specific) binding. These curves mimic those of E2 binding to insulin (Figure 1).
Since we have demonstrated previously that IR and IGFR contain significant regions of homology with INS itself (53–56), the similarity between the binding constants for E2 to both INS, IR and IGFR suggested that the INS-like regions of the IR and IGFR might be the binding sites for E2. We again used UV spectrometry to investigate whether steroid hormones bind directly to INS-like peptides derived from the IR (characterization of IGFR will be reported in the future). All of the IR peptides used in these experiments are from extracellular regions of the IR, and all but one (IR peptide 897–916) are found in the alpha chain of the IR (see Table 2 for sequences and binding results). Some of the peptides tested have previously been shown to bind INS in the nanomolar range (55, 56) and are therefore likely to be involved in INS binding to the IR (see Table 2 for INS binding data). E2 bound to several of the IR peptides associated with INS binding, but at about 15- to 20-fold higher concentrations of E2 than was found for the intact rat IR preparations. E2 bound to IR peptides 284–300 (IR alpha chain) and 897–916 (IR beta chain) at 900 and 780 nM, respectively (Table 2).
We next hypothesized that E2 binding to INS and to the IR might alter the affinity of INS for the IR. In order to explore this possibility, we used a simple enzyme-linked adsorption assay employed in previous studies of INS binding to the IR (55, 56). Figure 8 shows that the presence of E2 decreased binding of INS–HRP to the IR by about half a log unit (fivefold) when incubated with either INS or with the IR. When E2 was incubated with both INS and the IR, an eightfold decrease in INS binding to the IR resulted (Figure 6). It is important to emphasize that in each of these cases, the concentration of E2 present was initially at the maximum that INS or IR could bind in aqueous solution (200 nM E2 as determined by capillary electrophoresis) but that because the E2 was diluted in tandem with the INS and/or IR, at the inflection points of the binding curves, the concentration of E2 was about 22 nM and therefore likely represents the maximum possible effects that E2 could have under naturally occurring hyperestrogenemic conditions.
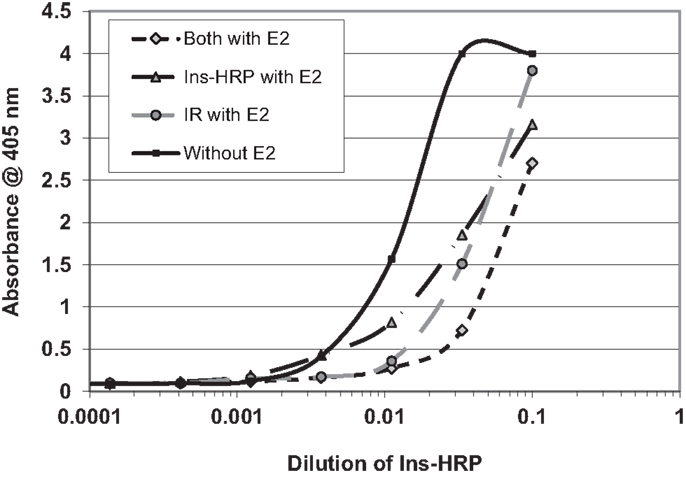
Figure 8. Curves showing the effects of estradiol (E2) on the binding of insulin conjugated to horse-radish peroxidase (INS–HRP) to isolated insulin receptor (IR). The beginning concentration of E2 was 200 nM as measured by capillary electrophoresis (data not shown), which was diluted in tandem with the INS–HRP. The mid-point concentration of E2 in each curve was therefore about 22 nM. E2 shifted the binding curve to the right about half a log unit (a fivefold decrease in binding) when incubated with either INS–HRP or the IR alone, and it shifted the binding curve to the right 0.8 log units (an eightfold decrease in binding) when both the IR and INS–HRP were exposed to E2.
Discussion
Figures 1–6 and Tables 1 and 2 demonstrate, using UV spectroscopy, capillary electrophoresis, and NMR, that E2 binds to INS. The UV spectroscopic studies (Figures 1–4; Tables 1 and 2) show clear E2-concentration-dependent shifts in absorbance in the nanomolar range, whereas most other estrogens, estrogen precursors, and SERMS such as raloxifene, tamoxifen, and the phytoestrogen genistein have negligible binding to INS. These data are consistent with the interpretation that E2 can cause INS resistance by direct binding to INS and IR and that the other compounds, including SERM studied here cannot produce such effects through this mechanism. In fact, SERMS are not associated clinically with development of diabetes or even INS resistance (57–59, 72–74) and phytoestrogens generally improve INS function (75–78).
The CE experiments (Figure 5) provide evidence that E2 binding to INS results in increased solubility of E2 in aqueous solution, as there is four times as much E2 in solution when INS is present as when it is absent. INS may therefore act as an E2 transporter at appropriate concentrations of the two compounds. E2-binding requires intact INS, as the oxidized INS A and B chains have very reduced affinity for E2. Comparing the INS sequence with those of the IR peptides that also bind E2 suggests that a single relatively conserved motif is necessary for E2 binding (Table 3). This motif consists of an arginine or lysine residue followed by a glycine or leucine and then a series of aromatics, usually a pair of phenylalanines and a tyrosine. At one or two amino acids removed, a hydrophilic residue such as lysine or asparagine often rounds out the motif.

Table 3. Homologies among the amino acid sequences of the peptides used in the present study.
The NMR study (Figures 6A,B) shows major shifts and quenching of specific peaks associated with aromatic residues such as tyrosine and phenylalanine suggesting that the mechanism of binding involves pi–pi bonding or charge transfer complexing between E2 and INS. The participation of tyrosine and phenylalanine residues in E2 binding is not only logical, given the polyaromatic structure of E2 itself, but also in light of the structure of the conserved E2-binding motif derived in Table 3.
E2 binds to INS with a Kd of about 12 nM, but other steroid hormones bind to INS with significantly less affinity so that only E2 is likely to have an impact on INS activity under physiological conditions. The impact of E2 binding to INS is evaluated in Table 4. Assuming standard binding constant kinetics [Kd = (INS)(E2)/(INS–E2 complex)], it follows that at 12 nM E2, half of the INS will be complexed with E2. Concentrations of E2 exceeding 12 nM E2 are observed in a number of syndromes associated with INS resistance including third-trimester pregnancy, GDM, and OHSS, so that in these syndromes, more than half of the available INS will likely be bound to E2 at any given time, decreasing INS binding to its receptor (Figure 8). Given that over half the INS in the body would have impaired function at any given time, it would be natural for the body to “over-produce” INS to compensate. Table 3 shows that increasing the concentration of INS threefold results in about a 2.5-fold increase in free INS. At concentrations of E2 that are typical of normal males and non-pregnant females (ca. 6 × 10−11 M), on the other hand, complexing of E2 to INS will be negligible [on the order of two-tenths of a percent, ignoring binding of E2 to sex hormone binding globulin (SHBG), which will decrease the amount of free E2 and therefore of E2–INS as well]. Thus, the huge variation in E2 concentrations observed in human beings results in very large and very significant differences in the amounts E2 bound to INS.
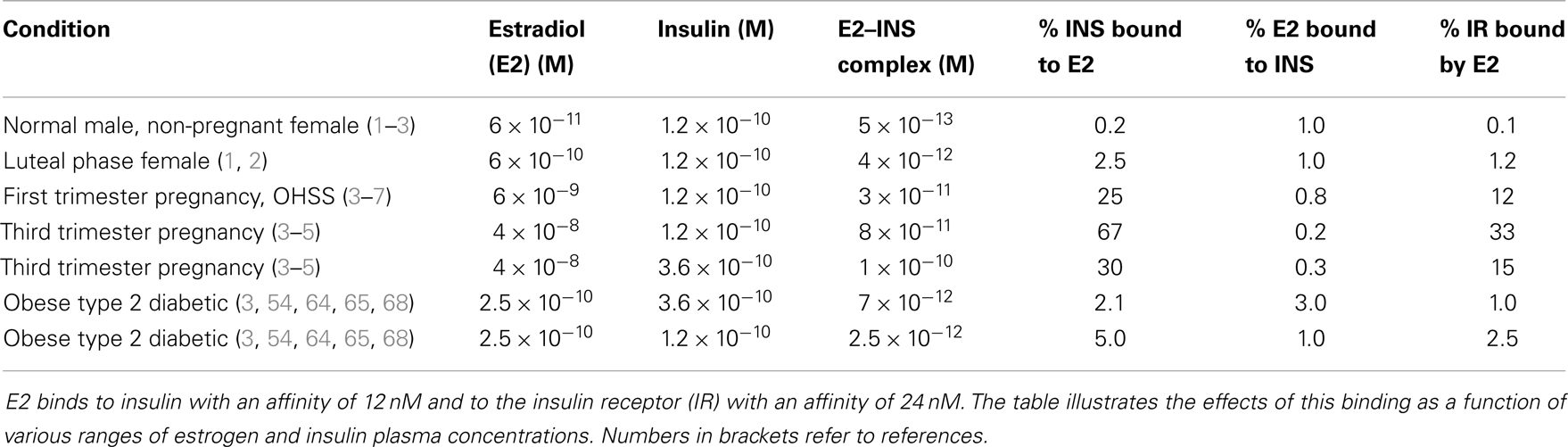
Table 4. Kinetic effects of estradiol (E2) binding to insulin.
As Table 3 illustrates, the impact of E2 and INS concentration variations occur between the normal baseline ranges and the extremes of pregnancy and over-production syndromes. Quadrupling E2 concentrations [which occurs frequently in obesity and is often exceeded (79)] without altering normal INS concentrations results in about 5% of the INS being bound to E2 at any given time. Compensating by tripling INS concentrations results in only 2% of the INS being bound to E2, but notably also results in 3% of the available E2 being tied up in the E2–INS complex. Each compensation, therefore, creates a negative functional effect on one or both of the hormones.
E2 binds to IR peptides as well as to INS. Tables 2 and 3 estimate the impact of E2 binding to the IR. As Table 2 demonstrates, E2 binds to INS-like peptides derived from the IR with Kd around 780–900 nM, validating the INS-dependent binding of E2 and suggesting possible locations on the IR at which E2 may bind. The binding constants for E2 binding to IR peptides reported clearly underestimate the actual binding affinity of E2 for the IR, which were determined by E2 binding to intact rat IR preparations and yielded a Kd of about 24 nM. There may be several reasons for this discrepancy. The peptides may not represent the main E2-binding sites on IR. The peptides may not be in the correct conformation for binding E2. Binding of E2 to IR may be a cooperative effect shared by several sites (as is, indeed, binding of INS to the IR). Evidence for cooperativity can be found in the fact that E2 has a much higher affinity for intact INS than for either the INS A or B chains by themselves. Finally, the binding measurements were done in solution, whereas the rat IR was membrane-bound; several studies have shown that binding of ligands to receptors improves as much as 100-fold when the receptor molecule is immobilized as the IR would be in nature (56, 67). Further investigation of the molecular determinants of E2 binding to the IR and IGFR may be warranted employing enzyme-linked E2 binding to peptides immobilized on ELISA plates, or some of the many genetically modified forms of the IR that have been produced by various laboratories in their studies of INS binding to the IR. Moreover, since the IR dimerizes with IGFR, it is possible that E2 will bind the IR:IGFR dimer with an affinity similar to the IR and IGFR affinities measured here.
E2 clearly inhibits binding of INS to IR in vitro. The impact of E2 binding to both INS and IR is additive, as Figure 8 demonstrates, so that the impact of increasing E2 concentrations is greater than the individual binding percentages shown in Table 3 indicated. Notably, the same effect has been observed in rats in vivo and in vitro. Hilf et al. (80) report that a 1 mg dose of E2 decreased INS binding in rats as much as 50% within 24 h while the same effect was seen in cell culture: “the addition of 10−8 M 17β-estradiol to the culture medium inhibited cell growth and decreased INS binding by ~20%.”
While it is clear that estradiol can produce effects on glucose regulation through a variety of indirect mechanisms, the results reported here lead us to suggest that direct binding of estradiol to INS and to the IR may be the main mechanism by which INS resistance is produced in hyperestrogenemic conditions such as gestational diabetes, OHSS and PCOS. As Table 4 illustrates, the concentrations of E2 that produce the severe INS resistance in GD, OHSS, and PCOS are in the same range as those calculated to be needed from the binding constants of E2 to INS and the IR. The fact that cell culture experiments (80), rat experiments (48–51), clinical observations of human patients undergoing sex-change therapies also demonstrate E2-concentration-dependent INS resistance or diabetes (27–29), third-trimester pregnancy (1–11), OHSS (16, 17), or PCOS (30–39, 47) are in the same concentration ranges as the in vitro effects suggests that the in vitro data can be extrapolated to in vivo cases.
We emphasize that direct binding of E2 to INS and IR is not the entire story behind hyperestrogenemia-associated INS resistance. Not all endogenous E2 is free in solution. It is assumed by the current literature that 80–90% of E2 is reversibly bound to SHBG, so that the effects of E2 on INS and IR may depend on the E2:SHBG ratio (12, 81). Women who develop gestational diabetes have 1.5–2.5 times higher E2:SHBG ratios than those who do not. (81). Further in vitro studies of how E2 is shared between SHBG, INS, and IR might clarify how this system is regulated. Other elements of the glucose regulatory and estrogen regulatory systems mentioned in the Section “Introduction,” such as GLUT and ERα expression, are certainly involved in the development of INS resistance as well. Additionally, IR activates IR complex 1 (IRC1), which interacts directly with ERα (61).
In vivo testing of this hypothesis will not be easy, since no direct techniques currently exist to demonstrate the binding of any hormone to another hormone or to a receptor in vivo. The demonstration of E2–INS and E2–IR complexes from cell cultures, animal or human plasma, or tissue samples is also fraught with difficulties, since any such experiment will require the use of purification techniques such as CE, HPLC, etc., to isolate such complexes from all of the other hormones and proteins in the original samples. Such purification is unlikely to leave complexes intact. One possible way to carry out such isolations would be to develop a form of E2 conjugated to a linker capable of cross-reacting irreversibly with any peptide or protein to which it binds. Mass spectrometry could then be used to identify the peptides or proteins to which the E2 has cross-reacted, and INS and the IR should be among those identified (in addition to, obviously, estrogen receptors). A more sensitive approach might be to create an OVEX knock-out mouse lacking estrogen receptors so that any binding to INS or IR is more readily identifiable. Development of such OVEX estrogen-receptor knockouts would also permit another type of experiment; treat the animal with radioactively labeled estradiol; then look for evidence of the accumulation of radioactivity in tissues rich in INS and IRs (e.g., the pancreas). One would expect an overlap at the microscopic level between some of the radioactivity and fluorescent antibodies against IR and INS in tissue slices from such animals.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Christopher Dobbelstein, MD, suggested that we explore the possibility that estrogens bind to insulin. Daniel Holmes, Ph.D., assisted with the NMR study. This research was funded by Maurine Bernstein as a gift to the Root-Bernstein Laboratory.
Abbreviations
E2, estradiol; ERα, ERβ, alpha and beta chains of estrogen receptor; ER, estrogen receptor; GDM, gestational diabetes mellitus; INS, insulin; INS–HRP, insulin conjugated to horse-radish peroxidase; IR, insulin receptor; IVF, in vitro fertilization; OHSS, ovarian hyperstimulation syndrome; PCOS, polycystic ovarian syndrome; SHBG, sex hormone binding globulin.
References
1. Stricker R, Eberhart R, Chevailler MC, Quinn FA, Bischof P, Stricker R. Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT analyzer. Clin Chem Lab Med (2006) 44(7):883–7. doi:10.1515/CCLM.2006.160
2. Wu CH, Motohashi T, Abdel-Rahman HA, Flickinger GL, Mikhail G. Free and protein-bound plasma estradiol-17 beta during the menstrual cycle. J Clin Endocrinol Metab (1976) 43(2):436–45. doi:10.1210/jcem-43-2-436
3. Maggio M, Lauretani F, Ceda GP, Bandinelli S, Basaria S, Paolisso G, et al. Estradiol and metabolic syndrome in older Italian men: the inCHIANTI study. J Androl (2010) 31(2):155–62. doi:10.2164/jandrol.108.006098
4. Kauppila A, Järvinen PA. Peripheral blood concentrations of progesterone and oestradiol during human pregnancy and delivery. Acta Physiol Hung (1985) 65(4):473–8.
5. Masuyama H, Hiramatsu Y. Potential role of estradiol and progesterone in insulin resistance through constitutive androstane receptor. J Mol Endocrinol (2011) 47(2):229–39. doi:10.1530/JME-11-0046
6. Vejrazkova D, Vcelak J, Vankova M, Lukasova P, Bradnova O, Halkova T, et al. Steroids and insulin resistance in pregnancy. J Steroid Biochem Mol Biol (2012) 139:122–9. doi:10.1016/j.jsbmb.2012.11.007
7. Kühl C. Etiology and pathogenesis of gestational diabetes. Diabetes Care (1998) 21(Suppl 2):B19–26.
8. Balani J, Hyer SL, Rodin DA, Shehata H. Pregnancy outcomes in women with gestational diabetes treated with metformin or insulin: a case-control study. Diabet Med (2009) 26(8):798–802. doi:10.1111/j.1464-5491.2009.02780.x
9. Sokup A, Swiatkowski M, Tyloch M, Szymanski W. Insulin Lispro as an alternative for insulin Humulin in the treatment of an obese gestational diabetic woman with allergy to Humulin U. Case report. Przegl Lek (2005) 62(4):260–1.
10. Mazor M, Hershkovitz R, Chaim W, Levy J, Sharony Y, Leiberman JR, et al. Human preterm birth is associated with systemic and local changes in progesterone/17 beta-estradiol ratios. Am J Obstet Gynecol (1994) 171(1):231–6. doi:10.1016/0002-9378(94)90474-X
11. Barros RPA, Morani A, Moriscot A, Machado UF. Insulin resistance of pregnancy involves estrogen-induced repression of GLUT-4. Mol Cell Endocrinol (2008) 295:24–31. doi:10.1016/j.mce.2008.07.008
12. Jin Z, Guan X, Gao H, Gao M, Su D, Li W. The change in sex hormone binding globulin and the influence by gestational diabetes in fetal period. Gynecol Endocrinol (2009) 25(10):647–52. doi:10.1080/09513590903015437
13. Jia MC, Zhou LY, Ren S, Dong L, Xiao B. Serum SHBG levels during normal menstrual cycle and after insertion of levonorgestrel-releasing IUD. Adv Contracept (1992) 8(1):33–40. doi:10.1007/BF01849346
14. Ryan EA, O’Sullivan MJ, Skyler JS. Insulin action during pregnancy. Studies with the euglycemic clamp technique. Diabetes (1985) 34(4):380–9. doi:10.2337/diab.34.4.380
15. Seely EW, Solomon CG. Insulin resistance and its role in pregnancy induced hypertension. J Clin Endocrinol Metab (2003) 88(6):2393–8. doi:10.1210/jc.2003-030241
16. Joo BS, Park SH, An BM, Kim KS, Moon SE, Moon HS. Serum estradiol levels during controlled ovarian hyperstimulation influence the pregnancy outcome of in vitro fertilization in a concentration-dependent manner. Fertil Steril (2010) 93(2):442–6. doi:10.1016/j.fertnstert.2009.02.066
17. Vanderlelie J, Bell K, Perkins AV. The serum concentration of estradiol after embryo transfer and the decline from preovulatory levels may influence the success of IVF treatment. Horm Res (2003) 59(2):95–9. doi:10.1159/000068578
18. Diamanti-Kandarakis E, Baillargeon JP, Iuorno MJ, Jakubowicz DJ, Nestler JE. Controversies in endocrinology, a modern medical quandary: polycystic ovary syndrome, insulin resistance, and oral contraceptive pills. J Clin Endocrinol Metab (2003) 88(5):1927–32. doi:10.1210/jc.2002-021528
19. Pandit MK, Burke J, Gustafson AB, Minocha A, Peiris AN. Drug-induced disorders of glucose tolerance. Ann Int Med (1993) 118(7):529–38. doi:10.7326/0003-4819-118-7-199304010-00008
20. Kalkhoff RK. Effects of oral contraceptive agents and sex steroids on carbohydrate metabolism. Annu Rev Med (1972) 23:429–38. doi:10.1146/annurev.me.23.020172.002241
21. Godsland IF, Walton C, Felton C, Proudler A, Patel A, Wynn V. Insulin resistance, secretion, and metabolism in users of oral contraceptives. J Clin Endocrinol Metab (1992) 74(1):64–71. doi:10.1210/jcem.74.1.1530790
22. Sitruk-Ware R, Nath A. Metabolic effects of contraceptive steroids. Rev Endocr Metab Disord (2011) 12(2):63–75. doi:10.1007/s11154-011-9182-4
23. Rimm EB, Manson JE, Stampfer MJ, Colditz GA, Willett WC, Rosner B, et al. Oral contraceptive use and the risk of Type 2 (non-insulin-dependent) diabetes mellitus in a large prospective study of women. Diabetologia (1992) 35:967–72. doi:10.1007/BF00401427
24. Wynn V, Godsland R, Niththyananthan R, Adams PW, Melrose J, Oakley NW, et al. Comparison of effects of different combined oral-contraceptive formulations on carbohydrate and lipid metabolism. Lancet (2003) 313:1045–9. doi:10.1016/S0140-6736(79)92949-0
25. Alberti KG, Zimmet P, Shaw J, IDF Epidemiology Task Force Consensus Group. The metabolic syndrome – a new worldwide definition. Lancet (2005) 366:1059–62. doi:10.1016/S0140-6736(05)67402-8
26. Cohen JC, Hickman R. Insulin resistance and diminished glucose tolerance in power lifters ingesting anabolic steroids. J Clin Endocrinol Metab (1987) 64:960–3. doi:10.1210/jcem-64-5-960
27. Polderman KH, Gooren LJG, Asscheman H, Bakker A, Heine RJ. Induction of insulin resistance by androgens and estrogens. J Clin Endocrinol Metab (1994) 79:265–71. doi:10.1210/jcem.79.1.8027240
28. Seed M, Godsland IF, Wynn V, Jacobs HS. The effects of cyproterone acetate and ethinyl estradiol on carbohydrate metabolism. Clin Endocrinol (1984) 23(6):689–99. doi:10.1111/j.1365-2265.1984.tb01411.x
29. Elbers JMH, Giltay EJ, Teerlink T, Scheffert PG, Asscheman H, Seidell JC, et al. Effects of sex steroids on components of the insulin resistance syndrome in transsexual subjects. Clin Endocrinol (2003) 58:562–71. doi:10.1046/j.1365-2265.2003.01753.x
30. Brettenthaler N, DeGeyter C, Huber PR, Keller U. Effect of the insulin sensitizer pioglitazone on insulin resistance, hyperandrogenism, and ovulatory dysfunction in women with polycystic ovary syndrome. J Clin Endocrinol Metab (2004) 89:3835–40. doi:10.1210/jc.2003-031737
31. Moghetti P, Tosi F, Castello R, Magnani CM, Negri C, Brun E, et al. The insulin resistance in women with hyperandrogenism is partially reversed by antiandrogen treatment: evidence that androgens impair insulin action in women. J Clin Endocrinol Metab (1996) 81(3):953–61.
32. Poretsky L. On the paradox of insulin-induced hyperandrogenism in insulin-resistant states. Endocr Rev (1991) 12(1):3–13. doi:10.1210/edrv-12-1-3
33. Taylor SI, Dons RF, Heranadez E, Roth J, Gorden P. Insulin resistance associated with androgen excess in women with androgen excess in women with autoantibodies to the insulin receptor. Ann Intern Med (1982) 97(6):851–4. doi:10.7326/0003-4819-97-6-851
34. Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab (1980) 50:113–6. doi:10.1210/jcem-50-1-113
35. Azziz R. Diagnostic criteria for polycystic ovary syndrome: a reappraisal. Fertil Steril (2005) 83(5):1343–6. doi:10.1016/j.fertnstert.2005.01.085
36. Ovalle F, Azziz R. Insulin resistance, PCOS, and type 2 diabetes. Fertil Steril (2002) 77(6):1098–105. doi:10.1016/S0015-0282(02)03111-4
37. Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J. Prevelance of impaired glucose tolerance in women with polycystic ovary syndrome. Diabetes Care (1999) 22(1):141–6. doi:10.2337/diacare.22.1.141
38. Duniaf A. Insulin resistance and polycystic ovary syndrome: mechanisms and implications for pathogenesis. Endocr Rev (1997) 18(6):774–800. doi:10.1210/edrv.18.6.0318
39. Elkind-Hirsch KE, Valdes CT, Malinak LR. Insulin resistance improves in hyperandrogenic women treated with Lupron. Fertil Steril (1993) 60:634–41.
40. Barros RPA, Machado UF, Gustafsson J-A. Estrogen receptors: new players in diabetes mellitus. Trends Mol Med (2006) 12(9):425–31. doi:10.1016/j.molmed.2006.07.004
41. Barros RPA, Machado UF, Warner M, Gustafsson J-A. Muscle GLUT4 regulation by estrogen receptors ERβ and Erα. Proc Natl Acad Sci USA (2006) 103(5):1605–8. doi:10.1073/pnas.0510391103
42. Gorres BK, Bomhoff GL, Morris JK, Geiger PC. In vivo stimulation of oestrogen receptor α increases insulin-stimulated skeletal muscle glucose uptake. J Physiol (2011) 598(8):2041–54. doi:10.1113/jphysiol.2010.199018
43. Alonso-Magdalena P, Ropero AB, Carrera MP, Cederroth CR, Baquie M, Gauthier BR, et al. Pancreatic insulin content regulation by the estrogen receptor Erα. PLoS One (2008) 3(4):e2069. doi:10.1371/journal.pone.0002069
44. Olde B, Leeb-Lundberg LMF. GPR30/GPER1: searching for a role in estrogen physiology. Trends Endocrinol Metab (2009) 20(8):409–16. doi:10.1016/j.tem.2009.04.006
45. Manrique C, Lastra G, Habibi J, Mugerfeld I, Garro M, Sowers JR. Loss of estrogen receptor α signaling leads to insulin resistance and obesity in young and adult female mice. Cardiorenal Med (2012) 2:200–10. doi:10.1159/000339563
46. Oh YS, Lee TS, Cheon GJ, Jang I-S, Jun H-S, Park SC. Modulation of insulin sensitivity and caveolin-1 expression by orchidectomy in a nonobese type 2 diabetes animal model. Mol Med (2011) 17(2):4–11. doi:10.2119/molmed.2009.00105
47. Livingstone C, Collison M. Sex steroids and insulin resistance. Clin Sci (2002) 102:151–66. doi:10.1042/CS20010197
48. Gonzalez C, Alonso A, Alvarez N, Diaz F, Martinez M, Fernandez S, et al. Role of 17β-estradiol and/or progesterone on insulin sensitivity in the rat: implications during pregnancy. J Endocrinol (2000) 166:283–91. doi:10.1677/joe.0.1660283
49. Gonzalez C, Alonso A, Grueso NA, Diaz F, Esteban MM, Fernandez S, et al. Role of 17β-estradiol administration on insulin sensitivity in the rat: implications for the insulin receptor. Steroids (2002) 67:993–1005. doi:10.1016/S0039-128X(02)00073-9
50. Gonzalez C, Alonso A, Diaz F, Patterson AM. Dose- and time-dependent effects of 17β-oestradiol on insulin sensitivity in insulin-dependent tissues of rat: implications of IRS-1. J Endocrinol (2003) 176:367–79. doi:10.1677/joe.0.1760367
51. Alonso A, Ordonez P, Fernandez R, Moreno M, Llaneza P, Patterson AM, et al. 17β-estradiol treatment is unable to reproduce p85α redistribution associated with gestational insulin resistance in rats. J Steroid Biochem Mol Biol (2009) 116:160–70. doi:10.1016/j.jsbmb.2009.05.010
52. Root-Bernstein RS, Dillon PF. Small molecule complementarity as a source of novel pharmaceutical agents and combination therapies. Curr Pharm Des (2008) 14(1):55–62. doi:10.2174/138161208783330727
53. Root-Bernstein RS. Molecular complementarity III. Molecular complementarity as the basis for peptide hormone evolution. A bioinformatic case study of insulin, glucagon, and gastrin. J Theor Biol (2002) 218:71–84. doi:10.1006/jtbi.2002.3056
54. Root-Bernstein RS. Peptide self-aggregation and peptide complementarity as bases for the evolution of peptide receptors: a review. J Mol Recognit (2005) 18:40–9. doi:10.1002/jmr.690
55. Root-Bernstein RS, Vonck J. The insulin receptor binds glucose altering the mutual affinity of insulin for its receptor. Cell Mol Life Sci (2009) 6(16):2721–32. doi:10.1007/s00018-009-0065-8
56. Root-Bernstein RS, Vonck J. Modularity in receptor evolution. Insulin- and glucagon-like peptide modules as binding sites for insulin and glucose in the insulin receptor. J Receptor Ligand Channel Res (2010) 3:87–96. doi:10.2147/JRLCR.S6737
57. Andersson B, Johannsson G, Holm G, Bengtsson BA, Sashegyi A, Pavo I, et al. Raloxifene does not affect insulin sensitivity or glycemic control in postmenopausal women with type 2 diabetes mellitus: a randomized clinical trial. J Clin Endocrinol Metab (2002) 87(1):122–8. doi:10.1210/jcem.87.1.8168
58. Cucinelli F, Soranna L, Romualdi D, Muzj G, Mancuso S, Lanzone A. The effect of raloxifene on glyco-insulinemic homeostasis in healthy postmenopausal women: a randomized placebo-controlled study. J Clin Endocrinol Metab (2002) 87(9):4186–92. doi:10.1210/jc.2001-011302
59. Cagnacci A, Paoletti AM, Zanni A, Arangino S, Ibba G, Orrù M, et al. Raloxifene does not modify insulin sensitivity and glucose metabolism in postmenopausal women. J Clin Endocrinol Metab (2003) 87(9):4117–21.
60. Nadal A, Alonso-Magdalena P, Soriano S, Ropero AB, Quesada I. The role of oestrogens in the adaptation of islets to insulin resistance. J Physiol (2009) 587(Pt 21):5031–7. doi:10.1113/jphysiol.2009.177188
61. Sisci D, Morelli C, Cascio S, Lanzino M, Garofalo C, Reiss K, et al. The estrogen receptor α: insulin receptor substrate 1 complex in breast cancer: structure-function relationships. Ann Oncol (2007) 18(Suppl 6):vi81–6. doi:10.1093/annonc/mdm232
62. Surath S, Banerjee K, Rupley JA. Binding of oligosaccharides to lysozyme temperature and pH dependence of the interaction. J Biol Chem (1973) 248:2117–24.
63. Matsumoto I, Jinbo A, Kitigaki H, Golovtchenko-Matsumoto AM, Seno N. Detection of lectin-sugar interaction by ultraviolet difference spectroscopy. J Biochem (1980) 88:1093–6.
64. Harris WR, Pecoraro VL. Thermodynamic binding constants for gallium transferring. Biochemistry (1983) 22:292–9. doi:10.1021/bi00285a030
65. Khaled MA, Mullins DW, Swindle M, Lacey JC Jr. Complexes of polyadenylic acid and the methyl esters of amino acids. Orig Life (1983) 13:87–96. doi:10.1007/BF00928886
66. Root-Bernstein RS, Dobbelstein C. Insulin binds to glucagon forming a complex that is hyper-antigenic and inducing complementary antibodies having an idiotype-antiidiotype relationship. Autoimmunity (2001) 33:153–69. doi:10.3109/08916930109008044
67. Dillon PF, Root-Bernstein RS, Lieder CM. Ascorbate enhancement of H1 histamine receptor coincides with ascorbate oxidation inhibition by histamine receptors. Am J Physiol Cell Physiol (2006) 291:C977–84. doi:10.1152/ajpcell.00613.2005
68. Dillon PF, Root-Bernstein RS, Lieder CM. Anti-oxidant independent ascorbate enhancement of catecholamine-induced contractions of vascular smooth muscle. Am J Physiol Heart Circ Physiol (2004) 286:H2353–60. doi:10.1152/ajpheart.00968.2003
69. Dillon PF, Root-Bernstein RS, Robinson NE, Abraham WM, Berney C. Ascorbate binding to the adrenergic receptor enhances in vivo and in vitro beta-adrenergic sensitivity. PLoS One (2010) 5(12):e15130. doi:10.1371/journal.pone.0015130
70. Dillon PF, Root-Bernstein RS, Sears PR, Olson LK. Natural electrophoresis of norepinephrine-ascorbic acid complex. J Biophys (2000) 79:370–6. doi:10.1016/S0006-3495(00)76298-9
71. Dillon PF, Lieder CM, Root-Bernstein RS. Molecular shielding of electric field complex dissociation. Biophys J (2006) 90:1432–8. doi:10.1529/biophysj.105.071969
72. Christodoulakos G, Lambrinoudaki I, Panoulis C, Sioulas V, Rizos D, Caramalis G, et al. Serum androgen levels and insulin resistance in postmenopausal women: association with hormone therapy, tibolone and raloxifene. Maturitas (2005) 50(4):321–30. doi:10.1016/j.maturitas.2004.08.002
73. Nagamani M, Szymajda A, Sepilian V, Urban RJ, Gilkison C. Effects of raloxifene on insulin sensitivity, beta-cell function, and hepatic insulin extraction in normal postmenopausal women. Fertil Steril (2008) 89(3):614–9. doi:10.1016/j.fertnstert.2007.03.083
74. Johansson H, Gandini S, Guerrieri-Gonzaga A, Iodice S, Ruscica M, Bonanni B, et al. Effect of fenretinide and low-dose tamoxifen on insulin sensitivity in premenopausal women at high risk for breast cancer. Cancer Res (2008) 68(22):9512–8. doi:10.1158/0008-5472.CAN-08-0553
75. Lee CC, Bloem CJ, Kasa-Vubu JZ, Liang LJ. Effect of oral phytoestrogen on androgenicity and insulin sensitivity in postmenopausal women. Diabetes Obes Metab (2012) 14(4):315–9. doi:10.1111/j.1463-1326.2011.01532.x
76. Gilbert ER, Liu D. Anti-diabetic functions of soy isoflavone genistein: mechanisms underlying its effects on pancreatic β-cell function. Food Funct (2013) 4(2):200–12. doi:10.1039/c2fo30199g
77. Choi JS, Koh IU, Song J. Genistein reduced insulin resistance index through modulating lipid metabolism in ovariectomized rats. Nutr Res (2012) 32(11):844–55. doi:10.1016/j.nutres.2012.10.002
78. Prasannarong M, Saengsirisuwan V, Piyachaturawat P, Suksamrarn A. Improvements of insulin resistance in ovariectomized rats by a novel phytoestrogen from Curcuma comosa Roxb. BMC Complement Altern Med (2012) 12:28. doi:10.1186/1472-6882-12-28
79. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest (2000) 106(4):473–85. doi:10.1172/JCI10842
80. Hilf R, Hissin PJ, Shafie SM. Regulatory interrelationships for insulin and estrogen action in mammary tumors. Cancer Res (1978) 48:4076–85.
Keywords: polycystic ovarian syndrome, gestational diabetes mellitus, insulin resistance, sex hormone binding globulin, ovarian hyperstimulation syndrome, obesity, metabolic syndrome, molecular complementarity
Citation: Root-Bernstein R, Podufaly A and Dillon PF (2014) Estradiol binds to insulin and insulin receptor decreasing insulin binding in vitro. Front. Endocrinol. 5:118. doi: 10.3389/fendo.2014.00118
Received: 29 April 2014; Accepted: 04 July 2014;
Published online: 21 July 2014.
Edited by:
Catherine Chan, University of Alberta, CanadaReviewed by:
Sarah Louise Gray, University of Northern British Columbia, CanadaCatherine Chan, University of Alberta, Canada
Copyright: © 2014 Root-Bernstein, Podufaly and Dillon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Root-Bernstein, Department of Physiology, Michigan State University, 2174 Biomedical and Physical Science Building, East Lansing, MI 48824, USA e-mail: rootbern@msu.edu