 Mariangela Zane1,2†
Mariangela Zane1,2† Veronica Catalano1†
Veronica Catalano1† Emanuela Scavo1
Emanuela Scavo1 Marco Bonanno1
Marco Bonanno1 Maria Rosa Pelizzo2
Maria Rosa Pelizzo2 Matilde Todaro1
Matilde Todaro1 Giorgio Stassi1*
Giorgio Stassi1*- 1Department of Surgical and Oncological Sciences, University of Palermo, Palermo, Italy
- 2Department of Surgical, Oncological and Gastroenterological Sciences, University of Padua, Padua, Italy
Recent discoveries highlight the emerging role of estrogens in the initiation and progression of different malignancies through their interaction with stem cell (SC) compartment. Estrogens play a relevant role especially for those tumors bearing a gender disparity in incidence and aggressiveness, as occurs for most thyroid diseases. Although several experimental lines suggest that estrogens promote thyroid cell proliferation and invasion, their precise contribution in SC compartment still remains unclear. This review underlines the interplay between hormones and thyroid function, which could help to complete the puzzle of gender discrepancy in thyroid malignancies. Defining the association between estrogen receptors’ status and signaling pathways by which estrogens exert their effects on thyroid cells is a potential tool that provides important insights in pathogenetic mechanisms of thyroid tumors.
Introduction
The endocrine system consists of a network of glands secreting hormones, which are chemical messengers that cooperate in growth, development, metabolism, and reproductive functions. The largest endocrine organ in the human body is the thyroid gland, whose function is the systemic metabolic regulation through thyroid hormones (THs) produced by follicular cells, and calcitonin produced by parafollicular cells. Different malignancy histotypes can arise from these cells: papillary (PTC), follicular (FTC), and anaplastic thyroid carcinomas (ATC) originate from follicular cells, while medullary thyroid carcinomas (MTC) derive from parafollicular cells (1). Notably, more than 95% of thyroid carcinomas (TCs) arise from follicular cells. These malignancies are indolent tumors treated by surgical resection with or without radioactive-iodine ablation since they maintain their distinct potential to concentrate Iodine. The loss of typical thyroid cell characteristics and functions, including expression of the thyroid-stimulating hormone (TSH) receptor (TSH-R), thyroglobulin (Tg), thyroid peroxidase (TPO), and sodium iodide symporter (NIS), defines the hallmark of ATCs, which are lethal malignancies with no effective therapy (1–3).
Besides genetic alterations in mitogen-activated protein kinase (MAPK), PI-3 kinase (PI3K), and TSH signaling pathways, thyroid carcinogenesis is fostered by the microenvironment, growth factors (GFs), and various hormones, including estrogens (4). Hormones can set off a cascade of signaling pathways, enhancing or contrasting specific effects triggered by other factors. Based on this scenario, the role of estrogens has been proposed in the pathogenesis of thyroid proliferative and neoplastic disorders. This hypothesis is supported by data regarding gender incidence, which reported a frequency of thyroid nodules about three to four times higher in women than in men with a peak rate occurring earlier in women (5, 6). Furthermore, the clarification of the estrogen-driven pathogenesis could be crucial in explaining why PTC constitutes the seventh most common cancer in the female gender (7, 8). An in vivo study reported that circulating estrogens are directly responsible for the increased female susceptibility to thyroid disease, through PI3K pathway activation and repressing p27 expression. The authors also observed a significant estrogen role in the transcriptional regulation of TPO, DUOX1, and NIS genes (9). Although several studies have demonstrated a direct action by estrogens on thyroid growth and function (7, 10–12), the precise mechanism underlying the proliferative and neoplastic disorders still remains undefined. In particular, it would be interesting to explore the role of hormones in TC initiation.
The cellular origin of TCs has been explained by different models (Figure 1). The multistep carcinogenesis model predicts that TC originates from follicular cells as a consequence of multiple mutations accumulated throughout their life-span. These events are characterized by a dedifferentiation process with a marked epithelial-to-mesenchymal transition (EMT), in which well-differentiated TC cells transform into a more undifferentiated phenotype (1). The fetal cell carcinogenesis model hypothesizes that TC cells would be generated by transforming three types of fetal thyroid cells, stem cells (SCs), thyroblasts, and prothyrocytes, which result in ATC, PTC, and FTC, respectively (13, 14). The heterogeneity of tumor bulk had led to a cancer stem cells (CSCs) model to propose TC as an SC disease. The growing body of experimental evidence has revealed that an accumulation of genetic abnormalities in tissue-resident SCs or in their more committed progenies, concomitant with the niche epigenetic alterations, result in their malignant transformation (15, 16).
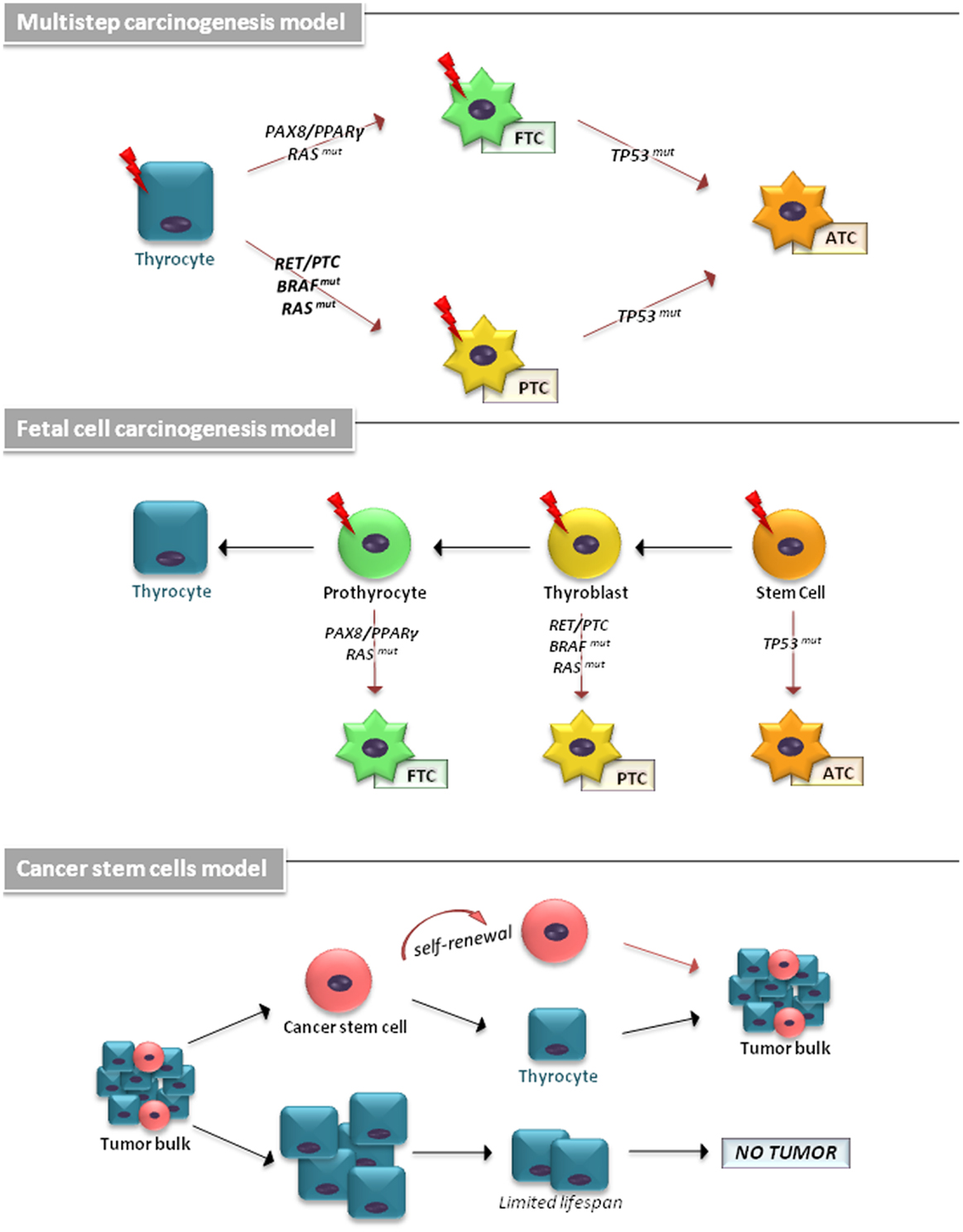
Figure 1. The cellular origin of thyroid carcinomas is shown. According to the multistep carcinogenesis model, TC originates from follicular cells as a consequence of multiple mutations accumulated throughout their life-span. Thyrocytes could give rise to PTC by RAS and BRAF mutations or RET/PTC and NTRK1 rearrangements and to FTC by point mutations of the RAS gene and PAX8/PPARγ rearrangement. ATC derive from PTC and FTC after deregulation of the p53 and the Wnt/β-catenin pathway. In fetal cell carcinogenesis model, three types of fetal thyroid cells were proposed to generate different forms of thyroid cancer. Fetal thyroid stem cells, characterized by expression of the oncofetal fibronectin (OF), generate ATC, thyroblasts, which express OF and the differentiation marker Tg, are proposed to be the cellular origin of PTC. The more differentiated prothyrocytes, expressing Tg, give rise to FTC. The cancer stem cells model proposes TC as an SC disease. The accumulation of mutations in differentiated thyrocytes leads to their transformation. A subset of these cells may (in more aggressive tumor types) dedifferentiate and assume CSC characteristics.
The “cell-of-origin” concept explains how a normal cell acquires the first alteration able to trigger tumor initiation (tumor-initiating cells, TICs) (17). Wnt pathway plays a crucial role in SC/progenitor compartment maintenance, and has been described in several tumors, including TC, resulting in nuclear β-catenin-induced proliferation (18–20).
In this review, the most current findings supporting the carcinogenesis effects of estrogens and THs will be addressed. A special emphasis will be given to the role of exogenous and endogenous GFs affecting thyroid proliferative pathways in SC compartment.
Estrogens
As recently published by Morrison’s research group, estrogens are involved in increasing hematopoietic SC self-renewal in female subjects and more specifically during pregnancy (21). It is likely that normal and tumor thyroid tissues, which express estrogen receptors (ER), could be subject to the same mechanism of estrogen action (10, 22–24).
Involved in cellular processes such as growth, cell motility, and apoptosis, in reproductive tissues and other organs, including endocrine glands, estrogens are mainly produced by the adrenal cortex and ovary, but also by the thyroid (25, 26). They are present in women and men with a notable increase in women at reproductive age. The three principal estrogens, estrone (E1), estradiol (E2), and estriol (E3), are processed in metabolites with different estrogenic abilities, which create a different risk in developing cancer (27–29).
Estradiol is the most potent estrogen since it has the highest affinity to its receptors. Estrogens perform their function by binding to ER alpha and beta (ER-α, ER-β), and a transmembrane intracellular non-classical ER G-protein-coupled receptor 30 (GPR30) (Figure 2). ER-α and ER-β are soluble intracellular nuclear receptors, belonging to a ligand-dependent nuclear receptor superfamily of transcription factors (TFs) (25, 26). ER-α is the key factor of E2-induced proliferation with an anti-apoptosis effect. In females of reproductive age, ER-α levels are higher in PTC compared to nodular goiter patients, showing a positive correlation between ER-α and Ki-67 expression levels. In contrast, ER-β is associated with apoptosis and growth inhibition, providing a negative correlation with mutant P53 (30). PPARγ also interacts with ER-α inhibiting each other, and with ER-β enhancing their inhibitory effect on cell proliferation and migration (31). In light of this, the ER-α/ER-β ratio could be helpful to elucidate the TC pathophysiology (25, 32).
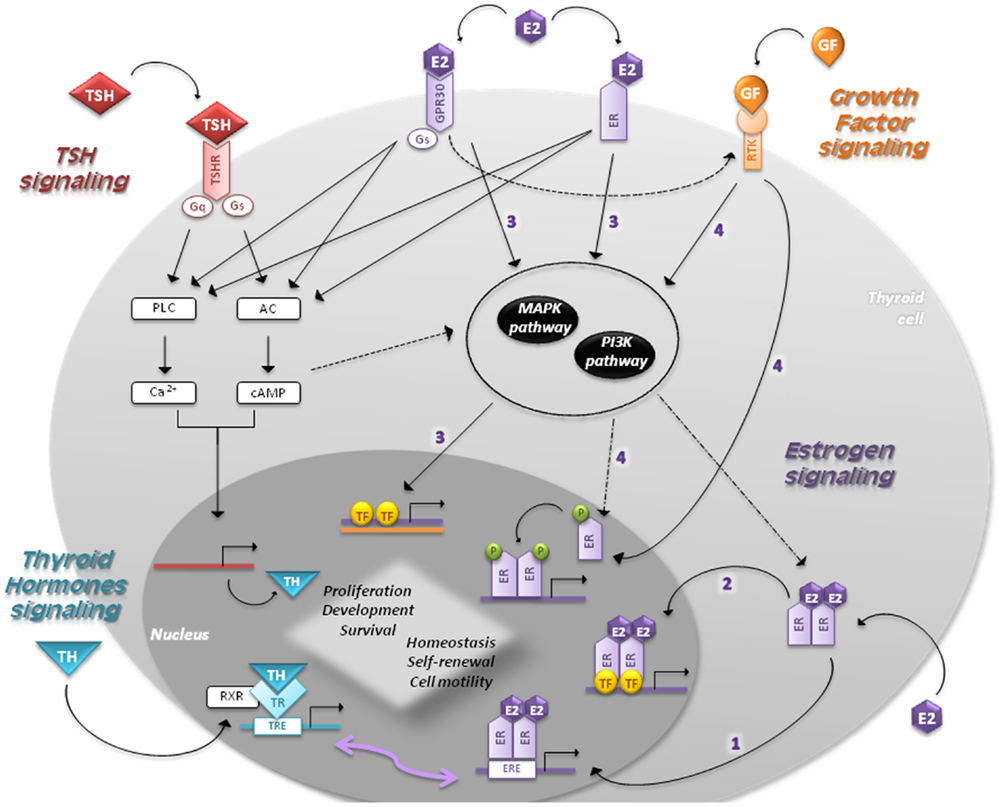
Figure 2. Signaling pathways in follicular cells are shown. The main regulators of thyroid proliferation and function act through TSH signaling and GF pathway. THs control the secretion of TSH, which binds to TSH-R and induces the coupling of G-proteins stimulating AC and PLC. TSH also acts via PI3K pathway. GFs act via MAPK and PI3K pathways regulating the expression of genes involved in survival, cell cycle progression, and proliferation. Estrogens regulate proliferation, cell motility, differentiation, and apoptosis through four different mechanisms: (1) genomic (or classical) estrogen-signaling: E2–ER complex translocates into the nucleus, where it binds to ERE-sequences; (2) ERE-independent genomic actions (TFs cross-talk): genes lacking in ERE-sequences are activated by other TFs in the nucleus through protein–protein interactions; (3) non-genomic (or membrane-initiated) estrogen-signaling: E2 activation of plasma membrane-associated ER and GPR30 trigger the activation of MAPK and PI3K pathways and/or increases the Ca2+ levels; (4) ligand-independent signaling: in absence of E2, GFs can stimulate ERs directly or indirectly through MAPK and/or PI3K pathways. THs play a critical role in development and homeostasis. Nuclear TRs activate gene expression by binding to RXR, which in turn bind to TRE-sequences. Given that EREs share a similar nucleotide sequence with TREs, ERs and TRs can interact and regulate several transcriptional responses. The cross-talk between genomic and non-genomic pathways and other integrative signaling lead to a synergic cell response.
The interaction between estrogens and ERs signals through different pathways:
• Genomic (or classical) estrogen-signaling: after accessing the cell through passive diffusion, E2 binds to ER, which changes its conformation and homo- or heterodimerizes (E2–ER). This complex translocates into the nucleus, where it binds to the 15-bp palindromic estrogen response element (ERE) located in the regulatory regions of target genes. This interaction leads to a co-activators recruitment, which in turn allows expression of genes involved in proliferation (33, 34).
• Estrogen response element-independent genomic actions (TFs cross-talk): ERE-lacking genes can be activated by modulating other TFs through protein–protein interactions. This molecular mechanism induces chromatin remodeling, histone unwinding, and interaction with the basal transcription machinery complex (35–37).
• Non-genomic (or membrane-initiated) estrogen-signaling: E2 activation of plasma membrane-associated ER and GPR30 promotes the MAPK and PI3K signaling pathways and/or increases the Ca2+ levels (10, 38–40). They can also activate G-proteins resulting in cAMP production, similar to TSH signaling in thyrocytes, and assist the activation of metalloproteinases (MMPs) and the GF pathway (5).
• Ligand-independent signaling: in absence of E2, GFs can stimulate ERs directly or indirectly through MAPK and/or PI3K pathways (41).
The cross-talk between genomic and non-genomic pathways, as well as the integrative signaling by E2 in different cell compartments, leads to a synergy that provides plasticity in cell response. Estrogens dispatch their proliferative role also by increasing T3 levels and stimulating the iodine-uptake and TPO activity (42).
Furlanetto et al. (43) reported that E2 increases proliferation of thyroid cells down-regulating NIS. These data underline the pivotal role of estrogens in the SC compartment maintenance. In normal and tumor thyroid cell lines, Rajoria et al. documented that E2 is associated with increased proliferation, adhesion, invasion, and migration via β-catenin (7) and MMP-9 modulation (44). Likewise, E-cadherin down-regulation and β-catenin translocation sustain the metastatic activity of TC cells (24). These results confirmed the findings by Kouzmenko et al., which reported the first evidence of cross-talk between estrogens and Wnt pathways through functional interaction of β-catenin with ER-α (45).
Xu et al. (8) analyzed whether differentiated and SC/progenitors could be target of estrogen action in thyroid. SCs isolated from goiter tissue enhanced their sphere-forming ability in presence of E2. Moreover, thyroid-sphere cells showed ER-α mRNA levels eight times higher than those of more differentiated thyrocytes. This suggests the gender discrepancy in TC incidence and a difference in terms of aggressiveness and survival.
Thyroid Hormones
Thyroid hormones control the secretion of thyrotropin-releasing hormone (TRH) from the hypothalamus and TSH from the anterior pituitary through negative feedback loops (1). Thyroid homeostasis and function are regulated by a concert of signals accumulated from TSH and GF pathways. TSH binds to TSH-R and induces the coupling of different G-proteins, stimulating adenylate cyclase (AC) and phospholipase C (PLC) (Figure 2). This promotes iodide uptake and TG, TPO, and NIS expression, producing thyroxine (T4) and triiodothyronine (T3) (19, 46). On the contrary, intracellular Ca2+ and PLC regulate iodine release, H2O2 production, and Tg iodination (47, 48). Although cAMP is the main mediator of TSH stimulation in thyroid cell growth, TSH via PI3K increases cyclin E levels leading to cell cycle progression (49, 50). TSH-R is also associated to the MAPK pathway through its desensitization and internalization apparatus (51).
Gain-of-function mutations in TSH-R or Gs genes result in increased cAMP accumulation and TSH-independent proliferation, which account for hyperfunctioning nodules in patients with multinodular goiters (52, 53). These alterations result insufficient for the malignant transformation of thyroid cells (54, 55). Hence, it is likely that other factors intervene in the SC compartment, which is assumed to be the target of neoplastic transformation. Alterations of the Wnt pathway effectors are involved in cancer initiation and progression (56). In particular, TSH-mediated Wnt-1 over-expression and GSK-3β inhibition promote thyroid cell proliferation (57, 58).
Thyroid hormones play a critical role in the tissue development and homeostasis by direct transcriptional regulation or modulation of different pathways (59). Although T4 is the predominant hormone produced by the thyroid, T3 is the active form that mediates gene regulation binding with a higher affinity to thyroid receptors (TRs) (60). Nuclear TRs activate gene expression by binding with the retinoid X receptors (RXRs) to TH response elements (TRE), located on the promoters of target genes (Figure 2) (61). Given that EREs share a similar nucleotide sequence with TREs, ERs and TRs can interact and regulate several transcriptional responses to environmental stimuli (5). Interestingly, ERE can act as a peroxisome proliferator responsive elements (PPRE), binding PPARγ/RXR. It can henceforth inhibit ER transactivation through a competition for ERE binding (62). In line with this cross-interaction, the proliferative effect of estrogens on human NPA-87-1 PTC cell line is TSH-independent (63). Lima et al. demonstrated a more direct proliferative effect since E2 administration to prepubertal and adult rats enhances thyroid weight without significant changes in T3, T4, and TSH hematopoietic levels (42).
Recent studies in human cancers and mouse models provide strong evidence that the loss of TRs function contributes to cancer initiation and progression (64). While the TRα1 trigger directly promotes transcription of CTNNB1 (65, 66), the effect generated by the TRα2 stimulation in SC compartment is still unknown. Cross-talk between THs-TRα1 and Wnt pathway has been confirmed by the up-regulation of several SC markers (67). Furthermore, it was reported that aberrant nuclear localization of β-catenin-induced by CTNNB1 mutations contributes to the progression of ATCs (68). Data reported by Todaro et al. showed that E-cadherin down-regulation together with β-catenin activation confers an invasive capacity and higher metastatic rate to thyroid CSCs (18).
Growth Factors
In thyroid, GFs exert their proliferative effects by inducing the RTK dimerization that activates the downstream PI3K pathway and the MAPK cascade via G-proteins (Figure 2). Alterations in genes involved in the MAPK pathway led to its constitutive activation, which represents a typical feature of TC (1). In particular, mutations in RET and NTRK and alterations in RAS and BRAF intracellular signal-transducers are clearly implicated in PTC pathogenesis (69). RAS point mutations and PAX8/PPARγ rearrangement have been frequently implicated in FTC pathogenesis (70, 71). The inactivation of RASAL1 (encoding a RAS GTPase-activating protein) by hypermethylation and mutations provides a new genetic background for FTCs and ATCs (72). Besides nuclear β-catenin accumulation and p53 inactivation, oncogenic activation of MAPK and PI3K/Akt/Foxo3a are frequently found in ATCs (2, 73, 74). The acquisition of a TERT promoter mutation was recently associated with clinical–pathological aggressiveness in FTCs and BRAF mutation-positive PTCs (72, 75).
The mesenchymal tissue is involved in thyroid development being that it releases Pro-epidermal growth factor (EGF) and basic fibroblast growth factor-2 (FGF-2), promoting cell proliferation and repressing differentiation (76, 77). Estrogens play a pivotal role in this context by inducing the production of EGF and other TFs, such as TGF-α (5).
After EGF binding, RTKs of the ErbB family (EGFR/ErbB1, ErbB2, ErbB3, and ErbB4) achieve activation through the arrangement in homo- and/or heterodimeric complexes (78, 79). In thyroid, TSH increases the expression of EGFRs that in turn promote the EGF mitogenic effect and contribute to gland homeostasis. The combination of specific EGFRs regulates the stimulation intensity, inducing transformation. Indeed, an increased expression of EGFRs in TCs compared to normal tissue has been reported (80). EGFR/ErbB1 over-expression and its constitutive phosphorylation have been observed on ATC samples and cell lines (81). Their expression has been retrieved in 90% of the PTC samples examined by Song (82). In combination with the repression of VEGF, EGF inhibitors could be a promising therapy for ATCs as demonstrated by in vitro studies (83, 84). EGF is also supplemented in the serum-free culture medium, which is used to isolate SCs and CSCs in vitro (18, 85–90).
Similarly, the cell response to FGF is regulated by FGF RTKs (FGFRs 1–4). FGF-2 exerts autocrine and paracrine stimulatory effects on thyroid growth, since the basement membrane of thyrocytes is able to produce FGF itself. FGF is also used in vitro for the maintenance of SC niche (18, 91); in particular, it could have an inhibitory effect on thyroid function through cAMP inhibition and TSH’s activity weakening (79). In TC, increased FGF-2 levels and FGFR2 over-expression are critical in tumor progression and neovascularization (92, 93). Therefore, the differential expression in normal and malignant conditions could make this receptor a potential diagnostic marker for TCs (94).
Growth factors also affect development and metabolic processes through insulin-like growth factor (IGF). After binding of their ligands, IGF receptors (IGF-Rs) autophosphorylate their intracellular domain and activate the MAPK and PI3K cascade (95). Consistently, IGF enhances the TSH mitogenic effect on follicular cells (96); on the other hand, it also cooperates with FGF-2 in establishing and maintaining the SC niche in vitro (96). Indeed, IGF pathway effectors are over-expressed in CSCs: IGFR2 is involved in an autocrine loop that sustains SC renewal, and IGF increases the expression of Oct-4 and Nanog when added to the culture medium (87, 97, 98).
Estrogen-Growth Factors Interacting Proteins
Recently, there has been a focus on importance of the ER-GFs interacting proteins on cancer cell proliferation and invasivity. An example is mediator of ERbB2-driven cell motility (MEMO), which enhances ER-α extra-nuclear functions through the interaction with IGFR1 and ERbB2, activating MAPK and PI3K signaling (99).
Concluding Remarks
Since the theory of fetal carcinogenesis has initially been postulated, thyroid CSCs have been studied for their potential role as TICs. It has been hypothesized that various factors could be involved in the malignant transformation, such as aberrant molecular events converging to RTK, MAPK, and PI3K pathway activation. Besides the oncogenes contribution, it is likely that a network of various hormones and GFs could maintain the SC niche and enhance the proliferation of progenitors sustaining tumor bulk growth. Indeed, recent studies demonstrate that sexual hormones could exert a supportive role in the propagation of SCs and progenitors, as suggested by the cross-talk between estrogen-signaling and Wnt pathway. Furthermore, the latter pathway has also been observed interacting with THs in SC compartment and so accelerating tumorigenic processes. This mechanism could be benefited by the interaction between different cascades, which enhances or contrasts specific cellular response in tumor conditions. In conclusion, an in-depth study on the concert between estrogens, THs, and GFs could be helpful to elucidate hormones-driven thyroid carcinogenesis. Gaining more insight into this interaction could also explain the gender imbalance in tumor incidence for the purpose of identifying a more targeted approach in TC therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from AIRC to Giorgio Stassi (IG12819). Mariangela Zane is Ph.D., student in Oncology and Surgical Oncology at University of Padua. Emanuela Scavo and Marco Bonanno are Ph.D., students at University of Palermo, in International Immunopharmacology and in Cellular and Developmental Biology, respectively. We would like to thank Tatiana Terranova for her thoroughness and passion in editing this review.
References
1. Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer (2006) 6:292–306. doi:10.1038/nrc1836
2. Nikiforova MN, Nikiforov YE. Molecular genetics of thyroid cancer: implications for diagnosis, treatment and prognosis. Expert Rev Mol Diagn (2008) 8:83–95. doi:10.1586/14737159.8.1.83
3. Smallridge RC, Marlow LA, Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr Relat Cancer (2009) 16:17–44. doi:10.1677/ERC-08-0154
4. Rahbari R, Zhang L, Kebebew E. Thyroid cancer gender disparity. Future Oncol (2010) 6:1771–9. doi:10.2217/fon.10.127
5. Rajoria S, Suriano R, George AL, Shanmugam A, Jussim C, Shin EJ, et al. Estrogen activity as a preventive and therapeutic target in thyroid cancer. Biomed Pharmacother (2012) 66:151–8. doi:10.1016/j.biopha.2011.11.010
6. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63:11–30. doi:10.3322/caac.21166
7. Rajoria S, Suriano R, Shanmugam A, Wilson YL, Schantz SP, Geliebter J, et al. Metastatic phenotype is regulated by estrogen in thyroid cells. Thyroid (2010) 20:33–41. doi:10.1089/thy.2009.0296
8. Xu S, Chen G, Peng W, Renko K, Derwahl M. Oestrogen action on thyroid progenitor cells: relevant for the pathogenesis of thyroid nodules? J Endocrinol (2013) 218:125–33. doi:10.1530/JOE-13-0029
9. Antico-Arciuch VG, Dima M, Liao XH, Refetoff S, Di Cristofano A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene (2010) 29:5678–86. doi:10.1038/onc.2010.308
10. Zeng Q, Chen GG, Vlantis AC, Van Hasselt CA. Oestrogen mediates the growth of human thyroid carcinoma cells via an oestrogen receptor-ERK pathway. Cell Prolif (2007) 40:921–35. doi:10.1111/j.1365-2184.2007.00471.x
11. Ceresini G, Milli B, Morganti S, Maggio M, Bacchi-Modena A, Sgarabotto MP, et al. Effect of estrogen therapy for 1 year on thyroid volume and thyroid nodules in postmenopausal women. Menopause (2008) 15:326–31. doi:10.1097/gme.0b013e318148b83e
12. Vaiman M, Olevson Y, Sandbank J, Habler L, Zehavi S, Kessler A. Are estrogen receptors alpha detectable in normal and abnormal thyroid tissue? Eur Arch Otorhinolaryngol (2010) 267:1753–7. doi:10.1007/s00405-010-1269-0
13. Takano T, Amino N. Fetal cell carcinogenesis: a new hypothesis for better understanding of thyroid carcinoma. Thyroid (2005) 15:432–8. doi:10.1089/thy.2005.15.432
14. Takano T. Fetal cell carcinogenesis of the thyroid: theory and practice. Semin Cancer Biol (2007) 17:233–40. doi:10.1016/j.semcancer.2006.02.001
15. Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells – old concepts, new insights. Cell Death Differ (2008) 15:947–58. doi:10.1038/cdd.2008.20
16. Lin RY. Thyroid cancer stem cells. Nat Rev Endocrinol (2011) 7:609–16. doi:10.1038/nrendo.2011.127
18. Todaro M, Iovino F, Eterno V, Cammareri P, Gambara G, Espina V, et al. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res (2010) 70:8874–85. doi:10.1158/0008-5472.CAN-10-1994
19. Sastre-Perona A, Santisteban P. Role of the wnt pathway in thyroid cancer. Front Endocrinol (Lausanne) (2012) 3:31. doi:10.3389/fendo.2012.00031
20. Van Camp JK, Beckers S, Zegers D, Van Hul W. Wnt signaling and the control of human stem cell fate. Stem Cell Rev (2014) 10:207–29. doi:10.1007/s12015-013-9486-8
21. Nakada D, Oguro H, Levi BP, Ryan N, Kitano A, Saitoh Y, et al. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature (2014) 505:555–8. doi:10.1038/nature12932
22. Manole D, Schildknecht B, Gosnell B, Adams E, Derwahl M. Estrogen promotes growth of human thyroid tumor cells by different molecular mechanisms. J Clin Endocrinol Metab (2001) 86:1072–7. doi:10.1210/jcem.86.3.7283
23. Lee ML, Chen GG, Vlantis AC, Tse GM, Leung BC, Van Hasselt CA. Induction of thyroid papillary carcinoma cell proliferation by estrogen is associated with an altered expression of Bcl-xL. Cancer J (2005) 11:113–21. doi:10.1097/00130404-200503000-00006
24. Dong W, Zhang H, Li J, Guan H, He L, Wang Z, et al. Estrogen induces metastatic potential of papillary thyroid cancer cells through estrogen receptor alpha and beta. Int J Endocrinol (2013) 2013:941568. doi:10.1155/2013/941568
25. Santin AP, Furlanetto TW. Role of estrogen in thyroid function and growth regulation. J Thyroid Res (2011) 2011:875125. doi:10.4061/2011/875125
26. Arciuch Antico VG, Di Cristofano A. Estrogen signaling and thyrocyte proliferation. In: Ward DL, editor. Thyroid and Parathyroid Diseases – New Insights into Some Old and Some New Issues. InTech (2012). 318 p. doi:10.5772/35913
27. Thiruvengadam A, Govindarajulu P, Aruldhas MM. Modulatory effect of estradiol and testosterone on the development of N-nitrosodiisopropanolamine induced thyroid tumors in female rats. Endocr Res (2003) 29:43–51. doi:10.1081/ERC-120018675
28. Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev (2007) 87:905–31. doi:10.1152/physrev.00026.2006
29. Zahid M, Goldner W, Beseler CL, Rogan EG, Cavalieri EL. Unbalanced estrogen metabolism in thyroid cancer. Int J Cancer (2013) 133:2642–9. doi:10.1002/ijc.28275
30. Huang Y, Dong W, Li J, Zhang H, Shan Z, Teng W. Differential expression patterns and clinical significance of estrogen receptor-alpha and beta in papillary thyroid carcinoma. BMC Cancer (2014) 14:383. doi:10.1186/1471-2407-14-383
31. Chu R, Van Hasselt A, Vlantis AC, Ng EK, Liu SY, Fan MD, et al. The cross-talk between estrogen receptor and peroxisome proliferator-activated receptor gamma in thyroid cancer. Cancer (2014) 120:142–53. doi:10.1002/cncr.28383
32. Leitman DC, Paruthiyil S, Vivar OI, Saunier EF, Herber CB, Cohen I, et al. Regulation of specific target genes and biological responses by estrogen receptor subtype agonists. Curr Opin Pharmacol (2010) 10:629–36. doi:10.1016/j.coph.2010.09.009
33. Klinge CM. Estrogen receptor interaction with co-activators and co-repressors. Steroids (2000) 65:227–51. doi:10.1016/S0039-128X(99)00107-5
34. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of estrogen action. Physiol Rev (2001) 81:1535–65.
35. Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol (2005) 19:833–42. doi:10.1210/me.2004-0486
36. Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor alpha and beta: impact on human health. Mol Aspects Med (2006) 27:299–402. doi:10.1016/j.mam.2006.07.001
37. Osmanbeyoglu HU, Lu KN, Oesterreich S, Day RS, Benos PV, Coronnello C, et al. Estrogen represses gene expression through reconfiguring chromatin structures. Nucleic Acids Res (2013) 41:8061–71. doi:10.1093/nar/gkt586
38. Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett (2006) 238:1–14. doi:10.1016/j.canlet.2005.06.018
39. Chen JQ, Brown TR, Yager JD. Mechanisms of hormone carcinogenesis: evolution of views, role of mitochondria. Adv Exp Med Biol (2008) 630:1–18. doi:10.1007/978-0-387-78818-0_1
40. He YY, Cai B, Yang YX, Liu XL, Wan XP. Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci (2009) 100:1051–61. doi:10.1111/j.1349-7006.2009.01148.x
41. Roman-Blas JA, Castaneda S, Largo R, Herrero-Beaumont G. Osteoarthritis associated with estrogen deficiency. Arthritis Res Ther (2009) 11:241. doi:10.1186/ar2791
42. Lima LP, Barros IA, Lisboa PC, Araujo RL, Silva AC, Rosenthal D, et al. Estrogen effects on thyroid iodide uptake and thyroperoxidase activity in normal and ovariectomized rats. Steroids (2006) 71:653–9. doi:10.1016/j.steroids.2006.03.007
43. Furlanetto TW, Nguyen LQ, Jameson JL. Estradiol increases proliferation and down-regulates the sodium/iodide symporter gene in FRTL-5 cells. Endocrinology (1999) 140:5705–11. doi:10.1210/endo.140.12.7197
44. Rajoria S, Suriano R, Wilson YL, George AL, Geliebter J, Schantz SP, et al. Estradiol-mediated tumor neo-vascularization. Oncol Lett (2011) 2:453–7. doi:10.3892/ol.2011.283
45. Kouzmenko AP, Takeyama K, Ito S, Furutani T, Sawatsubashi S, Maki A, et al. Wnt/beta-catenin and estrogen signaling converge in vivo. J Biol Chem (2004) 279:40255–8. doi:10.1074/jbc.C400331200
46. Postiglione MP, Parlato R, Rodriguez-Mallon A, Rosica A, Mithbaokar P, Maresca M, et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci U S A (2002) 99:15462–7. doi:10.1073/pnas.242328999
47. Medina DL, Santisteban P. Thyrotropin-dependent proliferation of in vitro rat thyroid cell systems. Eur J Endocrinol (2000) 143:161–78. doi:10.1530/eje.0.1430161
48. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev (2001) 22:631–56. doi:10.1210/edrv.22.5.0444
49. Cass LA, Summers SA, Prendergast GV, Backer JM, Birnbaum MJ, Meinkoth JL. Protein kinase A-dependent and -independent signaling pathways contribute to cyclic AMP-stimulated proliferation. Mol Cell Biol (1999) 19:5882–91.
50. Roger PP, Van Staveren WC, Coulonval K, Dumont JE, Maenhaut C. Signal transduction in the human thyrocyte and its perversion in thyroid tumors. Mol Cell Endocrinol (2010) 321:3–19. doi:10.1016/j.mce.2009.11.015
51. Kursawe R, Paschke R. Modulation of TSHR signaling by posttranslational modifications. Trends Endocrinol Metab (2007) 18:199–207. doi:10.1016/j.tem.2007.05.002
52. Tallini G. Molecular pathobiology of thyroid neoplasms. Endocr Pathol (2002) 13:271–88. doi:10.1385/EP:13:4:271
53. Krohn K, Fuhrer D, Bayer Y, Eszlinger M, Brauer V, Neumann S, et al. Molecular pathogenesis of euthyroid and toxic multinodular goiter. Endocr Rev (2005) 26:504–24. doi:10.1210/er.2004-0005
54. Matsuo K, Friedman E, Gejman PV, Fagin JA. The thyrotropin receptor (TSH-R) is not an oncogene for thyroid tumors: structural studies of the TSH-R and the alpha-subunit of Gs in human thyroid neoplasms. J Clin Endocrinol Metab (1993) 76:1446–51. doi:10.1210/jcem.76.6.8501149
55. Spambalg D, Sharifi N, Elisei R, Gross JL, Medeiros-Neto G, Fagin JA. Structural studies of the thyrotropin receptor and Gs alpha in human thyroid cancers: low prevalence of mutations predicts infrequent involvement in malignant transformation. J Clin Endocrinol Metab (1996) 81:3898–901. doi:10.1210/jcem.81.11.8923835
56. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature (2005) 434:843–50. doi:10.1038/nature03319
57. Kim WB, Lewis CJ, Mccall KD, Malgor R, Kohn AD, Moon RT, et al. Overexpression of Wnt-1 in thyrocytes enhances cellular growth but suppresses transcription of the thyroperoxidase gene via different signaling mechanisms. J Endocrinol (2007) 193:93–106. doi:10.1677/JOE-06-0025
58. Chen G, Jiang Q, You Z, Yao J, Mou L, Lin X, et al. Regulation of GSK-3 beta in the proliferation and apoptosis of human thyrocytes investigated using a GSK-3 beta-targeting RNAi adenovirus expression vector: involvement the Wnt/beta-catenin pathway. Mol Biol Rep (2010) 37:2773–9. doi:10.1007/s11033-009-9819-5
59. Pascual A, Aranda A. Thyroid hormone receptors, cell growth and differentiation. Biochim Biophys Acta (2013) 1830:3908–16. doi:10.1016/j.bbagen.2012.03.012
60. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev (2010) 31:139–70. doi:10.1210/er.2009-0007
61. Yen PM, Ando S, Feng X, Liu Y, Maruvada P, Xia X. Thyroid hormone action at the cellular, genomic and target gene levels. Mol Cell Endocrinol (2006) 246:121–7. doi:10.1016/j.mce.2005.11.030
62. Keller H, Givel F, Perroud M, Wahli W. Signaling cross-talk between peroxisome proliferator-activated receptor/retinoid X receptor and estrogen receptor through estrogen response elements. Mol Endocrinol (1995) 9:794–804. doi:10.1210/mend.9.7.7476963
63. Banu SK, Govindarajulu P, Aruldhas MM. Developmental profiles of TSH, sex steroids, and their receptors in the thyroid and their relevance to thyroid growth in immature rats. Steroids (2002) 67:137–44. doi:10.1016/S0039-128X(01)00144-1
64. Kim WG, Cheng SY. Thyroid hormone receptors and cancer. Biochim Biophys Acta (2013) 1830:3928–36. doi:10.1016/j.bbagen.2012.04.002
65. Plateroti M, Kress E, Mori JI, Samarut J. Thyroid hormone receptor alpha1 directly controls transcription of the beta-catenin gene in intestinal epithelial cells. Mol Cell Biol (2006) 26:3204–14. doi:10.1128/MCB.26.8.3204-3214.2006
66. Kress E, Samarut J, Plateroti M. Thyroid hormones and the control of cell proliferation or cell differentiation: paradox or duality? Mol Cell Endocrinol (2009) 313:36–49. doi:10.1016/j.mce.2009.08.028
67. Kress E, Skah S, Sirakov M, Nadjar J, Gadot N, Scoazec JY, et al. Cooperation between the thyroid hormone receptor TRalpha1 and the WNT pathway in the induction of intestinal tumorigenesis. Gastroenterology (2010) 138:1863–74. doi:10.1053/j.gastro.2010.01.041
68. Garcia-Rostan G, Camp RL, Herrero A, Carcangiu ML, Rimm DL, Tallini G. Beta-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am J Pathol (2001) 158:987–96. doi:10.1016/S0002-9440(10)64045-X
69. Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol (2011) 7:569–80. doi:10.1038/nrendo.2011.142
70. Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, et al. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science (2000) 289:1357–60. doi:10.1126/science.289.5483.1357
71. Garcia-Rostan G, Zhao H, Camp RL, Pollan M, Herrero A, Pardo J, et al. Ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol (2003) 21:3226–35. doi:10.1200/JCO.2003.10.130
72. Liu D, Yang C, Bojdani E, Murugan AK, Xing M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J Natl Cancer Inst (2013) 105:1617–27. doi:10.1093/jnci/djt249
73. Bellelli R, Castellone MD, Garcia-Rostan G, Ugolini C, Nucera C, Sadow PM, et al. FOXM1 is a molecular determinant of the mitogenic and invasive phenotype of anaplastic thyroid carcinoma. Endocr Relat Cancer (2012) 19:695–710. doi:10.1530/ERC-12-0031
74. Nehs MA, Nucera C, Nagarkatti SS, Sadow PM, Morales-Garcia D, Hodin RA, et al. Late intervention with anti-BRAF(V600E) therapy induces tumor regression in an orthotopic mouse model of human anaplastic thyroid cancer. Endocrinology (2012) 153:985–94. doi:10.1210/en.2011-1519
75. Landa I, Ganly I, Chan TA, Mitsutake N, Matsuse M, Ibrahimpasic T, et al. Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab (2013) 98:E1562–6. doi:10.1210/jc.2013-2383
76. De Felice M, Di Lauro R. Thyroid development and its disorders: genetics and molecular mechanisms. Endocr Rev (2004) 25:722–46. doi:10.1210/er.2003-0028
77. Fagman H, Nilsson M. Morphogenetics of early thyroid development. J Mol Endocrinol (2011) 46:R33–42. doi:10.1677/JME-10-0084
78. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol (2001) 2:127–37. doi:10.1038/35052073
79. Konturek A. Thyroid growth factors. In: Ward DL, editor. Thyroid and Parathyroid Diseases – New Insight into Some Old and Some New Issues. InTech (2012). 318 p. doi:10.5772/37570
80. Kato S, Kobayashi T, Yamada K, Nishii K, Sawada H, Ishiguro H, et al. Expression of erbB receptors mRNA in thyroid tissues. Biochim Biophys Acta (2004) 1673:194–200. doi:10.1016/j.bbagen.2004.04.016
81. Bergstrom JD, Westermark B, Heldin NE. Epidermal growth factor receptor signaling activates met in human anaplastic thyroid carcinoma cells. Exp Cell Res (2000) 259:293–9. doi:10.1006/excr.2000.4967
82. Song B. Immunohistochemical demonstration of epidermal growth factor receptor and ceruloplasmin in thyroid diseases. Acta Pathol Jpn (1991) 41:336–43.
83. Schiff BA, Mcmurphy AB, Jasser SA, Younes MN, Doan D, Yigitbasi OG, et al. Epidermal growth factor receptor (EGFR) is overexpressed in anaplastic thyroid cancer, and the EGFR inhibitor gefitinib inhibits the growth of anaplastic thyroid cancer. Clin Cancer Res (2004) 10:8594–602. doi:10.1158/1078-0432.CCR-04-0690
84. Hoffmann S, Glaser S, Wunderlich A, Lingelbach S, Dietrich C, Burchert A, et al. Targeting the EGF/VEGF-R system by tyrosine-kinase inhibitors – a novel antiproliferative/antiangiogenic strategy in thyroid cancer. Langenbecks Arch Surg (2006) 391:589–96. doi:10.1007/s00423-006-0104-y
85. Lan L, Cui D, Nowka K, Derwahl M. Stem cells derived from goiters in adults form spheres in response to intense growth stimulation and require thyrotropin for differentiation into thyrocytes. J Clin Endocrinol Metab (2007) 92:3681–8. doi:10.1210/jc.2007-0281
86. Zheng X, Cui D, Xu S, Brabant G, Derwahl M. Doxorubicin fails to eradicate cancer stem cells derived from anaplastic thyroid carcinoma cells: characterization of resistant cells. Int J Oncol (2010) 37:307–15. doi:10.3892/ijo_00000679
87. Malaguarnera R, Frasca F, Garozzo A, Giani F, Pandini G, Vella V, et al. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab (2011) 96:766–74. doi:10.1210/jc.2010-1255
88. Tseng LM, Huang PI, Chen YR, Chen YC, Chou YC, Chen YW, et al. Targeting signal transducer and activator of transcription 3 pathway by cucurbitacin I diminishes self-renewing and radiochemoresistant abilities in thyroid cancer-derived CD133+ cells. J Pharmacol Exp Ther (2012) 341:410–23. doi:10.1124/jpet.111.188730
89. Li W, Reeb AN, Sewell WA, Elhomsy G, Lin RY. Phenotypic characterization of metastatic anaplastic thyroid cancer stem cells. PLoS One (2013) 8:e65095. doi:10.1371/journal.pone.0065095
90. Ahn SH, Henderson YC, Williams MD, Lai SY, Clayman GL. Detection of thyroid cancer stem cells in papillary thyroid carcinoma. J Clin Endocrinol Metab (2014) 99:536–44. doi:10.1210/jc.2013-2558
91. Longmire TA, Ikonomou L, Hawkins F, Christodoulou C, Cao Y, Jean JC, et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell (2012) 10:398–411. doi:10.1016/j.stem.2012.01.019
92. Eggo MC, Hopkins JM, Franklyn JA, Johnson GD, Sanders DS, Sheppard MC. Expression of fibroblast growth factors in thyroid cancer. J Clin Endocrinol Metab (1995) 80:1006–11. doi:10.1210/jcem.80.3.7533768
93. Guo M, Liu W, Serra S, Asa SL, Ezzat S. FGFR2 isoforms support epithelial-stromal interactions in thyroid cancer progression. Cancer Res (2012) 72:2017–27. doi:10.1158/0008-5472.CAN-11-3985
94. Redler A, Di Rocco G, Giannotti D, Frezzotti F, Bernieri MG, Ceccarelli S, et al. Fibroblast growth factor receptor-2 expression in thyroid tumor progression: potential diagnostic application. PLoS One (2013) 8:e72224. doi:10.1371/journal.pone.0072224
95. Laron Z. Insulin-like growth factor 1 (IGF-1): a growth hormone. Mol Pathol (2001) 54:311–6. doi:10.1136/mp.54.5.311
96. Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature (2007) 448:1015–21. doi:10.1038/nature06027
97. Vella V, Pandini G, Sciacca L, Mineo R, Vigneri R, Pezzino V, et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metab (2002) 87:245–54. doi:10.1210/jcem.87.1.8142
98. Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (Lausanne) (2014) 5:10. doi:10.3389/fendo.2014.00010
Keywords: thyroid cancer, stem cells, cancer stem cells, estrogens, thyroid hormones, growth factors
Citation: Zane M, Catalano V, Scavo E, Bonanno M, Pelizzo MR, Todaro M and Stassi G (2014) Estrogens and stem cells in thyroid cancer. Front. Endocrinol. 5:124. doi: 10.3389/fendo.2014.00124
Received: 16 May 2014; Accepted: 11 July 2014;
Published online: 25 July 2014.
Edited by:
Terry Francis Davies, Icahn School of Medicine at Mount Sinai, USAReviewed by:
Carmelo Nucera, Harvard Medical School, USAAntonio Di Cristofano, Albert Einstein College of Medicine, USA
Copyright: © 2014 Zane, Catalano, Scavo, Bonanno, Pelizzo, Todaro and Stassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgio Stassi, Laboratory of Cellular and Molecular Pathophysiology, Department of Surgical and Oncological Sciences, University of Palermo, Via Liborio Giuffrè 5, Palermo 90127, Italy e-mail: giorgio.stassi@unipa.it
†Mariangela Zane and Veronica Catalano have contributed equally to this work.