 Wilhelm Kriz
Wilhelm Kriz Brunhilde Hähnel
Brunhilde Hähnel Hiltraud Hosser2,3
Hiltraud Hosser2,3- 1Institute of Transfusion Medicine and Immunology, Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany
- 2Institute of Neuroanatomy, Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany
- 3Pathologie Heidelberg, Heidelberg, Germany
- 4Global Non-Clinical Safety, Merck KGaA, Merck Serono, Darmstadt, Germany
Podocytes are lost by detachment from the GBM as viable cells; details are largely unknown. We studied this process in the rat after growth stimulation with FGF-2. Endothelial and mesangial cells responded by hyperplasia, podocytes underwent hypertrophy, but, in the long run, developed various changes that could either be interpreted showing progressing stages in detachment from the GBM or stages leading to a tighter attachment by foot process effacement (FPE). This occurred in microdomains within the same podocyte; thus, features of detachment and of reinforced attachment may simultaneously be found in the same podocyte. (1) Initially, hypertrophied podocytes underwent cell body attenuation and formed large pseudocysts, i.e., expansions of the subpodocyte space. (2) Podocytes entered the process of FPE starting with the retraction of foot processes (FPs) and the replacement of the slit diaphragm by occluding junctions, thereby sealing the filtration slits. Successful completion of this process led to broad attachments of podocyte cell bodies to the GBM. (3) Failure of sealing the slits led to gaps of varying width between retracting FPs facilitating the outflow of the filtrate from the GBM. (4) Since those gaps are frequently overarched by broadened primary processes, the drainage of the filtrate into the Bowman’s space may be hindered leading to the formation of small pseudocysts associated with bare areas of GBM. (5) The merging of pseudocysts created a system of communicating chambers through which the filtrate has to pass to reach Bowman’s space. Multiple flow resistances in series likely generated an expansile force on podocytes contributing to detachment. (6) Such a situation appears to proceed to complete disconnection generally of a group of podocytes owing to the junctional connections between them. (7) Since such groups of detaching podocytes generally make contact to parietal cells, they start the formation of tuft adhesions to Bowman’s capsule.
Introduction
Podocyte loss underlies the progression of glomerular diseases to end-stage renal failure, both in human beings (1) and in experimental animal models (2). How podocytes are lost is open to debate. Apoptosis is widely considered as a major mechanism, but this has been demonstrated largely in cell culture, and convincing evidence showing apoptosis of podocytes in situ by transmission electron microscopy (TEM) has never been presented. Also, necrosis of podocytes has rarely been shown and if it occurs, it is mostly in cells already detached from the GBM. On the other hand, viable podocytes in the urine have been found in several glomerular diseases (3, 4) and podocytes appearing to be viable detaching from the GBM in huge numbers were shown by TEM in various experimental models (5). This leads to the conclusion that detachment as viable cells is the major mechanism of podocyte loss and that the major causes of podocyte loss are likely to be mechanical forces challenging the attachment of podocytes to the GBM (6).
Recently, the hypothesis has been raised that the shear stress derived from filtrate flow through the filtration slits acting on the foot processes (FPs) may have a major impact on podocyte detachment (6, 7). Moreover, it has been suggested that glomerular hypertension, hyperfiltration, and excessive hypertrophy, which have previously been shown to underlie progression of glomerular diseases (8, 9), may account for increases in shear stress compromising the attachment of podocytes to the GBM.
Furthermore, it has been postulated that major changes of podocytes in glomerular diseases, such as foot process effacement (FPE) and cytoplasm shedding, which have been generally interpreted as pathological derangements of podocytes, actually show the “podocyte’s fight” against detachment, and thus are changes undertaken to secure survival, i.e., continued attachment to the GBM (5).
The present study analyzes the individual steps of podocyte detachment in a model of excessive glomerular hypertrophy that had been previously studied by our group (10). Daily administration of FGF-2 led to a long-term growth stimulation of glomeruli followed by the development of widespread focal and segmental glomerulosclerosis (FSGS). At the time of this study, in 1995, the idea that podocytes are lost as viable cells in the urine was not yet born. The focus of the study lay in the question of how, subsequent to podocyte loss, tuft adhesions develop and lead to the degeneration of the entire nephron. In the present study, we used the same material to structurally analyze earlier stages to describe how podocytes undergo detachment. We show that the process of detachment from the GBM is extremely gradual and frequently complicated by the fact that the sequence of steps to detachment may be mixed up with changes locally reinforcing the attachment to the GBM.
Materials and Methods
The material used in the present study was derived from previous experiments (10). Therefore, only a brief description of the methods is given here. The experiments complied with the “German law on the protection of animals.”
A total of 36 Sprague-Dawley rats (7 weeks old; half males, half females) were studied. Half received daily subcutaneous injections of 320 μg FGF-2/kg body weight and the other half served as controls, receiving the vehicle only. Two-thirds of the rats were studied after total body perfusion at 8 weeks; the remaining one-third was studied after 13 weeks. After anesthesia with Nembutal® (1.2 ml/kg body weight), the kidneys were retrogradely perfused via the abdominal aorta without prior flushing for 3 min at a pressure of 220 mm Hg. In 24 rats intended for ultrastructural studies, the fixative contained 1.5% glutaraldehyde and 1.5% formaldehyde in phosphate buffered saline (260 mOsm, pH 7.4) supplemented with 0.5 g/l picric acid. Blocks of cortical tissue were processed by standard procedures (i) for paraffin sections (4 μm; stained with hematoxylin eosin) and (ii) after postfixation with OsO4 or tannic acid (11), for Epon sections (1 μm, stained with methylene blue) to be studied by light micrograph and ultrathin sections stained with uranyl acetate and lead citrate to be studied by TEM. The remaining 12 rats were perfused with paraformaldehyde [see Ref. (10)].
Structural Studies
In addition to the structural assessments in the previous study (10), a quantification of glomerular injuries as defined in the present study (pseudocysts, podocyte detachment associated with small pseudocysts, podocyte clusters, and tuft adhesions; Figure 1A) was done. In three independent 1 μm Epon sections of each experimental animal (6 males and 6 females; values from 8 and 13 weeks were pooled) and of six controls, the glomerular profiles and the number of the above listed lesions found in these glomeruli were counted, figured up for each animal, expressed as percentage of the total number of profiles of each animal, and compared. In order to receive an overall weighted damage index, we graded large pseudocysts with 1, accumulations of small pseudocysts with 2, clusters of podocytes with 3, and adhesions with 4. Results are reported as mean ± SD; differences between the various groups were tested by one-way ANOVA using Sigma Plot.
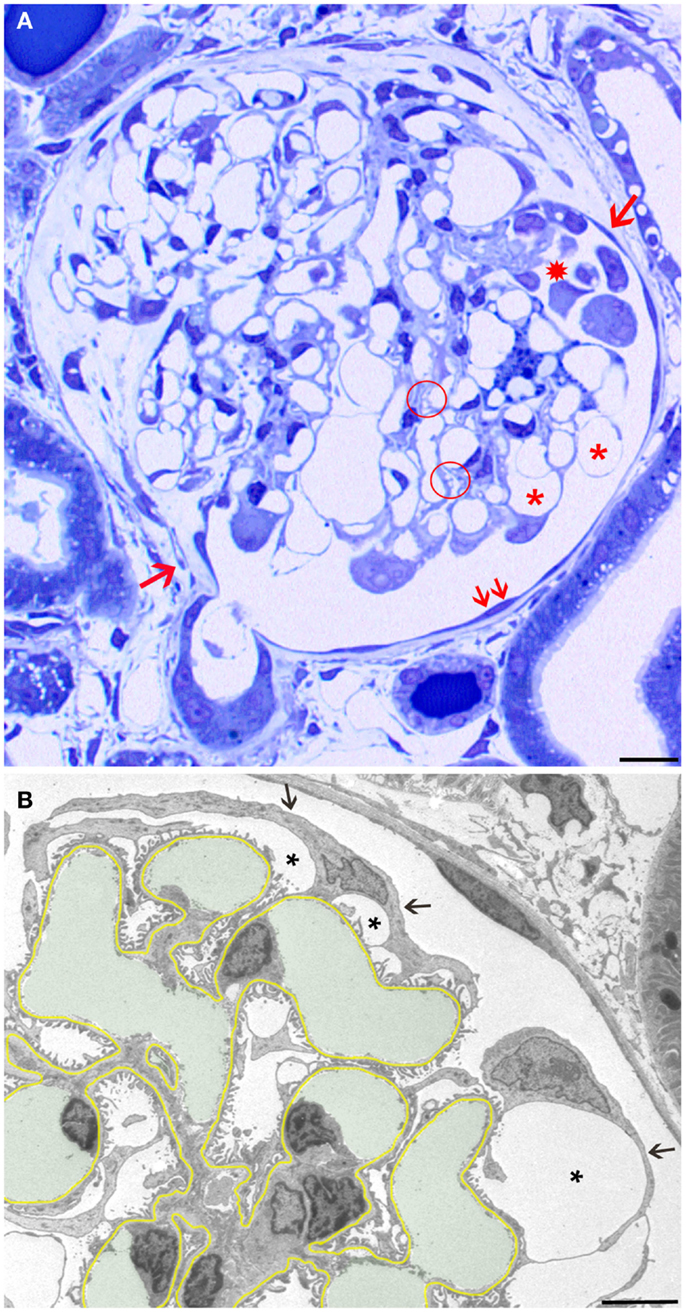
Figure 1. Overview and early changes. (A) Light micrograph (LM) of a glomerular profile showing the crucial injuries presented later in more detail in transmission electron micrographs (TEMs) and quantified in Table 1. Hypertrophied podocytes forming large pseudocysts (asterisks), areas of small pseudocysts generally associated with local detachments (enclosed by a hatched circle), detachment of podocytes forming a cluster in Bowman’s space (star), and a large tuft adhesion comprising almost two-thirds of the tuft (clockwise from arrow 1 to arrow 2) are seen. Note that outside the tuft adhesion, the parietal epithelium looks fully normal (two small arrows). (B) The GBM is highlighted in yellow, capillary lumens in green. The cell bodies of podocytes are attenuated (arrows) and pseudocysts (asterisks) have developed underneath them. Note that the foot process pattern is widely preserved including the floors of pseudocysts and, second, the parietal epithelium looks fully normal. Male rats, growth stimulation with FGF-2 for 13 weeks. (A) LM from 1 μm Epon section. Bar: 15 μm. (B) TEM. Bar: 5 μm.
Results
Essential stereological results of the previous paper (10) are summarized as follows. Prolonged daily treatment of rats with FGF-2 for 8 weeks led to increasing albuminuria (0.30 ± 0.15 versus 141.5 ± 132.4 mg/18 h) in males and (0.11 ± 0.02 versus 79.8 ± 150.3 mg/18 h) in females; the respective numbers after 13 weeks read 0.21 ± 0.09 versus 303.4 ± 78.4 and 0.13 ± 0.03 versus 85.1 ± 31.5. Serum creatinine increased significantly, indicating the development of chronic renal failure. No increase in body weight or in kidney weight compared with controls was found. However, a prominent significant increase in glomerular tuft volume (1651 ± 209 versus 3035 ± 596 μm3 × 103 in males, 1138 ± 85 versus 1998 ± 392 μm3 × 103 in females), and a numerical increase in mesangial and endothelial cells but no increase in podocyte number were found, resulting in a dramatic decrease in podocyte density (N podo/μm2 × 103), more pronounced in males (0.86 ± 0.007 versus 0.48 ± 0.07) than females (1.05 ± 0.09 versus 0.67 ± 0.14).
The discrepant growth of glomeruli with podocytes growing by hypertrophy only, resulted in structural changes that terminated in the detachment of podocytes from the GBM. The present study focused on the structural details of how podocytes reinforce attachment to the GBM, undergo detachment from the GBM and on the fate of detached podocytes.
The first derangements of podocytes consisted of cell body attenuation and pseudocyst formation (Figure 1B), which, as shown previously in another model (12), were likely causally linked. Growing and expanding capillaries forced podocytes to cover increased surfaces, leading to stretching and broadening of podocytes’ cell bodies. This, in turn, resulted in narrowing of the outflow clefts from the subpodocyte spaces, causing the focal bulging of the attenuated cytoplasmic sheets, usually called pseudocysts. Of note, at this stage, podocytes generally showed a normal FP pattern that also covered the floor of pseudocysts (Figure 1B).
The structural changes that finally led to the detachment of podocytes clearly started after this stage at the FPs (not necessarily as a consequence). The description is complicated by the fact that clear signs of detachment are mixed up with structural responses of podocytes taken to counteract detachment, which are generally subsumed under the term “FPE” (5).
Let us first consider the changes that may be suggested to be protective, i.e., to counteract detachment. FPs broadened making the filtration slits narrower; finally, the slit diaphragms were replaced by occluding junctions, thus sealing the filtration slits (Figure 2A). This was associated with a retraction of FPs into short, irregularly shaped cell projections (Figure 2B). Further on, the development led to the completion of FPE, changing podocyte processes into broad, flattened, disk-like projections that finally fused with the cell bodies resulting in podocytes adhering to the GBM along their total basal aspect. Thus, podocytes had become largely separated from each other, meaning that individual podocytes covered a clearly delimited capillary area, frequently the total circumference of one capillary profile (Figure 2C). Concomitantly, in the basal parts of these podocytes, a microfilament-based cytoskeletal mat developed (13) that has been interpreted as a way to achieve stronger adherence of podocytes to the GBM (5). This appearance may be regarded to underlie the successful prevention of podocyte detachment.
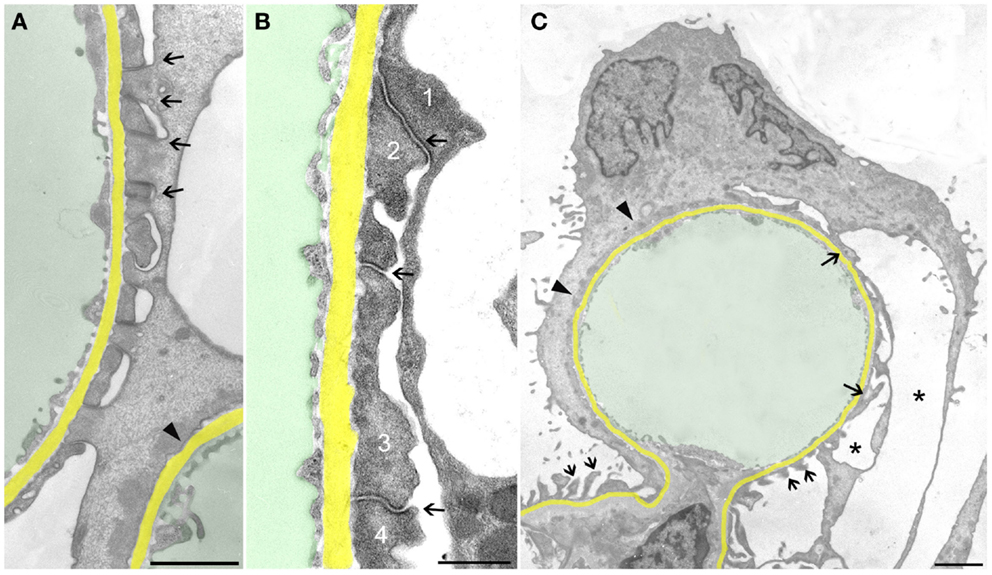
Figure 2. Sealing of the slits may proceed to complete foot process effacement (FPE). The GBM is highlighted in yellow, capillary lumens in green. (A) Early stage of FPE with broadening of FPs and sealing of the slits by occluding junctions (some are labeled by arrows). In the right upper corner part of a capillary, profile with completed FPE (arrowhead) is seen. (B) “Foot processes” have changed into broad plaques (1,2,3,4) that closely stick to the GBM. The sealing of the filtration slits by occluding junctions between them is maintained (arrows). (C) Capillary profile covered by a podocyte that is closely attached to the GBM by “effaced” basal cell portions stuffed with a prominent cytoplasmic mat (arrowheads). At the opposite side, broadened FPs (arrows) are undergoing retraction, whereas at the transition to the mesangial region, FPs appear intact (two small arrows). Pseudocyst formation is also seen (asterisks). (A) and (B) male, (C) female rats, growth stimulation with FGF-2 for 13 weeks. TEMs. Bars: (A) 1 μm, (B) 0.5 μm, (C) 2 μm.
Neighboring such developments, even at other aspects of the same podocyte, these protective responses may fail, starting the focal detachment of podocytes at seemingly random sites. The complete detachment of an entire podocyte results from the final coalescence of numerous discrete disconnected areas (Figure 3). The smallest and probably first signs of detachment consist of circumscribed bare areas of GBM encountered in between two FPs of the same podocyte, thus the former interposed FP was missing (Figure 3A). This phenomenon frequently occurs in series and may be derived from the retraction of a series of FPs arising from a common larger process. In such cases, the co-ordinated retraction of adjacent FPs after formation of occluding junctions between them (as described above) seems to have failed.
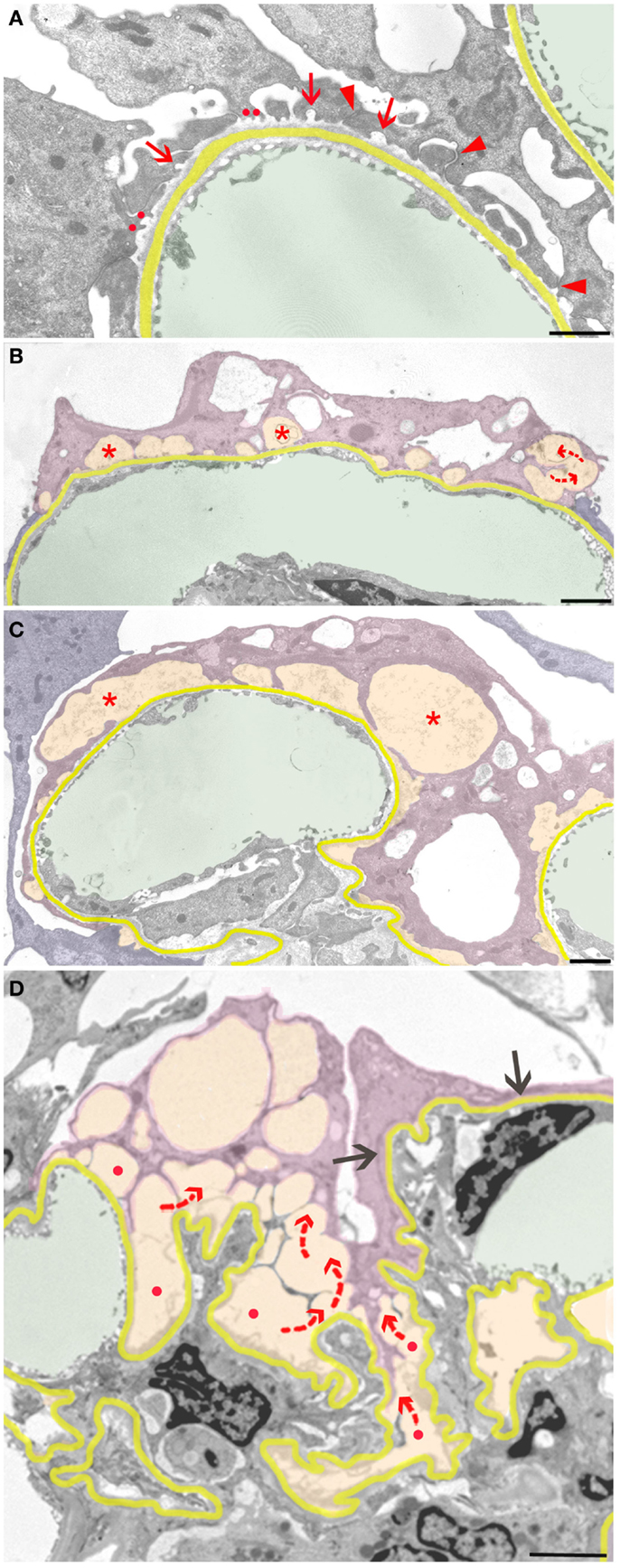
Figure 3. Sequence of podocyte disconnection from the GBM. The GBM is highlighted in yellow, capillary lumens in green, and spaces considered as pseodocysts connected to bare GBM in yellow-orange. Individual podocytes are highlighted in different shades of violet. (A) Early stage of podocyte detachment from the GBM. A sealing of filtration slits by occluding junctions replacing slit membranes was only partially successful (arrowheads). At many other sites, retraction of FPs has left behind circumscribed areas of bare GBM (arrows) that start to merge (red dots). (B) Advanced stage of podocyte detachment from the GBM. The dome-shaped spaces (asterisks) above bare areas of GBM have enlarged bulging toward the urinary space. Merging of these spaces leads to a communicating system of pseudocysts (arrows). (C) The pseudocysts (asterisks) increase in size by merging and bulging; they are related to large stretches of bare GBM and overarched by broadened and attenuated primary processes. (D) The final detachment of a podocyte is frequently characterized by the merging of innumerous interconnected (red arrows) pseudocysts that start over bare GBM (red dots) and drain into Bowman’s space (not seen in this section). Other portions of this podocyte may closely stick to the GBM with completed FPE (black arrows). Male rats, growth stimulation with FGF-2 for 13 weeks. TEMs. Bars: (A) 1 μm, (B) 2 μm, (C) 1 μm, (D) 2.5 μm.
Those areas of circumscribed detachments are frequently overarched by dome-shaped, flattened podocyte processes that are still attached to the GBM (Figure 3B). Thus, a new kind of small pseudocyst has developed that – in contrast to the above-described large pseudocysts – is underlain by sections of bare GBM. Progressive local detachment leads to the coalescence of these spaces (Figure 3C) and, finally, also to coalescence with large pseudocysts, creating a communicating system of extracellular spaces, through which the filtrate has to pass to reach Bowman’s space (Figure 3D). Podocytes associated with such assemblies of pseudocysts finally undergo detachment.
In general, podocytes detach in groups (Figure 4) forming clusters of cells within Bowman’s space, cells interconnected by occluding junctions. These interconnections are likely rooted in the early response to the danger of detachment, i.e., sealing the slits by replacing the slit diaphragms by occluding junctions. As mentioned above, sites of detachment from the GBM and sites of reinforced attachment including the attachment to adjacent podocytes may be found in the same podocyte early in the disease process. These interconnections were, at least partially, preserved when podocytes finally detached from the GBM as entire cells.
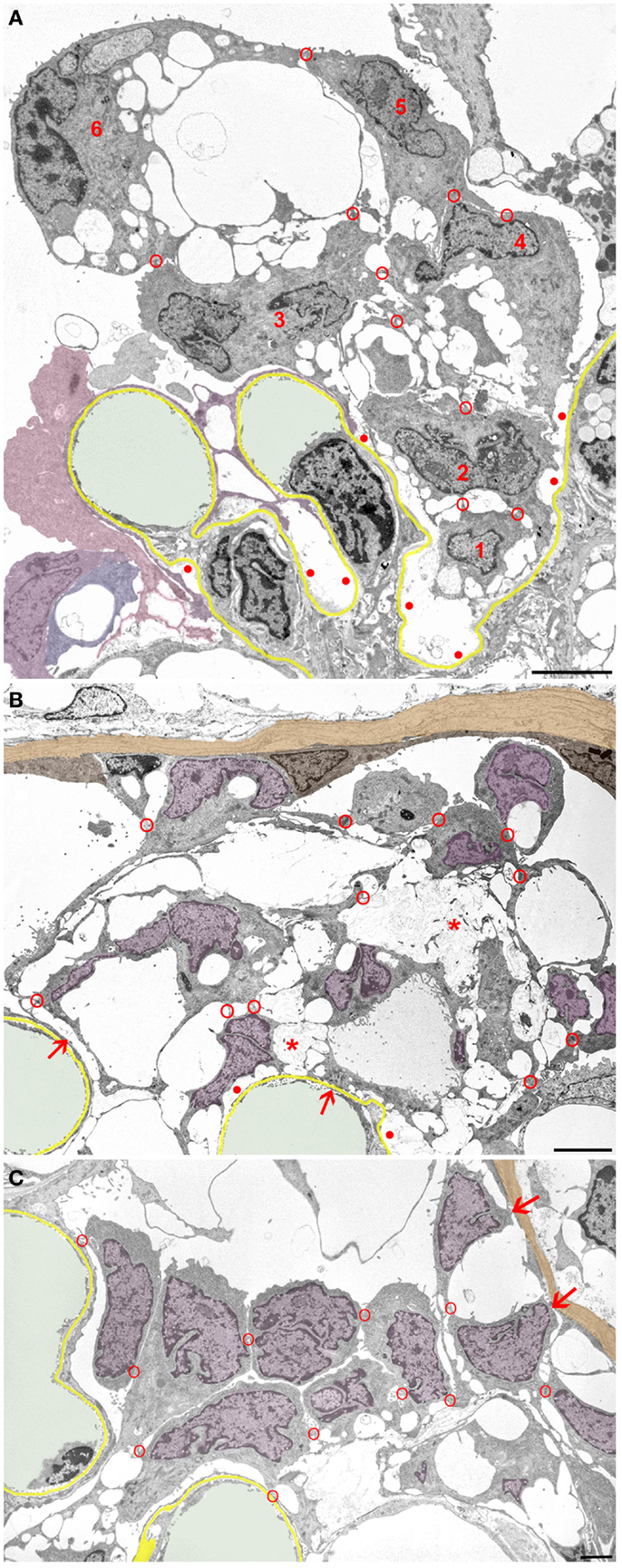
Figure 4. Clusters of detaching podocytes in Bowman’s space. Capillary lumens are highlighted in green, the GBM in yellow. In B and C cell nuclei of podocytes are shown in violet, the PBM in light brown. (A) A group of 6 podocytes in the process of detachment. Only podocytes 1 to 4 still have contact with the GBM, whereas 5 and 6 are attached to the podocytes beneath by junctions (red circles). Within this cluster of podocytes, a communicating system of pseudocysts is seen that starts at bare areas of GBM (red dots). It may readily be suggested that the filtrate entering through bare GBM has to pass these spaces to reach Bowman’s space; openings into Bowman’s space are not hit in this section. Note that additional detaching podocytes (colored in various shades of violet) are gathered in another cluster. (B) Cluster of some 10 detaching or detached podocytes forming a cell bridge between the tuft and Bowman’s capsule. The unlabeled cell within the parietal epithelium cannot be assigned by any plausible criterion to either parietal cells or to podocytes. The innermost cells are still partially attached to the GBM (arrows). The cells are interconnected to each other and still contain abundant pseudocysts that form a continuous chamber system starting from bare GBM (red dots) extending to the outermost layer. Note that the walls of many pseudocysts are undergoing shedding (asterisks). (C) Podocytes after almost complete detachment from the GBM are assembled as a cluster in Bowman’s space interconnected to each other by cell contacts (red circles). The inner row of podocytes shows assemblies of small pseudocysts, the outer row appears as rounded cells. The outermost cells have contacts to Bowman’s capsule (arrows). Male rats, growth stimulation with FGF-2 for 13 weeks. TEMs. Bars: (A) 5 μm, (B) 4 μm, (C) 2 μm.
Initially, those clusters of podocytes are connected to the GBM by the innermost cells (Figure 4A). In later stages, they made contact via the outermost cells with the parietal epithelium (Figures 4B,C and 5), thereby forming multicellular “bridges” between the GBM and the parietal epithelium. During this process, podocytes change in shape. Apparently by shedding of the pseudocysts, i.e., of their thin walls, the detached podocytes turned into groups of rounded cells without any matrix in between (Figures 4 and 5). Mitotic figures were never encountered in these cells.
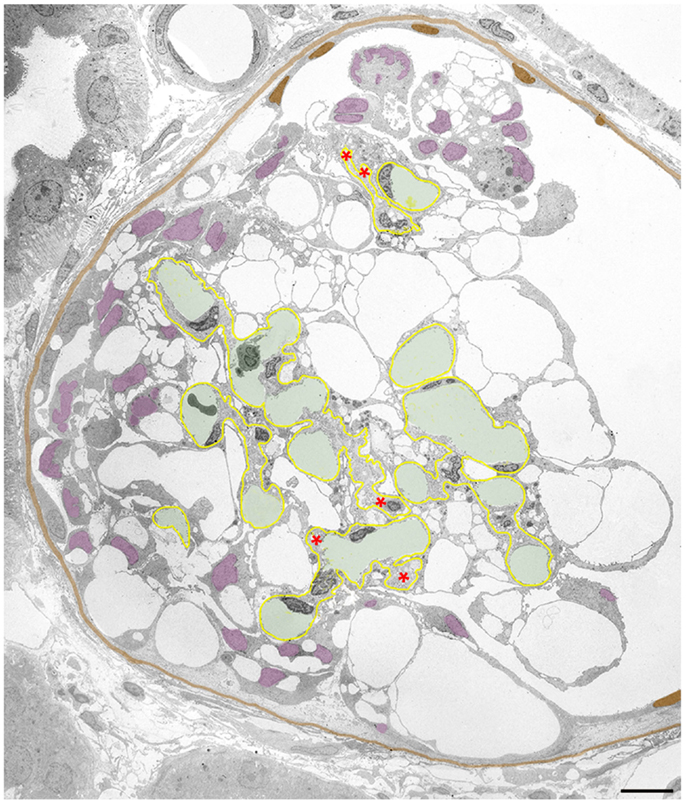
Figure 5. Clustering of podocytes in Bowman’s space is associated with shrinkage of the mesangio-capillary area. The GBM is highlighted in yellow, capillary lumens in green, the PBM and cell nuclei of parietal cells in brown, podocyte cell nuclei in violet. Glomerular profile with a tuft that is broadly connected to Bowman’s capsule by a large cellular crescent that surrounds the tuft from three sides. The mesangio-capillary area (delimited by the GBM) is small compared to the area occupied by podocytes. Note that podocyte cell bodies (visualized by their cell nuclei) have almost completely disappeared from central tuft areas being all contained within the crescent. Most of the podocytes have retained pseudocysts. The cluster of podocytes on top of the tuft does not show any contacts to the tuft nor to the PBM, whereas the major part of the crescent displays extensive contacts to the GBM as well as to Bowman’s capsule. Nuclei of cells, which by no plausible criterion can be assigned as podocytes or parietal cells are uncolored. Note that the GBM is heavily wrinkled at several sites and the mesangial area appears to be collapsed with disappearance of capillaries at several loci (asterisks). Male rat, growth stimulation with FGF-2 for 13 weeks. TEM. Bar: 10 μm.
In even later stages, these bridging clusters of detached podocytes initiate the formation of tuft adhesions to Bowman’s capsule, i.e., firm cell and matrix connections between the GBM and the parietal basement membrane, as described previously (10). Concomitantly, the tuft itself underwent considerable shrinkage, including wrinkling of the GBM, loss of capillaries, and mesangial cells (Figure 5; to be described in detail in another context).
The detachment of podocytes that have come to protrude into the urinary orifice has already been described in various models and interpreted as the consequence of high shear forces within the urinary orifice acting on the cell bodies (5, 14). Here, we found that the final changes accounting for detachment are likely also rooted in the destabilization of FP attachment to the GBM, as described above, and that these podocytes frequently detach in groups and travel in groups along the tubule (not shown).
Quantification of Glomerular Injuries
Crucial steps of the above-described sequence in damage progression were quantified in 1 μm sections. Figure 1A shows an entire glomerular profile indicating the injuries that were counted and compared. As seen in Table 1 and in agreement with the clinical data of the former study (10), the damage was significantly more severe in males than in females.
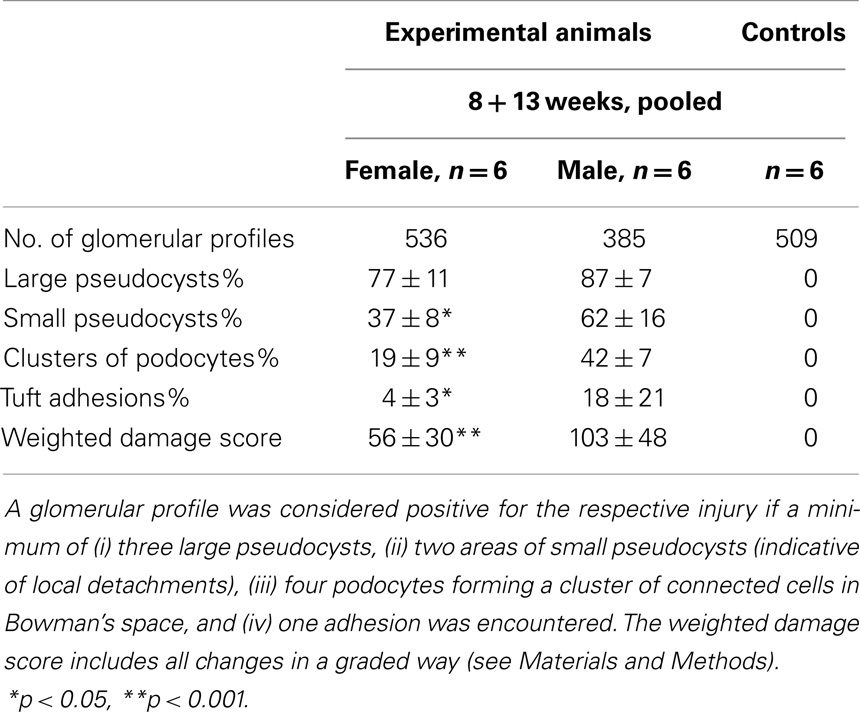
Table 1. Quantification of glomerular injuries.
Discussion
General
It is commonly agreed that the loss of podocytes underlies the development of progressive renal failure. Research of the past 20 years has shown that podocytes are lost by a unique mechanism, i.e., by detachment from the GBM as viable cells and excretion in the urine. This fact, in conjunction with their inability to replicate, puts podocytes in a precarious situation, unlike any other cell (5). Thus, the precise knowledge of why and how podocytes undergo detachment from the GBM is of great importance when searching for strategies to delay the progression to end-stage renal failure.
The present study has addressed this question in a model of excessive glomerular growth initiated by long-term stimulation with FGF-2 (10). Endothelial and mesangial cells proliferated in this condition; podocytes participating solely by hypertrophy could not cope with the overall glomerular growth, underwent structural derangements and, finally, detached from the GBM. This growth-stimulating rat model is similar to a recently published transgenic rat model (15) in which the ability of podocytes for hypertrophy was restricted resulting, after growth stimulation by uninephrectomy, in a mismatch of endothelial and mesangial cell proliferation on the one hand and podocyte hypertrophy on the other, followed by podocyte loss through detachment. Both models mirror what happens in degenerating progressive glomerular diseases including all forms of secondary FSGS.
The Detachment of Podocytes from the GBM
The difficulty in the interpretation of the changes encountered in conjunction with podocyte detachment consists of separating the sequence of changes progressing to detachment from the sequence terminating in complete FPE, thus reinforcing attachment to the GBM. Changes belonging to either of these developments may be found at different sites of the same podocyte. This emphasizes the most important insight of this study: podocyte damage is local, even in the sense that the same podocyte undergoes detachment in one part of the cell, whereas at another part, it successfully reinforced its attachment to the GBM. This fact supports the view that, at least in this model, overall derangements of podocyte cell function are not the crucial causes underlying detachment but local imbalances between the forces accounting for attachment and those for detachment appear to play the major role.
This triad of hypertrophy, hypertension, and hyperfiltration has been convincingly shown to account for the progression of glomerular diseases under a great variety of circumstances (8, 9). These three factors are causally interconnected with each other but it remains an open question whether one of them plays a dominant role. Likely all three are involved in a mutually reinforcing process (16, 17). An elegant study showing in two mouse models of glomerulosclerosis that the development of glomerular injuries was significantly curtailed after unilateral ligation of the ureter emphasizes the effects of changes in filtration dynamics (18), whereas prevention of glomerular growth in a model with established hyperfiltration and glomerular hypertension also resulted in a marked decrease in sclerosis development (19), showing a major impact of growth. Also, in human beings, glomerular hypertrophy was found to be the decisive cofactor in minimal change disease accounting for progression to FSGS (20). The present study suggests that excessive hypertrophy of podocytes may be a major factor in progression to what extent hyperfiltration and glomerular hypertension develop in this model is unknown.
Therefore, damage development in the FGF-2 model may be taken to reflect the combined effects of all three factors.
Let us look in detail to a hypothetical local scenario that may start the process of detachment. A certain podocyte has reached its ultimate limit of hypertrophy that allowed it to cover a correspondingly increased area of GBM with normal FPs. In case the rheological situation with high filtrate flows overtaxes the available slit area, the only protective option remaining consists of sealing the slits by occluding junctions. This represents a most crucial event. The formation of cell–cell junctions between FPs in glomerular disease models has been described previously by several groups, characterizing them as an “occluding” type (21–27). More recent work has shown that they are a certain type of tight junctions (28, 29). Moreover, it was shown that tight junctional proteins increase several-fold in puromycin-induced nephrosis (29).
If sealing the slits is successful, compensation by adjacent podocytes is required, with these being exposed to higher local filtrate flows. If these podocytes have some reserve capacity to hypertrophy, they may adequately manage the situation.
In the case that the sealing strategy fails, a fatal situation will develop. Damage to the slit membrane will increase the shear stress on the involved FPs and may initiate its local detachment, thereby increasing the bare area of GBM. This will lead to local increases in flow (local hyperfiltration) and worsen the unstable situation. Thus, as soon as any detachment has started locally, the final struggle against the detachment of the entire cell has begun. If there is any chance to reverse this situation, it probably consists of spreading of neighboring, uninvolved podocytes on the GBM covering the denuded area. If this maneuver also fails, detachment of the first podocyte will proceed and may include adjacent podocytes.
The process of detachment seems to follow a rather stereotyped structural sequence. Circumscribed bare areas of GBM in between two adherent FPs of the adjacent podocyte (as the result of initial local detachments) frequently form the floors of dome-like cavities overarched by attenuated cytoplasm of broadened primary processes. These spaces may be considered as a second type of pseudocyst associated, in contrast to the large pseudocysts, with bare GBM. They merge with each other, later with the large pseudocysts, finally forming a communicating system of extracellular spaces that starts above scattered areas of bare GBM and opens into Bowman’s space through the narrow openings of the subpodocyte spaces. In many previous studies, this system of empty spaces has been termed “podocyte vacuolation,” considered to consist of intracellular vacuoles (“blebs”) (23, 30–34), indicating a prenecrotic stage. Here, we show that for the most part, these are extracellular spaces bordered by intact cell membranes of viable podocytes.
It may readily be suggested that sites of bare GBM allow increased filtrate flows and are the sites of protein leakage, as frequently suggested and shown previously (22, 35–37). The filtrate has to find a way out of these spaces along routes offering some amount of resistance to the outflow. The resulting expansile forces on the walls of these spaces induce progressive bulging and detachment, leaving behind increasing areas of bare GBM. Consequently, the flow of the filtrate through the system of connected pseudocysts likely exerts expansile forces on the concerned podocyte, tending to lift it off the GBM (Figures 4 and 5).
Even if started locally at a single podocyte, the process of detachment generally comprises a group of podocytes. This may be another explanation for the observation by Ichikawa et al. (38) that damage to a podocyte always damages neighboring podocytes. In our view, the detachment of a podocyte, once started, will often take others along and may be difficult to arrest.
The interpretation of the sealing of filtration slits by “FPE” as a protective measure of podocytes against detachment conflicts with the widely held view that FPE represents the common initial derangement of podocytes leading to proteinuria potentially progressing to more severe glomerular damage (39). As discussed previously (5), FPE is necessarily accompanied by transient protein leakage as a consequence of the motility associated with the extensive local rearrangements. The transient character of this kind of proteinuria has recently been found in studies after Rac-1 stimulation (40–42) that is known to elicit FPE (39, 43). Progression of the damage to FSGS has not been found in these studies supporting our view of a protective impact.
The Fate of Detached Podocytes
In static structural studies, it is impossible to follow individual podocytes as they are translocated from one site to another. However, we can observe that along the course of the disease, podocytes are regularly encountered at other sites than where they are found under normal conditions. From such comparisons, we may deduce the routes how they came from site A to site B. We do not think that such movements of podocytes are effected by any active motility of the podocytes themselves, We think that detachment from the GBM exposes them to the rheological forces of filtrate flow pushing them toward Bowman’s capsule and the urinary orifice.
The fraction of podocytes that detach directly in front of the urinary orifice generally will be swept into the tubular lumen (5, 10, 14). Surprisingly, during their passage along the tubule, they did not undergo anoikosis but, as known from several studies (e.g., 3 and 4), may have reached the renal pelvis as living cells.
Podocytes detaching from most other sites of the tuft may form massive cell accumulations within Bowman’s space interconnected by occluding junctions with the innermost cells keeping contact to the GBM. During this gradual process of detachment, podocytes change in shape; they shed all the peripheral material forming the thin walls of pseudocysts, finally appearing as rounded cells. Frequently, such cell clusters establish, with their outermost cells, contacts to parietal epithelial cells, thus forming purely cellular crescents connecting the tuft and Bowman’s capsule.
These observations are in full agreement with recent in vivo studies by multiphoton microscopy in two other models with podocyte detachment, i.e., unilateral ureter ligation (UUO) and Adriamycin nephropathy (44). In these studies, the formation of multicellular clusters of podocytes in Bowman’s space was observed that made contact to Bowman’s capsule; also, podocytes in single file moving down the tubule were seen. The authors also observed that the podocyte clusters made contacts to parietal cells by cell projections and nano-tubes. We found by TEM abundant occluding junctions between podocytes as well as between podocytes and parietal epithelial cells suggesting that they are responsible for the mechanical cohesion. As also verified in serial sections, the outermost cells of the podocyte clusters are fully separated from the GBM but fixed to inner cells of the cluster by occluding junctions. It appears that as soon as they come into contact with parietal cells, they establish junctional contacts to parietal cells, as well.
The further evolution of such clusters of detached podocytes attaching to Bowman’s capsule, finally turning into tuft adhesions and crescents has been shown and discussed previously (10, 45, 46).
Relationships to pathologies seen in human renal diseases
It is quite obvious that the clustering of podocytes within Bowman’s space and the shrinkage and collapse of the mesangio-capillary area have equivalents in human renal diseases, known as “cellular crescent,” “cellular lesion,” and “collapsing glomerulopathy” (CGP). The origin of the abnormal cells in these pathologies is under debate (47–50). In the present degenerative animal model, the cells clustering on the outer aspect of the tuft within Bowman’s space are clearly derived from podocytes and do not proliferate. CGP combines the collapse of the mesangio-capillary compartment with hypertrophy and seemingly hyperplasia of the cells that take the place of podocytes on the outside of the GBM, assembled in clusters looking like crescents (51–54). The shrinkage of the mesangio-capillary area with wrinkling of the GBM and collapse of capillaries in CGP look identical to what is seen in the present animal model. Also, the clustering of cells in Bowman’s space in CGP may look very similar to the clustering of detached podocytes in our animal model.
The great number of such cells seen in a cross-sectional profile of CGP seems incompatible with detachment as the only mechanism. However, as far as we know, the total number of podocytes on the surface of a collapsed tuft in CGP has never been estimated by stereological methods. Though a fully reliable calculation is presently not possible, we tried the following approach applying the recently published method by Venkatareddy et al. (55) to estimate podocyte number from a single glomerular profile in a single section. Since we do not have own biopsy material from a case of CGP, we took the glomerulus shown in Figure 3 from the paper by Jhaveri et al. (56) and made the following calculation. In this glomerular profile, we counted 90 podocyte cell nuclei. From the listed magnification, the tuft diameter is about 100 μm (healthy tufts are usually in the range 180–220 μm, glomerular tufts are shrunken in CGP). The cross-sectional area is, therefore, 7854 μm2 and the area density of nuclei is 90/7854 = 0.01146. Using the estimated caliper diameter for human podocyte nuclei of 8.2 μm (given in 55), we obtain the volumetric podocyte density of 0.01146/8.2 = 0.001397 podocytes/μm3. The 100 μm tuft diameter yields an estimated tuft volume of 523,585 μm3 (much smaller than a typical tuft volume of 3,000,000 μm3). Multiplying the density by the tuft volume gives an estimate of 731 podocytes in the tuft. To the degree that any podocytes are binucleate, this will be an overestimate of cell number.
Thus, it seems possible that the seemingly great number of podocytes on a shrunken tuft in CGP is all derived from detachment, gathering on the shrunken surface without any cell proliferation. This is supported by the fact that mitotic figures are generally not found in these cell assemblies in CGP. Major support in favor of detachment comes from the work of Bariety et al. (57) and also from findings in a transgenic mouse model of HIV-associated nephropathy (58).
Conclusion
Loss of podocytes represents the central event underlying the glomerular disease progression (4, 46). Here, in a model of continuous growth stimulation by FGF-2, we show that loss of podocytes occurs by detachment from the GBM as viable cells. This is a gradual process that starts focally at randomly distributed sites. The local origin strongly supports the view that the forces starting detachment consists of local rheological disturbances. The detachment of a podocyte, once started, may be difficult to arrest. However, podocytes have developed protective mechanisms against detachment, most important the sealing of the filtration slits followed by FPE.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Kevin V. Lemley for extremely useful help, discussion, and insights. We gratefully acknowledge the continuous support of our work by the “Gotthard Schettler Gesellschaft fuer Herz-und Kreislaufforschung.”
Abbreviations
CGP, collapsing glomerulopathy; FP, foot process; FPE, foot process effacement; FSGS, focal segmental glomerulosclerosis; GBM, glomerular basement membrane; PBM, parietal basement membrane.
References
1. Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest (1997) 99:342–8. doi: 10.1172/JCI119163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. Podocyte depletion causes glomerulosclerosis: diphteria toxin-induced podocyte depletion in rats expressing human diphteria toxin receptor transgene. J Am Soc Nephrol (2005) 16:2941–52. doi:10.1681/ASN.2005010055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Vogelmann SU, Nelson WJ, Myers BD, Lemley KV. Urinary excretion of viable podocytes in health and renal disease. Am J Physiol Renal Physiol (2003) 285:F40–8. doi:10.1152/ajprenal.00404.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Wickman L, Afshinnia F, Wang SQ, Yang Y, Wang F, Chowdhury M, et al. Urine podocyte mRNAs, proteinuria, and progression in human glomerular diseases. J Am Soc Nephrol (2013) 24:2081–95. doi:10.1681/ASN.2013020173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Kriz W, Shirato I, Nagata M, Le Hir M, Lemley KV. The podocyte’s response to stress: the enigma of foot process effacement. Am J Physiol Renal Physiol (2013) 304:F333–47. doi:10.1152/ajprenal.00478.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol (2014). doi:10.1681/ASN.2014030278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Endlich N, Endlich KH. The challenge and response of podocytes to glomerular hypertension. Semin Nephrol (2012) 32:327–41. doi:10.1016/j.semnephrol.2012.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Hostetter TH, Olson JL, Rennke HG, Venkatachalam MA, Brenner BM. Hyperfiltration in remnant nephrons: a potentially adverse response to renal ablation. Am J Physiol (1981) 241:F85–93.
9. Brenner BM, Lawler EV, Mackenzie HS. The hyperfiltration theory: a paradigma shift in nephrology. Kidney Int (1996) 49:1774–7. doi:10.1038/ki.1996.265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Kriz W, Hähnel B, Rösener S, Elger M. Long-term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int (1995) 48:1435–50. doi:10.1038/ki.1995.433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Sakai T, Kriz W. The structural relationship between mesangial cells and basement membrane of the renal glomerulus. Anat Embryol (1987) 176:373–86. doi:10.1007/BF00310191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Nagata M, Kriz W. Glomerular damage after uninephrectomy in young rats. II. Mechanical stress on podocytes as a pathway to sclerosis. Kidney Int (1992) 42:148–60. doi:10.1038/ki.1992.272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Shirato I, Sakai T, Kimura K, Tomino Y, Kriz W. Cytoskeletal changes in podocytes associated with foot process effacement in Masugi nephritis. Am J Pathol (1996) 148:1283–96.
14. Friedrich C, Endlich N, Kriz W, Endlich KH. Podocytes are sensitive to fluid shear stress in vitro. Am J Physiol Renal Physiol (2006) 291:F856–65. doi:10.1152/ajprenal.00196.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Fukuda A, Chowdhury MA, Venkatareddy MP, Wang SQ, Nishizono R, Suzuki T, et al. Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol (2012) 23:1351–63. doi:10.1681/ASN.2012030271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Hostetter TH. Progression of renal disease and renal hypertrophy. Annu Rev Physiol (1995) 57:263–78. doi:10.1146/annurev.ph.57.030195.001403
17. Hostetter TH. Hypertrophy and hyperfunction of the diabetic kidney. J Clin Invest (2001) 107:161–2. doi:10.1172/JCI12066
18. Matsusaka T, Kobayashi K, Kon V, Pastan I, Fogo AB, Ichikawa I. Glomerular sclerosis is prevented during urinary tract obstruction due to podocyte protection. Am J Physiol Renal Physiol (2011) 300:F792–800. doi:10.1152/ajprenal.00570.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Yoshida Y, Fogo AB, Ichikawa I. Glomerular hemodynamic changes vs. hypertrophy in experimental glomerular sclerosis. Kidney Int (1989) 35:654–60. doi:10.1038/ki.1989.35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Fogo AB, Hawkins EP, Berry PL, Glick AD, Chiang ML, MacDonell RC Jr, et al. Glomerular hypertrophy in minimal change disease predicts subsequent progression to focal glomerular sclerosis. Kidney Int (1990) 38:115–23. doi:10.1038/ki.1990.175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Seiler MW, Rennke HG, Venkatachalam MA, Cotran RS. Pathogenesis of polycation-induced alterations (“fusion”) of glomerular epithelium. Lab Invest (1977) 36:48–62.
22. Venkatachalam MA, Karnovsky MJ, Cotran RS. Glomerular permeability – ultrastructural studies in experimental nephrosis using horseradish peroxidase as a tracer. J Exp Med (1969) 130:381–99. doi:10.1084/jem.130.2.381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Caulfield JP, Reid JJ, Farquhar MG. Alterations of the glomerular epithelium in acute aminonucleoside nephrosis. Evidence for formation of occluding junctions and epithelial cell detachment. Lab Invest (1976) 34:43–59.
24. Kerjaschki D. Polycation-induced dislocation of slit diaphragms and formation of cell junctions in rat kidney glomeruli. The effects of low temperature, divalent cations, colchicine, and cytochalasin B. Lab Invest (1978) 39:430–40.
25. Pricam C, Humbert F, Perrelet A, Amherdt M, Orci L. Intercellular junctions in podocytes of the nephrotic glomerulus as seen with freeze-fracture. Lab Invest (1975) 33:209–18.
26. Inokuchi S, Sakai T, Shirato I, Tomino Y, Koide H. Ultrastructural changes in glomerular epithelial cells in acute puromycin aminonucleoside nephrosis: a study by high-resolution scanning electron microscopy. Virchows Arch A Pathol Anat Histopathol (1993) 423:111–9. doi:10.1007/BF01606585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Kurihara H, Anderson JM, Kerjaschki D, Farquhar MG. The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am J Pathol (1992) 141:805–15.
28. Shono A, Tsukaguchi H, Yaoita E, Nameta M, Kurihara H, Qin XS, et al. Podocin participates in the assembly of tight junctions between foot processes in nephrotic podocytes. J Am Soc Nephrol (2007) 18:2525–33. doi:10.1681/ASN.2006101084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Fukasawa H, Bornheimer S, Kudlicka K, Farquhar MG. Slit diaphragms contain tight junction proteins. J Am Soc Nephrol (2009) 20:1491–503. doi:10.1681/ASN.2008101117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Wheeler J, Robertson H, Morley AR, Appleton DR. Anti-glomerular basement membrane glomerulonephritis (anti-GBM GN) in the mouse: BrdU-labelling indices and histological damage. Int J Exp Pathol (1993) 74:9–19.
31. Yoshikawa N, Hiroshi I, Akamatsu R, Hazikano H, Okada S, Matsuo T. Glomerular podocyte vacuolation in focal segmental glomerulosclerosis. Arch Pathol Lab Med (1986) 110:394–8.
32. Aizawa K, Takeda S, Tashiro Y, Yorozu K, Hirata M, Kanada H, et al. Renoprotection by continuous erythropoietin receptor activator in puromycin aminonucleoside-induced nephrotic syndrome. Am J Nephrol (2013) 36:419–26. doi:10.1159/000343493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Toth T, Takebayashi S. Glomerular podocyte vacuolation in idiopathic membranous glomerulonephritis. Nephron (1992) 61:16–20. doi:10.1159/000186828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Yamazaki T. Podocytic degeneration and regeneration in puromycin aminonucleoside nephropathy in the rat. Pathol Int (1995) 45:465–7. doi:10.1111/j.1440-1827.1995.tb03487.x
35. Whiteside C, Prutis K, Cameron R, Thompson J. Glomerular epithelial detachment, not reduced charge density, correlates with proteinuria in adriamycin and puromycin nephrosis. Lab Invest (1989) 61:650–60.
36. Fries JW, Sandstrom DJ, Meyer TW, Rennke HG. Glomerular hypertrophy and epithelial cell injury modulate progressive glomerulosclerosis in the rat. Lab Invest (1989) 60:205–18.
37. Olson JL, Hostetter TH, Rennke HG, Brenner BM, Venkatachalam MA. Altered glomerular permselectivity and progressive sclerosis following extreme ablation of renal mass. Kidney Int (1982) 22:112–26. doi:10.1038/ki.1982.143
38. Ichikawa I, Ma J, Motojima M, Matsusaka T. Podocyte damage damages podocytes: autonomous vicious cycle that drives local spread of glomerular sclerosis. Curr Opin Nephrol Hypertens (2005) 14:205–10.
39. Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol (2012) 74:299–323. doi:10.1146/annurev-physiol-020911-153238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int (2013) 84:920–30. doi:10.1038/ki.2013.175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Yu H, Suleiman H, Kim AH, Miner JH, Dani A, Shaw AS, et al. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol Cell Biol (2013) 33:4755–64. doi:10.1128/MCB.00730-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Asao R, Asanuma K, Kodama F, Takagi M, Hosoe Y, Tanaka E, et al. Rac1 in podocytes protects glomeruli against the formation of sclerosis. J Am Soc Nephrol FR-PO851. (2013).
43. Schaldecker T, Kim S, Tarabanis C, Tian D, Hakroush S, Castonguay P, et al. Inhibition of the TRPC5 ion channel protects the kidney filter. J Clin Invest (2013) 123:5298–309. doi:10.1172/JCI71165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Hackl M, Burford JL, Villanueva K, Lam L, Susztak K, Schermer B, et al. Tracking the fate of glomerular epithelial cells in vivo using serial multiphoton imaging in new mouse models with fluorescent lineage tags. Nat Med (2013) 19:1661–6. doi:10.1038/nm.3405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Le Hir M, Keller C, Eschmann V, Hähnel B, Hosser H, Kriz W. Podocyte bridges between the tuft and Bowman’s capsule: an early event in experimental crescentic glomerulonephritis. J Am Soc Nephrol (2001) 12:2060–71.
46. Kriz W, Le Hir M. Pathways to nephron loss starting from glomerular diseases – insights from animal models. Kidney Int (2005) 67:404–19. doi:10.1111/j.1523-1755.2005.67097.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Schwartz MM, Lewis EJ. Focal segmental glomerular sclerosis: the cellular lesion. Kidney Int (1985) 28:968–74. doi:10.1038/ki.1985.225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Schwartz MM, Evans J, Bain RP, Korbet SM. Focal segmental glomerulosclerosis: prognostic implications of the cellular lesion. J Am Soc Nephrol (1999) 10:1900–7.
49. Wang S, Kim JH, Moon KC, Hong HK, Lee HS. Cell-cycle mechanisms involved in podocyte proliferation in cellular lesion of focal segmental glomerulosclerosis. Am J Kidney Dis (2004) 43:19–27. doi:10.1053/j.ajkd.2003.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Taneda S, Honda K, Uchida K, Nitta K, Yumura W, Oda H, et al. Histological heterogeneity of glomerular segmental lesions in focal segmental glomerulosclerosis. Int Urol Nephrol (2012) 44:183–96. doi:10.1007/s11255-011-9932-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Barisoni L, Schnaper HW. A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol (2007) 2:529–42. doi:10.2215/CJN.04121206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Stokes MB, Valeri AM, Markowitz GS, d’Agati VD. Cellular focal segmental glomerulosclerosis: clinical and pathologic features. Kidney Int (2006) 70:1783–92. doi:10.1038/sj.ki.5001903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Laurinavicius A, Rennke HG. Collapsing glomerulopathy – a new pattern of renal injury. Semin Diagn Pathol (2002) 19:106–15.
54. Markowitz GS, Appel GB, Fine PL, Fenves AZ, Loon NR, Jagannath S, et al. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J Am Soc Nephrol (2001) 12:1164–72.
55. Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, et al. Estimating podocyte number and density using a single histologic section. J Am Soc Nephrol (2014) 25:1118–29. doi:10.1681/ASN.2013080859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Jhaveri KD, Shah HH, Calderon K, Campenot ES, Radhakrishnan J. Glomerular diseases seen with cancer and chemotherapy: a narrative review. Kidney Int (2013) 84:34–44. doi:10.1038/ki.2012.484
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Bariety J, Nochy D, Mandet C, Jacquot C, Glotz D, Meyrier A. Podocytes undergo phenotypic changes and express macrophagic-associated markers in idiopathic collapsing glomerulopathy. Kidney Int (1998) 53:918–25. doi:10.1111/j.1523-1755.1998.00845.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Barisoni L, Bruggeman LA, Mundel P, d’Agati VD, Klotman PE. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int (2000) 58:173–81. doi:10.1046/j.1523-1755.2000.00152.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: podocyte hypertrophy, foot process effacement, sealing of filtration slits, pseudocysts, podocyte detachment, crescents
Citation: Kriz W, Hähnel B, Hosser H, Rösener S and Waldherr R (2014) Structural analysis of how podocytes detach from the glomerular basement membrane under hypertrophic stress. Front. Endocrinol. 5:207. doi: 10.3389/fendo.2014.00207
Received: 17 September 2014; Paper pending published: 06 October 2014;
Accepted: 17 November 2014; Published online: 12 December 2014.
Edited by:
Barbara Lewko, Medical University of Gdansk, PolandReviewed by:
Kimberly Jean Reidy, Albert Einstein College of Medicine, USABarbara Lewko, Medical University of Gdansk, Poland
Copyright: © 2014 Kriz, Hähnel, Hosser, Rösener and Waldherr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wilhelm Kriz, Zentrum für Biomedizin und Medizintechnik Mannheim, Institut für Neuroanatomie, Ludolf-Krehl-Str. 13-17, Tridomus C, Ebene 6, Mannheim D68167, Germany e-mail: wilhelm.kriz@urz.uni-heidelberg.de