 Minwoo Nam
Minwoo Nam Marcus P. Cooper
Marcus P. Cooper- Division of Cardiovascular Medicine, Department of Medicine, University of Massachusetts Medical School, Worcester, MA, USA
In murine and human brown adipose tissue (BAT), mitochondria are powerful generators of heat that safely metabolize fat, a feature that has great promise in the fight against obesity and diabetes. Recent studies suggest that the actions of mitochondria extend beyond their conventional role as generators of heat. There is mounting evidence that impaired mitochondrial respiratory capacity is accompanied by attenuated expression of Ucp1 and other BAT-selective genes, implying that mitochondria exert transcriptional control over the brown fat gene program. In this review, we discuss the current understanding of brown fat mitochondria, their potential role in transcriptional control of the brown fat gene program, and potential strategies to treat obesity in humans by leveraging thermogenesis in brown adipocytes.
Introduction
Brown fat is composed of thermogenic adipocytes that convert chemical energy to heat. Found in homeothermic animals such as mammals, brown fat protects against cold stress. In neonates, brown fat plays a critical role in thermoregulation. The thermogenic capacity of brown fat is attributed to abundant mitochondria, which dissipates heat via uncoupling (1).
There are two types of thermogenic adipocytes: classical brown adipocytes and beige/brite adipocytes. Classical brown adipocytes exhibit constitutive thermogenic capacity with large numbers of mitochondria. Developmentally programed brown fat, such as the rodent interscapular depot, consists of these cells. Another type of thermogenic adipocytes is found in white fat. These cells are recruited by thermogenic stimuli and in turn display comparable thermogenic capacity to classical brown adipocytes. Due to these properties, they have been named beige/brite (brown-in-white) adipocytes (1, 2).
Brown fat has unique genetic signatures that support its thermogenic function. Uncoupling protein 1 (Ucp1), a key thermogenic protein, is highly expressed in brown fat. Cell death-inducing DFFA-like effector (Cidea), a modulator of UCP1, and Type II iodothyronine deiodinase (Dio2), an enzyme that converts T4 to active T3 within the tissue locally, are among major thermogenic genes (3–5). Fatty acid oxidation (FAO) genes and electron transport chain (ETC) subunit genes are also enriched (6). This transcriptional signature is in part determined by PR domain containing 16 (Prdm16), a determinant of brown fat, and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a), a key coactivator of peroxisome proliferator-activated receptor alpha (Ppara) and gamma (Pparg) (7, 8). The transcriptional network of the brown fat genes have been reviewed elsewhere (9). Since many of the brown fat-selective genes regulate thermogenesis, hereafter “the thermogenic gene program” and “the brown fat gene program” are reciprocally used.
Mitochondria play a central role in providing an energetic basis for thermogenesis in brown fat. Brown fat mitochondria fulfill their duty as cellular powerhouses in the non-stimulated state. In sympathetically stimulated brown fat, UCP1 is rapidly activated and uncouples electron transit and ATP production. Instead, the chemical energy stored in the proton gradient is dissipated as heat (10). Interestingly, several studies have demonstrated that impaired mitochondrial respiratory capacity is accompanied by attenuated expression of Ucp1 and other brown adipose tissue (BAT)-selective genes in classical brown adipocytes (11, 12). In addition to a conventional role as generators of heat, this suggests that brown fat mitochondria have an unappreciated role in regulating genes involved in thermogenesis depending on the status of respiratory capacity. Critical evaluation of these studies will broaden our thoughts on mitochondria in brown fat. This review summarizes the features of brown fat mitochondria and discusses their potential role in transcriptional control of the brown fat gene program and its therapeutic implications in humans.
Features of Brown Fat Mitochondria
Morphological Features
Brown fat mitochondria are dynamic organelles that meet the thermogenic needs of the organism by regulating their number and networking as well as their biochemical and ultrastructural profile. Acute cold exposure (or activation with norepinephrine) immediately promotes mitochondrial fission, an event that precedes and augments thermogenesis (13). Brown fat mitochondria exhibit unique morphological features. Notably, brown fat mitochondria are enlarged and densely packed with respiratory units, resulting in dense cristae (14). The morphological features of brown fat also differ across gender with females having larger mitochondria and denser cristae (15). The density of brown fat mitochondria is among the highest of any tissue (16). Even so, chronic cold exposure increases mitochondrial mass even further. This is mediated by catecholamines via β-adrenergic signaling, which increases PGC-1α, a transcriptional coactivator that induces ERRα and NRF-1, culminating in increased mitochondrial mass (17–19).
Biochemical Features
Early biochemical and functional studies on brown fat mitochondria in rodents revealed high cellular respiration but low ATP synthase activity (20). This implied that pathway involving proton leakage must underlie the basis of thermogenesis. Biochemical studies identified that UCP1 constitutes the molecular basis for enhanced proton leak (21). Interestingly, in larger mammals such as lambs the abundance of ATP synthase is higher (22), presumably because larger animals are less dependent on non-shivering thermogenesis due to their smaller surface-to-volume ratio and due to their increased capacity for shivering.
Long-chain free fatty acids (LCFAs) activate UCP1, while purine nucleotides inhibit UCP1 (10). Intuitively, mobilization of free fatty acids via β-adrenergic activation is coupled to UCP1 activation and heat production. Fedorenko et al. (23) reported that UCP1 does not exhibit constitutive proton transport activity. Instead, there is obligatory binding of LCFAs to UCP1, a process that transfers protons associated with LCFA into the matrix via a conformational change of UCP1. LCFAs can also overcome inhibition by purine nucleotides. This is important because in the basal state, there is no lipolysis and inhibition of UCP1 by purine nucleotides will promote coupled ATP-generating respiration.
In theory, uncoupling should de-energize brown fat mitochondria, culminating in an ATP crisis. ATP, however, is required for the activation of fatty acids during uncoupling. Brown fat mitochondria circumvent this by increasing glycolysis as well as the TCA cycle. Arsenite, an inhibitor of pyruvate dehydrogenase complex (PDC) and α-ketoglutarate dehydrogenase, depleted ATP in norepinephrine-stimulated brown adipocytes, implying that the TCA cycle is critical for maintaining ATP during thermogenesis (24). Although succinyl-CoA synthetase primarily generates GTP, this TCA enzyme complex can also generate ATP, a process which may explain how the TCA cycle is critical for maintaining ATP in brown fat (discussed further in Proteomical Features). Glycolysis is an important source of ATP, too. Notably, hexokinase activity increases fourfold in cold acclimated rats, achieving glycolytic activity similar to liver (25).
Proteomical Features
Mass spectrometric analysis of brown fat mitochondria has revealed striking proteomic difference compared with white fat mitochondria (26). In fact, the proteomic profile of brown fat mitochondria was most similar to that of skeletal muscle. Compared with white fat mitochondria, there was an enrichment of catabolic pathways including ETC, TCA cycle, and fatty acid metabolism in brown fat mitochondria. Complexes I–IV are present at higher levels, whereas complex V is present at lower levels, a pattern favorable for thermogenesis. There is robust expression of enzymes involved in the TCA cycle–ADP-forming succinyl-CoA synthetase β subunit (A-SCS-β), pyruvate dehydrogenase kinase 4 (PDK4), and pyruvate dehydrogenase phosphatase regulatory subunit (PDPr). SCS converts succinyl-CoA to succinate. This reaction is coupled to the formation of ATP or GTP, which is determined by two different β subunits, ADP-forming and GDP-forming. In mouse, rat, and human, metabolically active tissues, such as brain and heart, express high levels of ADP-forming subunits compared with GDP-forming subunits (27). Likewise, brown fat mitochondria may preferentially use A-SCS-β to supply ATP, a feature that matches a role of substrate-level phosphorylation in stimulated brown adipocytes. During cold exposure, lipid uptake and lipogenesis replenish fat stores that have been oxidized (28). Control of lipogenesis during cold exposure is complex and partly regulated by pyruvate metabolism (29, 30). Pyruvate can be targeted for complete oxidation by converting it into acetyl-CoA via the enzymatic action of PDC (31). Alternatively, inhibition of PDC by PDK4 diverts pyruvate into glycerol, which is the backbone for free fatty acid (FFA) esterification (31). An enzymatic complex consisting of PDPr counteracts the action of PDK4, and thus, targets pyruvate for complete oxidation (32, 33). In summary, opposing regulation by PDK4 and PDP may play a critical role in whether or not the brown adipocyte uses pyruvate for lipogenesis (PDK4-mediated) or complete oxidation (PDPr-mediated).
Fatty acids serve as major substrates for thermogenesis and they activate UCP1 (10). In brown fat, there is a high expression of enzymes involved in FAO, including short-, medium-, long-chain acyl-CoA dehydrogenases and 3-ketoacyl-CoA thiolase (26). Long-chain fatty acids require a carnitine palmitoyltransferase 1B (CPT1B)-mediated carnitine shuttle for oxidation in mitochondria. Supporting a role for robust oxidative capacity in brown fat, CPT1B is 50-fold higher in brown fat compared with white fat (26). Brown fat may also exhibit metabolic flexibility in fuel utilization. Highly expressed in brown fat, acetyl-CoA synthetase 2-like (ACSS1) permits oxidation of ketone bodies during starvation (26, 34, 35). Indeed, activity of ketone body oxidizing enzymes in brown fat parallels that of the heart (36).
Role of Energy Metabolism in the Brown Fat Gene Program
Although manipulating regulators of mitochondrial biogenesis, such as PGC-1 coactivators and ERRα, affects Ucp1 expression, those factors are also known to directly regulate transcription of respiratory subunits (37), making it challenging to delineate a direct relationship between respiratory capacity and the thermogenic gene program per se. Here, we discuss approaches to directly manipulate mitochondrial respiratory capacity and the attendant effects on the brown fat genes, which are summarized in Table 1.
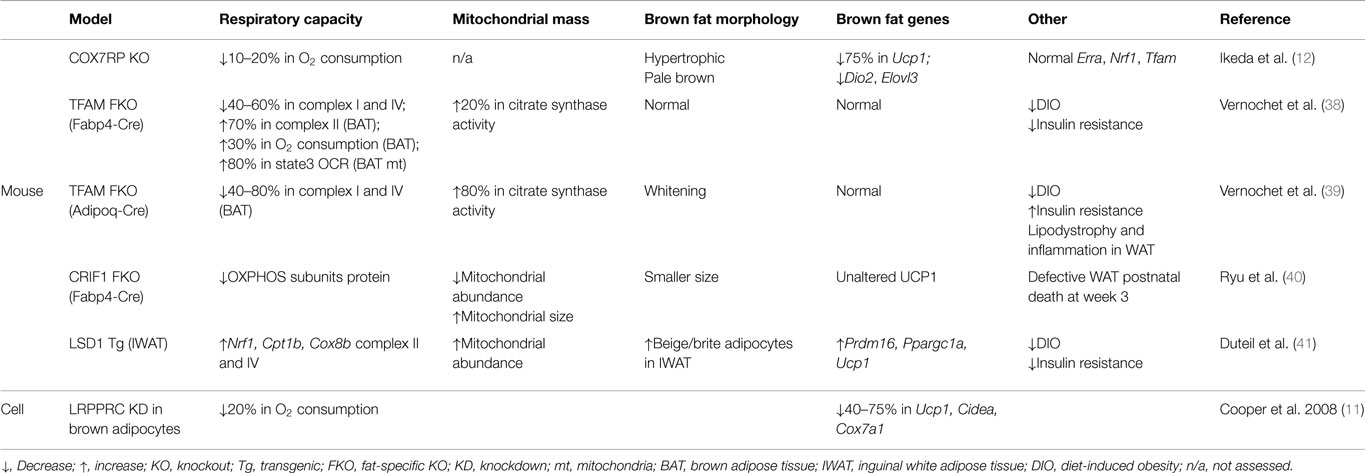
Table 1. Effects of manipulating mitochondrial respiratory capacity on the brown fat gene program.
Leucine-Rich Pentatricopeptide Repeat Containing Motif (LRPPRC; also Called Leucine-Rich Protein 130 kDa, LRP130)
A potential role of mitochondrial respiratory capacity in the brown fat gene program was reported in a study using LRPPRC-deficient brown adipocytes (11). LRPPRC was originally identified as a causal protein in a rare neurological disorder called Leigh Syndrome French Canadian variant (42). Initial studies using human fibroblasts identified defects in cytochrome c oxidase deficiency; however, later studies using mouse models revealed that LRPPRC affects the expression of all mitochondrially encoded subunits of the ETC but their differential effects on respiratory complex activity related to cell type (43–46). Brown adipocytes with depleted LRPPRC were notable for impaired oxygen consumption but intact mitochondrial density and PGC-1 coactivators, indicating a specific impairment of respiratory capacity without altering mitochondrial biogenesis (11).
LRPPRC-deficient brown adipocytes had a marked reduction in brown fat-selective genes, including Ucp1 and Cidea, suggesting a link between respiratory capacity and a basal expression of certain brown fat genes (11). While LRRPRC is weakly expressed in the nucleus, recent data show that the majority of LRPPRC is localized to mitochondria and regulates mtDNA-encoded transcripts across various species, suggesting that a nuclear role of LRPPRC in the regulation of brown fat genes may be modest and that the predominant effect is mediated by impaired cellular respiration (44, 47–49). In addition, cAMP-mediated induction of Ucp1 was unaffected in LRPPRC-deficient brown adipocytes (11). Given that PGC-1α is responsible for this cAMP effect, it is less likely that LRPPRC is an essential part of PGC-1α coactivator complexes necessary for Ucp1 expression. Furthermore, in human fibroblasts, deficiency of LRPPRC did not affect expression of PGC-1α target genes (50), implying that signals secondary to impaired cellular respiration, not reduced nuclear expression of LRPPRC, are likely important.
Cytochrome C Oxidase Subunit VIIa Polypeptide 2-Like (COX7RP)
Recent work using COX7RP knockout mice provides direct evidence for the role of respiratory capacity in the brown fat gene program (12). In this study, COX7RP was identified as a novel assembly factor for respiratory chain supercomplexes in mitochondria. ETC complexes are known to form supercomplexes, consisting mainly of complex I, III, and IV (so-called respirasome), which enhances respiratory activity (51). With reduced oxygen consumption at a whole-body level, COX7RP KO mice revealed hypertrophic and pale brown fat, generally indicative of defective brown fat. More importantly, Ucp1 was severely reduced in this dysmorphic brown fat. Microarray analysis also showed a downregulation of several brown fat-enriched genes including Dio2 and Elovl3. Expression of PGC-1 coactivators was decreased but their downstream targets, such as Erra, Nrf1, and Tfam, were unaltered, implying no significant impact on the PGC-1 coactivator network. All together, this study strongly suggests that respiratory capacity dictates a retrograde signaling from mitochondria to the nucleus regulating the brown fat gene program.
Transcription Factor A, Mitochondrial (TFAM)
One approach to genetically manipulate respiratory capacity is to target components of the basal transcriptional machinery of mitochondrial DNA (mtDNA). Among them is TFAM, a key player in mtDNA transcription and maintenance (52). Fabp4-Cre-driven loss of TFAM led to diminished respiratory activity and a drop in mtDNA copy number in brown and white fat (38). Paradoxically, in those mice, brown fat showed enhanced respiratory capacity as evidenced by increased oxygen consumption, FAO, and citrate synthase activity. Although reduced in weight, this brown fat had normal expression of brown fat genes. Similarly, brown fat markers were intact in brown fat with Adipoq-Cre-driven loss of TFAM, which was accompanied by increased citrate synthase activity (39). Therefore, it is likely that the unaltered brown fat gene program in TFAM-deficient fat is ascribed to a compensatory increase in respiratory capacity. Although not decisive, observations from adipose-specific TFAM knockout mice imply that whole-cell respiratory capacity is monitored by an innate sensor to dictate the brown fat gene program.
CR6-Interacting Factor 1 (CRIF1)
CRIF1 is a mitochondrial protein that controls the translation and insertion of mitochondrially encoded respiratory subunits into the inner membrane (53). The activities of Complexes I, III, and IV are abrogated by CRIF1 deficiency in mouse embryonic fibroblasts (53). The severity of impaired respiratory capacity by ablation of CRIF1 is evident in brain-specific and cardiac muscle-specific knockout mice in which severe neurodegeneration and premature death develop, respectively (53, 54). Adipose-specific CRIF1 knockout mice (driven by Fabp4-Cre) show a developmental defect in white fat, reduced body weight, and postnatal death at week 3 (40). Brown fat in these mice is smaller in size; however, histology is unremarkable and UCP1 expression is normal. Because mice die by 3 weeks of age, it was not possible to assess the chronic effect of respiratory defects in brown fat. Although mice with Adipoq-Cre-driven deletion of CRIF1 were viable, data regarding the brown fat genes were not shown (40). Even so, these data could suggest that impaired mitochondrial function does not influence brown adipocyte development. Future studies will be necessary to address if mitochondrial function is critical for the maintenance of the brown fat program. Finally, as mentioned for TFAM, the method by which cellular respiration is disrupted may have differential effects on mitochondrial signaling and subsequent transcriptional events in the nucleus.
LSD1 (Lysine-Specific Demethylase 1)
LSD1 demethylates mono- and di-methylated lysines (particularly lysine 4 and 9 of histone H3) via the cofactor flavin adenosine dinucleotide (FAD) (55). Ubiquitously expressed, LSD1 is essential for embryogenesis and tissue-specific differentiation (56). In the study by Duteil et al. (41), LSD1 was newly identified as a cold-, and β3-adrenergic signaling-inducible protein in mouse white fat. Ectopic expression of LSD1 further revealed that it was sufficient to induce respiratory capacity through nuclear respiratory factor 1 (NRF1) in white adipose cell lines. In addition, there was an activation of the brown fat gene program including Prdm16, Ppargc1a, and Ucp1 in LSD1-overexpressing white adipocytes. LSD1 transgenic mice confirmed these in vitro findings. Interestingly, this browning effect was more robust in subcutaneous white fat where beige adipocytes reside. However, in brown fat of LSD1 transgenic mice, respiratory activity was modestly increased and there were no significant changes in the brown fat markers. These data suggest that augmented respiratory capacity may promote browning by stimulating beige adipocytes.
Therapeutic Implications
There is an association between impaired brown fat function and various animal models of obesity and diabetes, including ob/ob mice, db/db mice, SHR/N-cp rats, and high-fat/high sucrose (HFHS)-fed mice (57–61). A similar association has been reported for human obesity (62–64). In the aforementioned murine models of obesity and diabetes, there is “de-browning” of brown fat, characterized by concomitant reduction in UCP1 protein, and respiratory complex activities (40, 53–56). Similar to classical brown adipocytes, beige adipocytes exhibit this type of de-browning in mice fed a high-fat diet, implying a shared mechanism for all types of thermogenic adipocytes (65). As detailed earlier, several models and systems have established a causal relationship between mitochondrial respiratory capacity and transcription of genes involved in thermogenesis. Impaired respiration in certain models of obesity and diabetes may explain impaired transcription of the thermogenic genes. In the future, altering respiratory capacity may prove promising in restoring brown fat function in certain forms of obesity and diabetes. Even if there are no effects on transcriptional programs involved in thermogenesis, augmenting respiratory capacity per se could still effectively protect against obesity and diabetes by way of increased respiratory capacity, which in turn would increase thermogenic capacity.
In humans and mice, cold exposure and β3-adrenergic agonists activate brown fat to promote energy expenditure (10, 66–68). Many adrenergic receptor (AR) agonists, including pan-adrenergic (ephedrine) and β3-adrenergic (CL-316,243, etc.) agonists, have been unsuccessful in humans due to either undesirable cardiovascular effects, poor oral availability, or in the case of CL 316,243, weak agonism for the human β3-AR (69). Recently, a clinical study using a new class of β3-AR agonist increased energy expenditure via brown fat-meditated thermogenesis (70). Based on our review of the literature, the status of mitochondrial respiratory capacity may powerfully influence the outcome of β3-adrenergic agents and should be considered as adjunctive therapies to either restore or enhance the brown fat gene program.
Concluding Remarks
The putative role of mitochondrial respiratory capacity in transcriptional programs that regulate thermogenesis raises an intriguing question (Figure 1). Why is respiratory capacity per se linked to the regulation of the thermogenic gene program in brown fat? And what are the signals from mitochondria that govern this control? From a teleological perspective, impaired respiratory capacity would impair thermogenesis, making it a futile process. There seems to be interesting retrograde signaling from the mitochondrion to the nucleus, signaling, which may inactivate programs that expend energy and may promote storing that energy as lipid. Upon recovery of respiratory capacity, such signaling is turned off and energy expending programs are re-activated.
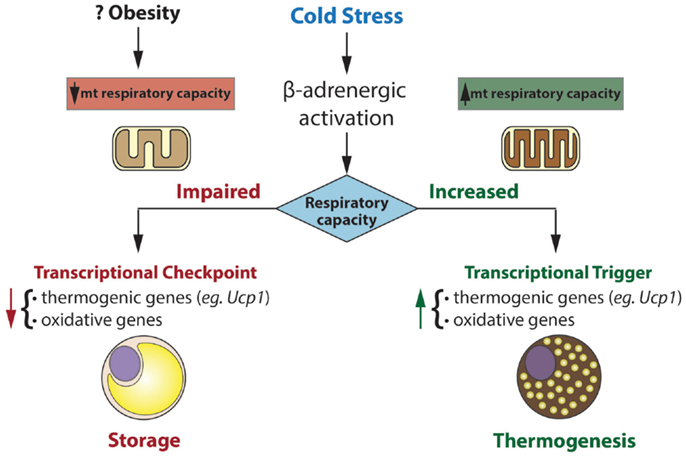
Figure 1. Mitochondria exert transcriptional control over gene programs involved in oxidative metabolism and thermogenesis. Transcriptional control is dependent on mitochondrial respiratory capacity. In this model, impaired respiratory capacity acts as a transcriptional checkpoint, whereas augmented respiratory capacity acts as a transcriptional trigger. In humans and mice, obesity is associated with reduced brown fat function. Our model may explain the inverse association between respiratory capacity and thermogenic gene expression in brown fat of certain obese mice (eg. ob/ob) and perhaps obese humans.
Control of nuclear genes based on the status of mitochondrial function (or stress) has been termed “mitohormesis” and is readily apparent in yeast and mammalian cells (71). In brown fat, a highly specialized tissue for thermogenesis, such mitohormesis may be crucial in matching functional and metabolic capacity to genetic programs that influence them.
In the future, it will be of great interest to unravel the signaling pathways by which respiratory capacity regulates the thermogenic gene program. Elucidating these signals and downstream pathways may inform the search for diagnostic and therapeutic interventions important for obesity and diabetes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Thomas E. Akie for critical reading and helpful comments. This work was supported by NIDDK grant 5R01DK089185 (MC) and by a predoctoral fellowship (13PRE13700002) from the American Heart Association (MN).
References
1. Kajimura S, Saito M. A new era in brown adipose tissue biology: homeostasis. Annu Rev Physiol (2014) 76:225–49. doi: 10.1146/annurev-physiol-021113-170252
2. Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev (2013) 27(3):234–50. doi:10.1101/gad.211649.112
3. Ricquier D. Uncoupling protein 1 of brown adipocytes, the only uncoupler: a historical perspective. Front Endocrinol (2011) 2:85. doi:10.3389/fendo.2011.00085
4. Zhou Z, Yon TS, Chen Z, Guo K, Ng CP, Ponniah S, et al. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat Genet (2003) 35(1):49–56. doi:10.1038/ng1225
5. de Jesus LA, Carvalho SD, Ribeiro MO, Schneider M, Kim SW, Harney JW, et al. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest (2001) 108(9):1379–85. doi:10.1172/JCI13803
6. Shinoda K, Luijten IH, Hasegawa Y, Hong H, Sonne SB, Kim M, et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat Med (2015) 21(4):389–94. doi:10.1038/nm.3819
7. Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab (2007) 6(1):38–54. doi:10.1016/j.cmet.2007.06.001
8. Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab (2006) 3(5):333–41. doi:10.1016/j.cmet.2006.04.002
9. Kajimura S, Seale P, Spiegelman BM. Transcriptional control of brown fat development. Cell Metab (2010) 11(4):257–62. doi:10.1016/j.cmet.2010.03.005
10. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev (2004) 84(1):277–359. doi:10.1152/physrev.00015.2003
11. Cooper MP, Uldry M, Kajimura S, Arany Z, Spiegelman BM. Modulation of PGC-1 coactivator pathways in brown fat differentiation through LRP130. J Biol Chem (2008) 283(46):31960–7. doi:10.1074/jbc.M805431200
12. Ikeda K, Shiba S, Horie-Inoue K, Shimokata K, Inoue S. A stabilizing factor for mitochondrial respiratory supercomplex assembly regulates energy metabolism in muscle. Nat Commun (2013) 4:2147. doi:10.1038/ncomms3147
13. Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J (2014) 33(5):418–36. doi:10.1002/embj.201385014
14. Justo R, Oliver J, Gianotti M. Brown adipose tissue mitochondrial subpopulations show different morphological and thermogenic characteristics. Mitochondrion (2005) 5(1):45–53. doi:10.1016/j.mito.2004.09.003
15. Rodriguez-Cuenca S, Pujol E, Justo R, Frontera M, Oliver J, Gianotti M, et al. Sex-dependent thermogenesis, differences in mitochondrial morphology and function, and adrenergic response in brown adipose tissue. J Biol Chem (2002) 277(45):42958–63. doi:10.1074/jbc.M207229200
16. Loncar D. Convertible adipose tissue in mice. Cell Tissue Res (1991) 266(1):149–61. doi:10.1007/BF00678721
17. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell (1998) 92(6):829–39. doi:10.1016/S0092-8674(00)81410-5
18. Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J Biol Chem (2003) 278(11):9013–8. doi:10.1074/jbc.M212923200
19. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell (1999) 98(1):115–24. doi:10.1016/S0092-8674(00)80611-X
20. Nicholls DG. The bioenergetics of brown adipose tissue mitochondria. FEBS Lett (1976) 61(2):103–10. doi:10.1016/0014-5793(76)81014-9
21. Nicholls DG, Bernson VS, Heaton GM. The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Experientia Suppl (1978) 32:89–93.
22. Cannon B, Romert L, Sundin U, Barnard T. Morphology and biochemical properties of perirenal adipose tissue from lamb (Ovis aries). a comparison with brown adipose tissue. Comp Biochem Physiol B (1977) 56(1):87–99.
23. Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell (2012) 151(2):400–13. doi:10.1016/j.cell.2012.09.010
24. Williamson JR. Control of energy metabolism in hamster brown adipose tissue. J Biol Chem (1970) 245(8):2043–50.
25. Cooney GJ, Newsholme EA. The maximum capacity of glycolysis in brown adipose tissue and its relationship to control of the blood glucose concentration. FEBS Lett (1982) 148(2):198–200. doi:10.1016/0014-5793(82)80807-7
26. Forner F, Kumar C, Luber CA, Fromme T, Klingenspor M, Mann M. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metab (2009) 10(4):324–35. doi:10.1016/j.cmet.2009.08.014
27. Lambeth DO, Tews KN, Adkins S, Frohlich D, Milavetz BI. Expression of two succinyl-CoA synthetases with different nucleotide specificities in mammalian tissues. J Biol Chem (2004) 279(35):36621–4. doi:10.1074/jbc.M406884200
28. Christoffolete MA, Linardi CC, de JL, Ebina KN, Carvalho SD, Ribeiro MO, et al. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes (2004) 53(3):577–84. doi:10.2337/diabetes.53.3.577
29. McCormack JG, Denton RM. Evidence that fatty acid synthesis in the interscapular brown adipose tissue of cold-adapted rats is increased in vivo by insulin by mechanisms involving parallel activation of pyruvate dehydrogenase and acetyl-coenzyme A carboxylase. Biochem J (1977) 166(3):627–30.
30. Yu XX, Lewin DA, Forrest W, Adams SH. Cold elicits the simultaneous induction of fatty acid synthesis and beta-oxidation in murine brown adipose tissue: prediction from differential gene expression and confirmation in vivo. FASEB J (2002) 16(2):155–68. doi:10.1096/fj.01-0568com
31. Cadoudal T, Distel E, Durant S, Fouque F, Blouin JM, Collinet M, et al. Pyruvate dehydrogenase kinase 4: regulation by thiazolidinediones and implication in glyceroneogenesis in adipose tissue. Diabetes (2008) 57(9):2272–9. doi:10.2337/db08-0477
32. Yan J, Lawson JE, Reed LJ. Role of the regulatory subunit of bovine pyruvate dehydrogenase phosphatase. Proc Natl Acad Sci U S A (1996) 93(10):4953–6. doi:10.1073/pnas.93.10.4953
33. Huang B, Gudi R, Wu P, Harris RA, Hamilton J, Popov KM. Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J Biol Chem (1998) 273(28):17680–8. doi:10.1074/jbc.273.28.17680
34. Sakakibara I, Fujino T, Ishii M, Tanaka T, Shimosawa T, Miura S, et al. Fasting-induced hypothermia and reduced energy production in mice lacking acetyl-CoA synthetase 2. Cell Metab (2009) 9(2):191–202. doi:10.1016/j.cmet.2008.12.008
35. Fujino T, Kondo J, Ishikawa M, Morikawa K, Yamamoto TT. Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J Biol Chem (2001) 276(14):11420–6. doi:10.1074/jbc.M008782200
36. Agius L, Williamson DH. The utilization of ketone bodies by the interscapular brown adipose tissue of the rat. Biochim Biophys Acta (1981) 666(1):127–32. doi:10.1016/0005-2760(81)90098-9
37. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest (2006) 116(3):615–22. doi:10.1172/JCI27794
38. Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Rardin MJ, et al. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab (2012) 16(6):765–76. doi:10.1016/j.cmet.2012.10.016
39. Vernochet C, Damilano F, Mourier A, Bezy O, Mori MA, Smyth G, et al. Adipose tissue mitochondrial dysfunction triggers a lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications. FASEB J (2014) 28(10):4408–19. doi:10.1096/fj.14-253971
40. Ryu MJ, Kim SJ, Kim YK, Choi MJ, Tadi S, Lee MH, et al. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS Genet (2013) 9(3):e1003356. doi:10.1371/journal.pgen.1003356
41. Duteil D, Metzger E, Willmann D, Karagianni P, Friedrichs N, Greschik H, et al. LSD1 promotes oxidative metabolism of white adipose tissue. Nat Commun (2014) 5:4093. doi:10.1038/ncomms5093
42. Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A (2003) 100(2):605–10. doi:10.1073/pnas.242716699
43. Merante F, Petrova-Benedict R, MacKay N, Mitchell G, Lambert M, Morin C, et al. A biochemically distinct form of cytochrome oxidase (COX) deficiency in the Saguenay-Lac-Saint-Jean region of Quebec. Am J Hum Genet (1993) 53(2):481–7.
44. Liu L, Sanosaka M, Lei S, Bestwick ML, Frey JH Jr, Surovtseva YV, et al. LRP130 protein remodels mitochondria and stimulates fatty acid oxidation. J Biol Chem (2011) 286(48):41253–64. doi:10.1074/jbc.M111.276121
45. Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Camara Y, et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J (2012) 31(2):443–56. doi:10.1038/emboj.2011.392
46. Mourier A, Ruzzenente B, Brandt T, Kuhlbrandt W, Larsson NG. Loss of LRPPRC causes ATP synthase deficiency. Hum Mol Genet (2014) 23(10):2580–92. doi:10.1093/hmg/ddt652
47. Bratic A, Wredenberg A, Gronke S, Stewart JB, Mourier A, Ruzzenente B, et al. The bicoid stability factor controls polyadenylation and expression of specific mitochondrial mRNAs in Drosophila melanogaster. PLoS Genet (2011) 7(10):e1002324. doi:10.1371/journal.pgen.1002324
48. Barkan A, Rojas M, Fujii S, Yap A, Chong YS, Bond CS, et al. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet (2012) 8(8):e1002910. doi:10.1371/journal.pgen.1002910
49. Sterky FH, Ruzzenente B, Gustafsson CM, Samuelsson T, Larsson NG. LRPPRC is a mitochondrial matrix protein that is conserved in metazoans. Biochem Biophys Res Commun (2010) 398(4):759–64. doi:10.1016/j.bbrc.2010.07.019
50. Gohil VM, Nilsson R, Belcher-Timme CA, Luo B, Root DE, Mootha VK. Mitochondrial and nuclear genomic responses to loss of LRPPRC expression. J Biol Chem (2010) 285(18):13742–7. doi:10.1074/jbc.M109.098400
51. Vartak R, Porras CA, Bai Y. Respiratory supercomplexes: structure, function and assembly. Protein Cell (2013) 4(8):582–90. doi:10.1007/s13238-013-3032-y
52. Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev (2008) 88(2):611–38. doi:10.1152/physrev.00025.2007
53. Kim SJ, Kwon MC, Ryu MJ, Chung HK, Tadi S, Kim YK, et al. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab (2012) 16(2):274–83. doi:10.1016/j.cmet.2012.06.012
54. Shin J, Lee SH, Kwon MC, Yang DK, Seo HR, Kim J, et al. Cardiomyocyte specific deletion of Crif1 causes mitochondrial cardiomyopathy in mice. PLoS One (2013) 8(1):e53577. doi:10.1371/journal.pone.0053577
55. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell (2004) 119(7):941–53. doi:10.1016/j.cell.2004.12.012
56. Pedersen MT, Helin K. Histone demethylases in development and disease. Trends Cell Biol (2010) 20(11):662–71. doi:10.1016/j.tcb.2010.08.011
57. Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, et al. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest (2006) 116(10):2791–8. doi:10.1172/JCI28570
58. Shimizu I, Aprahamian T, Kikuchi R, Shimizu A, Papanicolaou KN, MacLauchlan S, et al. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest (2014) 124(5):2099–112. doi:10.1172/JCI71643
59. Goodbody AE, Trayhurn P. GDP binding to brown-adipose-tissue mitochondria of diabetic – obese (db/db) mice. decreased binding in both the obese and pre-obese states. Biochem J (1981) 194(3):1019–22.
60. Masaki T, Yoshimatsu H, Chiba S, Sakata T. Impaired response of UCP family to cold exposure in diabetic (db/db) mice. Am J Physiol Regul Integr Comp Physiol (2000) 279(4):R1305–9.
61. Atgie C, Marette A, Desautels M, Tulp O, Bukowiecki LJ. Specific decrease of mitochondrial thermogenic capacity in brown adipose tissue of obese SHR/N-cp rats. Am J Physiol (1993) 265(6 Pt 1):C1674–80.
62. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med (2009) 360(15):1500–8. doi:10.1056/NEJMoa0808718
63. Vijgen GH, Bouvy ND, Teule GJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD. Brown adipose tissue in morbidly obese subjects. PLoS One (2011) 6(2):e17247. doi:10.1371/journal.pone.0017247
64. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360(15):1509–17. doi:10.1056/NEJMoa0810780
65. Rong JX, Qiu Y, Hansen MK, Zhu L, Zhang V, Xie M, et al. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes (2007) 56(7):1751–60. doi:10.2337/db06-1135
66. Cypess AM, Chen YC, Sze C, Wang K, English J, Chan O, et al. Cold but not sympathomimetics activates human brown adipose tissue in vivo. Proc Natl Acad Sci U S A (2012) 109(25):10001–5. doi:10.1073/pnas.1207911109
67. van der Lans AA, Hoeks J, Brans B, Vijgen GH, Visser MG, Vosselman MJ, et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest (2013) 123(8):3395–403. doi:10.1172/JCI68993
68. Carey AL, Formosa MF, Van EB, Bertovic D, Eikelis N, Lambert GW, et al. Ephedrine activates brown adipose tissue in lean but not obese humans. Diabetologia (2013) 56(1):147–55. doi:10.1007/s00125-012-2748-1
69. Arch JR. Challenges in β (3)-adrenoceptor agonist drug development. Ther Adv Endocrinol Metab (2011) 2(2):59–64. doi:10.1177/2042018811398517
70. Cypess AM, Weiner LS, Roberts-Toler C, Elia EF, Kessler SH, Kahn PA, et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab (2015) 21(1):33–8. doi:10.1016/j.cmet.2014.12.009
Keywords: brown fat, mitochondria, respiratory capacity, thermogenesis, brown fat gene program
Citation: Nam M and Cooper MP (2015) Role of energy metabolism in the brown fat gene program. Front. Endocrinol. 6:104. doi: 10.3389/fendo.2015.00104
Received: 08 May 2015; Accepted: 15 June 2015;
Published: 30 June 2015
Edited by:
Jun Wu, University of Michigan, USACopyright: © 2015 Nam and Cooper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcus P. Cooper, Division of Cardiovascular Medicine, Department of Medicine, University of Massachusetts Medical School, 368 Plantation Street, Albert Sherman Center, 7th Floor West, AS7-1053, Worcester, MA 01605, USA, marcus.cooper@umassmed.edu