 Munirah Mohamad Santosa
Munirah Mohamad Santosa Blaise Su Jun Low
Blaise Su Jun Low Nicole Min Qian Pek
Nicole Min Qian Pek Adrian Kee Keong Teo
Adrian Kee Keong Teo- 1Stem Cells and Diabetes Laboratory, Discovery Research Division, Institute of Molecular and Cell Biology, Singapore
- 2School of Biological Sciences, Nanyang Technological University, Singapore
- 3Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
- 4Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore
In the field of stem cell biology and diabetes, we and others seek to derive mature and functional human pancreatic β cells for disease modeling and cell replacement therapy. Traditionally, knowledge gathered from rodents is extended to human pancreas developmental biology research involving human pluripotent stem cells (hPSCs). While much has been learnt from rodent pancreas biology in the early steps toward Pdx1+ pancreatic progenitors, much less is known about the transition toward Ngn3+ pancreatic endocrine progenitors. Essentially, the later steps of pancreatic β cell development and maturation remain elusive to date. As a result, the most recent advances in the stem cell and diabetes field have relied upon combinatorial testing of numerous growth factors and chemical compounds in an arbitrary trial-and-error fashion to derive mature and functional human pancreatic β cells from hPSCs. Although this hit-or-miss approach appears to have made some headway in maturing human pancreatic β cells in vitro, its underlying biology is vaguely understood. Therefore, in this mini-review, we discuss some of these late-stage signaling pathways that are involved in human pancreatic β cell differentiation and highlight our current understanding of their relevance in rodent pancreas biology. Our efforts here unravel several novel signaling pathways that can be further studied to shed light on unexplored aspects of rodent pancreas biology. New investigations into these signaling pathways are expected to advance our knowledge in human pancreas developmental biology and to aid in the translation of stem cell biology in the context of diabetes treatments.
Introduction
In the field of stem cells and diabetes, many scientists are actively pursuing the generation of insulin-secreting pancreatic β cells from human pluripotent stem cells (hPSCs) for β cell transplantation/replacement and treatment of diabetes (1–3). While insights from rodent pancreas developmental biology has guided the generation of PDX1+ pancreatic progenitors from hPSCs, the specific developmental principles thereafter remain murky. Henceforth, research groups have relied upon the transplantation of pancreatic progenitors, derived in vitro, into rodents for in vivo maturation (4–6). However, there has been considerable progress toward the generation of mature and functional human pancreatic β cells in vitro in the recent years. These β cells purportedly co-express cardinal β cell markers, such as PDX1, NKX6.1, musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA), prohormone-processing enzymes, insulin, and C-peptide. Importantly, they are also monohormonal and glucose responsive.
Developmental biologists believe that there is much to be learnt from rodent developmental biology to guide hPSC-based generation of clinically useful cell types, such as pancreatic β cells. Owing to such efforts, the progression of definitive endoderm (DE) germ layer to PDX1+ pancreatic progenitors has been well-explored. However, the investigations on the later steps of pancreatic endocrine development and β cell maturation have not been quite fruitful. The most substantial advances in stem cell biology have relied upon an arbitrary approach of iterative trial-and-error testing to achieve mature and functional pancreatic β cells in vitro (7). Therefore, several pertinent questions remain: why were we not able to extrapolate rodent developmental principles and apply them on hPSCs to derive mature and functional pancreatic β cells? Are there differences between rodent and human pancreas development that prevent such an application? In this review, we look at signaling pathways that have been activated or repressed in stem cell biology and retrospectively revisit existing knowledge about rodent pancreas biology. Our efforts highlight novel aspects of signaling pathways that can be further investigated in our translational efforts for diabetes.
Inhibition of Transforming Growth Factor-β Signaling in the Later Stages of Pancreatic Differentiation
The transforming growth factor-β (TGF-β) superfamily of proteins regulates pancreas development and function (8). TGF-β1, TGF-β2, and TGF-β3 are expressed in pancreatic epithelial cells at E12.5 in mice. Thereafter, they become localized in the acinar cells (9). TGF-β1 can promote the development of mouse pancreatic β cells from pancreatic buds (10). Perplexingly, it also indirectly inhibits the formation of mouse pancreatic epithelial cells (11). In tandem, TGF-β2 has been demonstrated to inhibit Hnf1β and Pdx1 gene expression. Hence, TGF-β can purportedly restrain the specification of pancreatic cell fate (12). TGF-β signaling effector SMAD3 can bind the Ins gene promoter to suppress its expression. In agreement, Smad3-deficient islets exhibit an active insulin signaling pathway (13). Collectively, these evidences suggest the requirement to inhibit TGF-β signaling for the derivation of mature and functional pancreatic β cells (Figure 1A).
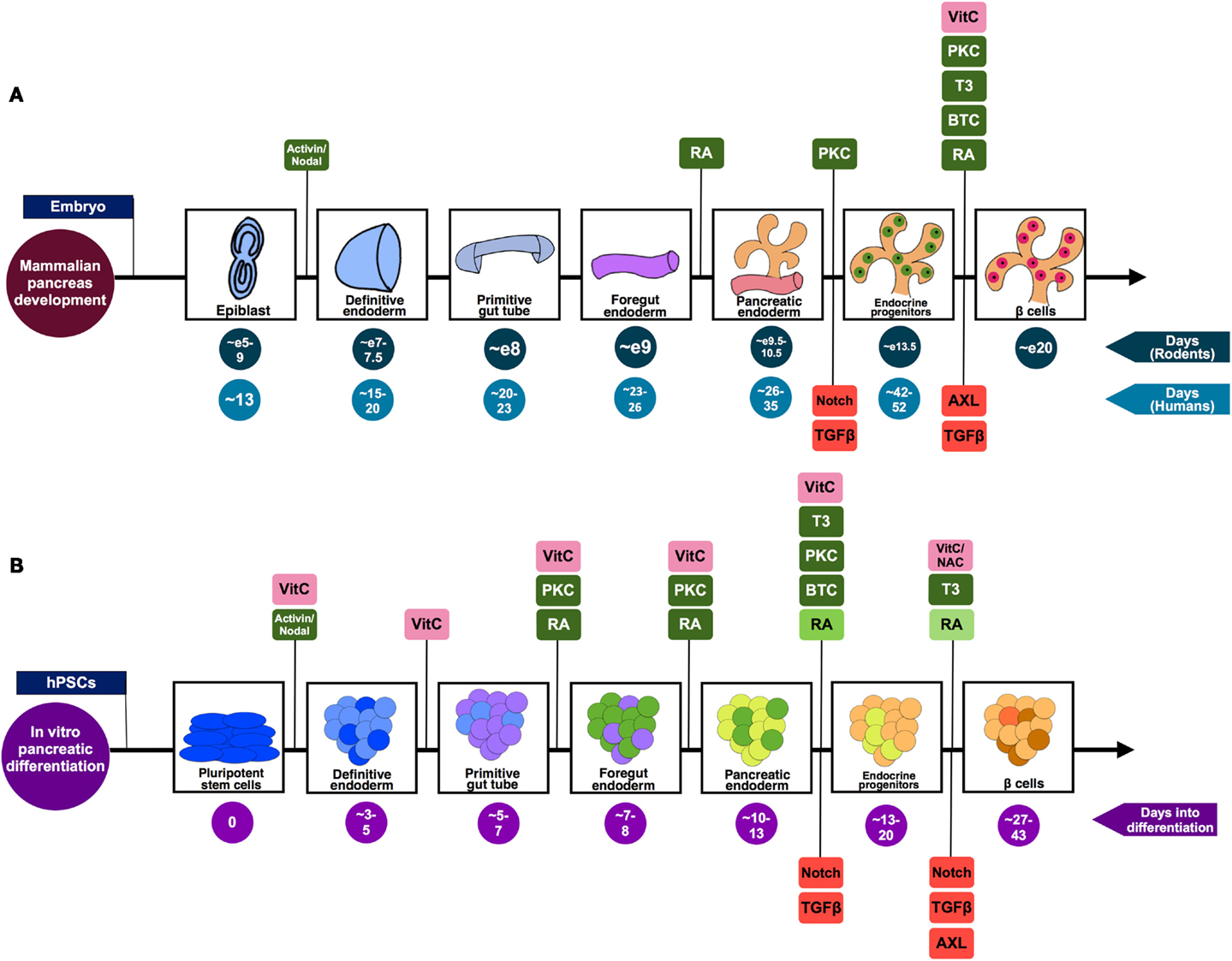
Figure 1. A summary of the pivotal stages of pancreatic differentiation. (A) Mammalian pancreas development based on knowledge from both rodent and human studies. The signaling pathways suggested to be positively regulating the differentiation process are highlighted in green, while those found to be inhibited in order to drive differentiation toward pancreatic lineage are shown in red. (B) An in vitro pancreatic differentiation timecourse generating β-like cells from hPSCs. The signaling pathways (green) and antioxidants (pink) that are positively regulating the differentiation process at each phase of development are highlighted. Stage-specific signaling pathways that are inhibited to drive differentiation toward pancreatic lineage are shown in red. The decreasing doses of RA used during the differentiation process [as described by Rezania et al. (6) and Pagliuca et al. (7)] are represented by the decreasing shades of green. The differing colors co-existing in an aggregate illustrates the heterogeneity of cells prevalent in such a differentiation scheme. While some of the cells will transit from being endocrine progenitors (light orange) to Pdx1+ insulin-producing β-like cells (brown), the end-product will include an assortment of maturing endocrine cell types (represented by orange and light orange).
In 2011, Nostro et al. used small molecule SB431542 (14), an Activin/TGF-β receptor antagonist, in their pancreatic differentiation protocol. SB431542 inhibits activin receptor-like kinases (ALK) 4/5/7 and the downstream TGF-β/Activin/Nodal signaling. SB431542 treatment was demonstrated to increase INS gene expression and the development of C-peptide+ cells (15). Similarly, Cho et al. also utilized SB431542, in the presence of retinoic acid (RA), for pancreatic differentiation (16). Alternatively, Schulz et al. used TGF-βRI kinase inhibitor IV to obtain pancreatic progenitors from CyT49 hPSCs (17). Rezania et al. identified that the use of 2-(3-[6-Methylpyridin-2-yl]-1H-pyrazol-4-yl)-1,5-naphthyridine (ALK5iII) can effectively induce the expression of NGN3, NEUROD1, INS, and GCG transcripts to promote pancreatic endocrine specification (18). Rezania et al. further demonstrated that 1 μM ALK5iII is necessary for the induction of NEUROD1+ cells, but it suppressed the proportion of NKX6.1+ cells (4), a hallmark of functional β cells (19). Most recently, Rezania et al. compared the effects of several ALK5 inhibitors at a later phase of differentiation of hPSCs and found that only ALK5iII downregulated NGN3 while increasing INS, GCG, and SST transcripts (6). Furthermore, 10 μM ALK5iII induced the expression of nuclear v-maf MAFA transcript, a critical mature β cell transcription factor, in diabetic rodents (20–22). Rezania et al. (6) concluded that ALK5iII was the most effective and specific inhibitor as it inhibited ALK5 but had minimal inhibition of other kinases. Similarly, Pagliuca et al. also employed 10 μM Alk5iII to derive mature and functional human pancreatic β cells from hPSCs (7) (Figure 1B; Table 1).
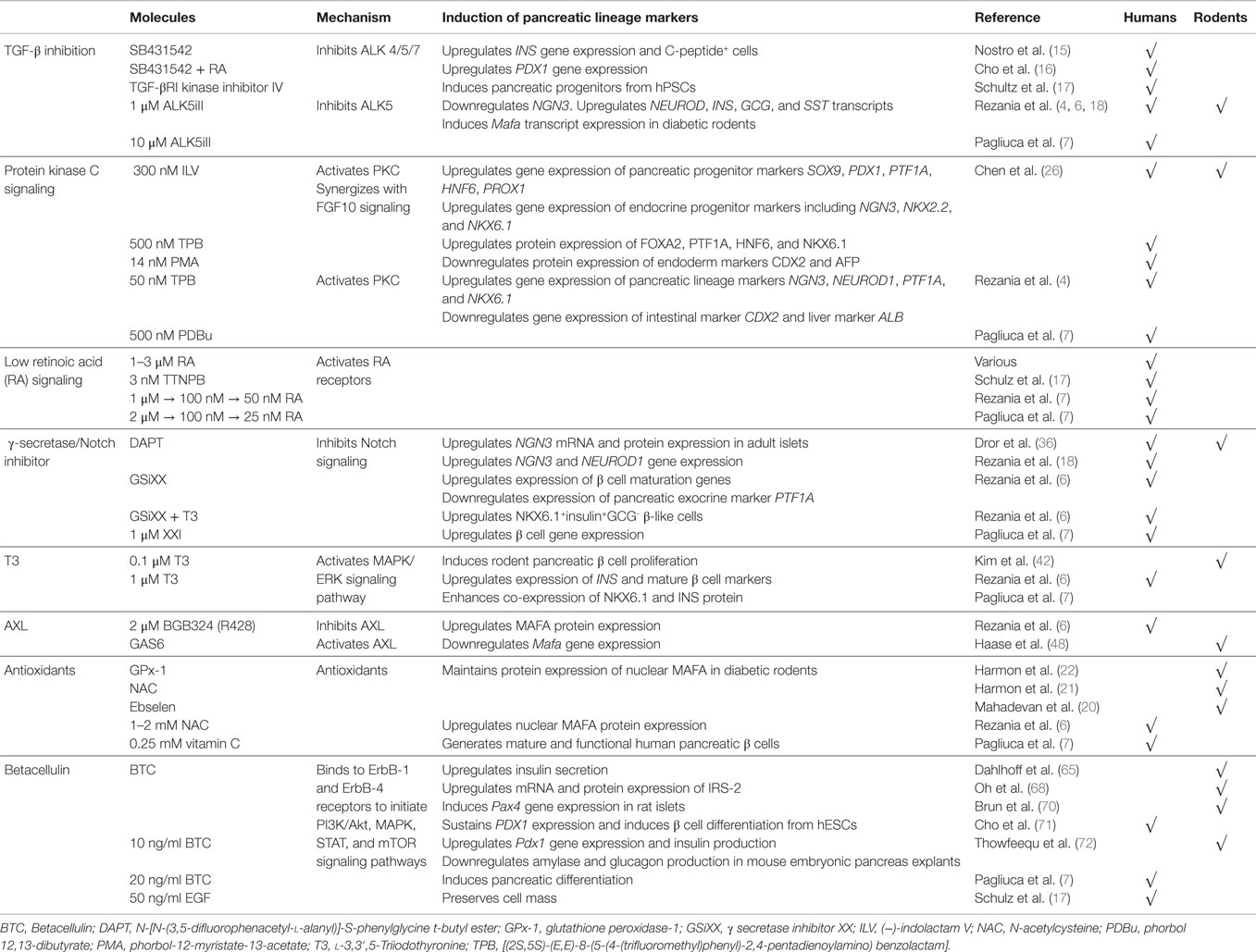
Table 1. Summary of some novel signaling pathways perturbed during pancreatic differentiation of hPSCs.
Overall, the inhibition of ALK5/TGF-βRI with ALK5iII appears to be more desirable as compared to the general inhibition of TGF-β signaling via the use of SB431542. Further studies are certainly required to investigate the intricacies of TGF-β signaling during pancreas development and β cell maturation.
Protein Kinase C Signaling Enhancement
Protein kinase C (PKC) is a family of serine/threonine kinases that are involved in diverse cellular processes, including survival, apoptosis, cell cycle regulation, proliferation, migration, and differentiation (23). In maturing neonatal rat islets, PKCα was only found in β cells, PKCγ in α cells, and PKCϵ in δ cells (24). This differential expression of PKC isoenzymes (25) hints that PKC signaling may play a role in the functional maturation of pancreatic endocrine progenitors (Figure 1A).
Chen et al. (26) was the first to demonstrate that 300 nM (−)-indolactam V (ILV) or PKC agonists {500 nM [(2S,5S)-(E,E)-8-(5-(4-(trifluoromethyl)phenyl)-2,4-pentadienoylamino) benzolactam (TPB)] or 14 nM phorbol-12-myristate-13-acetate (PMA)} can efficiently increase the formation of PDX1+ pancreatic progenitors from hPSCs via the activation of PKC signaling. ILV treatment resulted in an increased gene expression of several pancreatic progenitor markers, including SOX9, PDX1, PTF1A, HNF6, and PROX1, and endocrine progenitor markers, including NGN3, NKX2.2, and NKX6.1. The protein expression of pancreatic progenitor markers FOXA2, PTF1A, HNF6, and NKX6.1 were increased, whereas the expression of intestinal marker CDX2 and liver marker AFP were suppressed (26). In addition, they also found that ILV and PKC agonists, TPB or PMA, can synergize with FGF10 signaling to promote the proliferation of PDX1+ cells derived from hPSCs. Interestingly, ILV treatment also works on mouse embryonic stem cells, suggesting a conservation of signaling pathway in both mouse and human cells (26) (Table 1).
Subsequently, Rezania et al. (4) employed two PKC activators in the mid-late stage of hPSC differentiation; 50 nM TPB (safer profile) and phorbol 12,13-dibutyrate (PDBu) (27), demonstrating that the activation of PKC signaling induces the gene expression of pancreatic lineage markers PTF1A, NGN3, NEUROD1, and NKX6.1 while suppressing the expression of intestinal (CDX2) and liver (ALB) markers (4). This demonstrated that PKC signaling enriches the development of pancreatic progenitors while inhibiting intestinal and hepatic lineages. Similarly, Pagliuca et al. used 500 nM PDBu in their pancreatic differentiation protocol (7) (Figure 1B; Table 1). Although these data are encouraging, more studies remain to be done to thoroughly clarify the role of PKC signaling and the specific mechanisms in the maturation of pancreatic endocrine progenitors.
Lower Level of Retinoic Acid Signaling as Pancreatic Differentiation Progresses
It is well-established that RA signaling plays critical roles in the early and late stages of pancreas development (28). RA is a lipid-soluble vitamin A derivative synthesized from the oxidation of retinaldehyde via enzymes retinaldehyde dehydrogenase 1 (RALDH1), RALDH2, and RALDH3. RA produced at the splanchnic lateral plate mesoderm and Raldh2 expressed in the dorsal pancreatic mesenchyme promote Pdx1 induction in the dorsal foregut endoderm (Figure 1A). Raldh2 mutant mice exhibit dorsal pancreatic bud agenesis (29) as they fail to form pancreatic progenitors, indicated by the loss of Pdx1, Prox1, altered Isl1, and reduced Hlxb9 expression (29, 30).
Many existing protocols differentiating hPSCs into pancreatic cells utilize 1–3 μM RA. As an alternative, Schulz et al. replaced RA with 3 nM TTNPB/arotinoid acid – a more stable retinoid analog that can selectively activate RA receptors (RARs) (17). Interestingly, Rezania et al. started out using 1 μM RA during the early posterior foregut differentiation, subsequently reducing to 100 nM RA during pancreatic endoderm phase and further reducing to 50 nM during the pancreatic endocrine phase (6). Pagliuca et al. also reported a similar pancreatic differentiation protocol in which a decreasing dose of RA was used, starting with 2 μM followed by 100 nM RA at later stages, which was eventually reduced to 25 nM (7) (Figure 1B; Table 1).
While it is widely accepted that RA is crucial for pancreatic specification, using progressively lower doses of RA as practiced by Rezania et al. (6) and Pagliuca et al. (7) raises questions about the importance of RA concentrations during pancreas development both in vitro and in vivo. RA signals by binding RARs and retinoid X receptors (RXRs); so it is postulated that a decrease in RA signaling may be more conducive for subsequent pancreatic endocrine specification since retinoid receptors are upregulated in the pancreatic exocrine (31). However, given the lack of understanding in this regard, the significance of the dose of RA during human pancreas development warrants further studies.
Inhibiting γ-Secretase/Notch for Accelerated Pancreatic Endocrine Differentiation
Notch signaling is essential for the proper development of pancreatic endocrine progenitors as it regulates their decision between differentiation and proliferation (32). The reduction of Notch signaling is known to promote accelerated pancreatic endocrine differentiation (33). Similarly, the inhibition of Notch signaling via γ-secretase (an intra-membrane protease) inhibitor can downregulate the expression of Notch target Hes-1, an inhibitor of pro-endocrine gene Ngn3 (34) (Figure 1A). Conversely, the activation of Notch in PDX1+ pancreatic progenitors prevents pancreatic differentiation (35).
N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine t-butyl ester (DAPT) is a commonly used γ-secretase inhibitor with an IC50 in the nM range. The inhibition of Notch signaling with DAPT can increase Ngn3 mRNA and protein expression in adult islets (36). Likewise, DAPT increases NGN3 and NEUROD1 gene expression in hPSC-derived pancreatic progenitors (18). In recent protocols developed by Pagliuca et al. (7) and Rezania et al. (6), other γ-secretase inhibitors have been employed to retard Notch signaling. Rezania et al. used γ-secretase inhibitor XX (GSiXX) that has an IC50 in the low nM range. They showed that GSiXX can induce the expression of β cell maturation genes but inhibit the expression of PTF1A, a marker of pancreatic exocrine lineage (6). GSiXX can also act in concert with triiodothyronine (T3) to increase the percentage of NKX6.1+INS+GCG− β-like cells (6). Alternatively, Pagliuca et al. employed the use of XXI at 1 μM (7), which has an IC50 in the picomolar range, and demonstrated that XXI worked with other factors to improve β cell gene expression (Figure 1B; Table 1). However, it remains unclear whether there are differences between DAPT, GSIXX, or XXI in the induction or suppression of key pancreatic transcription factors for the eventual promotion of pancreatic β cell formation.
Triiodothyronine Could Promote Pancreatic β Cell Maturation
Studies in the 1980s suggest that thyroid hormones regulate insulin secretion, possibly via control over glucose oxidation and calcium uptake rates (37). T3, a thyroid hormone, can potentiate insulin signaling and increase insulin synthesis in diabetic rodents (38), in rodent islets (39), and in a rodent pancreatic β cell line (40) (Figure 1A). Mechanistically, T3 phosphorylates and activates AKT in pancreatic β cells, improving their survival (39, 41); 0.1 μM of T3 can increase rodent pancreatic β cell proliferation via the MAPK/extracellular signal-regulated kinase (ERK) signaling pathway (42). Interestingly, T3 apparently induces the transdifferentiation of human pancreatic ductal cell line (hPANC-1) into β-like cells, with an increased expression of INS transcripts (43). T3 can also increase both the mRNA expression of pro-endocrine gene Ngn3 and the number of β cells, indirectly inducing endocrine differentiation from exocrine cells; possibly via Akt signaling (44).
Of late, T3 has been shown to promote pancreatic β cell maturation and proliferation in rats (45). Based on these findings, Rezania et al. went on to demonstrate that 1 μM of T3 can actually induce the expression of INS and mature β cell markers, and enhance the co-expression of NKX6.1 and INSULIN protein (6). Similarly, Pagliuca et al. employed the same dose of 1 μM T3 in the later stages of their pancreatic differentiation protocol to generate human pancreatic β cells from hPSCs (7) (Figure 1B; Table 1). While the biology and role of T3 in pancreatic β cell maturation remains to be explored further, its inclusion in pancreatic differentiation protocols appears to serve a positive function.
Inhibition of Tyrosine Kinase Receptor Axl Induces Mature Pancreatic β Cell Marker (Mafa) Expression
AXL is a member of the Tyro3-Axl-Mer (TAM) trans-membrane receptor tyrosine kinase (RTK) family that plays an important role in essential cellular processes, such as cell survival, growth, proliferation, and differentiation. Its ligand, growth arrest specific 6 (Gas6), binds AXL to activate downstream signaling, including the phosphoinositide 3-kinase (PI3K), ERK, and signal transducer and activator of transcription 3 (STAT3) signaling (46). Interestingly, Rezania et al. performed small molecule and growth factor library screening to identify compounds that can induce mature β cell marker MAFA from hPSC-derived pancreatic progenitors and found that 2 μM BGB324 (R428), an inhibitor of AXL, can induce MAFA protein expression (6) (Figure 1B; Table 1). However, there is little information linking AXL to pancreas development and β cell maturation.
In 1999, Augustine et al. reported that the overexpression of AXL results in diabetes in mice. Furthermore, the administration of exogenous Gas6 exacerbated the condition (47). Haase et al. recently confirmed that GAS6 is expressed in pancreatic tissues and found that GAS6 reduced Mafa gene expression in rodents (48), likely due to the activation of AXL (Figure 1A). This corresponds with the increase in MAFA expression observed by Rezania et al. after the inhibition of AXL signaling (6). While there seems to be a bona fide association between AXL signaling and pancreas development, the cellular mechanism(s) remain a mystery.
Antioxidants may Benefit Pancreatic Differentiation
Excessive levels of reactive oxygen species (ROS) have been implicated in glucotoxicity-induced pancreatic β cell destruction and dysfunction. In this regard, antioxidants play important defensive roles against ROS. In the endocrine pancreas, the antioxidant vitamin C is known to be an effective co-factor for the peptidyl α-amidation of several biologically active peptides and is necessary for optimal insulin secretion from pancreatic β cells (Figure 1A). In fact, high concentrations of ascorbic acid (vitamin C) were found in neonatal rat endocrine pancreas (49). Mechanistic studies involving other antioxidants, such as glutathione peroxidase-1 (GPx-1), N-acetylcysteine (NAC), and ebselen, were reported to maintain the protein expression of mature β cell marker MAFA (20–22) (Table 1).
Interestingly, Rezania et al. used 1–2 mM NAC during their pancreatic differentiation and found that it also increased nuclear MAFA protein expression (6). However, this was not replicated with another antioxidant, vitamin E. Pagliuca et al. also relied upon the use of 0.25 mM of vitamin C throughout S1–S5 phase of their pancreatic differentiation protocol to generate mature and functional human pancreatic β cells (7) (Figure 1B; Table 1). While the metabolism of vitamins C and E are altered before the onset of diabetes in rats, their contribution to the pancreas is unclear (50). Antioxidant treatments may preserve β cell function, exerting positive effects in diabetes (51), but their role in pancreas development and β cell maturation certainly remains elusive.
Betacellulin Directs a Pancreatic β Cell Fate
Betacellulin (BTC) is a member of the epidermal growth factor (EGF) family that plays a role in the differentiation of pancreatic β cells (52) (Figure 1A). It is largely expressed in the liver, kidney, small intestine, and pancreas (53), and is specifically expressed in 9- to 24-week-old human fetal pancreas (54). BTC binds to ErbB-1 and ErbB-4 receptors (55) to initiate downstream signaling pathways involving PI3K/Akt, MAPK, STAT, and mTOR signaling pathways (56).
Betacellulin appears to direct a pancreatic β cell fate. It can convert exocrine cells (57) and α cells (58) into insulin-secreting cells. It can also induce β cell neogenesis from ductal cells in diabetic mice (59). Li et al. demonstrated that exogenous BTC can promote β cell regeneration in 90% pancreatectomized rats (60) and convert δ to β cells in STZ-induced diabetic mice (61). Also, Yamamoto et al. observed that long-term administration of BTC reverses STZ-induced hyperglycemia in mice (62). Surprisingly, the loss of BTC in mice yielded no overt defect (63) despite their active roles in the pancreas. This could be explained by compensatory effects exhibited by the other EGFR ligands (64). The overexpression of BTC in transgenic islets does not affect islet structure, endocrine cell ratio, or β cell mass but enhances glucose-stimulated insulin secretion (65). However, ubiquitous overexpression of BTC in mice results in various pathologies (66). Intriguingly, gene variants and polymorphisms in the BTC gene have also been found to be associated with types 1 and 2 diabetes (54, 67).
The induction of β cell development/differentiation by BTC (52) could be an outcome of the downstream increase in insulin receptor substrate-2 (IRS-2) expression (68), an important mediator of β cell function (69). BTC can induce Pax4 gene expression in rat islets, promoting β cell functionality (70). It can also sustain PDX1 expression and induce β cell differentiation from hPSCs (71). Ten ng/ml BTC are sufficient to increase Pdx1 gene expression, insulin production, and to inhibit amylase and glucagon production in mouse embryonic pancreas explants (72). Lately, Pagliuca et al. employed 20 ng/ml BTC in the later stages of their pancreatic differentiation protocol (7). Similarly, 50 ng/ml EGF was added to preserve cell mass (17) (Figure 1B; Table 1). These data strongly indicate that BTC and/or other EGF ligands are of importance in pancreas development and β cell maturation. Nonetheless, more detailed molecular mechanisms remain to be uncovered.
Concluding Remarks
Relating to the current most advanced human pancreatic β cell differentiation protocols, the biological mechanisms involved in the later stages of β cell development and maturation still remain elusive. The fact that the postnatal stage in rodents is grossly equivalent to the third trimester in human pancreas development (73) invites new approaches to study this aspect of β cell biology. In this review, we revisited some of the least-understood developmental signaling pathways in rodent pancreas biology. Our efforts unraveled interesting aspects of these signaling pathways that demand to be thoroughly elucidated at the mechanistic level. Future studies should seek to highlight how immature β cells transit into mature and functional β cells. This would certainly advance our knowledge of human pancreas developmental biology and boost translational efforts in the use of stem cells for diabetes treatment.
Author Contributions
MS: reviewed the literature, wrote and edited the paper. BL: edited the paper. NP: edited the paper and prepared the figure. AT: conceptualized the review topic and contents, wrote, edited, and approved the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Teo laboratory, including Chang Siang Lim, Hwee Hui Lau, and Larry Sai Weng Loo for their comments on the manuscript.
Funding
AT is supported by the Institute of Molecular and Cell Biology (IMCB), A*STAR, NHG-KTPH SIG/14033, the NUHS-CG Metabolic In Vitro Core Seed Funding and the JCO Career Development Award (CDA) 15302FG148, A*STAR.
References
1. McCall M, Shapiro AM. Islet cell transplantation. Semin Pediatr Surg (2014) 23:83–90. doi: 10.1053/j.sempedsurg.2014.03.006
2. Teo AK, Gupta MK, Doria A, Kulkarni RN. Dissecting diabetes/metabolic disease mechanisms using pluripotent stem cells and genome editing tools. Mol Metab (2015) 4:593–604. doi:10.1016/j.molmet.2015.06.006
3. Teo AK, Wagers AJ, Kulkarni RN. New opportunities: harnessing induced pluripotency for discovery in diabetes and metabolism. Cell Metab (2013) 18:775–91. doi:10.1016/j.cmet.2013.08.010
4. Rezania A, Bruin JE, Riedel MJ, Mojibian M, Asadi A, Xu J, et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes (2012) 61:2016–29. doi:10.2337/db11-1711
5. Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol (2008) 26:443–52. doi:10.1038/nbt1393
6. Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol (2014) 32:1121–33. doi:10.1038/nbt.3033
7. Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic beta cells in vitro. Cell (2014) 159:428–39. doi:10.1016/j.cell.2014.09.040
8. Rane SG, Lee JH, Lin HM. Transforming growth factor-beta pathway: role in pancreas development and pancreatic disease. Cytokine Growth Factor Rev (2006) 17:107–19. doi:10.1016/j.cytogfr.2005.09.003
9. Crisera CA, Rose MI, Connelly PR, Li M, Colen KL, Longaker MT, et al. The ontogeny of TGF-beta1, -beta2, -beta3, and TGF-beta receptor-II expression in the pancreas: implications for regulation of growth and differentiation. J Pediatr Surg (1999) 34:689–93; discussion 693–4. doi:10.1016/S0022-3468(99)90357-3
10. Sanvito F, Herrera PL, Huarte J, Nichols A, Montesano R, Orci L, et al. TGF-beta 1 influences the relative development of the exocrine and endocrine pancreas in vitro. Development (1994) 120:3451–62.
11. Jiang FX, Stanley EG, Gonez LJ, Harrison LC. Bone morphogenetic proteins promote development of fetal pancreas epithelial colonies containing insulin-positive cells. J Cell Sci (2002) 115:753–60.
12. Wandzioch E, Zaret KS. Dynamic signaling network for the specification of embryonic pancreas and liver progenitors. Science (2009) 324:1707–10. doi:10.1126/science.1174497
13. Lin HM, Lee JH, Yadav H, Kamaraju AK, Liu E, Zhigang D, et al. Transforming growth factor-beta/Smad3 signaling regulates insulin gene transcription and pancreatic islet beta-cell function. J Biol Chem (2009) 284:12246–57. doi:10.1074/jbc.M805379200
14. Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol (2002) 62:65–74. doi:10.1124/mol.62.1.65
15. Nostro MC, Sarangi F, Ogawa S, Holtzinger A, Corneo B, Li X, et al. Stage-specific signaling through TGFbeta family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development (2011) 138:861–71. doi:10.1242/dev.065904
16. Cho CH, Hannan NR, Docherty FM, Docherty HM, Joao Lima M, Trotter MW, et al. Inhibition of activin/nodal signalling is necessary for pancreatic differentiation of human pluripotent stem cells. Diabetologia (2012) 55:3284–95. doi:10.1007/s00125-012-2687-x
17. Schulz TC, Young HY, Agulnick AD, Babin MJ, Baetge EE, Bang AG, et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS One (2012) 7:e37004. doi:10.1371/journal.pone.0037004
18. Rezania A, Riedel MJ, Wideman RD, Karanu F, Ao Z, Warnock GL, et al. Production of functional glucagon-secreting alpha-cells from human embryonic stem cells. Diabetes (2011) 60:239–47. doi:10.2337/db10-0573
19. Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep (2013) 4:1262–75. doi:10.1016/j.celrep.2013.08.010
20. Mahadevan J, Parazzoli S, Oseid E, Hertzel AV, Bernlohr DA, Vallerie SN, et al. Ebselen treatment prevents islet apoptosis, maintains intranuclear Pdx-1 and MafA levels, and preserves beta-cell mass and function in ZDF rats. Diabetes (2013) 62:3582–8. doi:10.2337/db13-0357
21. Harmon JS, Stein R, Robertson RP. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem (2005) 280:11107–13. doi:10.1074/jbc.M410345200
22. Harmon JS, Bogdani M, Parazzoli SD, Mak SS, Oseid EA, Berghmans M, et al. beta-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology (2009) 150:4855–62. doi:10.1210/en.2009-0708
23. Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer (2007) 7:281–94. doi:10.1038/nrc2110
24. Fletcher DJ, Ways DK. Age-dependent expression of protein kinase C isoforms in rat islets. Diabetes (1991) 40:1496–503. doi:10.2337/diabetes.40.11.1496
25. Biden TJ, Schmitz-Peiffer C, Burchfield JG, Gurisik E, Cantley J, Mitchell CJ, et al. The diverse roles of protein kinase C in pancreatic beta-cell function. Biochem Soc Trans (2008) 36:916–9. doi:10.1042/BST0360916
26. Chen S, Borowiak M, Fox JL, Maehr R, Osafune K, Davidow L, et al. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat Chem Biol (2009) 5:258–65. doi:10.1038/nchembio.154
27. Kozikowski AP, Nowak I, Petukhov PA, Etcheberrigaray R, Mohamed A, Tan M, et al. New amide-bearing benzolactam-based protein kinase C modulators induce enhanced secretion of the amyloid precursor protein metabolite sAPPalpha. J Med Chem (2003) 46:364–73. doi:10.1021/jm020350r
28. Rhinn M, Dolle P. Retinoic acid signalling during development. Development (2012) 139:843–58. doi:10.1242/dev.065938
29. Martin M, Gallego-Llamas J, Ribes V, Kedinger M, Niederreither K, Chambon P, et al. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev Biol (2005) 284:399–411. doi:10.1016/j.ydbio.2005.05.035
30. Molotkov A, Molotkova N, Duester G. Retinoic acid generated by Raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev Dyn (2005) 232:950–7. doi:10.1002/dvdy.20256
31. Kadison A, Kim J, Maldonado T, Crisera C, Prasadan K, Manna P, et al. Retinoid signaling directs secondary lineage selection in pancreatic organogenesis. J Pediatr Surg (2001) 36:1150–6. doi:10.1053/jpsu.2001.25734
32. Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, et al. Control of endodermal endocrine development by Hes-1. Nat Genet (2000) 24:36–44. doi:10.1038/71657
33. Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, et al. Notch signalling controls pancreatic cell differentiation. Nature (1999) 400:877–81. doi:10.1038/23716
34. Miralles F, Lamotte L, Couton D, Joshi RL. Interplay between FGF10 and Notch signalling is required for the self-renewal of pancreatic progenitors. Int J Dev Biol (2006) 50:17–26. doi:10.1387/ijdb.052080fm
35. Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A (2003) 100:14920–5. doi:10.1073/pnas.2436557100
36. Dror V, Nguyen V, Walia P, Kalynyak TB, Hill JA, Johnson JD. Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia (2007) 50:2504–15. doi:10.1007/s00125-007-0835-5
37. Cortizo AM, Gomez Dumm CL, Gagliardino JJ. Effect of thyroid hormone levels upon pancreatic islet function. Acta Physiol Pharmacol Latinoam (1985) 35:181–91.
38. Lin Y, Sun Z. Thyroid hormone potentiates insulin signaling and attenuates hyperglycemia and insulin resistance in a mouse model of type 2 diabetes. Br J Pharmacol (2011) 162:597–610. doi:10.1111/j.1476-5381.2010.01056.x
39. Verga Falzacappa C, Mangialardo C, Raffa S, Mancuso A, Piergrossi P, Moriggi G, et al. The thyroid hormone T3 improves function and survival of rat pancreatic islets during in vitro culture. Islets (2010) 2:96–103. doi:10.4161/isl.2.2.11170
40. Goulart-Silva F, Teixeira Sda S, Luchessi AD, Dos Santos LR, Rebelato E, Carpinelli AR, et al. Potential contribution of translational factors to triiodo-L-thyronine-induced insulin synthesis by pancreatic beta cells. Thyroid (2012) 22:637–42. doi:10.1089/thy.2011.0252
41. Verga Falzacappa C, Petrucci E, Patriarca V, Michienzi S, Stigliano A, Brunetti E, et al. Thyroid hormone receptor TRbeta1 mediates Akt activation by T3 in pancreatic beta cells. J Mol Endocrinol (2007) 38:221–33. doi:10.1677/jme.1.02166
42. Kim TK, Lee JS, Jung HS, Ha TK, Kim SM, Han N, et al. Triiodothyronine induces proliferation of pancreatic beta-cells through the MAPK/ERK pathway. Exp Clin Endocrinol Diabetes (2014) 122:240–5. doi:10.1055/s-0034-1367060
43. Misiti S, Anastasi E, Sciacchitano S, Verga Falzacappa C, Panacchia L, Bucci B, et al. 3,5,3’-Triiodo-L-thyronine enhances the differentiation of a human pancreatic duct cell line (hPANC-1) towards a beta-cell-like phenotype. J Cell Physiol (2005) 204:286–96. doi:10.1002/jcp.20293
44. Aiello V, Moreno-Asso A, Servitja JM, Martin M. Thyroid hormones promote endocrine differentiation at expenses of exocrine tissue. Exp Cell Res (2014) 322:236–48. doi:10.1016/j.yexcr.2014.01.030
45. Aguayo-Mazzucato C, Zavacki AM, Marinelarena A, Hollister-Lock J, El Khattabi I, Marsili A, et al. Thyroid hormone promotes postnatal rat pancreatic beta-cell development and glucose-responsive insulin secretion through MAFA. Diabetes (2013) 62:1569–80. doi:10.2337/db12-0849
46. Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res (2008) 100:35–83. doi:10.1016/S0065-230X(08)00002-X
47. Augustine KA, Rossi RM, Van G, Housman J, Stark K, Danilenko D, et al. Noninsulin-dependent diabetes mellitus occurs in mice ectopically expressing the human Axl tyrosine kinase receptor. J Cell Physiol (1999) 181:433–47. doi:10.1002/(SICI)1097-4652(199912)181:3<433::AID-JCP7>3.0.CO;2-Y
48. Haase TN, Rasmussen M, Jaksch CA, Gaarn LW, Petersen CK, Billestrup N, et al. Growth arrest specific protein (GAS) 6: a role in the regulation of proliferation and functional capacity of the perinatal rat beta cell. Diabetologia (2013) 56:763–73. doi:10.1007/s00125-012-2821-9
49. Zhou A, Thorn NA. High ascorbic acid content in the rat endocrine pancreas. Diabetologia (1991) 34:839–42. doi:10.1007/BF00408361
50. Behrens WA, Madere R. Vitamin C and vitamin E status in the spontaneously diabetic BB rat before the onset of diabetes. Metabolism (1991) 40:72–6. doi:10.1016/0026-0495(91)90195-3
51. Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes (1999) 48:2398–406. doi:10.2337/diabetes.48.12.2398
52. Huotari MA, Miettinen PJ, Palgi J, Koivisto T, Ustinov J, Harari D, et al. ErbB signaling regulates lineage determination of developing pancreatic islet cells in embryonic organ culture. Endocrinology (2002) 143:4437–46. doi:10.1210/en.2002-220382
53. Sasada R, Ono Y, Taniyama Y, Shing Y, Folkman J, Igarashi K. Cloning and expression of cDNA encoding human betacellulin, a new member of the EGF family. Biochem Biophys Res Commun (1993) 190:1173–9. doi:10.1006/bbrc.1993.1173
54. Silver K, Tolea M, Wang J, Pollin TI, Yao F, Mitchell BD. The exon 1 Cys7Gly polymorphism within the betacellulin gene is associated with type 2 diabetes in African Americans. Diabetes (2005) 54:1179–84. doi:10.2337/diabetes.54.4.1179
55. Beerli RR, Hynes NE. Epidermal growth factor-related peptides activate distinct subsets of ErbB receptors and differ in their biological activities. J Biol Chem (1996) 271:6071–6. doi:10.1074/jbc.271.11.6071
56. Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol (2009) 21:177–84. doi:10.1016/j.ceb.2008.12.010
57. Mashima H, Ohnishi H, Wakabayashi K, Mine T, Miyagawa J, Hanafusa T, et al. Betacellulin and activin A coordinately convert amylase-secreting pancreatic AR42J cells into insulin-secreting cells. J Clin Invest (1996) 97:1647–54. doi:10.1172/JCI118591
58. Watada H, Kajimoto Y, Miyagawa J, Hanafusa T, Hamaguchi K, Matsuoka T, et al. PDX-1 induces insulin and glucokinase gene expressions in alphaTC1 clone 6 cells in the presence of betacellulin. Diabetes (1996) 45:1826–31. doi:10.2337/diabetes.45.12.1826
59. Yamamoto K, Miyagawa J, Waguri M, Sasada R, Igarashi K, Li M, et al. Recombinant human betacellulin promotes the neogenesis of beta-cells and ameliorates glucose intolerance in mice with diabetes induced by selective alloxan perfusion. Diabetes (2000) 49:2021–7. doi:10.2337/diabetes.49.12.2021
60. Li L, Seno M, Yamada H, Kojima I. Promotion of beta-cell regeneration by betacellulin in ninety percent-pancreatectomized rats. Endocrinology (2001) 142:5379–85. doi:10.1210/en.142.12.5379
61. Li L, Seno M, Yamada H, Kojima I. Betacellulin improves glucose metabolism by promoting conversion of intraislet precursor cells to beta-cells in streptozotocin-treated mice. Am J Physiol Endocrinol Metab (2003) 285:E577–83. doi:10.1152/ajpendo.00120.2003
62. Yamamoto Y, Yamada S, Kodera T, Hara A, Motoyoshi K, Tanaka Y, et al. Reversal of streptozotocin-induced hyperglycemia by continuous supply of betacellulin in mice. Growth Factors (2008) 26:173–9. doi:10.1080/08977190802136854
63. Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, et al. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J (2003) 22:2704–16. doi:10.1093/emboj/cdg264
64. Normanno N, Bianco C, Strizzi L, Mancino M, Maiello MR, De Luca A, et al. The ErbB receptors and their ligands in cancer: an overview. Curr Drug Targets (2005) 6:243–57. doi:10.2174/1389450053765879
65. Dahlhoff M, Dames PM, Lechner A, Herbach N, van Burck L, Wanke R, et al. Betacellulin overexpression in transgenic mice improves glucose tolerance and enhances insulin secretion by isolated islets in vitro. Mol Cell Endocrinol (2009) 299:188–93. doi:10.1016/j.mce.2008.11.022
66. Schneider MR, Dahlhoff M, Herbach N, Renner-Mueller I, Dalke C, Puk O, et al. Betacellulin overexpression in transgenic mice causes disproportionate growth, pulmonary hemorrhage syndrome, and complex eye pathology. Endocrinology (2005) 146:5237–46. doi:10.1210/en.2005-0418
67. Silver KD, Magnuson VL, Tolea M, Wang J, Hagopian WA, Mitchell BD. Association of a polymorphism in the betacellulin gene with type 1 diabetes mellitus in two populations. J Mol Med (Berl) (2006) 84:616–23. doi:10.1007/s00109-006-0052-6
68. Oh YS, Shin S, Lee YJ, Kim EH, Jun HS. Betacellulin-induced beta cell proliferation and regeneration is mediated by activation of ErbB-1 and ErbB-2 receptors. PLoS One (2011) 6:e23894. doi:10.1371/journal.pone.0023894
69. Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, et al. Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J Clin Invest (2003) 112:1521–32. doi:10.1172/JCI18581
70. Brun T, Franklin I, St-Onge L, Biason-Lauber A, Schoenle EJ, Wollheim CB, et al. The diabetes-linked transcription factor PAX4 promotes {beta}-cell proliferation and survival in rat and human islets. J Cell Biol (2004) 167:1123–35. doi:10.1083/jcb.200405148
71. Cho YM, Lim JM, Yoo DH, Kim JH, Chung SS, Park SG, et al. Betacellulin and nicotinamide sustain PDX1 expression and induce pancreatic beta-cell differentiation in human embryonic stem cells. Biochem Biophys Res Commun (2008) 366:129–34. doi:10.1016/j.bbrc.2007.11.112
72. Thowfeequ S, Ralphs KL, Yu WY, Slack JM, Tosh D. Betacellulin inhibits amylase and glucagon production and promotes beta cell differentiation in mouse embryonic pancreas. Diabetologia (2007) 50:1688–97. doi:10.1007/s00125-007-0724-y
Keywords: pancreas, islet, beta cell, human, pluripotent stem cell
Citation: Santosa MM, Low BSJ, Pek NMQ and Teo AKK (2016) Knowledge Gaps in Rodent Pancreas Biology: Taking Human Pluripotent Stem Cell-Derived Pancreatic Beta Cells into Our Own Hands. Front. Endocrinol. 6:194. doi: 10.3389/fendo.2015.00194
Received: 30 October 2015; Accepted: 25 December 2015;
Published: 14 January 2016
Edited by:
Robert Kenneth Semple, University of Cambridge, UKReviewed by:
Eusebio Chiefari, University of Catanzaro, ItalyNeil Hanley, University of Manchester, UK
Copyright: © 2016 Santosa, Low, Pek and Teo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrian Kee Keong Teo, ateo@imcb.a-star.edu.sg, drainteo@gmail.com