 Isabel Huang-Doran
Isabel Huang-Doran Stephen Franks
Stephen Franks- Institute of Reproductive and Developmental Biology, Department of Surgery and Cancer, Imperial College London, Hammersmith Hospital, London, UK
Polycystic ovary syndrome (PCOS) is the most common endocrinopathy affecting women and a leading cause of female infertility worldwide. Defined clinically by the presence of hyperandrogenemia and oligomenorrhoea, PCOS represents a state of hormonal dysregulation, disrupted ovarian follicle dynamics, and subsequent oligo- or anovulation. The syndrome’s prevalence is attributed, at least partly, to a well-established association with obesity and insulin resistance (IR). Indeed, the presence of severe PCOS in human genetic obesity and IR syndromes supports a causal role for IR in the pathogenesis of PCOS. However, the molecular mechanisms underlying this causality, as well as the important role of hyperandrogenemia, remain poorly elucidated. As such, treatment of PCOS is necessarily empirical, focusing on symptom alleviation. The generation of knockout and transgenic rodent models of obesity and IR offers a promising platform in which to address mechanistic questions about reproductive dysfunction in the context of metabolic disease. Similarly, the impact of primary perturbations in rodent gonadotrophin or androgen signaling has been interrogated. However, the insights gained from such models have been limited by the relatively poor fidelity of rodent models to human PCOS. In this mini review, we evaluate the ovarian phenotypes associated with rodent models of obesity and IR, including the extent of endocrine disturbance, ovarian dysmorphology, and subfertility. We compare them to both human PCOS and other animal models of the syndrome (genetic and hormonal), explore reasons for their discordance, and consider the new opportunities that are emerging to better understand and treat this important condition.
Introduction
The association between obesity, insulin resistance (IR), type II diabetes (T2DM), cardiovascular disease, and non-alcoholic fatty liver disease is well-established in the literature, discussed commonly in the clinic, and subject to intensive investigation in laboratories worldwide (1, 2). Perhaps less recognized is obesity’s association with ovarian dysfunction, most commonly in the form of polycystic ovary syndrome (PCOS). Diagnostic criteria of PCOS incorporate three key features: biochemical and/or clinical evidence of androgen excess (including acne, hirsutism, and alopecia), ovarian dysfunction or anovulation (manifesting as absent or irregular menstruation), and the appearance of multiple peripheral cysts on ovarian ultrasonography (3). Rarer causes of raised androgen levels (such as an androgen-producing tumor) are first excluded. Metabolic dysfunction is common but not invariable in women with PCOS and so, although cross-sectional and longitudinal studies support a significant role for IR in the etiology of PCOS, diagnostic criteria do not currently incorporate metabolic parameters. Nevertheless, not only is PCOS the most common cause of anovulatory infertility and menstrual irregularity but (since it often manifests in the second and third decades) young women with PCOS also represent a large, identifiable group who may be at increased risk of metabolic (4–6) and cardiovascular diseases (7–10). Indeed, PCOS is a strong predictor of future T2DM (11). Women with PCOS therefore represent an important target for research and prevention.
The heterogeneous nature of PCOS, along with a lack of consensus over precise diagnostic criteria, has complicated efforts to understand its pathogenesis. Familial clustering studies and monozygotic twin concordance reveal an important genetic predisposition to the syndrome. Genetic variants identified from candidate gene screening and genome-wide association studies implicate insulin, growth factor, and gonadotrophin signaling, cellular proliferation, and DNA repair pathways; however, they so far account for less than 10 percent of the syndrome’s heritability (12). The presence of PCOS-like features in animals exposed prenatally to androgens suggests that PCOS may have important developmental origins (13). Genetic and developmental influences likely interact with environmental factors in adolescence and adulthood to produce the complex physiological dysregulation that characterizes this syndrome.
Hormonal models, in which rodents, sheep, and non-human primates are treated during development or postnatally with androgens (testosterone, DHT, or DHEA), estrogens, aromatase inhibitors, or antiprogestins, are widely employed in PCOS research (14–19). Genetic rodent models offer a complementary albeit underutilized strategy in this field, allowing the contribution of individual genes to be evaluated on “clean” genetic backgrounds and providing tractable and affordable models in which to interrogate disease pathways (14, 20–23). Their value, however, depends on the fidelity of the model to human physiology and disease and the relevance of single-gene perturbations. After summarizing some main concepts relating to the pathogenesis of PCOS (Figure 1), we describe key rodent models relevant to the study of ovarian dysfunction in metabolic diseases (Table 1) and explore why their interpretation may be more complicated than initially apparent.
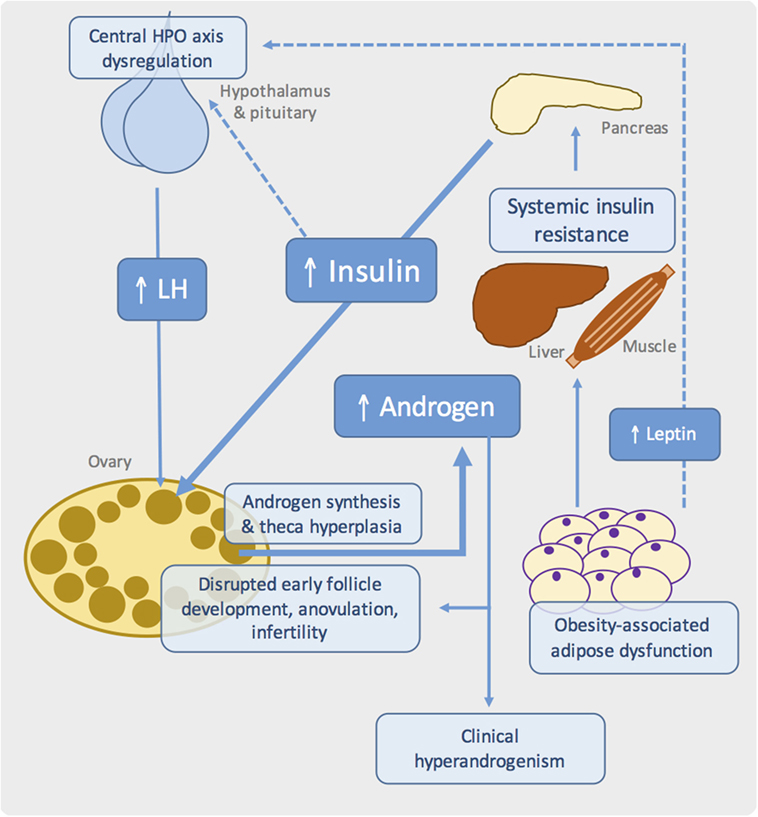
Figure 1. Proposed pathogenic mechanisms in obesity-associated ovarian dysfunction and subfertility. Schematic showing the major metabolic and reproductive pathways involved in PCOS. Systemic insulin resistance, commonly due to adipose tissue dysfunction in the context of obesity, results in compensatory hyperinsulinemia. At the ovary, insulin synergizes with luteinizing hormone (LH) to drive androgen synthesis. Disrupted insulin, growth factor, gonadotrophin, and sex steroid signaling in the ovary leads to failure of follicle development and ovulation. Genetic and developmental influences are also likely to play an important role.
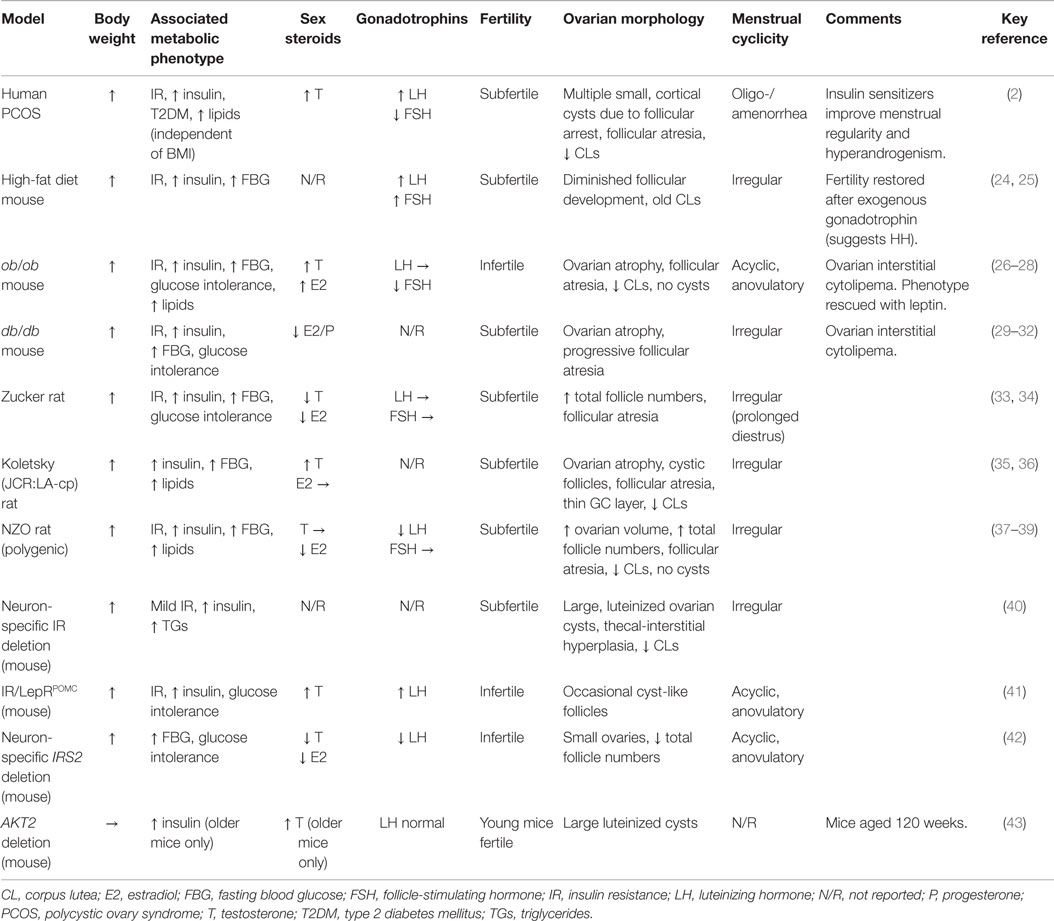
Table 1. Reproductive features of rodent models of obesity and insulin resistance.
Key Players in PCOS Pathogenesis
Metabolic Features of PCOS
While PCOS is robustly associated with impaired insulin sensitivity and hyperinsulinemia (Table 1), this is independent of body weight, and a significant proportion of insulin-resistant women with PCOS are lean (44, 45). However, it is recognized that increased body weight exacerbates hyperandrogenism, oligomenorrhoea, and metabolic risk in PCOS (46, 47), and genetic studies have revealed a role for obesity-associated genes (48, 49).
Several observations suggest that IR, and associated compensatory hyperinsulinemia, may play a key pathogenic role in PCOS. Firstly, IR is more common in women with both hyperandrogenism and anovulation, compared to weight-matched hyperandrogenemic women with normal ovulatory cycles (50). Second, interventions that increase insulin sensitivity improve, independent of weight loss, ovulatory function, menstrual cyclicity, fertility, and hyperandrogenism in lean and obese patients (51–55). Third, a severe PCOS-like syndrome is a prominent (often-presenting) feature in patients with severe, genetic forms of IR (56) and is also reportedly associated with pancreatic insulinomata and excessive exogenous insulin in type 1 diabetes (57, 58).
Importantly, PCOS likely represents a state of “partial” IR, in which preserved insulin signaling in ovarian theca cells causes excessive androgen synthesis and theca cell proliferation, with subsequent hyperandrogenemia (Figure 1) (59–62). Other potential effects of hyperinsulinemia include reduced hepatic synthesis of sex hormone-binding globulin, thereby increasing free testosterone, hypersecretion of pituitary luteinizing hormone (LH), and reduced insulin-like growth factor-binding protein (63–65). This latter effect potentially modulates the paracrine growth factor-dependent regulation of early follicle development and dominant follicle selection (Figure 1).
Ovarian Dysmorphology
The abnormal appearance of the ovarian cortex in PCOS represents inappropriate and excessive initiation of follicle growth from the primordial follicle pool, followed by developmental failure and growth arrest at the medium-sized antral stage (5–10 mm) (66–68). Loss of coordinated follicle development results in fewer or absent ovulations, and therefore subfertility. Histologically, the ovary contains a reduced number of corpora lutea (representing fewer ovulations), more atretic follicles, stromal hypertrophy, and increased ovarian weight. As mentioned, hyperthecosis is prominent, with in vitro evidence suggesting that abnormal thecal cell proliferation contributes to excessive androgen biosynthesis (62, 69).
Hormonal Dysregulation
While IR and hyperinsulinemia may play a central, and in some cases primary, role in PCOS pathogenesis, the importance of hyperandrogenism should be stressed. Not only is it a defining feature of the syndrome, both in ovulatory and anovulatory women, but other conditions associated with excessive androgen exposure (such as congenital adrenal hyperplasia and androgen-secreting tumors) also produce features of PCOS (70). Moreover, administration of androgens in rodents, sheep, and non-human primates results in pathophysiological changes that closely resemble features of PCOS in women. Androgens act at the ovary to disrupt follicular development and dominant follicle selection by promoting excessive early follicular growth, while systemic effects include development of IR and metabolic dysfunction (71–77). The role of androgens in PCOS may be particularly important during key developmental windows before the onset of IR (13). Prenatally, androgenized rhesus monkeys and sheep demonstrate ovarian hyperandrogenism and IR in adulthood, with increased follicle numbers, anovulation, and LH hypersecretion (78–81).
Dysregulation and reprograming of the hypothalamus–pituitary–ovarian (HPO) axis is common in PCOS, potentially driven by androgen exposure in utero and manifesting as hypersecretion of LH, persistently rapid LH pulse frequency, and below-normal levels of follicle-stimulating hormone (FSH) (82, 83). These alterations likely contribute to disrupted follicle development in PCOS, while high levels of LH also synergize with insulin to promote theca androgen production (Figure 1). However, it is noteworthy that many patients have normal LH levels, suggesting that elevated gonadotrophin levels is unlikely to be the primary defect in PCOS (84).
Ovarian Dysfunction in Genetic Models of Metabolic Disease
Rodent Models of Obesity
While there is no spontaneously occurring animal model of PCOS, transgenic and knockout rodent models widely used in metabolic research provide opportunities to study specifically the association between metabolic disease and ovarian dysfunction. However, it is important to note that key differences exist between human and rodent ovarian function. Whereas in humans, full follicular differentiation occurs in the later stages of fetal development, in rodents this occurs postnatally. The mouse estrus cycle lasts only 4–6 days, compared to 28 days in humans. Furthermore, rodents are polyovulatory, suggesting important differences in dominant follicle selection despite underlying similarities in the HPO axis.
In spite of these differences, various rodent models of obesity do display reproductive phenotypes comparable to PCOS (Table 1). Diet-induced obesity in wild-type mice is associated with disrupted estrus cyclicity, fewer corpora lutea, reduced fertility, and metabolic dysfunction, supporting the notion that obesity-associated metabolic dysfunction may contribute to PCOS (24, 25, 85). Among the genetic models, female ob/ob and db/db mice, which, due to loss-of-function mutations in leptin and leptin receptor, respectively, are hyperphagic, severely obese, hyperinsulinemic, and hyperglycemic are also infertile, acyclic, and anovulatory (Table 1). Morphologically, they show utero-ovarian atrophy, follicular atresia, apoptotic granulosa cells, deformed oocytes, absent corpora lutea, and no cystic structures (26, 27, 29–32). The endocrine profile of ob/ob mice includes elevated serum testosterone, estradiol, and progesterone, with reduced FSH but normal LH, while db/db mice have low estradiol and progesterone. The obese Koletsky and Zucker diabetic fat rats, both of which also lack functional leptin receptors, do exhibit estrus cycling (albeit irregularly) but are subfertile with increased follicle numbers, follicular atresia, and fewer corpora lutea. While androgen levels are elevated in the obese Koletsky, in Zucker, they are reportedly below normal (33–36). The New Zealand obese (NZO) mouse, notable for being a polygenic model of the human metabolic syndrome (37), also harbors leptin receptor variants and displays a reproductive phenotype similar to that of Zucker (Table 1) (38, 39).
In all of these models, reproductive dysfunction is at least partly attributable to loss of hypothalamic leptin signaling, rather than obesity per se. Genetic leptin deficiency in humans is associated with low gonadotrophins and pubertal failure, which are restored with leptin replacement (86). Fertility, litter size, and estrus cyclicity of ob/ob mice were similarly ameliorated by human recombinant leptin (87, 88) or transplantation with wild-type adipose tissue (89, 90). Along with other peripheral signals, leptin is believed to modulate the activity of gonadotrophin-releasing hormone (GnRH) releasing neurons – and thus the entire HPO axis – in response to nutritional status (91). Indeed, low body weight is known to interfere with reproductive function and pubertal timing (92). Failure of central leptin action in rodent models of obesity therefore leads to infertility due to hypogonadotrophic hypogonadism and follicle development (Figure 1). Indeed, human obesity is also associated with a degree of hypothalamic leptin resistance, which may contribute to HPO dysregulation in PCOS (93, 94). Reports of excess lipid accumulation in follicular cells of ob/ob mice and the obese Koletsky rat suggest an additional “lipotoxic” mechanism by which extreme obesity may produce ovarian dysfunction, although there are no reports of such a phenotype in PCOS (30, 36).
In these models, the relative contribution of IR-associated hyperinsulinemia and central leptin resistance is difficult to disentangle, particularly since hypothalamic insulin signaling also regulates GnRH release and thus reproduction function (40, 41, 95–97). Mice with neuron-specific deletion of the insulin receptor gene (Insr) or hypothalamic POMC neuron-specific deletion of both leptin and Insr were hyperphagic, insulin resistant, and subfertile due to impaired follicular development (Table 1) (40, 41, 98). The combined knockout was notable for high levels of LH, hyperandrogenemia, and cyst-like follicles. POMC-specific deletion of leptin receptor alone produced only a subtle reproductive phenotype (99). Counterintuitively, pituitary-specific Insr knockout reportedly rescued the PCOS-like phenotype associated with diet-induced obesity (24). These observations highlight complex interactions between leptin and insulin in their regulation of reproductive function. Indeed, studies in mammals and non-mammalian species reveal that nutritional status and reproductive capacity are tightly intertwined, ensuring that reproduction only proceeds if nutritional status is optimal (100).
Genetic Models of Insulin Resistance
In humans, rare loss-of-function mutations in INSR not only cause extreme hyperinsulinemia but also oligomenorrhoea, hyperandrogenism, and excessive development of sex hormone-dependent tissues (56). Common genetic defects in insulin signaling are suggested to contribute to PCOS heritability (101, 102), and cellular studies reveal abnormalities in insulin-mediated insulin receptor autophosphorylation, IRS expression, PI3-kinase activation, GLUT4 expression, and insulin-stimulated glucose uptake in adipocytes and skeletal muscle from women with PCOS (103–107). However, the results of such studies are variable and need further verification.
Mice lacking functional insulin receptor develop profound metabolic abnormalities at birth and die within days. Of the tissue-specific knockouts, only those targeting the brain have a reported reproductive phenotype (108). Similar to the neuron-specific Insr knockout, global deletion of Irs2 (but not Irs1) causes a combination of metabolic, reproductive, and ovarian features that likely result from disrupted central insulin and leptin action rather than abnormal systemic glucose metabolism (42) (Table 1). Thus, in addition to the impact of systemic hyperinsulinemia, the interpretation of global insulin signaling defects must consider the actions of insulin at the hypothalamus as well as disruption to the regulation of early follicle development by IGF1. There are no corresponding human syndromes of IRS dysfunction or deficiency with which to compare.
Downstream of IRS in the signaling pathway, non-functional mutations in human AKT2 result in ovarian hyperandrogenism in the context of partial lipodystrophy, severe IR, diabetes, metabolic dyslipidemia, and fatty liver (109). In mice, global AKT2 deletion produced a somewhat comparable ovarian phenotype, with increased androgenic steroidogenesis in the theca-interstitium, theca-interstitial hyperplasia, hyperandrogenemia, reduced corpora lutea, and ovarian cysts but normal LH levels (Table 1) (43). However, the large, luteinized, serous-filled cysts were quite distinct from the ovarian morphology characterizing human PCOS. For unclear reasons, reproductive features were absent in younger mice, although could be induced by treatment with LH, perhaps due to synergism with hyperinsulinemia.
Other human lipodystrophy syndromes (genetic or acquired) are similarly characterized by severe IR, ovarian hyperandrogenism, amenorrhea, and infertility (110–112). While genetic mouse models of generalized lipodystrophy manifest many metabolic features of the human diseases, “partial” lipodystrophy has been more challenging to model (113). Moreover, while the metabolic properties of these models have been interrogated in detail, their reproductive and ovarian phenotypes have not been reported widely. Studying these models may provide important new insights into the role of BMI-independent IR in PCOS-like ovarian dysfunction.
Genetic Models Targeting the HPO Axis
To better understand PCOS pathogenesis, rodent models of obesity and IR should be considered alongside those in which other implicated systems are targeted. Transgenic mice with chronically elevated gonadotrophin levels have a thickened theca cell layer, similar to PCOS, with correspondingly increased estrogen and testosterone levels (23, 114). However, unlike PCOS, their ovaries contain large, hemorrhagic cysts, as do those of mice lacking LH receptor (114, 115). Global or theca-specific deficiency of estrogen receptor subunits ERα or ERβ, or global deficiency of aromatase, produces chronically elevated gonadotrophins (due to lack of estradiol), arrested follicular growth, absent corpora lutea and anovulation (116–118). ERα knockout mice also show increased adiposity (without hyperphagia), IR, and diabetes (118, 119), whereas constitutive elevation of LH activity produces hyperphagic obesity with hyperleptinemia and hyperinsulinemia (120). These observations further illuminate the complexity of nutritional and reproductive cross talk in humans, again challenging the value of simplified rodent models.
Transgenic Rodents and PCOS – Not Fit for Purpose?
This discussion reveals that transgenic models of PCOS are complex, heterogeneous, and even the best examples deviate in important ways from the human syndrome. Models of obesity and IR have not typically been studied comprehensively from a reproductive perspective. Even when a reproductive deficit is noted, the ovarian and endocrinological phenotyping is often incomplete, with concerns raised over timing of the studies (relative to time of day, phase of the estrous cycle, and age of the animal), the rigor of morphological analyses, and the variability of ovarian appearances described as “cystic.” Furthermore, as outlined above, important differences exist between human and rodent ovarian function. Such differences may explain why the reproductive consequences of androgen exposure are less consistent in rodents than in sheep or primates, and emphasize that results from rodent-based studies (genetic or hormonal) need to be extrapolated with caution to human PCOS (13, 23).
Emerging from this discussion is an important reminder that reproductive capacity and nutritional status are intertwined tightly through feedback and cross talk between reproductive and metabolic pathways. Across a wide range of species, including Caenorhabditis elegans and Drosophila, conserved mechanisms operate to regulate reproduction and energy homeostasis (121–123). In rodent models of obesity, the same lesions that produce hyperphagia also directly impact on the HPO axis, thereby complicating their interpretation. The bidirectional interaction between reproductive and nutritional signaling also operates systemically: while estrogen drives adipogenesis, and while testosterone drives food intake, both steroids in excess produce IR, hyperinsulinemia, high levels of circulating leptin, and reduced levels of adiponectin, all of which impact on the HPO axis and ovarian function. The hope of mimicking this complex network by perturbing single or a few genes is perhaps ambitious. Indeed, the notion that PCOS is precipitated by a single etiological factor is undoubtedly too simple. While monogenic perturbations in insulin signaling or adipose function in humans do produce PCOS-like syndromes, differences between human and rodent metabolism and reproduction mean that PCOS will not necessarily emerge from equivalent defects in mice. As in all complex human disease, the role of genetic, developmental, and environmental factors likely contribute heavily to the heterogeneity of human PCOS.
Future Opportunities
Complementary strategies are required to better understand this growing health problem. The combined use of hormonal treatments in transgenic animals may afford interesting, clinically relevant insights. Primary follicular cell and whole follicle cultures, including from transgenic animals, facilitate the study of tightly regulated paracrine and autocrine networks in early follicle development that become disordered in PCOS (124). The ease and efficiency of CRISPR-Cas9-based gene editing technologies will doubtless prove invaluable, particularly to explore new susceptibility loci emerging from large GWAS studies (48, 49, 101, 102, 125). Many of these loci implicate genes of largely unknown function. As they are investigated over the coming years, prudent selection of appropriate cell and animal systems will be imperative. The study of candidate genes in non-ovarian cell types is questionable, yet primary cultures are difficult to acquire and maintain, and ovarian cell lines are too atypical in their properties to be useful. Therefore, in spite of reservations highlighted above, transgenic rodent models will likely play an ongoing role in our effort to better understand and manage this challenging condition.
Conclusion
A clear relationship exists between obesity, metabolic dysregulation, and ovarian dysfunction. However, the mechanisms of this association are poorly understood. Without detailed knowledge of the etiology of PCOS, management is limited to empirical and symptomatic treatment. While hormonal models of PCOS demonstrate an important role for hyperandrogenemia, the reported genetic models incompletely replicate the PCOS phenotype. Their study has offered important insights into the interaction between metabolism and reproduction, but clear conclusions about PCOS pathogenesis have not been forthcoming. Nevertheless, specific models may prove useful for answering reductionist questions about aspects of the condition, such as disordered folliculogenesis or disruption of the HPO axis. Future efforts will benefit from ongoing combined study of humans, mouse models, and cells, driven by insights emerging from human genetic studies. These studies will continue to advance our understanding of this important condition and, with time, support new approaches to addressing both the metabolic and reproductive problems faced by affected women.
Author Contributions
Both IH-D and SF contributed to the content, writing, and editing of this manuscript.
Conflict of Interest Statement
The authors declare that this article was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
IH-D is supported by the National Institute for Health Research. SF is supported by the Medical Research Council (program grant G0802702, project grant MR/M012638/1) and Wellbeing of Women (project grant RG1853).
Abbreviations
FSH, follicle-stimulating hormone; GnRH, gonadotrophin-releasing hormone; IR, insulin resistance; LH, luteinizing hormone; PCOS, polycystic ovary syndrome; T2DM, type 2 diabetes mellitus.
References
1. Franks S. Polycystic ovary syndrome. N Engl J Med (1995) 333(13):853–61. doi: 10.1056/NEJM199509283331307
2. Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol (2011) 7(4):219–31. doi:10.1038/nrendo.2010.217
3. Dunaif A, Fauser BCJM. Renaming PCOS – a two-state solution. J Clin Endocrinol Metab (2013) 98(11):4325–8. doi:10.1210/jc.2013-2040
4. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes (1989) 38(9):1165–74. doi:10.2337/diab.38.9.1165
5. Dunaif A, Graf M, Mandeli J, Laumas V, Dobrjansky A. Characterization of groups of hyperandrogenic women with acanthosis nigricans, impaired glucose tolerance, and/or hyperinsulinemia. J Clin Endocrinol Metab (1987) 65(3):499–507. doi:10.1210/jcem-65-3-499
6. Lo Dico G, Alongi G, Savatteri L, Rini GB, Mascellino MR, Di Fede G, et al. Polycystic ovary syndrome: obesity, insulin resistance, hyperandrogenism. Acta Eur Fertil (1989) 20(5):309–13.
7. Orio F, Vuolo L, Palomba S, Lombardi G, Colao A. Metabolic and cardiovascular consequences of polycystic ovary syndrome. Minerva Ginecol (2008) 60(1):39–51.
8. Baldani DP, Skrgatic L, Ougouag R. Polycystic ovary syndrome: important underrecognised cardiometabolic risk factor in reproductive-age women. Int J Endocrinol (2015) 2015:786362. doi:10.1155/2015/786362
9. Daan NM, Louwers YV, Koster MP, Eijkemans MJ, de Rijke YB, Lentjes EW, et al. Cardiovascular and metabolic profiles amongst different polycystic ovary syndrome phenotypes: who is really at risk? Fertil Steril (2014) 102(5):1444.e–51.e. doi:10.1016/j.fertnstert.2014.08.001
10. Bhathena RK. Insulin resistance and the long-term consequences of polycystic ovary syndrome. J Obstet Gynaecol (2011) 31(2):105–10. doi:10.3109/01443615.2010.539722
11. Capula C, Chiefari E, Vero A, Foti DP, Brunetti A, Vero R. Prevalence and predictors of postpartum glucose intolerance in Italian women with gestational diabetes mellitus. Diabetes Res Clin Pract (2014) 105(2):223–30. doi:10.1016/j.diabres.2014.05.008
12. Dunaif A. Perspectives in polycystic ovary syndrome: from hair to eternity. J Clin Endocrinol Metab (2016) 101(3):759–68. doi:10.1210/jc.2015-3780
13. Franks S, Berga SL. Does PCOS have developmental origins? Fertil Steril (2012) 97(1):2–6. doi:10.1016/j.fertnstert.2011.11.029
14. Walters KA, Allan CM, Handelsman DJ. Rodent models for human polycystic ovary syndrome. Biol Reprod (2012) 86(5):1–12. doi:10.1095/biolreprod.111.097808
15. Familiari G, Toscano V, Motta PM. Morphological studies of polycystic mouse ovaries induced by dehydroepiandrosterone. Cell Tissue Res (1985) 240(3):519–28. doi:10.1007/BF00216340
16. Bogovich K. Induction of follicular cysts in rat ovaries by prolonged administration of human chorionic gonadotropin. Adv Exp Med Biol (1987) 219:659–63. doi:10.1007/978-1-4684-5395-9_38
17. Poretsky L, Clemons J, Bogovich K. Hyperinsulinemia and human chorionic gonadotropin synergistically promote the growth of ovarian follicular cysts in rats. Metabolism (1992) 41(8):903–10. doi:10.1016/0026-0495(92)90175-A
18. Beloosesky R, Gold R, Almog B, Sasson R, Dantes A, Land-Bracha A, et al. Induction of polycystic ovary by testosterone in immature female rats: modulation of apoptosis and attenuation of glucose/insulin ratio. Int J Mol Med (2004) 14(2):207–15. doi:10.3892/ijmm.14.2.207
19. Kafali H, Iriadam M, Ozardali I, Demir N. Letrozole-induced polycystic ovaries in the rat: a new model for cystic ovarian disease. Arch Med Res (2004) 35(2):103–8. doi:10.1016/j.arcmed.2003.10.005
20. McNeilly AS, Colin Duncan W. Rodent models of polycystic ovary syndrome. Mol Cell Endocrinol (2013) 373(1–2):2–7. doi:10.1016/j.mce.2012.10.007
21. Oakley O, Lin P. Animal models for the study of polycystic ovarian syndrome. Endocrinol Metab (2011) 26(3):193–202. doi:10.3803/EnM.2011.26.3.193
22. Shi D, Vine DF. Animal models of polycystic ovary syndrome: a focused review of rodent models in relationship to clinical phenotypes and cardiometabolic risk. Fertil Steril (2012) 98(1):185–93. doi:10.1016/j.fertnstert.2012.04.006
23. van Houten ELAF, Visser JA. Mouse models to study polycystic ovary syndrome: a possible link between metabolism and ovarian function? Reprod Biol (2014) 14(1):32–43. doi:10.1016/j.repbio.2013.09.007
24. Brothers KJ, Wu S, DiVall SA, Messmer MR, Kahn CR, Miller RS, et al. Rescue of obesity-induced infertility in female mice due to a pituitary-specific knockout of the insulin receptor. Cell Metab (2010) 12(3):295–305. doi:10.1016/j.cmet.2010.06.010
25. Tortoriello DV, McMinn J, Chua SC. Dietary-induced obesity and hypothalamic infertility in female DBA/2J mice. Endocrinology (2004) 145(3):1238–47. doi:10.1210/en.2003-1406
26. Hamm ML, Bhat GK, Thompson WE, Mann DR. Folliculogenesis is impaired and granulosa cell apoptosis is increased in leptin-deficient mice. Biol Reprod (2004) 71(1):66–72. doi:10.1095/biolreprod.104.027292
27. Olatinwo MO, Bhat GK, Stah CD, Mann DR. Impact of gonadotropin administration on folliculogenesis in prepubertal ob/ob mice. Mol Cell Endocrinol (2005) 245(1–2):121–7. doi:10.1016/j.mce.2005.11.003
28. Barash IA, Cheung CC, Weigle DS, Ren H, Kabigting EB, Kuijper JL, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology (1996) 137(7):3144–7. doi:10.1210/en.137.7.3144
29. Garris DR. Effects of estradiol and progesterone on diabetes-associated utero-ovarian atrophy in C57BL/KsJ (db/db) mutant mice. Anat Rec (1989) 225(4):310–7. doi:10.1002/ar.1092250407
30. Garris DR, Garris BL. Diabetes (db/db) mutation-induced ovarian involution: progressive hypercytolipidemia. Exp Biol Med (Maywood) (2003) 228(9):1040–50.
31. Garris DR. Ovarian hypercytolipidemia induced by obese (ob/ob) and diabetes (db/db) mutations: basis of female reproductive tract involution II. Tissue Cell (2004) 36(3):157–69. doi:10.1016/j.tice.2004.01.001
32. Garris DR, Williams SK, West L. Morphometric evaluation of diabetes-associated ovarian atrophy in the C57BL/KsJ mouse: relationship to age and ovarian function. Anat Rec (1985) 211(4):434–43. doi:10.1002/ar.1092110410
33. Chelich AM, Edmonds ES. Copulatory behavior and reproductive capacity of the genetically obese female Zucker rat. Physiol Behav (1981) 27(2):331–5. doi:10.1016/0031-9384(81)90276-6
34. Honnma H, Endo T, Kiya T, Shimizu A, Nagasawa K, Baba T, et al. Remarkable features of ovarian morphology and reproductive hormones in insulin-resistant Zucker fatty (fa/fa) rats. Reprod Biol Endocrinol (2010) 8(1):73. doi:10.1186/1477-7827-8-73
35. Wu-Peng XS, Chua SC Jr, Okada N, Liu SM, Nicolson M, Leibel RL. Phenotype of the obese Koletsky (f) rat due to Tyr763Stop mutation in the extracellular domain of the leptin receptor (Lepr): evidence for deficient plasma-to-CSF transport of leptin in both the Zucker and Koletsky obese rat. Diabetes (1997) 46(3):513–8. doi:10.2337/diab.46.3.513
36. Shi D, Dyck MK, Uwiera RR, Russell JC, Proctor SD, Vine DF. A unique rodent model of cardiometabolic risk associated with the metabolic syndrome and polycystic ovary syndrome. Endocrinology (2009) 150(9):4425–36. doi:10.1210/en.2008-1612
37. Kluge R, Scherneck S, Schürmann A, Joost HG. Pathophysiology and genetics of obesity and diabetes in the New Zealand obese mouse: a model of the human metabolic syndrome. Methods Mol Biol (2012) 933:59–73. doi:10.1007/978-1-62703-068-7_5
38. Radavelli-Bagatini S, Blair AR, Proietto J, Spritzer PM, Andrikopoulos S. The New Zealand obese mouse model of obesity insulin resistance and poor breeding performance: evaluation of ovarian structure and function. J Endocrinol (2011) 209(3):307–15. doi:10.1530/JOE-11-0022
39. Igel M, Becker W, Herberg L, Joost HG. Hyperleptinemia, leptin resistance, and polymorphic leptin receptor in the New Zealand obese mouse. Endocrinology (1997) 138(10):4234–9. doi:10.1210/endo.138.10.5428
40. Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science (2000) 289(5487):2122–5. doi:10.1126/science.289.5487.2122
41. Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab (2010) 11(4):286–97. doi:10.1016/j.cmet.2010.03.002
42. Burks DJ, Font de Mora J, Schubert M, Withers DJ, Myers MG, Towery HH, et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature (2000) 407(6802):377–82. doi:10.1038/35030105
43. Restuccia DF, Hynx D, Hemmings BA. Loss of PKB/Akt2 predisposes mice to ovarian cyst formation and increases the severity of polycystic ovary formation in vivo. Dis Model Mech (2012) 5(3):403–11. doi:10.1242/dmm.008136
44. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev (1997) 18(6):774–800. doi:10.1210/er.18.6.774
45. Silfen ME, Denburg MR, Manibo AM, Lobo RA, Jaffe R, Ferin M, et al. Early endocrine, metabolic, and sonographic characteristics of polycystic ovary syndrome (PCOS): comparison between nonobese and obese adolescents. J Clin Endocrinol Metab (2003) 88(10):4682–8. doi:10.1210/jc.2003-030617
46. Dahlgren E, Johansson S, Lindstedt G, Knutsson F, Odén A, Janson PO, et al. Women with polycystic ovary syndrome wedge resected in 1956 to 1965: a long-term follow-up focusing on natural history and circulating hormones. Fertil Steril (1992) 57(3):505–13. doi:10.1016/S0015-0282(16)54892-4
47. Wild S, Pierpoint T, McKeigue P, Jacobs H. Cardiovascular disease in women with polycystic ovary syndrome at long-term follow-up: a retrospective cohort study. Clin Endocrinol (Oxf) (2000) 52(5):595–600. doi:10.1046/j.1365-2265.2000.01000.x
48. Barber TM, Bennett AJ, Groves CJ, Sovio U, Ruokonen A, Martikainen H, et al. Association of variants in the fat mass and obesity associated (FTO) gene with polycystic ovary syndrome. Diabetologia (2008) 51(7):1153–8. doi:10.1007/s00125-008-1028-6
49. Day FR, Hinds DA, Tung JY, Stolk L, Styrkarsdottir U, Saxena R, et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat Commun (2015) 6:8464. doi:10.1038/ncomms9464
50. Robinson S, Kiddy D, Gelding SV, Willis D, Niththyananthan R, Bush A, et al. The relationship of insulin insensitivity to menstrual pattern in women with hyperandrogenism and polycystic ovaries. Clin Endocrinol (Oxf) (1993) 39(3):351–5. doi:10.1111/j.1365-2265.1993.tb02376.x
51. Yilmaz M, Biri A, Karakoç A, Törüner F, Bingöl B, Cakir N, et al. The effects of rosiglitazone and metformin on insulin resistance and serum androgen levels in obese and lean patients with polycystic ovary syndrome. J Endocrinol Invest (2005) 28(11):1003–8. doi:10.1007/BF03345339
52. Pasquali R, Gambineri A, Biscotti D, Vicennati V, Gagliardi L, Colitta D, et al. Effect of long-term treatment with metformin added to hypocaloric diet on body composition, fat distribution, and androgen and insulin levels in abdominally obese women with and without the polycystic ovary syndrome. J Clin Endocrinol Metab (2000) 85(8):2767–74. doi:10.1210/jcem.85.8.6738
53. Pasquali R, Antenucci D, Casimirri F, Venturoli S, Paradisi R, Fabbri R, et al. Clinical and hormonal characteristics of obese amenorrheic hyperandrogenic women before and after weight loss. J Clin Endocrinol Metab (1989) 68(1):173–9. doi:10.1210/jcem-68-1-173
54. Gambineri A, Patton L, Vaccina A, Cacciari M, Morselli-Labate AM, Cavazza C, et al. Treatment with flutamide, metformin, and their combination added to a hypocaloric diet in overweight-obese women with polycystic ovary syndrome: a randomized, 12-month, placebo-controlled study. J Clin Endocrinol Metab (2006) 91(10):3970–80. doi:10.1210/jc.2005-2250
55. Baillargeon JP, Jakubowicz DJ, Iuorno MJ, Jakubowicz S, Nestler JE. Effects of metformin and rosiglitazone, alone and in combination, in nonobese women with polycystic ovary syndrome and normal indices of insulin sensitivity. Fertil Steril (2004) 82(4):893–902. doi:10.1016/j.fertnstert.2004.02.127
56. Semple RK, Savage DB, Cochran EK, Gorden P, O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev (2011) 32(4):498–514. doi:10.1210/er.2010-0020
57. Murray RD, Davison RM, Russell RC, Conway GS. Clinical presentation of PCOS following development of an insulinoma: case report. Hum Reprod (2000) 15(1):86–8. doi:10.1093/humrep/15.1.86
58. Barber TM, Franks S. The link between polycystic ovary syndrome and both Type 1 and Type 2 diabetes mellitus: what do we know today? Womens Health (Lond Engl) (2012) 8(2):147–54. doi:10.2217/whe.11.94
59. Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab (1998) 83(6):2001–5. doi:10.1210/jc.83.6.2001
60. Willis D, Mason H, Gilling-Smith C, Franks S. Modulation by insulin of follicle-stimulating hormone and luteinizing hormone actions in human granulosa cells of normal and polycystic ovaries. J Clin Endocrinol Metab (1996) 81(1):302–9. doi:10.1210/jc.81.1.302
61. Musso C, Shawker T, Cochran E, Javor ED, Young J, Gorden P. Clinical evidence that hyperinsulinaemia independent of gonadotropins stimulates ovarian growth. Clin Endocrinol (Oxf) (2005) 63(1):73–8. doi:10.1111/j.1365-2265.2005.02302.x
62. Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N Engl J Med (1996) 335(9):617–23. doi:10.1056/NEJM199608293350902
63. Nestler JE, Strauss JF. Insulin as an effector of human ovarian and adrenal steroid metabolism. Endocrinol Metab Clin North Am (1991) 20(4):807–23.
64. Adashi EY, Hsueh AJ, Yen SS. Insulin enhancement of luteinizing hormone and follicle-stimulating hormone release by cultured pituitary cells. Endocrinology (1981) 108(4):1441–9. doi:10.1210/endo-108-4-1441
65. Kelly CJ, Stenton SR, Lashen H. Insulin-like growth factor binding protein-1 in PCOS: a systematic review and meta-analysis. Hum Reprod Update (2011) 17(1):4–16. doi:10.1093/humupd/dmq027
66. Webber LJ, Stubbs S, Stark J, Trew GH, Margara R, Hardy K, et al. Formation and early development of follicles in the polycystic ovary. Lancet (2003) 362(9389):1017–21. doi:10.1016/S0140-6736(03)14410-8
67. Maciel GA, Baracat EC, Benda JA, Markham SM, Hensinger K, Chang RJ, et al. Stockpiling of transitional and classic primary follicles in ovaries of women with polycystic ovary syndrome. J Clin Endocrinol Metab (2004) 89(11):5321–7. doi:10.1210/jc.2004-0643
68. Barber TM, Alvey C, Greenslade T, Gooding M, Barber D, Smith R, et al. Patterns of ovarian morphology in polycystic ovary syndrome: a study utilising magnetic resonance imaging. Eur Radiol (2010) 20(5):1207–13. doi:10.1007/s00330-009-1643-8
69. Dunaif A, Hoffman AR, Scully RE, Flier JS, Longcope C, Levy LJ, et al. Clinical, biochemical, and ovarian morphologic features in women with acanthosis nigricans and masculinization. Obstet Gynecol (1985) 66(4):545–52.
70. Barnes RB, Rosenfield RL, Ehrmann DA, Cara JF, Cuttler L, Levitsky LL, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab (1994) 79(5):1328–33. doi:10.1210/jc.79.5.1328
71. Rincon J, Holmäng A, Wahlström EO, Lönnroth P, Björntorp P, Zierath JR, et al. Mechanisms behind insulin resistance in rat skeletal muscle after oophorectomy and additional testosterone treatment. Diabetes (1996) 45(5):615–21. doi:10.2337/diab.45.5.615
72. Diamond MP, Grainger D, Diamond MC, Sherwin RS, Defronzo RA. Effects of methyltestosterone on insulin secretion and sensitivity in women. J Clin Endocrinol Metab (1998) 83(12):4420–5. doi:10.1210/jcem.83.12.5333
73. Corbould A. Effects of androgens on insulin action in women: is androgen excess a component of female metabolic syndrome? Diabetes Metab Res Rev (2008) 24(7):520–32. doi:10.1002/dmrr.872
74. Cohen JC, Hickman R. Insulin resistance and diminished glucose tolerance in powerlifters ingesting anabolic steroids. J Clin Endocrinol Metab (1987) 64(5):960–3. doi:10.1210/jcem-64-5-960
75. Corbould A. Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J Endocrinol (2007) 192(3):585–94. doi:10.1677/joe.1.07070
76. Mannerås L, Cajander S, Holmäng A, Seleskovic Z, Lystig T, Lönn M, et al. A new rat model exhibiting both ovarian and metabolic characteristics of polycystic ovary syndrome. Endocrinology (2007) 148(8):3781–91. doi:10.1210/en.2007-0168
77. Demissie M, Lazic M, Foecking EM, Aird F, Dunaif A, Levine JE. Transient prenatal androgen exposure produces metabolic syndrome in adult female rats. Am J Physiol Endocrinol Metab (2008) 295(2):E262–8. doi:10.1152/ajpendo.90208.2008
78. Bruns CM, Baum ST, Colman RJ, Eisner JR, Kemnitz JW, Weindruch R, et al. Insulin resistance and impaired insulin secretion in prenatally androgenized male rhesus monkeys. J Clin Endocrinol Metab (2004) 89(12):6218–23. doi:10.1210/jc.2004-0918
79. Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord (2007) 8(2):127–41. doi:10.1007/s11154-007-9046-0
80. Abbott DH, Zhou R, Bird IM, Dumesic DA, Conley AJ. Fetal programming of adrenal androgen excess: lessons from a nonhuman primate model of polycystic ovary syndrome. Endocr Dev (2008) 13:145–58. doi:10.1159/000134831
81. Abbott DH, Dumesic DA, Eisner JR, Colman RJ, Kemnitz JW. Insights into the development of polycystic ovary syndrome (PCOS) from studies of prenatally androgenized female rhesus monkeys. Trends Endocrinol Metab (1998) 9(2):62–7. doi:10.1016/S1043-2760(98)00019-8
82. Taylor AE, McCourt B, Martin KA, Anderson EJ, Adams JM, Schoenfeld D, et al. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab (1997) 82(7):2248–56. doi:10.1210/jc.82.7.2248
83. Haisenleder DJ, Dalkin AC, Ortolano GA, Marshall JC, Shupnik MA. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology (1991) 128(1):509–17. doi:10.1210/endo-128-1-509
84. Adams JM, Taylor AE, Crowley WF, Hall JE. Polycystic ovarian morphology with regular ovulatory cycles: insights into the pathophysiology of polycystic ovarian syndrome. J Clin Endocrinol Metab (2004) 89(9):4343–50. doi:10.1210/jc.2003-031600
85. Jungheim ES, Schoeller EL, Marquard KL, Louden ED, Schaffer JE, Moley KH. Diet-induced obesity model: abnormal oocytes and persistent growth abnormalities in the offspring. Endocrinology (2010) 151(8):4039–46. doi:10.1210/en.2010-0098
86. Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med (1999) 341(12):879–84. doi:10.1056/NEJM199909163411204
87. Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet (1996) 12(3):318–20. doi:10.1038/ng0396-318
88. Mounzih K, Lu R, Chehab FF. Leptin treatment rescues the sterility of genetically obese ob/ob males. Endocrinology (1997) 138(3):1190–3. doi:10.1210/en.138.3.1190
89. Klebanov S, Astle CM, DeSimone O, Ablamunits V, Harrison DE. Adipose tissue transplantation protects ob/ob mice from obesity, normalizes insulin sensitivity and restores fertility. J Endocrinol (2005) 186(1):203–11. doi:10.1677/joe.1.06150
90. Zhang Y, Hu M, Ma H, Qu J, Wang Y, Hou L, et al. The impairment of reproduction in db/db mice is not mediated by intraovarian defective leptin signaling. Fertil Steril (2012) 97(5):1183–91. doi:10.1016/j.fertnstert.2012.01.126
91. Moschos S, Chan JL, Mantzoros CS. Leptin and reproduction: a review. Fertil Steril (2002) 77(3):433–44. doi:10.1016/S0015-0282(01)03010-2
93. El-Haschimi K, Pierroz DD, Hileman SM, Bjørbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest (2000) 105(12):1827–32. doi:10.1172/JCI9842
94. Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med (1995) 1(12):1311–4. doi:10.1038/nm1295-1311
95. Salvi R, Castillo E, Voirol MJ, Glauser M, Rey JP, Gaillard RC, et al. Gonadotropin-releasing hormone-expressing neurons immortalized conditionally are activated by insulin: implication of the mitogen-activated protein kinase pathway. Endocrinology (2006) 147(2):816–26. doi:10.1210/en.2005-0728
96. Okamoto H, Nakae J, Kitamura T, Park BC, Dragatsis I, Accili D. Transgenic rescue of insulin receptor-deficient mice. J Clin Invest (2004) 114(2):214–23. doi:10.1172/JCI200421645
97. Hill JW, Elmquist JK, Elias CF. Hypothalamic pathways linking energy balance and reproduction. Am J Physiol Endocrinol Metab (2008) 294(5):E827–32. doi:10.1152/ajpendo.00670.2007
98. Marino JS, Iler J, Dowling AR, Chua S, Bruning JC, Coppari R, et al. Adipocyte dysfunction in a mouse model of polycystic ovary syndrome (PCOS): evidence of adipocyte hypertrophy and tissue-specific inflammation. PLoS One (2012) 7(10):e48643. doi:10.1371/journal.pone.0048643
99. Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron (2004) 42(6):983–91. doi:10.1016/j.neuron.2004.06.004
100. Roa J, Tena-Sempere M. Connecting metabolism and reproduction: roles of central energy sensors and key molecular mediators. Mol Cell Endocrinol (2014) 397(1–2):4–14. doi:10.1016/j.mce.2014.09.027
101. Du J, Wang J, Sun X, Xu X, Zhang F, Wang B, et al. Family-based analysis of INSR polymorphisms in Chinese PCOS. Reprod Biomed Online (2014) 29(2):239–44. doi:10.1016/j.rbmo.2014.03.028
102. Chen ZJ, Zhao H, He L, Shi Y, Qin Y, Shi Y, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet (2011) 43(1):55–9. doi:10.1038/ng.732
103. Dunaif A, Xia J, Book CB, Schenker E, Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest (1995) 96(2):801–10. doi:10.1172/JCI118126
104. Wu S, Divall S, Nwaopara A, Radovick S, Wondisford F, Ko C, et al. Obesity-induced infertility and hyperandrogenism are corrected by deletion of the insulin receptor in the ovarian theca cell. Diabetes (2014) 63(4):1270–82. doi:10.2337/db13-1514
105. Corbould A. Insulin resistance in skeletal muscle and adipose tissue in polycystic ovary syndrome: are the molecular mechanisms distinct from type 2 diabetes? Panminerva Med (2008) 50(4):279–94.
106. Rajkhowa M, Brett S, Cuthbertson DJ, Lipina C, Ruiz-Alcaraz AJ, Thomas GE, et al. Insulin resistance in polycystic ovary syndrome is associated with defective regulation of ERK1/2 by insulin in skeletal muscle in vivo. Biochem J (2009) 418(3):665–71. doi:10.1042/BJ20082176
107. Corbould A, Kim YB, Youngren JF, Pender C, Kahn BB, Lee A, et al. Insulin resistance in the skeletal muscle of women with PCOS involves intrinsic and acquired defects in insulin signaling. Am J Physiol Endocrinol Metab (2005) 288(5):E1047–54. doi:10.1152/ajpendo.00361.2004
108. Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, et al. PPAR gamma mediates high-fat diet–induced adipocyte hypertrophy and insulin resistance. Mol Cell (1999) 4:597–609. doi:10.1016/S1097-2765(00)80210-5
109. Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. J Clin Invest (2009) 119:315–22. doi:10.1172/JCI37432
110. Joy TR, Hegele RA. Prevalence of reproductive abnormalities among women with familial partial lipodystrophy. Endocr Pract (2008) 14(9):1126–32. doi:10.4158/EP.14.9.1126
111. Pahuja I, De P, Sharma N, Kulshreshtha B. Polycystic ovarian syndrome in patients with lipodystrophy: report of 2 cases with review of literature. Indian J Endocrinol Metab (2012) 16(6):1022–5. doi:10.4103/2230-8210.103031
112. Vantyghem MC, Vincent-Desplanques D, Defrance-Faivre F, Capeau J, Fermon C, Valat AS, et al. Fertility and obstetrical complications in women with LMNA-related familial partial lipodystrophy. J Clin Endocrinol Metab (2008) 93(6):2223–9. doi:10.1210/jc.2007-2521
113. Savage DB. Mouse models of inherited lipodystrophy. Dis Model Mech (2009) 2(11–12):554–62. doi:10.1242/dmm.002907
114. Huhtaniemi I, Ahtiainen P, Pakarainen T, Rulli SB, Zhang FP, Poutanen M. Genetically modified mouse models in studies of luteinising hormone action. Mol Cell Endocrinol (2006) 252(1–2):126–35. doi:10.1016/j.mce.2006.03.026
115. Danilovich N, Ram Sairam M. Recent female mouse models displaying advanced reproductive aging. Exp Gerontol (2006) 41(2):117–22. doi:10.1016/j.exger.2005.10.010
116. Fisher CR, Graves KH, Parlow AF, Simpson ER. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci U S A (1998) 95(12):6965–70. doi:10.1073/pnas.95.12.6965
117. Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci U S A (1998) 95(26):15677–82. doi:10.1073/pnas.95.26.15677
118. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A (1993) 90(23):11162–6. doi:10.1073/pnas.90.23.11162
119. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A (2000) 97(23):12729–34. doi:10.1073/pnas.97.23.12729
120. Kero JT, Savontaus E, Mikola M, Pesonen U, Koulu M, Keri RA, et al. Obesity in transgenic female mice with constitutively elevated luteinizing hormone secretion. Am J Physiol Endocrinol Metab (2003) 285(4):E812–8. doi:10.1152/ajpendo.00367.2002
121. Tissenbaum HA, Ruvkun G. An insulin-like signaling pathway affects both longevity and reproduction in Caenorhabditis elegans. Genetics (1998) 148(2):703–17.
122. Hsin H, Kenyon C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature (1999) 399(6734):362–6. doi:10.1038/20694
123. Böhni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, et al. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell (1999) 97(7):865–75. doi:10.1016/S0092-8674(00)80799-0
124. Franks S, Mason H, Willis D. Follicular dynamics in the polycystic ovary syndrome. Mol Cell Endocrinol (2000) 163(1–2):49–52. doi:10.1016/S0303-7207(99)00239-7
Keywords: androgen, fertility, insulin resistance, mouse models, obesity, PCOS
Citation: Huang-Doran I and Franks S (2016) Genetic Rodent Models of Obesity-Associated Ovarian Dysfunction and Subfertility: Insights into Polycystic Ovary Syndrome. Front. Endocrinol. 7:53. doi: 10.3389/fendo.2016.00053
Received: 01 April 2016; Accepted: 17 May 2016;
Published: 07 June 2016
Edited by:
Nicholas Michael Morton, The University of Edinburgh, UKReviewed by:
Eusebio Chiefari, University “Magna Græcia” of Catanzaro, ItalyPhilippa Saunders, The Edinburgh Cancer Research Centre, UK
Copyright: © 2016 Huang-Doran and Franks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel Huang-Doran, ih240@cam.ac.uk
†Present address: Isabel Huang-Doran, University of Cambridge Metabolic Research Laboratories, Wellcome Trust-MRC Institute of Metabolic Science, Addenbrooke’s Hospital, Cambridge, UK