 Kai Wang1,2* Andrew C. Edmondson3,4 Mingyao Li5 Fan Gao2 Atif N. Qasim3 Joseph M. Devaney6 Mary Susan Burnett6 Dawn M. Waterworth7 Vincent Mooser7 Struan F. A. Grant1,8 Stephen E. Epstein6 Muredach P. Reilly3 Hakon Hakonarson1,8 Daniel J. Rader3,4
Kai Wang1,2* Andrew C. Edmondson3,4 Mingyao Li5 Fan Gao2 Atif N. Qasim3 Joseph M. Devaney6 Mary Susan Burnett6 Dawn M. Waterworth7 Vincent Mooser7 Struan F. A. Grant1,8 Stephen E. Epstein6 Muredach P. Reilly3 Hakon Hakonarson1,8 Daniel J. Rader3,4
- 1 Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA, USA
- 2 Zilkha Neurogenetic Institute, University of Southern California, Los Angeles, CA, USA
- 3 Cardiovascular Institute, University of Pennsylvania,Philadelphia, PA, USA
- 4 Institute for Translational Medicine and Therapeutics, University of Pennsylvania, Philadelphia, PA, USA
- 5 Department of Biostatistics and Epidemiology, University of Pennsylvania, Philadelphia, PA, USA
- 6 Cardiovascular Research Institute, MedStar Health Research Institute, Washington Hospital Center, Washington, DC, USA
- 7 Genetics Division and Drug Discovery, GlaxoSmithKline, King of Prussia, PA, USA
- 8 Department of Pediatrics, University of Pennsylvania, Philadelphia, PA, USA
Pathway-based association methods have been proposed to be an effective approach in identifying disease genes, when single-marker association tests do not have sufficient power. The analysis of quantitative traits may be benefited from these approaches, by sampling from two extreme tails of the distribution. Here we tested a pathway association approach on a small genome-wide association study (GWAS) on 653 subjects with extremely high high-density lipoprotein cholesterol (HDL-C) levels and 784 subjects with low HDL-C levels. We identified 102 genes in the sterol transport and metabolism pathways that collectively associate with HDL-C levels, and replicated these association signals in an independent GWAS. Interestingly, the pathways include 18 genes implicated in previous GWAS on lipid traits, suggesting that genuine HDL-C genes are highly enriched in these pathways. Additionally, multiple biologically relevant loci in the pathways were not detected by previous GWAS, including genes implicated in previous candidate gene association studies (such as LEPR, APOA2, HDLBP, SOAT2), genes that cause Mendelian forms of lipid disorders (such as DHCR24), and genes expressing dyslipidemia phenotypes in knockout mice (such as SOAT1, PON1). Our study suggests that sampling from two extreme tails of a quantitative trait and examining genetic pathways may yield biological insights from smaller samples than are generally required using single-marker analysis in large-scale GWAS. Our results also implicate that functionally related genes work together to regulate complex quantitative traits, and that future large-scale studies may benefit from pathway-association approaches to identify novel pathways regulating HDL-C levels.
Introduction
Genome-wide association studies (GWAS) have been very successful in identifying genes or loci that are associated with complex diseases or quantitative traits. However, for most GWAS, the collection of known loci explains only a minor fraction of disease heritability (Maher, 2008; Manolio et al., 2009). Since most GWAS are not powered to detect the majority of genuinely associated loci, researchers could expand sample size indefinitely in an attempt to find more associated loci; however, this approach has its limitations as the GWAS are likely to have diminishing returns. That is, after the first few loci (presumably more readily detectable with larger effect sizes) have been identified and replicated, the remaining loci will be more and more difficult to find as they will have much smaller effect sizes (Goldstein, 2009). An alternative approach to detect trait-associated loci in GWAS, which can provide complementary information to single-marker analysis, is a pathway-based approach, which examines whether test statistics for related genes in the same pathway are consistently associated with a disease trait under study (Wang et al., 2010).
There have been a few published examples demonstrating the power and effectiveness of pathway-wide association approaches for several diseases. We have previously developed a pathway association approach (Wang et al., 2007), by adopting the Gene Set Enrichment Analysis (Subramanian et al., 2005) strategies, but with appropriate modifications to account for the linkage disequilibrium between SNP markers, the different sizes of genes and pathways. This approach has been successfully applied to uncover association between the IL12/IL23 pathway and Crohn’s disease (Wang et al., 2009a), the involvement of cell adhesion molecules in autism spectrum disorders (Wang et al., 2009b), as well as the WNT-signaling pathway in Type 2 Diabetes (T2D) (Perry et al., 2009). Additionally, Lesnick et al. (2007) have demonstrated that SNPs in the candidate pathway (axon guidance) collectively predisposed to a Parkinson’s disease, even though no individual SNP reaches genome-wide significance level. Dinu et al. (2007) applied a pathway approach and discovered that multiple complement factor pathway genes, in additional to complement factor H (a well known susceptibility locus), are associated with risk for developing age-related macular degeneration (AMD). Torkamani et al. (2008) applied a pathway-based approach to analyze the Wellcome Trust Case Control Consortium (WTCCC) data sets, and discovered numerous pathways implicated in disease predisposition, many of which have long been assumed to contain polymorphic genes that lead to disease predisposition. Askland et al. (2009) analyzed GWAS data sets on bipolar disorder using a pathway association approach, and implicated genes mediating ion channel activity and synaptic neurotransmission. Holmans et al. (2009) also described a pathway approach and applied it on meta-analysis of bipolar disorder, and implicated a collection of genes involved in the modulation of transcription and cellular activity. Furthermore, several recent studies have focused on incorporating biological network structure into the analysis of GWAS (Pan, 2008; Baranzini et al., 2009). Collectively, all these studies show that pathway-based approaches can be applied to GWAS data sets to gain insights into disease susceptibility mechanisms, and to provide enhanced or complementary information to the conventional single-marker analysis.
Unlike previous disease studies, our current study focused on a quantitative trait, plasma levels of high-density lipoprotein cholesterol (HDL-C), to examine whether pathway-based approaches can be effective in enriching a group of truly associated genes. HDL-C, together with low-density lipoprotein cholesterol (LDL-C) and triglyceride (TG), are three major lipid phenotypes in the human serum. Lipid levels are heritable traits that are strongly associated with cardiovascular risk, therefore they are widely measured in clinical practice. Lipid measures represent excellent secondary phenotypes for genetic investigation by many GWAS, and relatively large sample sizes can be obtained to study these traits. Three large-scale GWAS have been reported to identify genetic loci associated with lipid traits: Kathiresan et al. (2009) conducted GWAS screens in 19,840 individuals and replication in up to 20,623 individuals, and identified 30 loci (36 trait-loci associations) controlling HDL-C, LDL-C, or TG levels. Sabatti et al. (2009) have assayed a birth cohort from a founder population with ∼4,500 subjects, and identified 17 trait-loci associations for lipid levels. Aulchenko et al. (2009) analyzed 16 European population cohorts with ∼20,000 subjects and identified 22 trait-loci associations on lipid traits. More recently, Teslovich et al. (2010) have published an even larger meta-analysis on lipid traits involving over 100,000 subjects. Despite these early successes, a more complete ensemble of lipids genes is needed for better understanding of the gene networks involved, and the current successes probably merely represent the start of a long road (Edmondson and Rader, 2008; Holleboom et al., 2008) for the following reasons: First, among genes that cause Mendelian disorders of lipid traits, only a subset of them have been identified in GWAS (Edmondson and Rader, 2008). Second, multiple candidate genes believed to affect HDL-C levels based on preclinical models have not yet been identified in GWAS for HDL-C. Finally, the collection of HDL-C genes identified in GWAS only explains a minor fraction of inter-individual variability in lipoprotein levels (Kathiresan et al., 2009). Therefore, it is likely that many more genes regulating HDL-C levels remain to be discovered.
In this study, we applied a pathway-based association approach on a GWAS for the purpose of identifying genes regulating HDL-C levels. The study is distinct from previous GWAS on lipids in that it was originally designed and ascertained as a case/control study, so the study participants have either extremely high or low HDL-C levels but without syndromic dyslipidemia features. Although the initial single-marker association analysis identified only one well known locus that significantly associates with HDL-C, the pathway-based association approach resulted in ∼100 loci that potentially associate with HDL-C, including multiple previously known lipid loci. We further validated some of the novel findings through literature survey of previously known Mendelian forms of lipid disorders, previously published candidate gene association studies, as well as the examination of metabolic phenotypes of gene knockout mice maintained by the Jackson laboratory. Our study has significant implications for the study of quantitative traits and the analysis of GWAS data: First, it demonstrated that a modest case/control data set sampling from two extreme tails of a quantitative trait can be very useful in identifying genuinely associated genes. Second, it illustrated that pathway-wide association approaches could provide biological insights that are not readily available from single-marker analysis on orders of magnitude larger sample sizes.
Materials and Methods
Study Participants
Discovery cohort
Cases and control subjects were ascertained for the purpose of identifying genetic variation regulating HDL-C levels. All study participants were self-declared to be of European ancestry, and were genotyped on the Affymetrix Genome-wide Human SNP 6.0 arrays. The “cases” were defined as persons with HDL-C levels >94th percentile for age and gender. The control subjects were defined as persons with HDL-C <25th percentile for age and gender. Based on the whole-genome genotyping data, we performed multi-dimensional scaling analysis on a set of independent SNP markers (Figure S1 in Supplementary Material), to ensure that only subjects of genetically inferred European ancestry were used in analysis. We also removed subjects with call rates of SNPs less than 95%. Additionally, we have removed pairs of subjects with cryptic relatedness, as defined by identity-by-descent score higher than 0.2. The final data set for association analysis included a total of 653 cases and 784 control subjects with 681,050 SNP markers. The genomic control inflation factor was 1.057, indicating that cases and control subjects were well matched and that no systematic batch effects were present.
Replication cohort
The replication cohort was compiled from PennCATH and MedStar, two ongoing GWAS on coronary artery disease, which were previously described in detail (Myocardial Infarction Genetics Consortium et al., 2009). The PennCATH study is a University of Pennsylvania Medical Center based angiographic study of over 3,800 subjects that has been used previously for replication of novel genes and risk factors for atherosclerotic cardiovascular disease and T2D (Helgadottir et al., 2006; Steinthorsdottir et al., 2007). The MedStar study is a Washington Hospital Center based angiographic study of 1,500 subjects specifically designed for biomarker and genetic association studies of acute and chronic coronary atherosclerosis. Since the PennCATH and MedStar samples were genotyped together at the same site and were analyzed by identical procedures, to improve power to detect association, we combined the two data sets together. To define HDL-C case–control status, the subjects were dichotomized by sex-specific cutoff points: those subjects with HDL-C higher than 80% percentile were regarded as “cases” and those with HDL-C levels lower than 20% percentile were regarded as control subjects. The thresholds differ from the discovery cohort, because the discovery cohort is specifically designed and ascertained for studying high HDL levels, whereas the PennCATH and MedStar were not ascertained in this manner. Subjects who participated in the discovery cohort were not included in the replication cohort. Through these criteria, a total of 496 cases and 373 control subjects were included in the analysis.
Ethics statement
All subjects have been enrolled in a University of Pennsylvania or Washington Hospital Institutional Review Board approved protocol, and all subjects gave written informed consent.
Pathway Association Analysis
The method for pathway-based association test was previously described in detail elsewhere (Wang et al., 2007, 2009a). Briefly, we assign each SNP to the overlapping gene/genes, or its closest gene (subject to a distance constraint) in the genome if it does not overlap with a gene. For each gene, we assigned the highest statistic value among all SNPs mapped to the gene as the statistic value of the gene. For all the N genes that are represented by SNPs in the GWAS, we sorted their statistic values from the largest to the smallest, denoted by r(1), …, r(N). For any given gene set S composed of NH genes, we then calculated a weighted Kolmogorov–Smirnov-like running sum statistic(Hollander and Wolfe, 1999) that reflects the overrepresentation of genes within the set S at the top of the entire ranked list of genes in the genome,
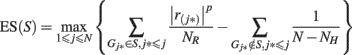
where 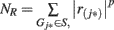 and p (default value = 1) is a parameter that gives higher weight to genes with extreme statistic values. To adjust for differences in gene size (hence the different number of SNPs located within/nearby each gene), as well as the linkage disequilibrium between SNPs within the same gene, we conducted a two-step correction procedure. In the first step, we permuted the disease labels of all samples ensuring the same number of individuals in each phenotype group for case–control studies. During each permutation (denoted by π), we repeated the calculation of enrichment score as described above as ES(S, π). In the second-step, we calculated normalized enrichment score (NES) as a Z-score, defined as [ES(S) − mean(ES(S, π))]/SD[ES(S, π)], so that different gene sets are directly comparable to each other. The P-value can be calculated from the π permutations, and a false discovery rate (FDR) procedure can be used to control the fraction of expected false positive findings below a certain threshold (Reiner et al., 2003). For a gene set, let NES* denote the NES in the observed data. The FDR is calculated as
and p (default value = 1) is a parameter that gives higher weight to genes with extreme statistic values. To adjust for differences in gene size (hence the different number of SNPs located within/nearby each gene), as well as the linkage disequilibrium between SNPs within the same gene, we conducted a two-step correction procedure. In the first step, we permuted the disease labels of all samples ensuring the same number of individuals in each phenotype group for case–control studies. During each permutation (denoted by π), we repeated the calculation of enrichment score as described above as ES(S, π). In the second-step, we calculated normalized enrichment score (NES) as a Z-score, defined as [ES(S) − mean(ES(S, π))]/SD[ES(S, π)], so that different gene sets are directly comparable to each other. The P-value can be calculated from the π permutations, and a false discovery rate (FDR) procedure can be used to control the fraction of expected false positive findings below a certain threshold (Reiner et al., 2003). For a gene set, let NES* denote the NES in the observed data. The FDR is calculated as
(% of all (S, π) with NES(S, π) ≥ NES*)
/(% of observed S with NES(S) ≥ NES*)
Alternatively, we also calculated the FWER P-value as the fraction of all permutations whose highest NES score in all gene sets are higher than the NES* for a given gene set. For the discovery cohort, a total of 10,000 phenotype permutation cycles were used so that a more accurate P-value can be calculated for each pathway; for replication cohort, 1,000 phenotype permutation cycles were used.
The software implementing this method was made freely available at http://www.openbioinformatics.org/gengen. The current software uses chi-square test statistics for association, so it cannot incorporate covariates in the analysis. The software package included pathway definitions for the three pathway collections (KEGG, BioCarta, GO) used in the study, as well as library files mapping SNP identifiers to gene names for the Affymetrix Genome-wide Human SNP 6.0 array. The pathway collections were accessed and assembled on March 03, 2008. These default library files, as well as all default parameters in the software, were used in our pathway association analysis.
Results
Single-Marker and Pathway Association Analysis on HDL-C Levels
We examined a previously unpublished cohort on HDL-C levels (discovery cohort, Table 1), genotyped on the Affymetrix Genome-wide Human SNP 6.0 arrays. Unlike other disease studies that investigated HDL-C levels as an additional/secondary phenotype for investigation, this cohort consists of “cases” with extremely high HDL-C levels and “control” subjects with low HDL-C levels (see Material and Methods). One locus on 16q13 reached genome-wide significance levels of P < 5 × 10−8 (Figure 1), with the most significant SNP being rs1532625 (P = 6.8 × 10−12, OR = 1.68) within CETP (cholesteryl ester transfer protein). This locus was a well-established HDL-C locus before the GWAS era (Koizumi et al., 1985; Kurasawa et al., 1985; Inazu et al., 1990). Therefore, through single-marker association analysis, no novel lipid genes were identified that met genome-wide significance criterion, though we were able to replicate several loci from other published studies on lipid levels (Table S1 in Supplementary Material). Altogether, these single-marker association results testified the high quality of the phenotype data, and demonstrated the unique power of case/control data set that samples from extreme tails of normal distributions on a quantitative trait.
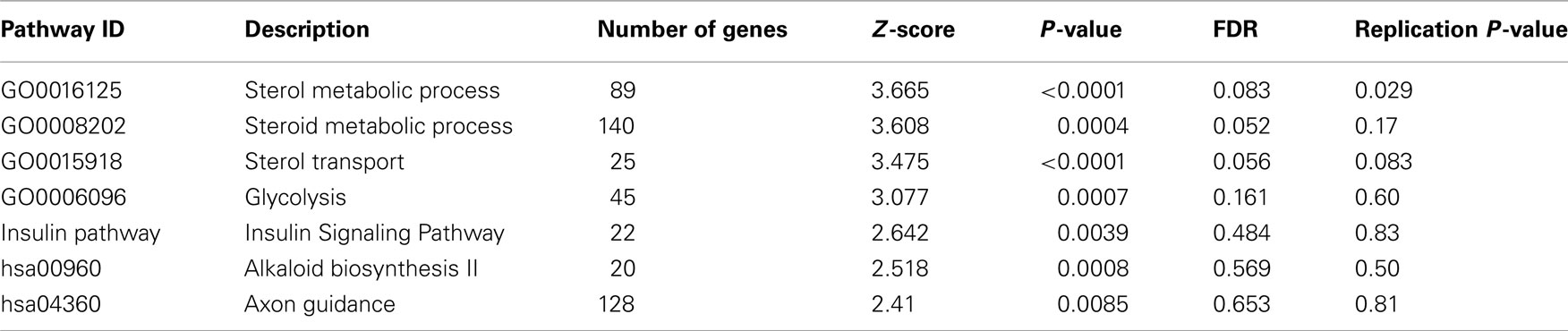
Table 1. The most significantly associated pathways in the discovery GWAS data set.
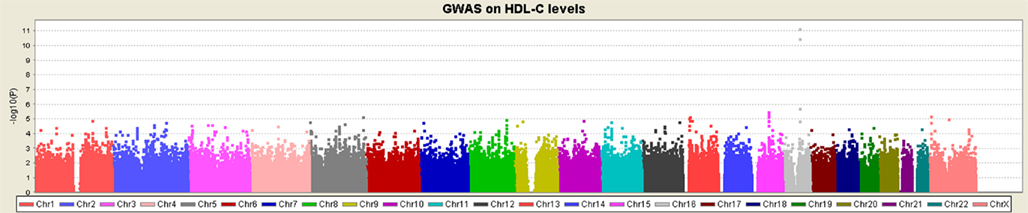
Figure 1. Manhattan plot of the single-marker association analysis on the discovery cohort. Except CETP on 16p13.3, single-marker analysis failed to identify any locus reaching genome-wide significance (P < 5 × 10−]).
To screen for novel biological pathways or collections of functionally related genes that associate with HDL-C levels, we next applied a pathway-based association approach (Wang et al., 2007) on this GWAS data set, with the same set of subjects and SNPs used in single-marker association analysis. This pathway association method permutes the case/control labels a large number of times, and for each permuted data set, it re-calculates the association test statistics for all SNP markers, and then assesses the significance of the pre-defined pathways or sets of genes by comparing the observed test statistics with the null distribution generated by the permutations. The collection of pathways or gene sets analyzed in the study was retrieved from the BioCarta (BioCarta, BioCarta-Charting pathways of life), KEGG (Kanehisa et al., 2010) and level 4 of the Gene Ontology (Ashburner et al., 2000) databases. We performed 10,000 permutations and assessed a total of 568 pathways passing size threshold (20–200 genes). We found that seven pathways were associated with HDL-C levels at a nominal P-value less than 0.01 (Table 1), slightly more than expected by chance (5.68). Among these pathways, four have false discovery rate (FDR) less than 25%. However, we note that all genes in the “sterol metabolic process” pathway belong to the “steroid metabolic process” pathway as both were from the Gene Ontology collection (Figure S2 in Supplementary Material), therefore we excluded the “steroid metabolic process” pathway from further discussion as it has lower Z-score and P-value than its subset. The full list of genes and representative SNPs for the sterol metabolism and transport pathways were given in Figure S2 in Supplementary Material. To further evaluate the sensitivity of the pathway analysis to the inclusion of CETP – a well known HDL-C gene that showed genome-wide significance in our single-marker association analysis – we removed CETP from the pathway analysis. This practice is similar to removing APOE from pathway analysis on Alzheimer’s disease (Jones et al., 2010) or removing NOD2 from pathway analysis on Crohn’s disease (Wang et al., 2009a). We still observed association signals for the “sterol metabolic process” and “sterol transport” pathways but with reduced significance levels (P = 0.0011 and P = 0.007, respectively), suggesting that the observed pathway associations above were not due to the effect of CETP alone.
Similar to single-marker association tests, pathway-based association tests may be susceptible to spurious associations, so we next sought to replicate the pathway findings using independent data sets. Since the pathway association method requires phenotype permutation of case/control status, we compiled a case–control dataset (496 cases, 373 controls) from ongoing GWAS at University of Pennsylvania which have fasting lipid measures on some study participants (see Materials and Methods). The replication P-values for sterol metabolic process pathway and sterol transport pathway are 0.029 and 0.083, respectively. These values do not reach stringent levels of statistical significance considering the need to test two hypotheses, and this may be due to the lack of power in the replication study.
Additional Evidence Supporting Pathway Association Signals
Although our study demonstrated that a group of genes were collectively associated with HDL-C levels, only one locus (CETP) truly showed a genome-wide significance signal in single-marker association analysis. The Gene Ontology pathways were curated from heterogeneous data sources such as electronic annotation from protein homology searches, so they may contain genes that are not genuinely associated with HDL-C levels. Therefore, we applied three complementary approaches, to search for additional evidence supporting the role of a subset of genes in the pathways in regulating HDL-C levels, and summarized the results in Table 2.
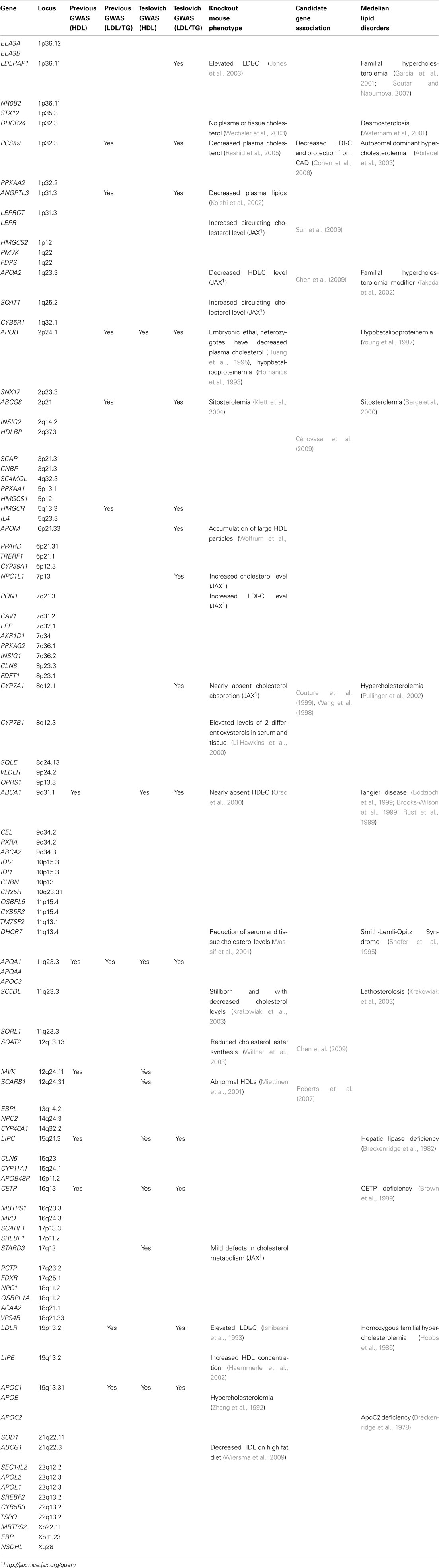
Table 2. List of genes in the sterol transport/metabolism pathway with additional evidence of association with lipid traits.
First, we examined the sterol transport and metabolism genes in three independent GWAS [the Kathiresan et al. (2009) meta-analysis, the Aulchenko et al. (2009) meta-analysis, the Sabatti et al. (2009) study on founder populations), as well as the large-scale Teslovich et al. (2010) meta-analysis. Among the 102 unique genes (96 unique loci), genes in the sterol transport and metabolism pathways, 9 and 15 loci were reported as genuine HDL-C loci and/or LDL-C/TG loci in at least one of these publications, respectively (Table 2). The abundance of previously known lipid genes in the identified pathway therefore served as de novo confirmation supporting the biological relevance of the pathways. Additionally, when examining previously published candidate gene association studies, we found that multiple genes in the sterol transport/metabolism pathway, such as LEPR, APOA2, SOAT2, HDLBP, were previously associated with lipid levels (Table 2), albeit with relatively small effect sizes and in some cases not replicated.
Second, we examined published literature on monogenic form of lipid disorders, and determined whether any of the sterol transport/metabolism genes have been previously associated with such diseases. Interestingly, multiple additional genes, such as DHCR24 and APOA2, have never been implicated by GWAS on lipid traits, but were identified as genuine lipid genes through the study of rare forms of lipid disorders (Table 2). It is possible that no common causal variants in these genes exist in European populations, or that the effect sizes of causal variants are too small to be detected by the sample sizes used in previous studies. Nevertheless, this fact implicates the complementary nature of pathway-based association approach to single-marker association approach, and supports the utility of the pathway-based approach in identifying biologically relevant genes.
Third, we examined previous functional studies conducted on model organisms, including mouse knockout studies. Unlike many complex human diseases where genetic models are difficult to replicate or study in animal models, the lipid traits are easily measurable and quantifiable, so they were almost always included in knockout experiments, facilitating the functional study of lipid genes. This analysis revealed multiple additional previously unreported genes, including SOAT1, LEPR, and PON1, that had a metabolic phenotype (increased or decreased cholesterol levels) in knockout experiments (Table 2).
Altogether, the three complementary approaches presented above resulted in the confirmation of a prioritized set of genes that have strong a prior evidence of playing a role in HDL-C regulation. Although the sterol transport/metabolism pathways contains 102 genes, this particular subset of genes may represent a prioritized subset that are promising candidates to be investigated in functional studies of lipid traits.
Discussion
In this study, we performed pathway-based genome-wide association analysis on two GWAS data sets, and demonstrated that a group of sterol transport and metabolism genes are collectively associated with HDL-C levels. We further validated a subset of these genes through examination of previously published candidate gene association studies, known Mendelian forms of lipid disorders, and known dyslipidemia phenotypes in knockout mice. Compared to previously published large-scale GWAS, our results supplied a richer list of additional candidate genes regulating HDL-C levels, and demonstrated the enhanced power of pathway-based association analysis on finding biologically relevant genes. Given the relatively small sample size, we were not able to identify previously unknown pathways as being involved in regulation of HDL-C levels; however, the fact that known pathways and genes are identified suggest that the approach may be applicable to larger sample sets to identify novel pathways.
The physiology of HDL metabolism results from the complex interplay of multiple gene products including apolipoproteins, enzymes, lipid transfer proteins, cellular transporters, and receptors. Although the pathways identified in the study are intuitive in a biological sense, caution should be exercised as some of the genes within the pathways probably play more important and direct roles than other genes in regulating HDL-C homeostasis. This explains the rationale of our three-tiered prioritization approach to identify subsets of HDL-C genes for future functional studies.
We also wish to discuss a few points related to sample ascertainment scheme. One particular point is that all individuals in the discovery cohort are ascertained from the general population with extreme lipids measures, yet the individuals in the replication cohort have CAD. HDL-C levels might be more disturbed by lipid medications or other clinical factors in CAD patients. Therefore, although it would be valid to use individuals with CAD for replication purposes, it would be most appropriate to make discoveries from general population, to ensure that the data ascertainment scheme does not bias the results (for example, pathways that associate with CAD status rather than lipids levels). Another point is that our study did not utilize a larger cohort and took a QTL approach for the association analysis. It is expected that under fixed genotyping budget, extreme phenotypes have greatly improved power to detect associations due to increased odds ratio measures. However, lipids measures are easily obtainable, so they represent excellent secondary phenotypes available from many GWAS on diverse phenotypes with large sample sizes. Therefore, we expect that a QTL approach for a very large cohort may also have sufficient power to identify the implicated pathways.
Although the name “pathway” was used in our analysis, we caution that the term merely refers to a collection of genes, rather than an inter-connected biological process. The pathway-based association analysis is borrowed from microarray data analysis, where sets of related genes are combined into a single group to examine the up or down-regulation of gene expression levels for a group of genes that are consistent. Therefore, “pathway” does not necessarily correspond to biologists’ understanding of biological process, but rather a functional classification of genes based on a set of pre-defined criteria. Our study used pathways compiled from three resources, but we note that other pathway collections could also be explored. Furthermore, we note that pathways analyses have the potential to move beyond the so-called “common-disease/common-variant” paradigm, and consider SNP and genes together rather than in isolation.
In conclusion, using two distinct GWAS data sets based on modestly-sized samples ascertained by extremes of a quantitative trait (HDL-C), we have applied pathway analysis to suggest that an ensemble of ∼100 genes is collectively associated with HDL-C levels. A fraction of these genes has already been suggested by previously published literature and by animal models, lending strong support to the validity of the pathway-based approach. Our results therefore confirm the validity and potential utility of the pathway association approach as applied to the extreme tails of a quantitative trait.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all participating subjects and families involved in the study. Recruitment of the high/low HDL cohort was supported by a Doris Duke Charitable Foundation Distinguished Clinical Scientist Award to Daniel J. Rader. Recruitment of the PennCATH cohort was supported by the Cardiovascular Institute of the University of Pennsylvania. Recruitment of the MedStar cohort was supported by a research grant from GlaxoSmithKline and from the MedStar Research Institute. Genotyping was done at the Center for Applied Genomics at the Children’s Hospital of Philadelphia. Genotyping of the high/low HDL cohort was supported by R01 HL089309 from the NHLBI and of the PennCATH and MedStar cohorts by GlaxoSmithKline through an Alternate Drug Discovery Initiative research alliance award (to Muredach P. Reilly and Daniel J. Rader) with the University of Pennsylvania School of Medicine. Andrew C. Edmondson was supported by a National Heart, Lung and Blood Institute Ruth L. Kirschstein National Research Service Award for Individual Predoctoral MD/PhD Fellows (F30). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Applied_Genetic_Epidemiology/10.3389/fgene.2011.00041/abstract
References
Abifadel, M., Varret, M., Rabes, J. P., Allard, D., Ouguerram, K., Devillers, M., Cruaud, C., Benjannet, S., Wickham, L., Erlich, D., Derré, A., Villéger, L., Farnier, M., Beucler, I., Bruckert, E., Chambaz, J., Chanu, B., Lecerf, J. M., Luc, G., Moulin, P., Weissenbach, J., Prat, A., Krempf, M., Junien, C., Seidah, N. G., and Boileau, C. (2003). Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., and Sherlock, G. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29.
Askland, K., Read, C., and Moore, J. (2009). Pathways-based analyses of whole-genome association study data in bipolar disorder reveal genes mediating ion channel activity and synaptic neurotransmission. Hum. Genet. 125, 63–79.
Aulchenko, Y. S., Ripatti, S., Lindqvist, I., Boomsma, D., Heid, I. M., Pramstaller, P. P., Penninx, B. W., Janssens, A. C., Wilson, J. F., Spector, T., Martin, N. G., Pedersen, N. L., Kyvik, K. O., Kaprio, J., Hofman, A., Freimer, N. B., Jarvelin, M. R., Gyllensten, U., Campbell, H., Rudan, I., Johansson, A., Marroni, F., Hayward, C., Vitart, V., Jonasson, I., Pattaro, C., Wright, A., Hastie, N., Pichler, I., Hicks, A. A., Falchi, M., Willemsen, G., Hottenga, J. J., de Geus, E. J., Montgomery, G. W., Whitfield, J., Magnusson, P., Saharinen, J., Perola, M., Silander, K., Isaacs, A., Sijbrands, E. J., Uitterlinden, A. G., Witteman, J. C., Oostra, B. A., Elliott, P., Ruokonen, A., Sabatti, C., Gieger, C., Meitinger, T., Kronenberg, F., Döring, A., Wichmann, H. E., Smit, J. H., McCarthy, M. I., van Duijn, C. M., Peltonen, L., ENGAGE Consortium. (2009). Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 41, 47–55.
Baranzini, S. E., Galwey, N. W., Wang, J., Khankhanian, P., Lindberg, R., Pelletier, D., Wu, W., Uitdehaag, B. M., Kappos, L., GeneMSA Consortium. Polman, C. H., Matthews, P. M., Hauser, S. L., Gibson, R. A., Oksenberg, J. R., and Barnes, M. R. (2009). Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet. 18, 2078–2090.
Berge, K. E., Tian, H., Graf, G. A., Yu, L., Grishin, N. V., Schultz, J., Kwiterovich, P., Shan, B., Barnes, R., and Hobbs, H. H. (2000). Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 290, 1771–1775.
Bodzioch, M., Orso, E., Klucken, J., Langmann, T., Bottcher, A., Diederich, W., Drobnik, W., Barlage, S., Büchler, C., Porsch-Ozcürümez, M., Kaminski, W. E., Hahmann, H. W., Oette, K., Rothe, G., Aslanidis, C., Lackner, K. J., and Schmitz, G. (1999). The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 22, 347–351.
Breckenridge, W. C., Little, J. A., Alaupovic, P., Wang, C. S., Kuksis, A., Kakis, G., Lindgren, F., and Gardiner, G. (1982). Lipoprotein abnormalities associated with a familial deficiency of hepatic lipase. Atherosclerosis 45, 161–179.
Breckenridge, W. C., Little, J. A., Steiner, G., Chow, A., and Poapst, M. (1978). Hypertriglyceridemia associated with deficiency of apolipoprotein C-II. N. Engl. J. Med. 298, 1265–1273.
Brooks-Wilson, A., Marcil, M., Clee, S. M., Zhang, L. H., Roomp, K., van Dam, M., Yu, L., Brewer, C., Collins, J. A., Molhuizen, H. O., Loubser, O., Ouelette, B. F., Fichter, K., Ashbourne-Excoffon, K. J., Sensen, C. W., Scherer, S., Mott, S., Denis, M., Martindale, D., Frohlich, J., Morgan, K., Koop, B., Pimstone, S., Kastelein, J. J., Genest, J. Jr., and Hayden, M. R. (1999). Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 22, 336–345.
Brown, M. L., Inazu, A., Hesler, C. B., Agellon, L. B., Mann, C., Whitlock, M. E., Marcel, Y. L., Milne, R. W., Koizumi, J., Mabuchi, H., Takeda, R., and Tall, A. R. (1989). Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature 342, 448–451.
Cánovasa, E., Quintanillab, R., Badaouia, B., Porredóna, C., Gallardoa, D., Pena, R. N., Díaz, I., and Amills, M. (2009). Pig HDL-binding protein (HDLBP) genotype is associated with intramuscular fat percentage. Livest. Sci. 126, 298–301.
Chen, S. N., Cilingiroglu, M., Todd, J., Lombardi, R., Willerson, J. T., Gotto, A. M. Jr., Ballantyne, C. M., and Marian, A. J. (2009). Candidate genetic analysis of plasma high-density lipoprotein-cholesterol and severity of coronary atherosclerosis. BMC Med. Genet. 10, 111. doi: 10.1186/1471-2350-10-111
Cohen, J. C., Boerwinkle, E., Mosley, T. H. Jr., and Hobbs, H. H. (2006). Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272.
Couture, P., Otvos, J. D., Cupples, L. A., Wilson, P. W., Schaefer, E. J., and Ordovas, J. M. (1999). Association of the A-204C polymorphism in the cholesterol 7alpha-hydroxylase gene with variations in plasma low density lipoprotein cholesterol levels in the Framingham Offspring Study. J. Lipid Res. 40, 1883–1889.
Dinu, V., Miller, P. L., and Zhao, H. (2007). Evidence for association between multiple complement pathway genes and AMD. Genet. Epidemiol. 31, 224–237.
Edmondson, A. C., and Rader, D. J. (2008). Genome-wide approaches to finding novel genes for lipid traits: the start of a long road. Circ. Cardiovasc. Genet. 1, 3–6.
Garcia, C. K., Wilund, K., Arca, M., Zuliani, G., Fellin, R., Maioli, M., Calandra, S., Bertolini, S., Cossu, F., Grishin, N., Barnes, R., Cohen, J. C., and Hobbs, H. H. (2001). Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 292, 1394–1398.
Goldstein, D. B. (2009). Common genetic variation and human traits. N. Engl. J. Med. 360, 1696–1698.
Haemmerle, G., Zimmermann, R., Strauss, J. G., Kratky, D., Riederer, M., Knipping, G., and Zechner, R. (2002). Hormone-sensitive lipase deficiency in mice changes the plasma lipid profile by affecting the tissue-specific expression pattern of lipoprotein lipase in adipose tissue and muscle. J. Biol. Chem. 277, 12946–12952.
Helgadottir, A., Manolescu, A., Helgason, A., Thorleifsson, G., Thorsteinsdottir, U., Gudbjartsson, D. F., Gretarsdottir, S., Magnusson, K. P., Gudmundsson, G., Hicks, A., Jonsson, T., Grant, S. F., Sainz, J., O’Brien, S. J., Sveinbjornsdottir, S., Valdimarsson, E. M., Matthiasson, S. E., Levey, A. I., Abramson, J. L., Reilly, M. P., Vaccarino, V., Wolfe, M. L., Gudnason, V., Quyyumi, A. A., Topol, E. J., Rader, D. J., Thorgeirsson, G., Gulcher, J. R., Hakonarson, H., Kong, A., and Stefansson, K. (2006). A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction. Nat. Genet. 38, 68–74.
Hobbs, H. H., Brown, M. S., Goldstein, J. L., and Russell, D. W. (1986). Deletion of exon encoding cysteine-rich repeat of low density lipoprotein receptor alters its binding specificity in a subject with familial hypercholesterolemia. J. Biol. Chem. 261, 13114–13120.
Holleboom, A. G., Vergeer, M., Hovingh, G. K., Kastelein, J. J., and Kuivenhoven, J. A. (2008). The value of HDL genetics. Curr. Opin. Lipidol. 19, 385–394.
Holmans, P., Green, E. K., Pahwa, J. S., Ferreira, M. A., Purcell, S. M., Sklar, P., Wellcome Trust Case-Control Consortium. Owen, M. J., O’Donovan, M. C., and Craddock, N. (2009). Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am. J. Hum. Genet. 85, 13–24.
Homanics, G. E., Smith, T. J., Zhang, S. H., Lee, D., Young, S. G., and Maeda, N. (1993). Targeted modification of the apolipoprotein B gene results in hypobetalipoproteinemia and developmental abnormalities in mice. Proc. Natl. Acad. Sci. U.S.A. 90, 2389–2393.
Huang, L. S., Voyiaziakis, E., Markenson, D. F., Sokol, K. A., Hayek, T., and Breslow, J. L. (1995). apo B gene knockout in mice results in embryonic lethality in homozygotes and neural tube defects, male infertility, and reduced HDL cholesterol ester and apo A-I transport rates in heterozygotes. J. Clin. Invest. 96, 2152–2161.
Inazu, A., Brown, M. L., Hesler, C. B., Agellon, L. B., Koizumi, J., Takata, K., Maruhama, Y., Mabuchi, H., and Tall, A. R. (1990). Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N. Engl. J. Med. 323, 1234–1238.
Ishibashi, S., Brown, M. S., Goldstein, J. L., Gerard, R. D., Hammer, R. E., and Herz, J. (1993). Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Invest. 92, 883–893.
Jones, C., Hammer, R. E., Li, W. P., Cohen, J. C., Hobbs, H. H., and Herz, J. (2003). Normal sorting but defective endocytosis of the low density lipoprotein receptor in mice with autosomal recessive hypercholesterolemia. J. Biol. Chem. 278, 29024–29030.
Jones, L., Holmans, P. A., Hamshere, M. L., Harold, D., Moskvina, V., Ivanov, D., Pocklington, A., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Pahwa, J. S., Jones, N., Stretton, A., Morgan, A. R., Lovestone, S., Powell, J., Proitsi, P., Lupton, M. K., Brayne, C., Rubinsztein, D. C., Gill, M., Lawlor, B., Lynch, A., Morgan, K., Brown, K. S., Passmore, P. A., Craig, D., McGuinness, B., Todd, S., Holmes, C., Mann, D., Smith, A. D., Love, S., Kehoe, P. G., Mead, S., Fox, N., Rossor, M., Collinge, J., Maier, W., Jessen, F., Schürmann, B., Heun, R., Kölsch, H., van den Bussche, H., Heuser, I., Peters, O., Kornhuber, J., Wiltfang, J., Dichgans, M., Frölich, L., Hampel, H., Hüll, M., Rujescu, D., Goate, A. M., Kauwe, J. S., Cruchaga, C., Nowotny, P., Morris, J. C., Mayo, K., Livingston, G., Bass, N. J., Gurling, H., McQuillin, A., Gwilliam, R., Deloukas, P., Al-Chalabi, A., Shaw, C. E., Singleton, A. B., Guerreiro, R., Mühleisen, T. W., Nöthen, M. M., Moebus, S., Jöckel, K. H., Klopp, N., Wichmann, H. E., Rüther, E., Carrasquillo, M. M., Pankratz, V. S., Younkin, S. G., Hardy, J., O’Donovan, MC., Owen, M. J., and Williams, J. (2010). Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS ONE 5, e13950. doi:10.1371/journal.pone.0013950
Kanehisa, M., Goto, S., Furumichi, M., Tanabe, M., and Hirakawa, M. (2010). KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 38, D355–D360.
Kathiresan, S., Willer, C. J., Peloso, G. M., Demissie, S., Musunuru, K., Schadt, E. E., Kaplan, L., Bennett, D., Li, Y., Tanaka, T., Voight, B. F., Bonnycastle, L. L., Jackson, A. U., Crawford, G., Surti, A., Guiducci, C., Burtt, N. P., Parish, S., Clarke, R., Zelenika, D., Kubalanza, K. A., Morken, M. A., Scott, L. J., Stringham, H. M., Galan, P., Swift, A. J., Kuusisto, J., Bergman, R. N., Sundvall, J., Laakso, M., Ferrucci, L., Scheet, P., Sanna, S., Uda, M., Yang, Q., Lunetta, K. L., Dupuis, J., de Bakker, P. I., O’Donnell, C. J., Chambers, J. C., Kooner, J. S., Hercberg, S., Meneton, P., Lakatta, E. G., Scuteri, A., Schlessinger, D., Tuomilehto, J., Collins, F. S., Groop, L., Altshuler, D., Collins, R., Lathrop, G. M., Melander, O., Salomaa, V., Peltonen, L., Orho-Melander, M., Ordovas, J. M., Boehnke, M., Abecasis, G. R., Mohlke, K. L., and Cupples, L. A. (2009). Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 41, 56–65.
Klett, E. L., Lu, K., Kosters, A., Vink, E., Lee, M. H., Altenburg, M., Shefer, S., Batta, A. K., Yu, H., Chen, J., Klein, R., Looije, N., Oude-Elferink, R., Groen, A. K., Maeda, N., Salen, G., and Patel, S. B. (2004). A mouse model of sitosterolemia: absence of Abcg8/sterolin-2 results in failure to secrete biliary cholesterol. BMC Med. 2, 5. doi:10.1186/1741-7015-2-5
Koishi, R., Ando, Y., Ono, M., Shimamura, M., Yasumo, H., Fujiwara, T., Horikoshi, H., and Furukawa, H. (2002). Angptl3 regulates lipid metabolism in mice. Nat. Genet. 30, 151–157.
Koizumi, J., Mabuchi, H., Yoshimura, A., Michishita, I., Takeda, M., Itoh, H., Sakai, Y., Sakai, T., Ueda, K., and Takeda, R. (1985). Deficiency of serum cholesteryl-ester transfer activity in patients with familial hyperalphalipoproteinaemia. Atherosclerosis 58, 175–186.
Krakowiak, P. A., Wassif, C. A., Kratz, L., Cozma, D., Kovarova, M., Harris, G., Grinberg, A., Yang, Y., Hunter, A. G., Tsokos, M., Kelley, R. I., and Porter, F. D. (2003). Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Hum. Mol. Genet. 12, 1631–1641.
Kurasawa, T., Yokoyama, S., Miyake, Y., Yamamura, T., and Yamamoto, A. (1985). Rate of cholesteryl ester transfer between high and low density lipoproteins in human serum and a case with decreased transfer rate in association with hyperalphalipoproteinemia. J. Biochem. 98, 1499–1508.
Lesnick, T. G., Papapetropoulos, S., Mash, D. C., Ffrench-Mullen, J., Shehadeh, L., de Andrade, M., Henley, J. R., Rocca, W. A., Ahlskog, J. E., and Maraganore, D. M. (2007). A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet. 3, e98. doi:10.1371/journal.pgen.0030098
Li-Hawkins, J., Lund, E. G., Turley, S. D., and Russell, D. W. (2000). Disruption of the oxysterol 7alpha-hydroxylase gene in mice. J. Biol. Chem. 275, 16536–16542.
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., McCarthy, M. I., Ramos, E. M., Cardon, L. R., Chakravarti, A., Cho, J. H., Guttmacher, A. E., Kong, A., Kruglyak, L., Mardis, E., Rotimi, C. N., Slatkin, M., Valle, D., Whittemore, A. S., Boehnke, M., Clark, A. G., Eichler, E. E., Gibson, G., Haines, J. L., Mackay, T. F., McCarroll, S. A., and Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753.
Miettinen, H. E., Rayburn, H., and Krieger, M. (2001). Abnormal lipoprotein metabolism and reversible female infertility in HDL receptor (SR-BI)-deficient mice. J. Clin. Invest. 108, 1717–1722.
Myocardial Infarction Genetics Consortium. Kathiresan, S., Voight, B. F., Purcell, S., Musunuru, K., Ardissino, D., Mannucci, P. M., Anand, S., Engert, J. C., Samani, N. J., Schunkert, H., Erdmann, J., Reilly, M. P., Rader, D. J., Morgan, T., Spertus, J. A., Stoll, M., Girelli, D., McKeown, PP., Patterson, C. C., Siscovick, D. S., O’Donnell, CJ, Elosua, R., Peltonen, L., Salomaa, V., Schwartz, S. M., Melander, O., Altshuler, D., Ardissino, D., Merlini, P. A., Berzuini, C., Bernardinelli, L., Peyvandi, F., Tubaro, M., Celli, P., Ferrario, M., Fetiveau, R., Marziliano, N., Casari, G., Galli, M., Ribichini, F., Rossi, M., Bernardi, F., Zonzin, P., Piazza, A., Mannucci, P. M., Schwartz, S. M., Siscovick, D. S., Yee, J., Friedlander, Y., Elosua, R., Marrugat, J., Lucas, G., Subirana, I., Sala, J., Ramos, R., Kathiresan, S., Meigs, J. B., Williams, G., Nathan, D. M., MacRae, CA, O’Donnell, CJ, Salomaa, V., Havulinna, A. S., Peltonen, L., Melander, O., Berglund, G., Voight, B. F., Kathiresan, S., Hirschhorn, J. N., Asselta, R., Duga, S., Spreafico, M., Musunuru, K., Daly, M. J., Purcell, S., Voight, B. F., Purcell, S., Nemesh, J., Korn, J. M., McCarroll, S. A., Schwartz, S. M., Yee, J., Kathiresan, S., Lucas, G., Subirana, I., Elosua, R., Surti, A., Guiducci, C., Gianniny, L., Mirel, D., Parkin, M., Burtt, N., Gabriel, S. B., Samani, N. J., Thompson, J. R., Braund, P. S., Wright, B. J., Balmforth, A. J., Ball, S. G., Hall, A. S., Wellcome Trust Case Control Consortium. Schunkert, H., Erdmann, J., Linsel-Nitschke, P., Lieb, W., Ziegler, A., König, I., Hengstenberg, C., Fischer, M., Stark, K., Grosshennig, A., Preuss, M., Wichmann, H. E., Schreiber, S., Schunkert, H., Samani, N. J., Erdmann, J., Ouwehand, W., Hengstenberg, C., Deloukas, P., Scholz, M., Cambien, F., Reilly, M. P., Li, M., Chen, Z., Wilensky, R., Matthai, W., Qasim, A., Hakonarson, H. H., Devaney, J., Burnett, M. S., Pichard, A. D., Kent, K. M., Satler, L., Lindsay, J. M., Waksman, R., Knouff, C. W., Waterworth, D. M., Walker, M. C., Mooser, V., Epstein, S. E., Rader, D. J., Scheffold, T., Berger, K., Stoll, M., Huge, A., Girelli, D., Martinelli, N., Olivieri, O., Corrocher, R., Morgan, T., Spertus, J. A., McKeown, P., Patterson, C. C., Schunkert, H., Erdmann, E., Linsel-Nitschke, P., Lieb, W., Ziegler, A., König, I. R., Hengstenberg, C., Fischer, M., Stark, K., Grosshennig, A., Preuss, M., Wichmann, H. E., Schreiber, S., Hólm, H., Thorleifsson, G., Thorsteinsdottir, U., Stefansson, K., Engert, J. C., Do, R., Xie, C., Anand, S., Kathiresan, S., Ardissino, D., Mannucci, P. M., Siscovick, D., O’Donnell, C. J., Samani, N. J., Melander, O., Elosua, R., Peltonen, L., Salomaa, V., Schwartz, S. M., and Altshuler, D. (2009). Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 41, 334–341.
Orso, E., Broccardo, C., Kaminski, W. E., Bottcher, A., Liebisch, G., Drobnik, W., Götz, A., Chambenoit, O., Diederich, W., Langmann, T., Spruss, T., Luciani, M. F., Rothe, G., Lackner, K. J., Chimini, G., and Schmitz, G. (2000). Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1-deficient mice. Nat. Genet. 24, 192–196.
Pan, W. (2008). Network-based model weighting to detect multiple loci influencing complex diseases. Hum. Genet. 124, 225–234.
Perry, J. R., McCarthy, M. I., Hattersley, A. T., Zeggini, E., Wellcome Trust Case Control Consortium. Weedon, M. N., and Frayling, T. M. (2009). Interrogating type 2 diabetes genome-wide association data using a biological pathway-based approach. Diabetes 58, 1463–1467.
Pullinger, C. R., Eng, C., Salen, G., Shefer, S., Batta, A. K., Erickson, S. K., Verhagen, A., Rivera, C. R., Mulvihill, S. J., Malloy, M. J., and Kane, J. P. (2002). Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Invest. 110, 109–117.
Rashid, S., Curtis, D. E., Garuti, R., Anderson, N. N., Bashmakov, Y., Ho, Y. K., Hammer, R. E., Moon, Y. A., and Horton, J. D. (2005). Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. U.S.A. 102, 5374–5379.
Reiner, A., Yekutieli, D., and Benjamini, Y. (2003). Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375.
Roberts, C. G., Shen, H., Mitchell, B. D., Damcott, C. M., Shuldiner, A. R., and Rodriguez, A. (2007). Variants in scavenger receptor class B type I gene are associated with HDL cholesterol levels in younger women. Hum. Hered. 64, 107–113.
Rust, S., Rosier, M., Funke, H., Real, J., Amoura, Z., Piette, J. C., Deleuze, J. F., Brewer, H. B., Duverger, N., Denèfle, P., and Assmann, G. (1999). Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 22, 352–355.
Sabatti, C., Service, S. K., Hartikainen, A. L., Pouta, A., Ripatti, S., Brodsky, J., Jones, C. G., Zaitlen, N. A., Varilo, T., Kaakinen, M., Sovio, U., Ruokonen, A., Laitinen, J., Jakkula, E., Coin, L., Hoggart, C., Collins, A., Turunen, H., Gabriel, S., Elliot, P., McCarthy, M. I., Daly, M. J., Järvelin, M. R., Freimer, N. B., and Peltonen, L. (2009). Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 41, 35–46.
Shefer, S., Salen, G., Batta, A. K., Honda, A., Tint, G. S., Irons, M., Elias, E. R., Chen, T. C., and Holick, M. F. (1995). Markedly inhibited 7-dehydrocholesterol-delta 7-reductase activity in liver microsomes from Smith-Lemli-Opitz homozygotes. J. Clin. Invest. 96, 1779–1785.
Soutar, A. K., and Naoumova, R. P. (2007). Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 4, 214–225.
Steinthorsdottir, V., Thorleifsson, G., Reynisdottir, I., Benediktsson, R., Jonsdottir, T., Walters, G. B., Styrkarsdottir, U., Gretarsdottir, S., Emilsson, V., Ghosh, S., Baker, A., Snorradottir, S., Bjarnason, H., Ng, M. C., Hansen, T., Bagger, Y., Wilensky, R. L., Reilly, M. P., Adeyemo, A., Chen, Y., Zhou, J., Gudnason, V., Chen, G., Huang, H., Lashley, K., Doumatey, A., So, W. Y., Ma, R. C., Andersen, G., Borch-Johnsen, K., Jorgensen, T., van Vliet-Ostaptchouk, J. V., Hofker, M. H., Wijmenga, C., Christiansen, C., Rader, D. J., Rotimi, C., Gurney, M., Chan, J. C., Pedersen, O., Sigurdsson, G., Gulcher, J. R., Thorsteinsdottir, U., Kong, A., and Stefansson, K. (2007). A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 39, 770–775.
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., Paulovich, A., Pomeroy, S. L., Golub, T. R., Lander, E. S., and Mesirov, J. P. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550.
Sun, Y. M., Wang, L. F., Li, J., Li, Z. Q., and Pan, W. (2009). The 223A>G polymorphism of the leptin receptor gene and lipid-lowering efficacy of simvastatin in Chinese patients with coronary heart disease. Eur. J. Clin. Pharmacol. 65, 157–161.
Takada, D., Emi, M., Ezura, Y., Nobe, Y., Kawamura, K., Iino, Y., Katayama, Y., Xin, Y., Wu, L. L., Larringa-Shum, S., Stephenson, S. H., Hunt, S. C., and Hopkins, P. N. (2002). Interaction between the LDL-receptor gene bearing a novel mutation and a variant in the apolipoprotein A-II promoter: molecular study in a 1135-member familial hypercholesterolemia kindred. J. Hum. Genet. 47, 656–664.
Teslovich, T. M., Musunuru, K., Smith, A. V., Edmondson, A. C., Stylianou, I. M., Koseki, M., Pirruccello, J. P., Ripatti, S., Chasman, D. I., Willer, C. J., Johansen, C. T., Fouchier, S. W., Isaacs, A., Peloso, G. M., Barbalic, M., Ricketts, S. L., Bis, J. C., Aulchenko, Y. S., Thorleifsson, G., Feitosa, M. F., Chambers, J., Orho-Melander, M., Melander, O., Johnson, T., Li, X., Guo, X., Li, M., Shin Cho, Y., Jin Go, M., Jin Kim, Y., Lee, J. Y., Park, T., Kim, K., Sim, X., Twee-Hee Ong, R., Croteau-Chonka, D. C., Lange, L. A., Smith, J. D., Song, K., Hua Zhao, J., Yuan, X., Luan, J., Lamina, C., Ziegler, A., Zhang, W., Zee, R. Y., Wright, A. F., Witteman, J. C., Wilson, J. F., Willemsen, G., Wichmann, H. E., Whitfield, J. B., Waterworth, D. M., Wareham, N. J., Waeber, G., Vollenweider, P., Voight, B. F., Vitart, V., Uitterlinden, A. G., Uda, M., Tuomilehto, J., Thompson, J. R., Tanaka, T., Surakka, I., Stringham, H. M., Spector, T. D., Soranzo, N., Smit, J. H., Sinisalo, J., Silander, K., Sijbrands, E. J., Scuteri, A., Scott, J., Schlessinger, D., Sanna, S., Salomaa, V., Saharinen, J., Sabatti, C., Ruokonen, A., Rudan, I., Rose, L. M., Roberts, R., Rieder, M., Psaty, B. M., Pramstaller, P. P., Pichler, I., Perola, M., Penninx, B. W., Pedersen, N. L., Pattaro, C., Parker, A. N., Pare, G., Oostra, B. A., O’Donnell, CJ, Nieminen, M. S., Nickerson, D. A., Montgomery, G. W., Meitinger, T., McPherson, R., McCarthy, MI, McArdle, W., Masson, D., Martin, N. G., Marroni, F., Mangino, M., Magnusson, P. K., Lucas, G., Luben, R., Loos, R. J., Lokki, M. L., Lettre, G., Langenberg, C., Launer, L. J., Lakatta, E. G., Laaksonen, R., Kyvik, K. O., Kronenberg, F., König, I. R., Khaw, K. T., Kaprio, J., Kaplan, L. M., Johansson, A., Jarvelin, M. R., Janssens, A. C., Ingelsson, E., Igl, W., Kees Hovingh, G., Hottenga, J. J., Hofman, A., Hicks, A. A., Hengstenberg, C., Heid, I. M., Hayward, C., Havulinna, A. S., Hastie, N. D., Harris, T. B., Haritunians, T., Hall, A. S., Gyllensten, U., Guiducci, C., Groop, L. C., Gonzalez, E., Gieger, C., Freimer, N. B., Ferrucci, L., Erdmann, J., Elliott, P., Ejebe, K. G., Döring, A., Dominiczak, A. F., Demissie, S., Deloukas, P., de Geus, E. J., de Faire, U., Crawford, G., Collins, F. S., Chen, Y. D., Caulfield, M. J., Campbell, H., Burtt, N. P., Bonnycastle, L. L., Boomsma, D. I., Boekholdt, S. M., Bergman, R. N., Barroso, I., Bandinelli, S., Ballantyne, C. M., Assimes, T. L., Quertermous, T., Altshuler, D., Seielstad, M., Wong, T. Y., Tai, E. S., Feranil, A. B., Kuzawa, C. W., Adair, L. S., Taylor, H.A. Jr., Borecki, I. B., Gabriel, S. B., Wilson, J. G., Holm, H., Thorsteinsdottir, U., Gudnason, V., Krauss, R. M., Mohlke, K. L., Ordovas, J. M., Munroe, P. B., Kooner, J. S., Tall, A. R., Hegele, R. A., Kastelein, J. J., Schadt, E. E., Rotter, J. I., Boerwinkle, E., Strachan, D. P., Mooser, V., Stefansson, K., Reilly, M. P., Samani, N. J., Schunkert, H., Cupples, L. A., Sandhu, M. S., Ridker, P. M., Rader, D. J., van Duijn, C. M., Peltonen, L., Abecasis, G. R., Boehnke, M., and Kathiresan, S. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713.
Torkamani, A., Topol, E. J., and Schork, N. J. (2008). Pathway analysis of seven common diseases assessed by genome-wide association. Genomics 92, 265–272.
Wang, J., Freeman, D. J., Grundy, S. M., Levine, D. M., Guerra, R., and Cohen, J. C. (1998). Linkage between cholesterol 7alpha-hydroxylase and high plasma low-density lipoprotein cholesterol concentrations. J. Clin. Invest. 101, 1283–1291.
Wang, K., Li, M., and Bucan, M. (2007). Pathway-based approaches for analysis of genomewide association studies. Am. J. Hum. Genet. 81, 1278–1283.
Wang, K., Li, M., and Hakonarson, H. (2010). Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 11, 843–854.
Wang, K., Zhang, H., Kugathasan, S., Annese, V., Bradfield, J. P., Russell, R. K., Sleiman, P. M., Imielinski, M., Glessner, J., Hou, C., Wilson, D. C., Walters, T., Kim, C., Frackelton, E. C., Lionetti, P., Barabino, A., Van Limbergen, J., Guthery, S., Denson, L., Piccoli, D., Li, M., Dubinsky, M., Silverberg, M., Griffiths, A., Grant, S. F., Satsangi, J., Baldassano, R., and Hakonarson, H. (2009a). Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn Disease. Am. J. Hum. Genet. 84, 399–405.
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., Salyakina, D., Imielinski, M., Bradfield, J. P., Sleiman, P. M., Kim, C. E., Hou, C., Frackelton, E., Chiavacci, R., Takahashi, N., Sakurai, T., Rappaport, E., Lajonchere, C. M., Munson, J., Estes, A., Korvatska, O., Piven, J., Sonnenblick, L. I., Alvarez Retuerto, A. I., Herman, E. I., Dong, H., Hutman, T., Sigman, M., Ozonoff, S., Klin, A., Owley, T., Sweeney, J. A., Brune, C. W., Cantor, R. M., Bernier, R., Gilbert, J. R., Cuccaro, M. L., McMahon, W. M., Miller, J., State, M. W., Wassink, T. H., Coon, H., Levy, S. E., Schultz, R. T., Nurnberger, J. I., Haines, J. L., Sutcliffe, J. S., Cook, E. H., Minshew, N. J., Buxbaum, J. D., Dawson, G., Grant, S. F., Geschwind, D. H., Pericak-Vance, M. A., Schellenberg, G. D., and Hakonarson, H. (2009b). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533.
Wassif, C. A., Zhu, P., Kratz, L., Krakowiak, P. A., Battaile, K. P., Weight, F. F., Grinberg, A., Steiner, R. D., Nwokoro, N. A., Kelley, R. I., Stewart, R. R., and Porter, F. D. (2001). Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith – Lemli – Opitz syndrome. Hum. Mol. Genet. 10, 555–564.
Waterham, H. R., Koster, J., Romeijn, G. J., Hennekam, R. C., Vreken, P., Andersson, H. C., FitzPatrick, D. R., Kelley, R. I., and Wanders, R. J. (2001). Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am. J. Hum. Genet. 69, 685–694.
Wechsler, A., Brafman, A., Shafir, M., Heverin, M., Gottlieb, H., Damari, G., Gozlan-Kelner, S., Spivak, I., Moshkin, O., Fridman, E., Becker, Y., Skaliter, R., Einat, P., Faerman, A., Björkhem, I., and Feinstein, E. (2003). Generation of viable cholesterol-free mice. Science 302, 2087.
Wiersma, H., Nijstad, N., de Boer, J. F., Out, R., Hogewerf, W., Van Berkel, T. J., Kuipers, F., and Tietge, U. J. (2009). Lack of Abcg1 results in decreased plasma HDL cholesterol levels and increased biliary cholesterol secretion in mice fed a high cholesterol diet. Atherosclerosis 206, 141–147.
Willner, E. L., Tow, B., Buhman, K. K., Wilson, M., Sanan, D. A., Rudel, L. L., and Farese, R. V. Jr. (2003). Deficiency of acyl CoA: cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 100, 1262–1267.
Wolfrum, C., Poy, M. N., and Stoffel, M. (2005). Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat. Med. 11, 418–422.
Young, S. G., Bertics, S. J., Curtiss, L. K., Dubois, B. W., and Witztum, J. L. (1987). Genetic analysis of a kindred with familial hypobetalipoproteinemia. Evidence for two separate gene defects: one associated with an abnormal apolipoprotein B species, apolipoprotein B-37; and a second associated with low plasma concentrations of apolipoprotein B-100. J. Clin. Invest. 79, 1842–1851.
Keywords: GWAS, lipid, HDL-C, pathway analysis, cholesterol, sterol transport, sterol metabolism, genetic association
Citation: Wang K, Edmondson AC, Li M, Gao F, Qasim AN, Devaney JM, Burnett MS, Waterworth DM, Mooser V, Grant SFA, Epstein SE, Reilly MP, Hakonarson H and Rader DJ (2011) Pathway-wide association study implicates multiple sterol transport and metabolism genes in HDL cholesterol regulation. Front. Gene. 2:41. doi: 10.3389/fgene.2011.00041
Received: 05 May 2011; Paper pending published: 26 May 2011;
Accepted: 21 June 2011; Published online: 05 July 2011.
Edited by:
Robert Klein, Memorial Sloan-Kettering Cancer Center, USAReviewed by:
John Charles Huber, Texas A&M Health Science Center School of Rural Public Health, USAYiran Guo, Children’s Hospital of Philadelphia, USA
Sarah Buxbaum, Jackson State University, USA
Peter Holmans, Cardiff University, UK
Copyright: © 2011 Wang, Edmondson, Li, Gao, Qasim, Devaney, Burnett, Waterworth, Mooser, Grant, Epstein, Reilly, Hakonarson and Rader. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Kai Wang, University of Southern California Los Angeles, 1501 San Pablo Street, ZNI 221, Los Angeles, CA 90089, USA. e-mail: kaichop@gmail.com