 Katrin Sikk1* Sulev Kõks2,3 Ursel Soomets3,4 Leonard C. Schalkwyk5 Cathy Fernandes5 Sulev Haldre1 Sten-Magnus Aquilonius6 Pille Taba1
Katrin Sikk1* Sulev Kõks2,3 Ursel Soomets3,4 Leonard C. Schalkwyk5 Cathy Fernandes5 Sulev Haldre1 Sten-Magnus Aquilonius6 Pille Taba1
- 1 Department of Neurology and Neurosurgery, University of Tartu, Tartu, Estonia
- 2 Department of Physiology, University of Tartu, Tartu, Estonia
- 3 Centre for Translational Research, University of Tartu, Tartu, Estonia
- 4 Department of Biochemistry, University of Tartu, Tartu, Estonia
- 5 Social, Genetic and Developmental Psychiatry Centre, Institute of Psychiatry, King’s College London, London, UK
- 6 Section of Neurology, Department of Neuroscience, Uppsala University, Uppsala, Sweden
Methcathinone (ephedrone) is relatively easily accessible for abuse. Its users develop an extrapyramidal syndrome and it is not known if this is caused by methcathinone itself, by side-ingredients (manganese), or both. In the present study we aimed to clarify molecular mechanisms underlying this condition. We used microarrays to analyze whole-genome gene expression patterns of peripheral blood from 20 methcathinone users and 20 matched controls. Gene expression profile data were analyzed by Bayesian modeling and functional annotation. Of 28,869 genes on the microarrays, 326 showed statistically significant differential expression with FDR adjusted p-values below 0.05. Quantitative real-time PCR confirmed differential expression for the most of the genes selected for validation. Functional annotation and network analysis indicated activation of a gene network that included immunological disease, cellular movement, and cardiovascular disease functions (enrichment score 42). As HIV and HCV infections were confounding factors, we performed additional stratification of subjects. A similar functional activation of the “immunological disease” category was evident when we compared subjects according to injection status (past versus current users, balanced for HIV and HCV infection). However, this difference was not large therefore the major effect was related to the HIV status of the subjects. Mn–methcathinone abusers have blood RNA expression patterns that mostly reflect their HIV and HCV infections.
Introduction
Methcathinone (mcat) is an illicit stimulant drug synthesized by oxidation of ephedrine or pseudoephedrine with potassium permanganate. Motor dysfunction, such as hypokinesia and impaired balance, in methcathinone users has been reported in several case series from different countries, and probably develops due to chronic manganese intoxication (Sikk et al., 2007; Selikhova et al., 2008; Stepens et al., 2008). As methcathinone is relatively easy to manufacture using household materials, this poses a significant public health risk. Methcathinone abuse and its preparation with potassium permanganate is quite common in eastern European countries, but there are already signs that the practice is spreading to western countries (De Bie et al., 2007).
Molecular mechanisms underlying combined Mn and methcathinone toxicity are poorly understood. Mn neurotoxicity is well known and proposed mechanisms for neuronal death are described in several review articles (Fitsanakis et al., 2006; Burton and Guilarte, 2009; Curran et al., 2009). However, methcathinone has a molecular structure similar to methamphetamine and its pharmacodynamic properties suggest potential toxicity as well (Glennon et al., 1995; Rockhold et al., 1997; McCann et al., 1998). There are no molecular studies on the effect of these substances in combination.
Gene expression profiling using whole-genome microarray screening is a well-established technology to obtain a snapshot of the complex response variables (transcriptome) in biological systems. Expression profiling has great potential for examining molecular pathogenesis and biomarkers of disease. It is a relatively easy to use technology which gives a genome-wide overview of the molecular changes during pathological conditions. Peripheral blood gene expression has been used as a surrogate fingerprint of cerebral neurological diseases (Sharp et al., 2006; Sullivan et al., 2006; Mohr and Liew, 2007; Davies et al., 2009). The resulting profile is a complex description of the molecular phenotype and could be used to analyze different acute and chronic conditions.
The aim of this study was to analyze gene expression profiles in chronic methcathinone abusers. In addition, we applied functional genomic analysis to identify gene networks related to this chronic condition.
Materials and Methods
Study Participants
Twenty illicit methcathinone users and 20 sex- and age-matched controls reporting to be non-drug users and not to be infected with HIV and HCV were enrolled in the present study. Methcathinone was the main drug of abuse. Subjects were divided according to self reported drug abuse history into current or past users (reported having discontinued at least 1 year previously). Main subject characteristics including the results of motor and mental function and quality of life assessment scales are summarized in Table 1 (Hoehn and Yahr, 1967; Schwab and England, 1969; Folstein et al., 1975; Fahn et al., 1987; Peto et al., 1995). All subjects were positive for hepatitis C virus (HCV). Written informed consent was obtained from all subjects. The study was approved by the ethics committee on human research at Tartu University.
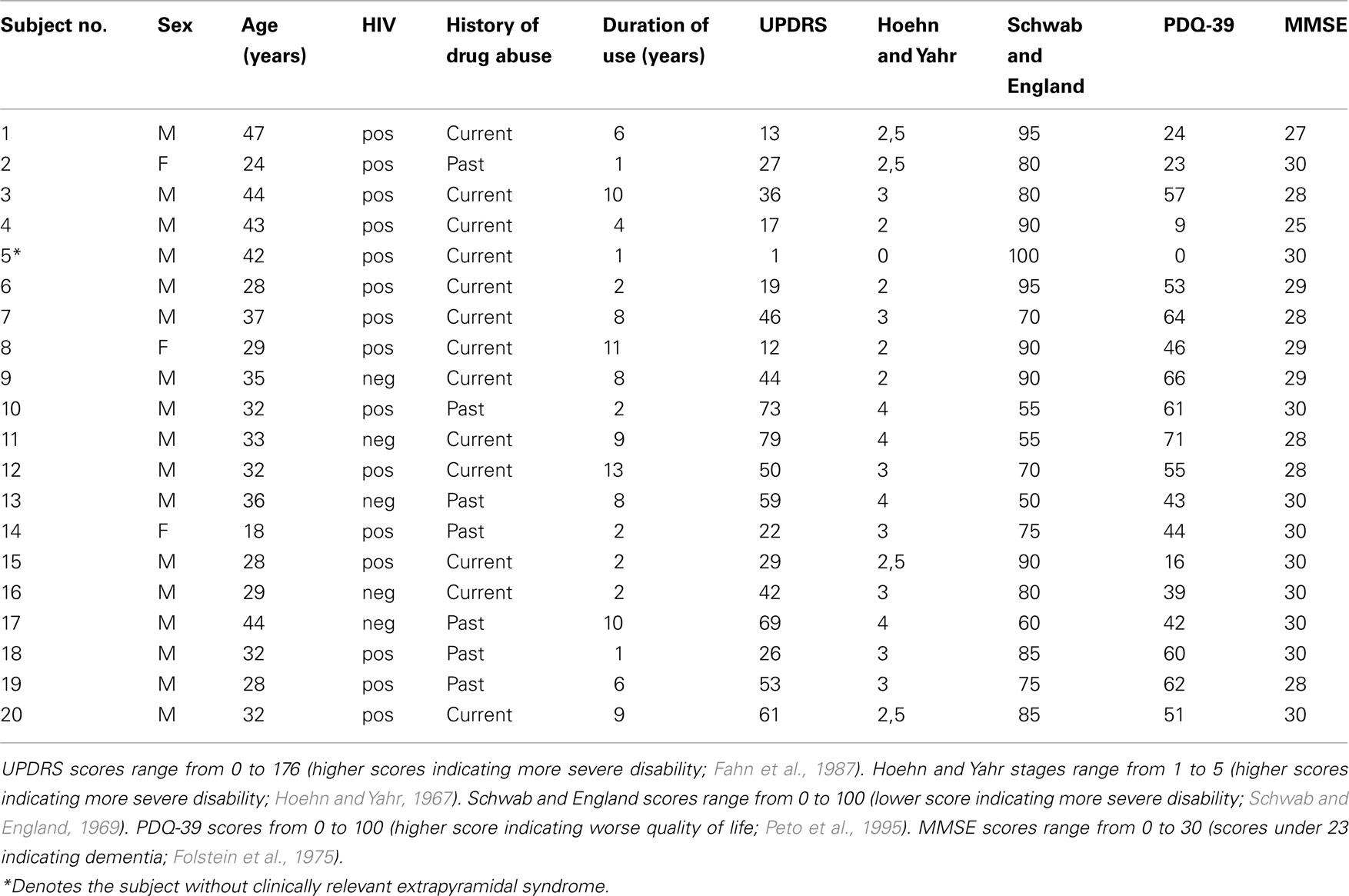
Table 1. Clinical and demographic characteristics.
Sample Collection and RNA Preparation
Blood samples from 20 subjects and 20 healthy controls were collected into Tempus tubes (Applied Biosystems, Foster City, USA). Blood was frozen and stored until further processing. RNA extraction from whole blood was performed according to the manufacturer’s protocol (PN 4379228C). After RNA extraction, alpha and beta globin mRNA was depleted with GlobinClear Whole Blood Globin Reduction kit (Ambion, Austin, USA). The quality of RNA was analyzed with a Bioanalyzer 2100 (Agilent, Santa Clara, USA) and gene expression profiling was performed with GeneChip Human Gene 1.0 ST Arrays (Affymetrix, Santa Clara, USA) containing probes for all exons of 28,869 genes. We analyzed all the genes on the array without any filtering for presence/absence cells.
Microarray Hybridization and Analysis
In order to label the RNA we used the Affymetrix GeneChip Whole Transcript (WT) Sense Target Labeling Assay (Affymetrix, Santa Clara, USA), which is designed to generate amplified and biotinylated sense-strand targets from the entire expressed genome without bias. Briefly, double-stranded cDNA was synthesized from 300 ng of total RNA by reverse transcription using random hexamers tagged with a T7 promotor primer sequence. The double-stranded cDNA was subsequently used as a template and amplified by T7 RNA polymerase producing many copies of antisense cRNA. In the second cycle of cDNA synthesis, random hexamers were used to prime reverse transcription of the cRNA from the first cycle to produce single-stranded DNA in the sense orientation. This DNA was fragmented with a combination of uracil DNA glycosylase (UDG) and apurinic/apyrimidinic endonuclease 1 (APE 1). DNA was labeled by terminal deoxynucleotidyl transferase (TdT) and hybridization was performed according to the manufacturer’s protocol. The arrays were subsequently washed, stained with phycoerythrin streptavidin and scanned according to standard Affymetrix protocol. Images were processed using the Affymetrix Expression Console and the MAS 5.0 algorithm was used to measure quality control parameters.
Statistical Analysis
The normalized, background subtracted, and modeled expression (Robust Multi-array Analysis, RMA) data (GEO accession number GSE28686) were further analyzed using a linear model combined with Bayesian moderation for standard errors implemented in the Bioconductor limma package of the statistical software R1 (Ihaka and Gentleman, 1996; Smyth, 2004). False discovery rate (FDR) was used to address multiple testing corrections (Benjamini and Hochberg, 1995; Storey and Tibshirani, 2003). Moderated model calculates B-value, which is the log-odd that the gene is differentially expressed. B-statistics of zero corresponds to a 50-50 chance that the gene is differentially expressed and B-statistics is automatically adjusted for multiple testing.
Quantitative Real-Time PCR Analysis
Eight genes with significant differences and with potentially interesting functions from the gene expression profiling data were further analyzed by means of real-time PCR (RT-PCR). These genes were: C15orf26, GPR15, IGFR1, SNRPN, IFI44L, IFI44, IFI27, and IFNG. RNA was converted into cDNA using the High Capacity cDNA Synthesis kit from Applied Biosystems. TaqMan assays and Gene Expression Master mix was used for the RT-PCR reaction in the SDS 7900 HT system (Applied Biosystems). Sample (20 controls and 20 methcathinone users in four replicates) comparisons were made using Welch’s t-test.
Functional Annotation of Differentially Expressed Genes
To define the functional networks of the differentially expressed genes, data were analyzed by ingenuity pathway analysis (IPA, Ingenuity Systems2). A data set containing Affymetrix probeset identifiers and corresponding fold change (log2) values was uploaded. Each gene identifier was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base to generate the list of focus genes. Networks of these focus genes were then algorithmically generated based on inter-gene relationships manually curated in the Ingenuity database from published studies. Ingenuity pathway analysis calculates a significance score for each network. The score is generated using a p-value calculation, and is displayed as the negative logarithm of that p-value. This score indicates the likelihood that the assembly of a set of focus genes into a network could be explained by chance alone (e.g. score of 2 indicates that there is a 1° in 100 chance that the focus genes are together in a network by random chance).
Results
Gene Expression Profiling with Microarrays
Comparison of the blood RNA samples isolated from methcathinone users and healthy controls revealed distinct gene expression profiles. Statistically significant differential expression was observed for 326 genes with p-values less than 0.05 (FDR adjusted for multiple testing, Table 2). The magnitudes of expression signal differences (fold change or logFC) between these two groups were generally low – 17 genes were upregulated in the methcathinone user more than 1.1-fold and only one gene was down-regulated more than 1.1-fold. However, the B-statistics values for the list of genes were very high suggesting real biological differences between these groups. A heatmap (Figure 1) from unsupervised hierarchical clustering indicates clear clustering of samples into drug users and non-users. However, in the middle of the heatmap, some overlap and mixture between groups occurs. These mcat samples are persons with HIV negative status. Therefore, HIV status has a significant impact on the gene expression profile and is a strong confounding factor.
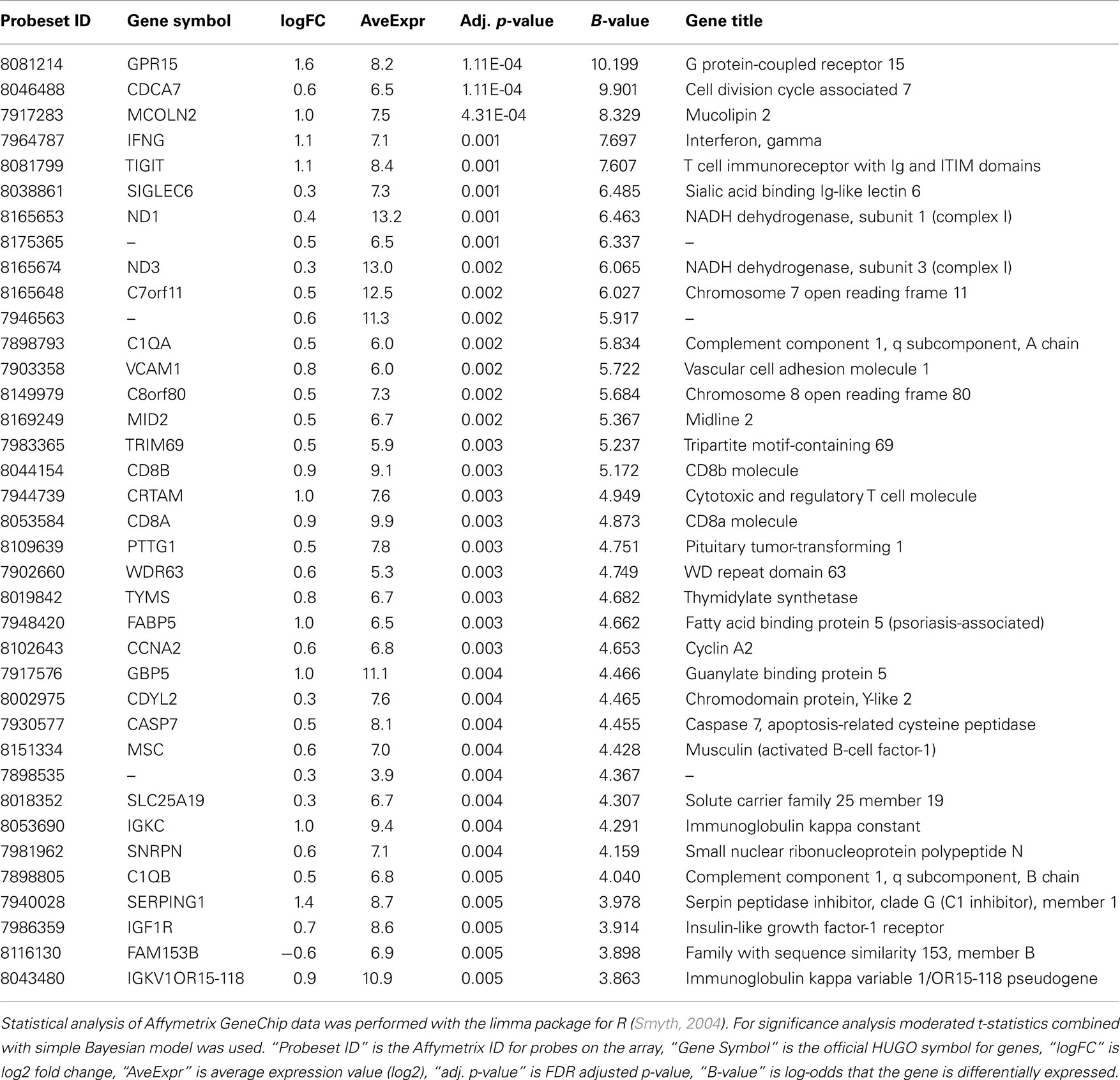
Table 2. Differential expression analysis of the blood RNA suggests significant differences between study groups.
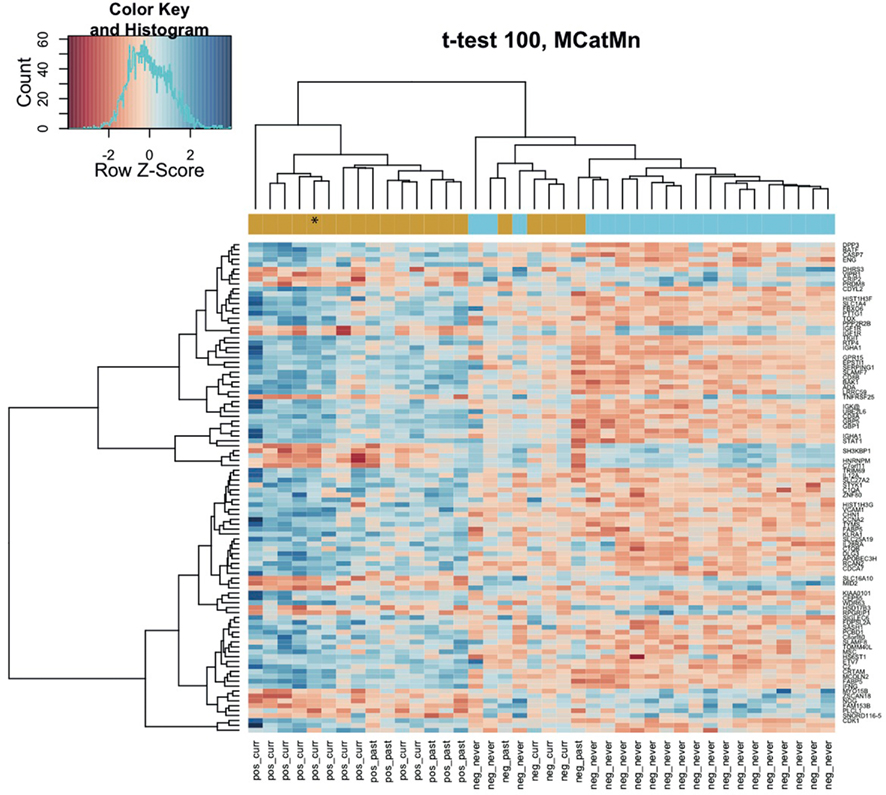
Figure 1. Methcathinone users group distinctly on the hierarchical clustering heatmap of blood gene expression levels. The top 100 genes from the decreasing ordered list of moderated t-values were clustered according to similarity in their gene expression patterns. Signals are scaled to Z-scores of the rows. The colored bar above the heatmap indicates the grouping variable – goldenrod for methcathinone user, blue for controls. Column labels at the bottom combine HIV status (positive or negative) with the drug user status (current, past, never). Row labels are gene symbols and the asterisk denotes the subject without clinically relevant extrapyramidal syndrome. Note some overlap between the two groups at the middle of the heatmap.
A functional annotation analysis was performed to define gene networks within the observed expression profiles. Relationships among the 326 genes significantly different in methcathinone users compared to control groups included two major networks. The first (with the highest score of 47; score indicates the −log of enrichment p-value) contained genes annotated with cell death, antigen presentation, or neurological disease functions (Table 3, Figure 2). A second network (score 42) was related to immunological disease, cellular movement, or cardiovascular disease. Enrichment or activation of these networks suggests that methcathinone users have changes in the genetic pathways related to the function of the nervous and immune systems.
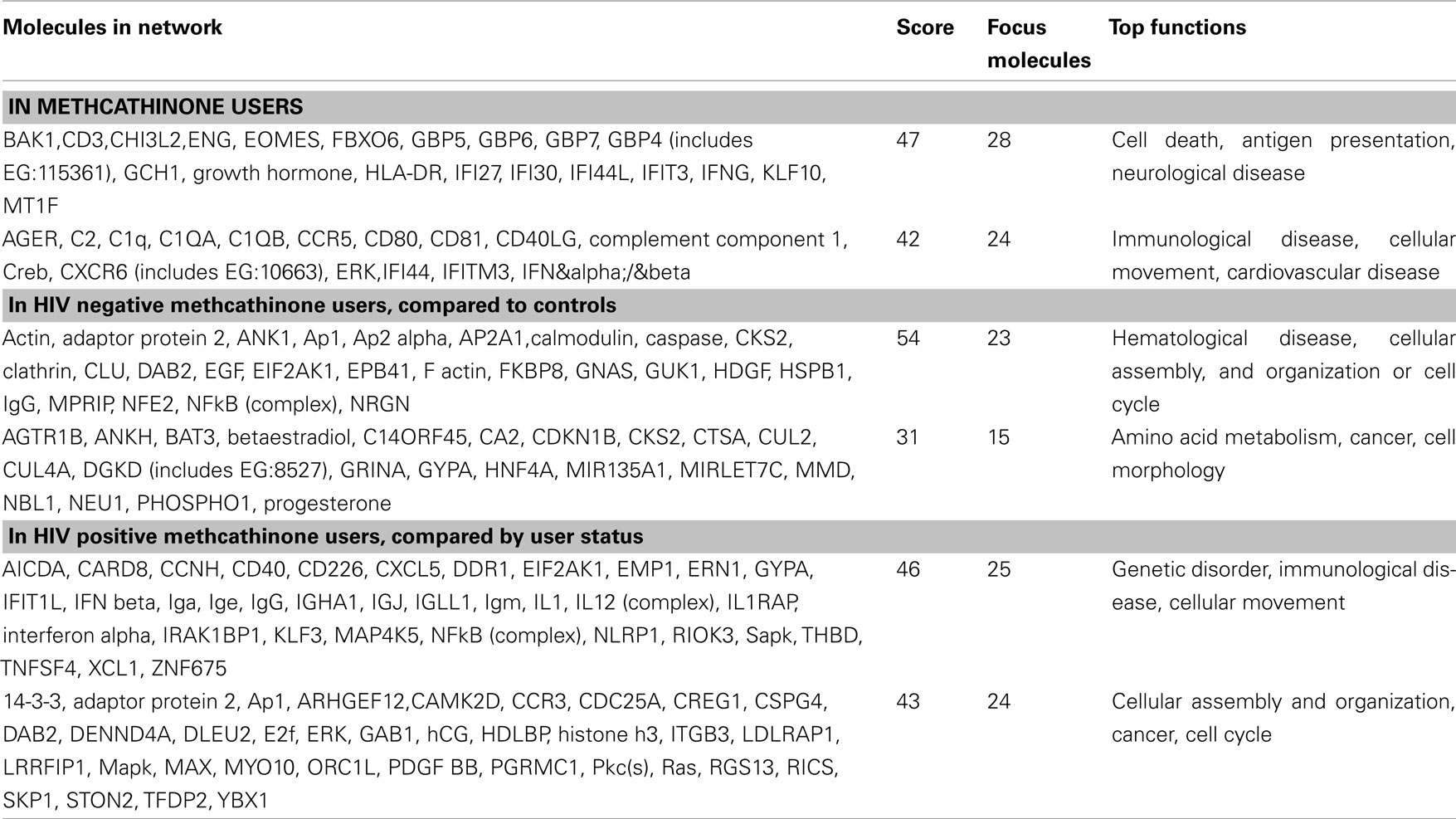
Table 3. Genetic networks significantly changed.
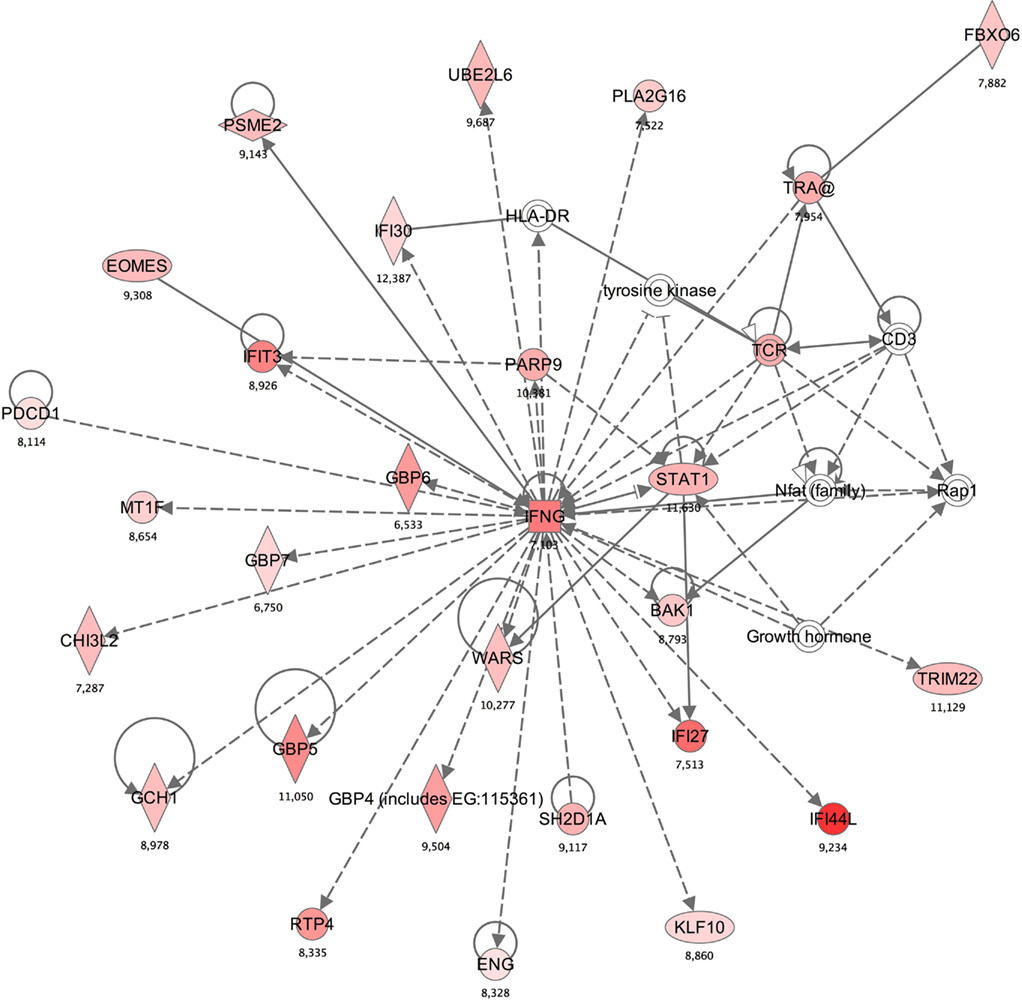
Figure 2. Annotation enrichment analysis indicated that a network including “cell death, antigen presentation, neurological disease” gene functions is significantly enriched in the subjects with methcathinone abuse [score 47, score is −log (p-value)]. Red nodes designate upregulated genes and the number indicates log2 fold change (0 is for equal expression). Uncolored nodes are genes in this network that were not in our list of differentially expressed genes.
Table 2 includes many genes that are related to immune response, and may be differentially expressed compared to healthy controls because most of drug users were HIV positive. We therefore compared the subgroup of HIV negative drug users to the healthy control group. In this comparison, no genes were significantly different at FDR less than 5%, but 93 genes were different at or below a 30% FDR threshold (Table A1 in Appendix). Annotation analysis for this dataset of HIV negative drug users indicated up-regulation (23 genes, score 54) in a network of genes with functions including hematological disease, cellular assembly, and organization or cell cycle (Table 3). In another subgroup analysis we compared HIV positive subjects according to their user status, current versus discontinued. This comparison should indicate the difference caused by active drug abuse (Table 3). In this case both groups are HIV and HCV positive, so infection status is balanced. The most significantly enriched network in this comparison included genes in the Genetic Disorder, Immunological Disease, or Cellular Movement categories (25 genes, enrichment score 46). This network supports the involvement of the immune system in the drug abuse induced pathologies.
Quantitative Real-Time PCR
To further verify changes found with the GeneChip experiment, we performed quantitative RT-PCR and analyzed gene expression levels of eight selected genes. For seven genes, statistically significant differential expression between 20 healthy controls and 20 methcathinone users was seen in the expected direction of change (C15orf26 p < 0.0001, GPR15 p < 0.0001, IGFR1 p = 0.0002, SNRPN p = 0.038, IFI44L p = 0.0264, IFI27 p = 0.0450, IFNG p < 0.0001). These results support the validity of our microarray gene expression analysis.
Discussion
In this study, we examined changes in gene expression profiles in peripheral blood induced by methcathinone abuse. All but one of the subjects had a clinically relevant extrapyramidal syndrome. RNA expression patterns of drug users clearly differed from those of healthy controls. Also, RT-PCR results confirmed differences between these two groups. The overlap between controls and HIV negative methcathinone users in the middle of the heatmap (Figure 1) suggested that most of the hierarchical clustering was driven by HIV infection status. During recent years several microarray studies of HIV induced alterations in host cell gene expression have been performed using in vitro infected primary cells, cell lines, or tissue samples (Giri et al., 2006). However, few in vivo studies in HIV infected humans have evaluated expression profiles of tissue samples (Masliah et al., 2004; Everall et al., 2005) or peripheral blood mononuclear cells (Motomura et al., 2004; Kottilil et al., 2009; Monaco et al., 2009). Several of the same apoptosis, antigen presentation, and immune response genes (IFIT3, IFI27, IFI44, IFNG, NFAT, STAT1, TCR, CCR5, ERK, IFITM3, LY6E, IFNG, NFkB, OAS1, PLSCR1) described in previous HIV microarray studies were upregulated in our analysis. Thus peripheral blood can be used for finding gene expression alterations in HIV infected humans. Besides cell death and antigen presentation the genetic network with the highest score was also related to neurological disease, consistent with known associations of HIV infection and neurodegeneration (Clifford et al., 2005).
Further minor subgroup analysis of HIV negative users who were all positive for HCV indicated that genes related to cell cycle, cellular growth and proliferation, cancer, and cellular development were upregulated compared to healthy controls. The same pathways have been shown to be involved in HCV induced hepatic changes like cirrhosis, dysplasia, or hepatocellular carcinoma (Wurmbach et al., 2007; De Giorgi et al., 2009).
As the aim of our study was to analyze the drug abuse effect, we decided to stratify our group of subjects. We analyzed only HIV and HCV positive subjects according to their injection status (past versus current) and a clinical measure of the severity of the neurological syndrome UPDRS. The most significantly enriched network after comparing HIV positive current and past users included genes annotated with functions in Genetic Disorder, Immunological Disease, or Cellular Movement categories (25 genes, enrichment score 46). This network confirms the involvement of the immune system in drug abuse induced pathologies. However, as both comparison groups had neurological symptoms we can not draw any conclusions on the potential causes of the extrapyramidal syndrome. On the other hand, injection status seems to influence immune system and this is one factor involved in the development of a neurodegenerative syndrome. Psychostimulant abuse has been shown to compromise immunological status (Everall et al., 2005). Chronic Mn exposure has similar effects on the immune system (Sengupta et al., 2007). Thus, both of these factors have potential to induce neurodegeneration.
The UPDRS score reflects the clinical severity of the extrapyramidal syndrome. No significant correlations were found between the duration of Mn–methcathinone use and UPDRS or between blood RNA profiles and UPDRS.
There are several limitations in our study. The main problem is related to the sample organization. Our sample contains many confounding factors – HIV and HCV infection, different duration of methcathinone use, concomitant consumption of other drugs, alcohol, and tobacco. We have not analyzed the clinical status of the HIV and HCV infections. In addition, several drug abusers were undernourished which makes identification of a relevant control group even more complicated. To overcome these limitations, we have applied complex bioinformatics analytical tools in relatively large samples (N = 20 in both groups) that allowed us to perform some cohort stratification. For further studies within methcathinone users more balanced control samples are needed. We have tried to collect similar balanced sample as a control group, but due to the specific and complex nature of our study group it is difficult and time consuming. Ideal controls would be either drug-free HIV and HCV positive subjects or addicts abusing other drugs. We fully understand the limitations caused by the complexity of the problem we analyze. On the other hand, the situation of a non-balanced control sample is quite common in clinical research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The financial support by the Estonian Science Foundation (grant 7433) is acknowledged.
Footnotes
References
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate – a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300.
Burton, N. C., and Guilarte, T. R. (2009). Manganese neurotoxicity: lessons learned from longitudinal studies in nonhuman primates. Environ. Health Perspect. 117, 325–332.
Clifford, D. B., Yang, Y., and Evans, S. (2005). Neurologic consequences of hepatitis C and human immunodeficiency virus coinfection. J. Neurovirol. 11(Suppl. 3), 67–71.
Curran, C. P., Park, R. M., Ho, S. M., and Haynes, E. N. (2009). Incorporating genetics and genomics in risk assessment for inhaled manganese: from data to policy. Neurotoxicology 30, 754–760.
Davies, M. N., Lawn, S., Whatley, S., Fernandes, C., Williams, R. W., and Schalkwyk, L. C. (2009). To what extent is blood a reasonable surrogate for brain in gene expression studies: estimation from mouse hippocampus and spleen. Front. Neurogenomics 1:1–6. doi: 10.3389/fnins.2011.00161
De Bie, R. M., Gladstone, R. M., Strafella, A. P., Ko, J. H., and Lang, A. E. (2007). Manganese-induced Parkinsonism associated with methcathinone (Ephedrone) abuse. Arch. Neurol. 64, 886–889.
De Giorgi, V., Monaco, A., Worchech, A., Tornesello, M., Izzo, F., Buonaguro, L., Marincola, F. M., Wang, E., and Buonaguro, F. M. (2009). Gene profiling, biomarkers and pathways characterizing HCV-related hepatocellular carcinoma. J. Transl. Med. 7, 85.
Everall, I., Salaria, S., Roberts, E., Corbeil, J., Sasik, R., Fox, H., Grant, I., and Masliah, E. (2005). Methamphetamine stimulates interferon inducible genes in HIV infected brain. J. Neuroimmunol. 170, 158–171.
Fahn, S., Elton, R. L., and Committee, U. D. (1987). Unified Parkinson’s Disease Rating Scale. Florham Park: Macmillan.
Fitsanakis, V. A., Au, C., Erikson, K. M., and Aschner, M. (2006). The effects of manganese on glutamate, dopamine and gamma-aminobutyric acid regulation. Neurochem. Int. 48, 426–433.
Folstein, M. F., Folstein, S. E., and Mchugh, P. R. (1975). “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198.
Giri, M. S., Nebozhyn, M., Showe, L., and Montaner, L. J. (2006). Microarray data on gene modulation by HIV-1 in immune cells: 2000-2006. J. Leukoc. Biol. 80, 1031–1043.
Glennon, R. A., Young, R., Martin, B. R., and Dal Cason, T. A. (1995). Methcathione (“cat”): an enantiomeric potency comparison. Pharmacol. Biochem. Behav. 50, 601–606.
Hoehn, M. M., and Yahr, M. D. (1967). Parkinsonism: onset, progression and mortality. Neurology 17, 427–442.
Ihaka, R., and Gentleman, R. (1996). R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5, 299–314.
Kottilil, S., Yan, M. Y., Reitano, K. N., Zhang, X., Lempicki, R., Roby, G., Daucher, M., Yang, J., Cortez, K. J., Ghany, M., Polis, M. A., and Fauci, A. S. (2009). Human immunodeficiency virus and hepatitis C infections induce distinct immunologic imprints in peripheral mononuclear cells. Hepatology 50, 34–45.
Masliah, E., Roberts, E. S., Langford, D., Everall, I., Crews, L., Adame, A., Rockenstein, E., and Fox, H. S. (2004). Patterns of gene dysregulation in the frontal cortex of patients with HIV encephalitis. J. Neuroimmunol. 157, 163–175.
McCann, U. D., Wong, D. F., Yokoi, F., Villemagne, V., Dannals, R. F., and Ricaurte, G. A. (1998). Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J. Neurosci. 18, 8417–8422.
Mohr, S., and Liew, C. C. (2007). The peripheral-blood transcriptome: new insights into disease and risk assessment. Trends. Mol. Med. 13, 422–432.
Monaco, A., Marincola, F. M., Sabatino, M., Pos, Z., Tornesello, M. L., Stroncek, D. F., Wang, E., Lewis, G. K., Buonaguro, F. M., and Buonaguro, L. (2009). Molecular immune signatures of HIV-1 vaccines in human PBMCs. FEBS Lett. 583, 3004–3008.
Motomura, K., Toyoda, N., Oishi, K., Sato, H., Nagai, S., Hashimoto, S., Tugume, S. B., Enzama, R., Mugewa, R., Mutuluuza, C. K., Mugyeyi, P., Nagatake, T., and Matsushima, K. (2004). Identification of a host gene subset related to disease prognosis of HIV-1 infected individuals. Int. Immunopharmacol. 4, 1829–1836.
Peto, V., Jenkinson, C., Fitzpatrick, R., and Greenhall, R. (1995). The development and validation of a short measure of functioning and well being for individuals with Parkinson’s disease. Qual. Life Res. 4, 241–248.
Rockhold, R. W., Carlton, F. B. Jr., Corkern, R., Derouen, L., Bennett, J. G., and Hume, A. S. (1997). Methcathinone intoxication in the rat: abrogation by dextrorphan. Ann. Emerg. Med. 29, 383–391.
Schwab, R. S., and England, A. C. (1969). Projection Technique for Evaluating Surgery in Parkinson’s Disease. Edinburgh: EandS Livingstone.
Selikhova, M., Fedoryshyn, L., Matviyenko, Y., Komnatska, I., Kyrylchuk, M., Krolicki, L., Friedman, A., Taylor, A., Jager, H. R., Lees, A., and Sanotsky, Y. (2008). Parkinsonism and dystonia caused by the illicit use of ephedrone – a longitudinal study. Mov. Disord. 23, 2224–2231.
Sengupta, A., Mense, S. M., Lan, C., Zhou, M., Mauro, R. E., Kellerman, L., Bentsman, G., Volsky, D. J., Louis, E. D., Graziano, J. H., and Zhang, L. (2007). Gene expression profiling of human primary astrocytes exposed to manganese chloride indicates selective effects on several functions of the cells. Neurotoxicology 28, 478–489.
Sharp, F. R., Xu, H., Lit, L., Walker, W., Apperson, M., Gilbert, D. L., Glauser, T. A., Wong, B., Hershey, A., Liu, D. Z., Pinter, J., Zhan, X., Liu, X., and Ran, R. (2006). The future of genomic profiling of neurological diseases using blood. Arch. Neurol. 63, 1529–1536.
Sikk, K., Taba, P., Haldre, S., Bergquist, J., Nyholm, D., Zjablov, G., Asser, T., and Aquilonius, S. M. (2007). Irreversible motor impairment in young addicts – ephedrone, manganism or both? Acta. Neurol. Scand. 115, 385–389.
Smyth, G. K. (2004). Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, 1–26.
Stepens, A., Logina, I., Liguts, V., Aldins, P., Eksteina, I., Platkajis, A., Martinsone, I., Terauds, E., Rozentale, B., and Donaghy, M. (2008). A Parkinsonian syndrome in methcathinone users and the role of manganese. N. Engl. J. Med. 358, 1009–1017.
Storey, J. D., and Tibshirani, R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445.
Sullivan, P. F., Fan, C., and Perou, C. M. (2006). Evaluating the comparability of gene expression in blood and brain. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B, :261–:268.
Wurmbach, E., Chen, Y. B., Khitrov, G., Zhang, W., Roayaie, S., Schwartz, M., Fiel, I., Thung, S., Mazzaferro, V., Bruix, J., Bottinger, E., Friedman, S., Waxman, S., and Llovet, J. M. (2007). Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 45, 938–947.
Keywords: ephedrone, gene expression profiling, HIV, intravenous drug abuse, methcathinone, manganese
Citation: Sikk K, Kõks S, Soomets U, Schalkwyk LC, Fernandes C, Haldre S, Aquilonius S-M and Taba P (2011) Peripheral blood RNA expression profiling in illicit methcathinone users reveals effect on immune system. Front. Gene. 2:42. doi: 10.3389/fgene.2011.00042
Received: 14 March 2011;
Paper pending published: 27 April 2011;
Accepted: 22 June 2011;
Published online: 19 August 2011.
Edited by:
Ruth Luthi-Carter, Ecole Polytechnique Fédérale de Lausanne, SwitzerlandReviewed by:
Don A. Baldwin, University of Pennsylvania, USAJohn Hogenesch, University of Pennsylvania, USA
Copyright: © 2011 Sikk, Kõks, Soomets, Schalkwyk, Fernandes, Haldre, Aquilonius and Taba. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Katrin Sikk, Department of Neurology and Neurosurgery, University of Tartu, L.Puusepa 8, Tartu 51014, Estonia. e-mail: katrin.sikk@regionaalhaigla.ee