
- 1 Division of Biostatistics and Epidemiology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
- 2 Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
- 3 Department of Pediatrics, University of Cincinnati School of Medicine, Cincinnati, OH, USA
- 4 Division of Cardiology, Cincinnati Children’s Hospital, Cincinnati, OH, USA
Objectives: To determine whether the contributions of genetics and bicuspid aortic valve (BAV) independently influence aortic (Ao) dimensions. Background: Ao dilation is a risk factor for aneurysm, dissection, and sudden cardiac death. Frequent association of BAV with Ao dilation implicates a common underlying defect possibly due to genetic factors. Methods: Families enriched for BAV underwent standardized transthoracic echocardiography. In addition to BAV status, echocardiographic measures of Ao (annulus to descending Ao), pulmonary artery, and mitral valve annulus (MVA) diameters were obtained. Using variance components analysis, heritability was estimated with and without BAV status. Additionally, bivariate genetic analyses between Ao dimensions and BAV were performed. Results: Our cohort was obtained from 209 families enriched for BAV. After adjusting for age, body surface area, and sex, individuals with BAV had a statistically significant increase in all echocardiographic measurements (p < 0.006) except descending Ao and MVA. Individuals with BAV were at greater odds of having Ao dilation (OR = 4.44, 95% CI 2.93–6.72) than family members without BAV. All echocardiographic measurements exhibited moderate to strong heritability (0.25–0.53), and these estimates were not influenced by inclusion of BAV as a covariate. Bivariate genetic analyses supported that the genetic correlation between BAV and echo measures were not significantly different from zero. Conclusion: We show for the first time that echocardiographic measurements of Ao, pulmonary artery and MVA diameters are quantitative traits that exhibit significant heritability. In addition, our results suggest the presence of BAV independently influences the proximal Ao and pulmonary artery measures but not those in the descending Ao or MVA.
Introduction
Thoracic aorta (Ao) dilation describes an enlargement of Ao dimensions; it is an important clinical problem because it may be a risk factor for an aortopathy characterized by aneurysm formation, Ao dissection, and sudden cardiac death (Hiratzka et al., 2010). Thoracic Ao dilation and aneurysm (TAA) is associated with known connective tissue disorders, e.g., Marfan syndrome (OMIM 154700) and non-syndromic familial clustering of isolated cases are well known, e.g., aortic aneurysm, familial thoracic; AAT1 (OMIM 607086; Hiratzka et al., 2010) and AAT6 (OMIM 611788; Guo et al., 2007). Bicuspid aortic valve (BAV, OMIM 109730) has also been associated with TAA (Hahn et al., 1992; Michelena et al., 2008; Elefteriades and Farkas, 2010); the TAA–BAV association persists even when the BAV is functionally normal (Hahn et al., 1992; Burks et al., 1998; Nistri et al., 1999; Cecconi et al., 2005; Beroukhim et al., 2006; Michelena et al., 2008).
Bicuspid aortic valve describes an aortic valve with two rather than three cusps. BAV is the most common cardiovascular malformation (CVM) in humans; a figure of 1–2% is usually cited (reviewed in Siu and Silversides, 2010). Previous studies have supported a strong underlying genetic basis of BAV (Andelfinger et al., 2002; Cripe et al., 2004; Garg et al., 2005; Martin et al., 2007; Hinton et al., 2009). Family-based studies also support a genetic basis for Ao dilation and TAA (Guo et al., 2001; Vaughan et al., 2001; Hasham et al., 2003). The frequent association of BAV with TAA and the observation of abnormal Ao tissue architecture in patients with BAV lead to the postulation of a common underlying defect (Mckusick, 1972; de Sa et al., 1999). Human genetic studies have demonstrated that Ao dilation is increased in first degree relatives of individuals with BAV (Huntington et al., 1997; Cripe et al., 2004; Biner et al., 2009; Elefteriades and Farkas, 2010), further implicating a common underlying genetic etiology (Loscalzo et al., 2007). However, the reasons for co-occurrence may not be due to common underlying genetic etiology; instead, other factors, e.g., altered aortic blood flow may act upon susceptible individuals to increase risk. Unfortunately, no studies have directly addressed whether Ao dimensions are associated with BAV due to genetics.
Given the heritability of BAV and the increased risk of Ao dilation in BAV family members, the objective of this study was to determine whether the contributions of heritability and BAV independently influence Ao dimensions. To accomplish this objective, we examined the heritability of echocardiographic (echo) measurements in a family-based cohort enriched for BAV, emphasizing Ao dimensions at multiple sites, including and excluding BAV as a covariate. Additionally, bivariate genetic analyses were performed to formally test whether Ao dimensions and BAV share underlying genetic factors.
Materials and Methods
Subjects
Previously (Hinton et al., 2007), we found that first degree relative recurrence risk of BAV was identical in kindreds ascertained by identifying probands with either BAV or hypoplastic left heart syndrome (HLHS OMIM 242550). Therefore, to obtain a family-based cohort enriched for BAV we utilized families ascertained independently by identifying probands with either BAV or HLHS (Hinton et al., 2007, 2009). The sampling methods were reported previously (Hinton et al., 2009), and thus will be described briefly. Probands with a known genetic syndrome (e.g., Turner syndrome) were excluded. All first degree relatives of the proband were invited to participate. To expand the pedigrees, we used a sequential sampling strategy as previously described (Cripe et al., 2004; Martin et al., 2007). This cohort includes 169 and 40 pedigrees recruited based on the proband having BAV and HLHS, respectively. In the BAV families, 5.1% of non-probands had BAV; while in the HLHS families, 3.6% of non-probands had BAV. This protocol was approved by the Institutional Review Board of Cincinnati Children’s Hospital Medical Center. Informed consent was obtained from all participants.
Echocardiography
For all participants, cardiovascular phenotype was determined by a cross-sectional 2-dimensional and Doppler transthoracic echocardiogram performed at our center and/or a detailed medical history including records describing cardiac catheterization, surgical procedures, or echocardiograms (Cripe et al., 2004; Martin et al., 2007). Commercially available echocardiographic systems (Hewlett–Packard Sonos 5500, GE Vivid 7 or a GE Vivid 5) were utilized. Aortic valve morphology was examined in both systole and diastole in the parasternal short axis view. All additional anatomic and hemodynamic abnormalities were recorded in probands and relatives. A single experienced echocardiographer (Linda H. Cripe) interpreted all echocardiograms performed on site. To eliminate effects that cardiac surgery may have on some aorta echo measures, individuals with a history of cardiac surgery were excluded from analyses utilizing aorta echo measures. As all HLHS participants undergo cardiac surgery in infancy, they were excluded from the quantitative analysis of echo measures.
Echo measurements of Ao diameters were obtained at six sites, including the aortic valve annulus (AoVA), aortic root (AoR), sinotubular junction (STJ), ascending aorta (AoA), transverse aorta (AoT), and descending aorta (AoD). Echo measures of the mitral valve annulus (MVA), pulmonary valve annulus (PVA), and main pulmonary artery (MPA) were also obtained (Figure 1). Ao diameters were measured from leading edge to leading edge in the parasternal long axis in end diastole, as previously described (Roman et al., 1989; Lang et al., 2006). By 2-dimensional imaging the MPA segment and the PVA were measured in the parasternal short axis view. The MV annulus was measured in the apical four chamber view using 2-dimensional images. Echocardiography variables were measured in triplicate. Individuals with a measurement of AoR, or AoA greater than published normal values were given a clinical diagnosis of Ao dilation (Roman et al., 1989).
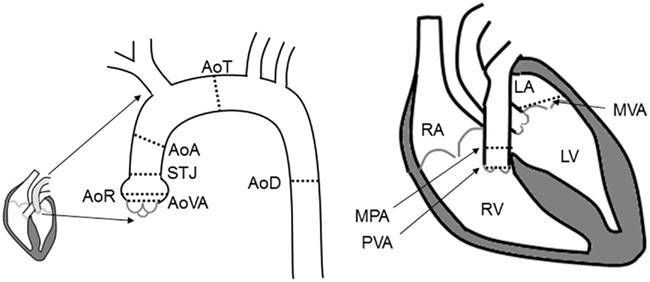
Figure 1. Illustration of the locations of the echocardiographic measures. Abbreviation definitions: AoR, aortic root; AoVA, aortic valve annulus; STJ, sinotubular junction; AoA, ascending aorta; AoT, transverse aorta; AoD, descending aorta; PVA, pulmonary valve annulus; MPA, main pulmonary artery; MVA, mitral valve annulus; RV, right ventricle; LV, left ventricle; RA, right atrium; LA, left atrium.
Derived Variables
Body surface area
We used the Mosteller formula to calculate body surface area (BSA) as described previously (Mosteller, 1987).
Statistical Analysis
Data cleaning
Echo measurements were first evaluated among individuals. Measures outside of 4 SD were reviewed and errors were excluded. Since the measures were in triplicate, we then evaluated intraobserver variability. Prior to data cleaning, average SD of triplicate measures was low [range 0.07 (AoA) to 0.31 (PVA)], suggesting that these variables were measured reproducibly. When the triplicate SD exceeded 4 SD units (based on the average of all triplicates across individuals), values within the triplicate, which exhibited >20% difference from the other measures within the triplicate were excluded. Less than 0.3% of all echo measures were excluded.
Phenotypic associations between echo measures and BAV and/or CVM
To investigate whether echo measures were associated with the presence or absence of BAV and or CVM, we first examined the effect of incorporating a covariate effect for BAV and CVM as separate terms using a variance components model. In this model the random effects are defined by familial relationships (Eq. 1) and covariates are included as fixed effects. Other covariates included age, age squared, sex, age by sex, age squared by sex, and BSA.
Genetic analysis
To investigate the heritability of echo measures, we applied a variance components model, which is a mixed model. In the variance components model, the fixed effects are the covariates of age, age squared, sex, age by sex, age squared by sex, and BSA. The adjustment of covariates allows removal of known variation prior to modeling the random effects. The random effects are defined by partitioning the covariance between relative pairs into additive genetic and error covariance. In this way, the covariance between relative pairs is defined as

where 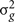 is the genetic variance due to residual additive genetic factors, and
is the genetic variance due to residual additive genetic factors, and 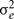 is the error variance (residual variance not explained by the additive genetic component). The additive genetic variance is multiplied by the structuring matrix for familial patterning (2Φ) which follows expected additive genetic transmission. In this matrix, parents and offspring would be expected to share 1/2 of their genes in common based on expected Mendelian transmission (Falconer, 1989). The error variance is multiplied by the identity matrix (I) which is simply a matrix whose diagonal elements are one and remaining elements are zero (permitting individual specific effects).
is the error variance (residual variance not explained by the additive genetic component). The additive genetic variance is multiplied by the structuring matrix for familial patterning (2Φ) which follows expected additive genetic transmission. In this matrix, parents and offspring would be expected to share 1/2 of their genes in common based on expected Mendelian transmission (Falconer, 1989). The error variance is multiplied by the identity matrix (I) which is simply a matrix whose diagonal elements are one and remaining elements are zero (permitting individual specific effects).
To solve for the variance terms, we used maximum-likelihood based variance decomposition implemented in the computer package SOLAR (Southwest Foundation for Biomedical Research, San Antonio, TX, USA), which allows us to simultaneously consider all possible relative pairs (Almasy and Blangero, 1998). Heritability is an estimate and is calculated as 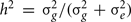 . Significance of the heritability estimates was assessed by likelihood ratio tests. The maximum likelihood for the model in which all parameters were estimated are compared to those for restricted models in which the value of the parameter to be tested will be held constant at some value (usually zero). Twice the difference in the natural logarithm likelihoods of the two models to be compared is distributed asymptotically, approximately as a 1/2:1/2 mixture of
. Significance of the heritability estimates was assessed by likelihood ratio tests. The maximum likelihood for the model in which all parameters were estimated are compared to those for restricted models in which the value of the parameter to be tested will be held constant at some value (usually zero). Twice the difference in the natural logarithm likelihoods of the two models to be compared is distributed asymptotically, approximately as a 1/2:1/2 mixture of 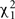 variable and a point mass at zero (Self and Liang, 1987). Values of p ≤ 0.05 were considered significant.
variable and a point mass at zero (Self and Liang, 1987). Values of p ≤ 0.05 were considered significant.
Bivariate analysis. In the multivariate model, the phenotype covariance between two traits is decomposed to include the genetic correlation between traits due to additive genetic effects and the correlation between traits due to shared (unmeasured) environmental effects,
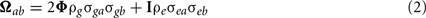
where a and b are the two traits of interest, ρg is the additive genetic correlation between the two traits and ρe is the correlation between unmeasured environmental effects. The genetic correlation estimates the proportion of genes shared in common between the two traits. This approach has been implemented in SOLAR (Comuzzie et al., 1994).
Quantitative definition of Ao dilation
Because echo measures vary by age, sex, and BSA (Roman et al., 1989), identification of individuals at the tails of the distribution in our cohort was challenging. Thus, to better understand the distribution of echo measures in individuals with and without BAV, we normalized the data adjusting for age, sex, and BSA. Each echo measure (y) in individuals without BAV was first analyzed using a regression model (see Eq. 3 below). Age, sex, and BSA showed significant effect (p ≤ 0.05), thus being included in the final model.
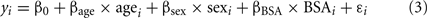
The mean (με_nonBAV) and the standard deviation (SDε_nonBAV) of the estimated residuals for each echo measure were calculated. We then applied Eq. 3 to the entire dataset. Estimated residuals were normalized to generate normalization-scores using Eq. 4.

Normalization-scores were then evaluated using t-tests to compare BAV to non-BAV participants, to ensure consistency with the family-based SOLAR analyses. Chi-square analysis was used to determine if individuals with BAV were more likely to have measures exceeding 2 SD units. We recognize that both the t-test and the chi-square analyses assume independent samples thus our use of these tests may be biased. However, as we are using these analyses to augment the family-based analyses in SOLAR, we are not concerned about incorrect inference.
Using the normalization-scores, we defined individuals as having Ao dilation when normalization-scores exceeded 2 units for AoAV, AoR, STJ, or AoA. This quantitative definition of Ao dilation is distinct from the usual clinical diagnosis of Ao dilation which is characterized by a measurement of AoR or AoA greater than published normal values (Roman et al., 1989). To compare frequency of Ao dilation between individuals with and without BAV, chi-square analysis was used.
In this study the reference samples are from family members without BAV; thus, the normalization-scores are not equivalent to population referent values. Since it is known that family members without BAV may have Ao dilation (Bella et al., 2002; Loscalzo et al., 2007; Biner et al., 2009), this approach may underestimate Ao dilation in family members with BAV.
Multiple testing correction
For all tests described above, we were evaluating nine echocardiographic measures. Thus when using the standard nominal significance of α = 0.05, we will have an inflated type I error rate. To correct for this, we apply Bonferroni correction (0.05/9 = 0.006) to achieve our significance threshold.
Results
Patient Characteristics
Our cohort was obtained from 209 BAV-enriched families (n = 1955 individuals). The number of individuals with BAV (n = 255) and intervention for Ao valve disease (n = 70) or mitral valve disease (n = 12) provide a description of the disease burden of this large cohort. A further characterization includes a clinical diagnosis of AoR dilation in 48 individuals including 5 individuals with AoR replacement and 3 individuals with combined Ao valve intervention and root replacement.
However, inclusion in the quantitative analyses presented in this study required measurements from the echocardiogram protocol performed at our center. Thus, from the larger cohort, 1143 individuals, consisting of 2723 relative pairs (Table 1), met this criterion and were included. Descriptive characteristics of this smaller cohort are provided in Table 2. Briefly, 175 individuals had BAV and 144 had CVM, e.g., atrial septal defect, ventricular septal defect including 48 individuals with both BAV and CVM. Participants spanned a broad age range (0.01–88.41 years, mean = 28.0 ± 18.9) with slightly over one-third of the cohort ≤18 years of age. To eliminate effects that cardiac surgery may have on some aorta echo measures, 47 individuals were excluded from the heritability analysis because of a history of cardiac surgery. The two cohorts with echo measures (1143 versus 1096) did not significantly differ on any demographic measures (not shown). Further, we compared the univariate effects of BSA on STJ and AoR to the univariate effects presented by Roman et al. (1989), and found no substantial differences (data not shown).

Table 1. Pedigree structure of the cohort.
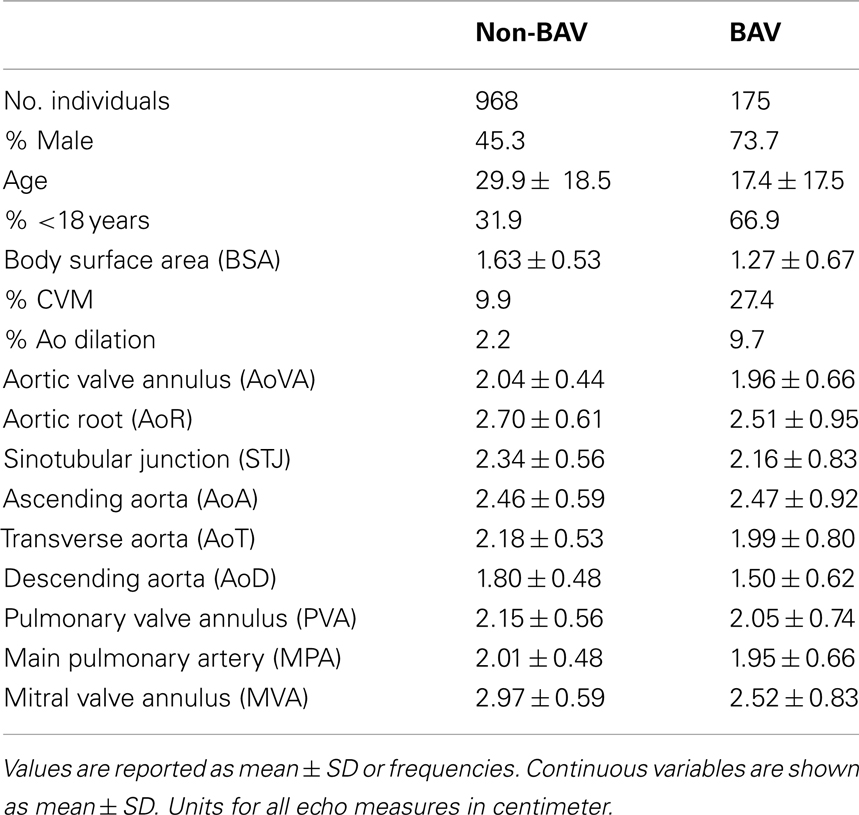
Table 2. Descriptive statistics of the cohort.
BAV Increases Some but not All Echo Measures
We performed a covariate analysis in SOLAR. As expected, age and BSA were associated with statistically significant increases in all echo measures. Further, as previously observed (Roman et al., 1989) females had smaller measures than males. After adjusting for age, sex, and BSA in the family-based cohort, presence of BAV is associated with a statistically significant increase in some but not all echo measures (p-value range 0.009–3.3 × 10−25; Table 3). Affected measures included AoVA, AoR, STJ, AoA, AoT, PVA, and MPA. AoD and MVA measures were not associated with BAV. CVM showed significant effects on AoR, STJ, AoA, and AoT. Importantly, while BAV and CVM have similar effect on some measures (STJ and AoT) other measures exhibit different magnitudes of effect (see beta values for BAV and CVM-Table 3).
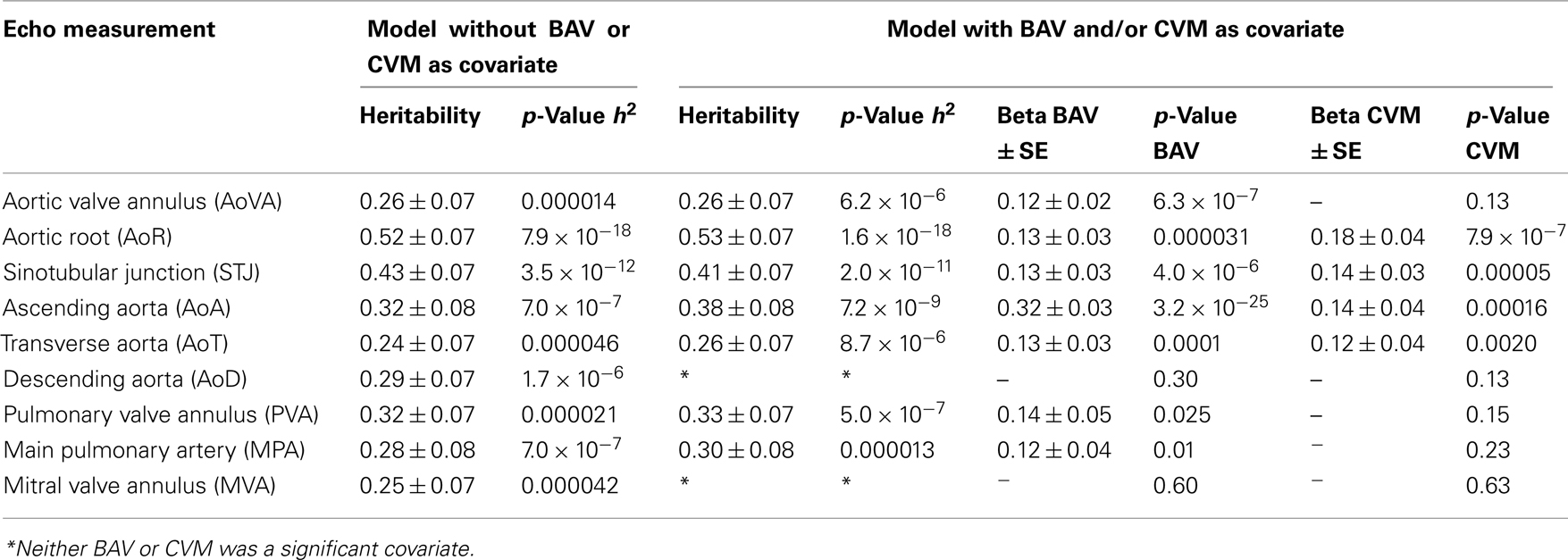
Table 3. Heritability estimates of aortic and pulmonary artery measures with and without the inclusion of BAV/CVM as covariates.
Echo Measures are Quantitative Traits that Exhibit Significant Heritability
After correcting for BSA, sex, and age, all Ao, pulmonary artery and mitral valve measures exhibited significant heritability (p < 4.6 × 10−5 for all) with moderate to strong estimates (h2 range 0.24 ± 0.07 to 0.52 ± 0.07; Table 3), identifying echo measures as quantitative traits. Heritability estimates were markedly similar when incorporating the influence of BAV and CVM, suggesting a genetic basis for echo measures, which are distinct from the underlying etiology of BAV or CVM.
Echo Measures Do not Share Underlying Genetic Effects with BAV
Given the consistency in heritability measures, we then tested whether there was evidence of underlying shared genetic effects using bivariate genetic analyses. The genetic correlation between two traits, ρG, can be interpreted to be an estimate of the proportion of genes shared in common between the two traits. No estimates of ρG were significantly different using a nominal significance threshold of α ≤ 0.05 (Table 4).
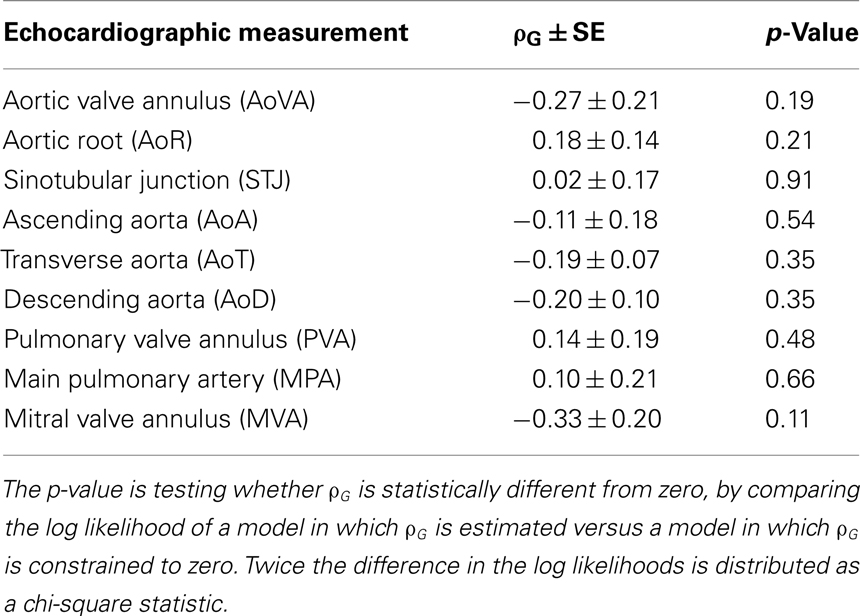
Table 4. Genetic correlations between aortic and pulmonary artery measures and BAV.
Normalized Echo Measures Exhibit Differences by BAV
To verify the impact of BAV on specific echo measures, we evaluated normalization-scores adjusted for age, gender, and BSA. When used as continuous measures (t-tests), we found that normalization-scores for some echo measures were increased in individuals with BAV (p < 0.005; Figure 2), but AoD and MVA were not (p > 0.93). When normalization-scores were dichotomized (chi-square), BAV was associated with an increased proportion of observations exceeding a threshold of 2 SD for AoVA, AoR, STJ, AoA, and AoT (p < 0.0001), suggesting all proximal thoracic Ao regions are prone to dilation. MPA showed a slight increase in observations exceeding 2 SD in individuals with BAV versus those without (5.5 versus 3.4%), but this difference was not statistically significant (p = 0.28).
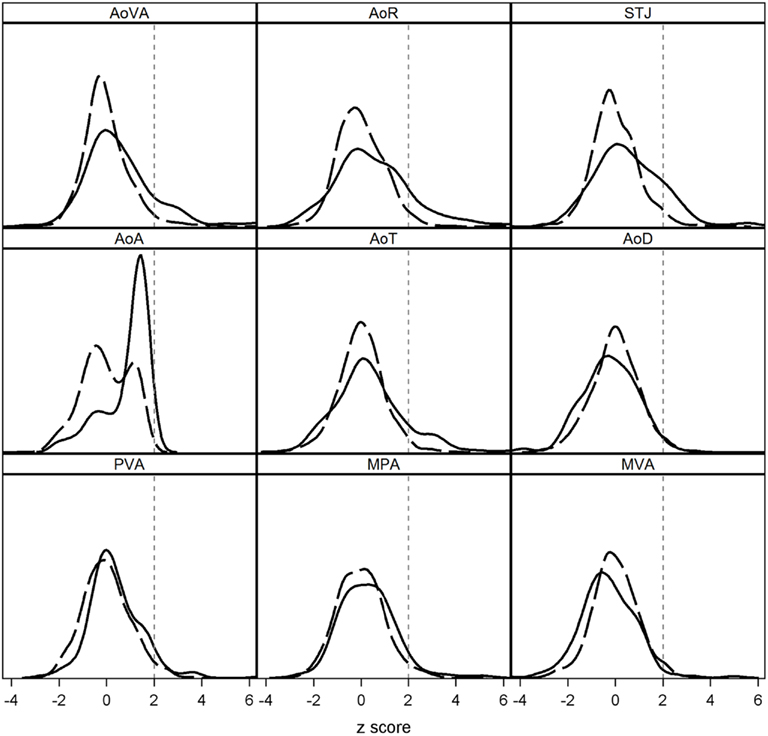
Figure 2. Distribution of echocardiographic normalization-scores (z-scores) in family members with BAV (solid line) and without BAV (dashed line). z-score measures >2 units (vertical dashed line) are considered abnormal. When used as continuous measures, we found that z-scores for some echo measures of AoVA, AoR, STJ, AoA, AoT, and MPA were increased in individuals with BAV (p < 0.005). BAV was associated with an increased proportion of Ao dilation, i.e., measures exceeding a threshold of 2 z-score units for AoVA, AoR, STJ, AoA, and AoT (p < 0.0001).
BAV Associated with Increased Frequency of Ao Dilation
Among the 1143 individuals with echo measures, a clinical diagnosis of Ao dilation or a history of AoR replacement was identified in 38 (3.1%) participants represented by 33 (15.8%) families: 17 had BAV and 21 had a normal Ao valve. While familial clustering of clinically significant Ao dilation was typically not present, individuals with BAV were at greater odds of having clinical Ao dilation (OR = 4.82, 95% CI = 2.49–9.33, p < 0.0001) than individuals without BAV. Using a quantitative definition of Ao dilation as exceeding 2 normalization-score units for any proximal Ao echo measures (AoVA, AoR, STJ, or AoA), we found that 133 (11.6%) individuals from 83 (39.7%) families met this criteria. Ao dilation thus defined in 84 individuals with BAV. Further, individuals with BAV were more likely than family members without BAV to exhibit Ao dilation (OR = 4.44, 95% CI 2.93–6.72). Importantly, only modest correlation was present between the normalization-scores of these four Ao measures (average pairwise Pearson correlation = 0.37 ± 0.27), suggesting that increased measurement at one Ao site was not very predictive of increased measures at other Ao sites. Indeed, among individuals, we identified different patterns of dilation, with some individuals showing increased measures across all proximal Ao echo sites, while others exhibited dilation at a single site (Figure 3).
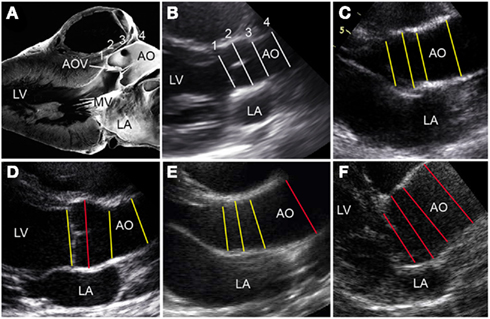
Figure 3. Variations in BAV-associated aortopathy. (A) Pathology oriented to show anatomy that correlates with the echocardiographic parasternal long axis view. (B–F) The proximal thoracic aorta dimensions are designated by numbered lines (1 = AoVA, 2 = AoR, 3 = STJ, 4 = AoA). (B) A normal heart is showing aortic dimensions with normalization-scores (z-scores) approximating 0 (white lines). (C) Hearts with BAV and high normal aortic dimensions at all sites (z-scores between +1 and +2; yellow lines); (D) BAV and dilation (z-score > 2) of AoR only (red line); (E) BAV and dilation of the AoA only (red line); and (F) BAV and dilation of all four sites (red lines). AOV, aortic valve; LA, left atrium; LV, left ventricle; MV, mitral valve. (A) Is a modified reproduction with permission (Edwards, 2008).
Discussion
In this study, we show for the first time that Ao dimensions are independently influenced by both genetics and BAV. To demonstrate these findings we first showed that the presence of BAV is associated with increased echo measures in the proximal portion of the Ao and pulmonary artery but not in the descending Ao or mitral valve. Second, this family-based study demonstrated that echo measures of the Ao, pulmonary artery, and MVA exhibit moderate heritability, meaning that some variability in the measures is determined by genetic effects, i.e., they are quantitative traits. Importantly, the heritability estimates were markedly stable when accounting for BAV and bivariate analyses did not support shared underlying genetic effects of BAV and Ao measures. Taken together these results suggest that the genetic basis for dimensions of all echo measures we studied is distinct (independent) from the underlying genetic etiology of BAV.
BAV is Associated with Increased Ao and Pulmonary Artery Measures
Our finding of BAV being associated with increased Ao measures was consistent with previous studies in children and adults (Hahn et al., 1992; Nistri et al., 1999; Keane et al., 2000b; Nkomo et al., 2003; Beroukhim et al., 2006; Warren et al., 2006; Yuan et al., 2010). Further, our estimates of Ao dimensions were similar to values reported previously (Beroukhim et al., 2006). In addition, we found that the pulmonary artery also exhibits increased diameter in BAV patients; this is not surprising given previous findings of abnormal histopathology and dilation of the pulmonary artery in BAV patients (de Sa et al., 1999). Abnormal pulmonary artery dimensions also imply common pathology in both great arteries, suggesting the durability of the Ross procedure may be limited in such cases. Similar pathology in the Ao and MPA is not surprising given their common developmental origin from the outflow tract endocardial cushions and the conotruncus. This may be clinically relevant in the context of individuals with BAV who require aortic valve replacement and may be candidates for the Ross procedure (placement of the pulmonary valve in the aortic position).
Echo Measures are Heritable and Persist when Controlling for BAV
These results demonstrate that echo measures are heritable, and this genetic effect is not attenuated with the inclusion of BAV as a covariate. These findings are consistent with a previous report showing a heritability of 0.51 for the AoR measured at the sinuses of Valsalva (Figure 1; AoR) in an adult cohort (Bella et al., 2002). Further, identification of linkage regions in families enriched for TAA (Guo et al., 2001; Hasham et al., 2003) also supports a genetic basis of the Ao measures. In addition to providing the first evidence of heritability across these echo measures, these results may provide insight into the relationship between the echo measures and BAV. Specifically, it has been proposed that BAV and Ao dilation have a common underlying genetic etiology (Loscalzo et al., 2007). If both BAV and Ao and pulmonary artery measures were influenced by the same genes, one would expect that adding BAV as a covariate in the genetic model would reduce the genetic signal. However, our heritability estimates were markedly similar before and after inclusion of BAV. Nonetheless, as heritability is a function of both the additive genetic and total phenotypic variance, heritability may remain constant if both the additive genetic and environmental variance are influenced by a covariate. Thus, taken alone these results provide only circumstantial evidence of the independence of the underlying genetics of BAV and echo measures.
No Evidence of Shared Underlying Genetic Effects between BAV and Echo Measures
Results from the bivariate genetic analyses do not support that there are shared underlying genetic effects between BAV and aortic echo measures. While no previous studies have directly examined bivariate genetic relationships between BAV and aortic echo measures, loci identified for TAA (Guo et al., 2001, 2007, 2009, 2011; Hasham et al., 2003) do not overlap with loci identified for BAV (Martin et al., 2007; Hinton et al., 2009). Thus, the evidence of minimal impact of BAV on heritability of Ao echo measures, non-significant genetic correlations in the bivariate analyses, and the lack of overlap in previously identified TAA and BAV linkage regions provides strong evidence that BAV and Ao dimensions do not share underlying genetic effects.
If underlying genetic factors are not the cause of an association between BAV and aortic echo measures, the question remains why are these two phenotypes associated? We can speculate that the association between BAV and Ao dilation may be due to degenerative changes in tissue media or hemodynamic factors. Several studies have examined aortic tissue from BAV patients. In patients with aortic valve disease, those with BAV have more severe degenerative changes in the media of the AoA and MPA, including a loss of elastic fibers (Roberts and Roberts, 1991), than patients with tricuspid aortic valves (de Sa et al., 1999). Specifically, Fedak et al. (2002, 2003) speculated that deficiency of fibrillin-1 in BAV patients might trigger matrix metalloproteinase (MMP) production, leading to maladaptive remodeling of extracellular matrix and ultimately Ao dilation. Further, there is evidence that hemodynamic changes may increase the risk of Ao dilation. Both computational fluid dynamics simulations and analysis of blood flow patterns in the AoA of patients with BAV demonstrate that asymmetric blood flow patterns and perturbations in flow velocity may contribute to aneurysm formation (Hope et al., 2010; Viscardi et al., 2010). Thus, previous work supports the plausibility of some shared pathogenesis in the context of distinct genetic etiologies for BAV and Ao dilation.
BAV is Associated with Aortic Dilation
We also found that BAV was associated with increased Ao dilation, using both a clinical and research diagnosis. Our findings are in agreement with previous studies, which report an increased prevalence of aortic aneurysm/dissection in BAV versus non-BAV individuals (Ando et al., 1998; Coady et al., 1999) or individuals with BAV (Burks et al., 1998; Keane et al., 2000a; Cecconi et al., 2005; Warren et al., 2006). Indeed, we found that individuals with BAV have a 4.44-fold increase in Ao dilation, which is a risk factor for aneurysm and ultimately dissection. Importantly, we confirmed the findings of previous studies, showing that the Ao valve annulus, AoR, STJ, and the ascending Ao are dilated in BAV individuals, but the descending Ao is not (Nistri et al., 1999; Warren et al., 2006; Siu and Silversides, 2010); this is not surprising given the different developmental origins and disease manifestations of ascending versus descending Ao (Appelbaum et al., 1976; Wasteson et al., 2008). While we found consistent association with BAV with the clinical and research diagnosis of Ao dilation, substantially more individuals were classified as dilated using the research than the clinical diagnosis (11.6 versus 3.1%, respectively). The increased proportion of the research diagnosis is likely due to the fact that the research diagnosis uses all four measures of the proximal Ao whereas the clinical diagnosis uses two. Further, our normalization-scores may underestimate the frequency and severity of Ao dilation – BAV association. First, the cohort we studied has a relatively young age and individuals with the most severe disease manifestations, e.g., individuals with a history of cardiac surgery for valve replacement or AoR replacement, were excluded from the quantitative analyses. Second, the use of family members without BAV as the comparison group may diminish the true difference between affected and unaffected individuals since family members without BAV may be Ao dilation (Bella et al., 2002; Loscalzo et al., 2007; Biner et al., 2009).
Future Considerations/Study Design
A unique component of our study is the use of family members without BAV as the comparison group. The use of family members to assess risk may have a decided advantage over the traditional case–control design for estimated increased risk of Ao dilation associated with BAV. Indeed, in the case–control design, it is likely that cases and controls may differ in their underlying genetic background. As BAV is determined in large part by genetics (Cripe et al., 2004; Mcbride et al., 2005; Martin et al., 2007) and similarly TAA has been shown to have a genetic basis (Guo et al., 2001, 2011; Vaughan et al., 2001; Hasham et al., 2003), differences in genetic backgrounds may create confounding as associations may be due to random segregation of genes. This is a problem because confounding can create spurious associations (Knowler et al., 1988). Nonetheless, the use of family-based controls may not be reflective of the general population. In future studies, investigators should be aware of the strengths and weaknesses of case–control versus family-based designs.
A challenge for comparing results of the many investigations in this area has been the varied research methodology. A strength of the results presented is the comprehensive evaluation of echo measures; examining only a single Ao region, e.g., the AoR, may dramatically underestimate the rates of aortopathy. Indeed, the clinical recognition of Ao dilation was dramatically lower than when comparing normalization-scores at multiple Ao sites, i.e., AoVA, AoR, STJ, and AoA.
One potential limitation of our study is that there was no data available for blood pressure in our sample. However, Roman et al. (1989) demonstrated that once adjusting for age, blood pressure had no independent effect on aortic size. Further, Biner et al. (2009) showed that AoR elastic properties, including distensibility and stiffness index, are abnormal in family members of BAV patients, regardless of a history of hypertension. These results suggest that the inclusion of hypertension data would not dramatically alter our findings.
Conclusion
We have shown genetics and the presence of BAV independently influence variation in Ao and pulmonary artery diameters. These results have implications for both BAV patients and their families. Specifically, BAV patients are at increased risk of developing valve disease and aortic dilation, underscoring the importance of monitoring for both aortic valve disease and aortopathy. Further, because there is evidence for distinct underlying genetic etiologies of BAV and TAA, family members of BAV patients may also be at risk for TAA, even if they don’t have BAV. Indeed, the ability to identify those individuals at risk for disease progression will be complicated and individuals with BAV may have different risk profiles. As the genetic bases of these diseases are elucidated and the phenotypic classification system is refined, the possibilities of identifying new therapeutics and modifying clinical recommendations will be realized.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the families who participated in the study. We would also like to thank the anonymous Reviewers who provided us guidance on the improvement of the manuscript.
Funding Sources
This work was supported by grants from the National Institutes of Health HL069712 (D. Woodrow Benson), HL074728 (Lisa J. Martin, D. Woodrow Benson), HL085122 (Robert B. Hinton), and MH059490 for SOLAR.
References
Almasy, L., and Blangero, J. (1998). Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62, 1198–1211.
Andelfinger, G., Tapper, A. R., Welch, R. C., Vanoye, C. G., George, A. L. Jr., and Benson, D. W. (2002). KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am. J. Hum. Genet. 71, 663–668.
Ando, M., Okita, Y., Morota, T., and Takamoto, S. (1998). Thoracic aortic aneurysm associated with congenital bicuspid aortic valve. Cardiovasc. Surg. 6, 629–634.
Appelbaum, A., Karp, R. B., and Kirklin, J. W. (1976). Ascending vs descending aortic dissections. Ann. Surg. 183, 296–300.
Bella, J. N., Maccluer, J. W., Roman, M. J., Almasy, L., North, K. E., Welty, T. K., Lee, E. T., Fabsitz, R. R., Howard, B. V., and Devereux, R. B. (2002). Genetic influences on aortic root size in American Indians: the Strong Heart Study. Arterioscler. Thromb. Vasc. Biol. 22, 1008–1011.
Beroukhim, R. S., Kruzick, T. L., Taylor, A. L., Gao, D., and Yetman, A. T. (2006). Progression of aortic dilation in children with a functionally normal bicuspid aortic valve. Am. J. Cardiol. 98, 828–830.
Biner, S., Rafique, A. M., Ray, I., Cuk, O., Siegel, R. J., and Tolstrup, K. (2009). Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J. Am. Coll. Cardiol. 53, 2288–2295.
Burks, J. M., Illes, R. W., Keating, E. C., and Lubbe, W. J. (1998). Ascending aortic aneurysm and dissection in young adults with bicuspid aortic valve: implications for echocardiographic surveillance. Clin. Cardiol. 21, 439–443.
Cecconi, M., Manfrin, M., Moraca, A., Zanoli, R., Colonna, P. L., Bettuzzi, M. G., Moretti, S., Gabrielli, D., and Perna, G. P. (2005). Aortic dimensions in patients with bicuspid aortic valve without significant valve dysfunction. Am. J. Cardiol. 95, 292–294.
Coady, M. A., Rizzo, J. A., Goldstein, L. J., and Elefteriades, J. A. (1999). Natural history, pathogenesis, and etiology of thoracic aortic aneurysms and dissections. Cardiol. Clin. 17, 615–635; vii.
Comuzzie, A. G., Blangero, J., Mahaney, M. C., Mitchell, B. D., Stern, M. P., and Maccluer, J. W. (1994). Genetic and environmental correlations among skinfold measures. Int. J. Obes. Relat. Metab. Disord. 18, 413–418.
Cripe, L., Andelfinger, G., Martin, L. J., Shooner, K., and Benson, D. W. (2004). Bicuspid aortic valve is heritable. J. Am. Coll. Cardiol. 44, 138–143.
de Sa, M., Moshkovitz, Y., Butany, J., and David, T. E. (1999). Histologic abnormalities of the ascending aorta and pulmonary trunk in patients with bicuspid aortic valve disease: clinical relevance to the ross procedure. J. Thorac. Cardiovasc. Surg. 118, 588–594.
Edwards, W. (2008). “Cardiac anatomy and examination of cardiac specimens,” in Moss and Adans heart disease in infants, children and adolescents, eds H. Allen, D. Driscoll, R. Shaddy, and T. Feltes (Philadelphia: Lipincott, Williams and WIlkins), 2–34.
Elefteriades, J. A., and Farkas, E. A. (2010). Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J. Am. Coll. Cardiol. 55, 841–857.
Falconer, D. (1989). Introduction to Quantitative Genetics. New York: Longman Scientific and Technical.
Fedak, P. W., de Sa, M. P., Verma, S., Nili, N., Kazemian, P., Butany, J., Strauss, B. H., Weisel, R. D., and David, T. E. (2003). Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. J. Thorac. Cardiovasc. Surg. 126, 797–806.
Fedak, P. W., Verma, S., David, T. E., Leask, R. L., Weisel, R. D., and Butany, J. (2002). Clinical and pathophysiological implications of a bicuspid aortic valve. Circulation 106, 900–904.
Garg, V., Muth, A. N., Ransom, J. F., Schluterman, M. K., Barnes, R., King, I. N., Grossfeld, P. D., and Srivastava, D. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature 437, 270–274.
Guo, D., Hasham, S., Kuang, S. Q., Vaughan, C. J., Boerwinkle, E., Chen, H., Abuelo, D., Dietz, H. C., Basson, C. T., Shete, S. S., and Milewicz, D. M. (2001). Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13-14. Circulation 103, 2461–2468.
Guo, D. C., Pannu, H., Tran-Fadulu, V., Papke, C. L., Yu, R. K., Avidan, N., Bourgeois, S., Estrera, A. L., Safi, H. J., Sparks, E., Amor, D., Ades, L., Mcconnell, V., Willoughby, C. E., Abuelo, D., Willing, M., Lewis, R. A., Kim, D. H., Scherer, S., Tung, P. P., Ahn, C., Buja, L. M., Raman, C. S., Shete, S. S., and Milewicz, D. M. (2007). Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 39, 1488–1493.
Guo, D. C., Papke, C. L., Tran-Fadulu, V., Regalado, E. S., Avidan, N., Johnson, R. J., Kim, D. H., Pannu, H., Willing, M. C., Sparks, E., Pyeritz, R. E., Singh, M. N., Dalman, R. L., Grotta, J. C., Marian, A. J., Boerwinkle, E. A., Frazier, L. Q., Lemaire, S. A., Coselli, J. S., Estrera, A. L., Safi, H. J., Veeraraghavan, S., Muzny, D. M., Wheeler, D. A., Willerson, J. T., Yu, R. K., Shete, S. S., Scherer, S. E., Raman, C. S., Buja, L. M., and Milewicz, D. M. (2009). Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 84, 617–627.
Guo, D. C., Regalado, E. S., Minn, C., Tran-Fadulu, V., Coney, J., Cao, J., Wang, M., Yu, R. K., Estrera, A. L., Safi, H. J., Shete, S. S., and Milewicz, D. M. (2011). Familial thoracic aortic aneurysms and dissections: identification of a novel locus for stable aneurysms with a low risk for progression to aortic dissection. Circ. Cardiovasc. Genet. 4, 36–42.
Hahn, R. T., Roman, M. J., Mogtader, A. H., and Devereux, R. B. (1992). Association of aortic dilation with regurgitant, stenotic and functionally normal bicuspid aortic valves. J. Am. Coll. Cardiol. 19, 283–288.
Hasham, S. N., Willing, M. C., Guo, D. C., Muilenburg, A., He, R., Tran, V. T., Scherer, S. E., Shete, S. S., and Milewicz, D. M. (2003). Mapping a locus for familial thoracic aortic aneurysms and dissections (TAAD2) to 3p24-25. Circulation 107, 3184–3190.
Hinton, R. B. Jr., Martin, L. J., Tabangin, M. E., Mazwi, M. L., Cripe, L. H., and Benson, D. W. (2007). Hypoplastic left heart syndrome is heritable. J. Am. Coll. Cardiol. 50, 1590–1595.
Hinton, R. B., Martin, L. J., Rame-Gowda, S., Tabangin, M. E., Cripe, L. H., and Benson, D. W. (2009). Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J. Am. Coll. Cardiol. 53, 1065–1071.
Hiratzka, L. F., Bakris, G. L., Beckman, J. A., Bersin, R. M., Carr, V. F., Casey, D. E. Jr., Eagle, K. A., Hermann, L. K., Isselbacher, E. M., Kazerooni, E. A., Kouchoukos, N. T., Lytle, B. W., Milewicz, D. M., Reich, D. L., Sen, S., Shinn, J. A., Svensson, L. G., and Williams, D. M. (2010). 2010 ACCF/AHA/AATS/ACR/ASA/SCA/ SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 121, e266–e369.
Hope, M. D., Hope, T. A., Meadows, A. K., Ordovas, K. G., Urbania, T. H., Alley, M. T., and Higgins, C. B. (2010). Bicuspid aortic valve: four-dimensional MR evaluation of ascending aortic systolic flow patterns. Radiology 255, 53–61.
Huntington, K., Hunter, A. G., and Chan, K. L. (1997). A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J. Am. Coll. Cardiol. 30, 1809–1812.
Keane, M. G., Wiegers, S. E., Plappert, T., Pochettino, A., Bavaria, J. E., and Sutton, M. G. (2000a). Bicuspid aortic valves are associated with aortic dilatation out of proportion to coexistent valvular lesions. Circulation 102, III35–III39.
Keane, M. G., Wiegers, S. E., Yang, E., Ferrari, V. A., St John Sutton, M. G., and Bavaria, J. E. (2000b). Structural determinants of aortic regurgitation in type A dissection and the role of valvular resuspension as determined by intraoperative transesophageal echocardiography. Am. J. Cardiol. 85, 604–610.
Knowler, W. C., Williams, R. C., Pettitt, D. J., and Steinberg, A. G. (1988). Gm3,5,13,14 and type 2 diabetes mellitus: an association in American Indians with genetic admixture. Am. J. Hum. Genet. 43, 520–526.
Lang, R. M., Bierig, M., Devereux, R. B., Flachskampf, F. A., Foster, E., Pellikka, P. A., Picard, M. H., Roman, M. J., Seward, J., Shanewise, J., Solomon, S., Spencer, K. T., St John Sutton, M., and Stewart, W. (2006). Recommendations for chamber quantification. Eur. J. Echocardiogr. 7, 79–108.
Loscalzo, M. L., Goh, D. L., Loeys, B., Kent, K. C., Spevak, P. J., and Dietz, H. C. (2007). Familial thoracic aortic dilation and bicommissural aortic valve: a prospective analysis of natural history and inheritance. Am. J. Med. Genet. A 143A, 1960–1967.
Martin, L. J., Ramachandran, V., Cripe, L. H., Hinton, R. B., Andelfinger, G., Tabangin, M., Shooner, K., Keddache, M., and Benson, D. W. (2007). Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum. Genet. 121, 275–284.
Mcbride, K. L., Pignatelli, R., Lewin, M., Ho, T., Fernbach, S., Menesses, A., Lam, W., Leal, S. M., Kaplan, N., Schliekelman, P., Towbin, J. A., and Belmont, J. W. (2005). Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. A 134, 180–186.
Mckusick, V. A. (1972). Association of congenital bicuspid aortic valve and erdheim’s cystic medial necrosis. Lancet 1, 1026–1027.
Michelena, H. I., Desjardins, V. A., Avierinos, J. F., Russo, A., Nkomo, V. T., Sundt, T. M., Pellikka, P. A., Tajik, A. J., and Enriquez-Sarano, M. (2008). Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation 117, 2776–2784.
Nistri, S., Sorbo, M. D., Marin, M., Palisi, M., Scognamiglio, R., and Thiene, G. (1999). Aortic root dilatation in young men with normally functioning bicuspid aortic valves. Heart 82, 19–22.
Nkomo, V. T., Enriquez-Sarano, M., Ammash, N. M., Melton, L. J. III, Bailey, K. R., Desjardins, V., Horn, R. A., and Tajik, A. J. (2003). Bicuspid aortic valve associated with aortic dilatation: a community-based study. Arterioscler. Thromb. Vasc. Biol. 23, 351–356.
Roberts, C. S., and Roberts, W. C. (1991). Dissection of the aorta associated with congenital malformation of the aortic valve. J. Am. Coll. Cardiol. 17, 712–716.
Roman, M. J., Devereux, R. B., Kramer-Fox, R., and O’loughlin, J. (1989). Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am. J. Cardiol. 64, 507–512.
Self, S. G., and Liang, K.-Y. (1987). Asymptototic properties of maximum likelihood estimators and likelihood ratio tests under non-standard conditions. J. Am. Stat. Assoc. 82, 605–610.
Siu, S. C., and Silversides, C. K. (2010). Bicuspid aortic valve disease. J. Am. Coll. Cardiol. 55, 2789–2800.
Vaughan, C. J., Casey, M., He, J., Veugelers, M., Henderson, K., Guo, D., Campagna, R., Roman, M. J., Milewicz, D. M., Devereux, R. B., and Basson, C. T. (2001). Identification of a chromosome 11q23.2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation 103, 2469–2475.
Viscardi, F., Vergara, C., Antiga, L., Merelli, S., Veneziani, A., Puppini, G., Faggian, G., Mazzucco, A., and Luciani, G. B. (2010). Comparative finite element model analysis of ascending aortic flow in bicuspid and tricuspid aortic valve. Artif. Organs 34, 1114–1120.
Warren, A. E., Boyd, M. L., O’connell, C., and Dodds, L. (2006). Dilatation of the ascending aorta in paediatric patients with bicuspid aortic valve: frequency, rate of progression and risk factors. Heart 92, 1496–1500.
Wasteson, P., Johansson, B. R., Jukkola, T., Breuer, S., Akyurek, L. M., Partanen, J., and Lindahl, P. (2008). Developmental origin of smooth muscle cells in the descending aorta in mice. Development 135, 1823–1832.
Keywords: genetics, heart malformations, aortopathy, echocardiography
Citation: Martin LJ, Hinton RB, Zhang X, Cripe LH and Benson DW (2011) Aorta measurements are heritable and influenced by bicuspid aortic valve. Front. Gene. 2:61. doi: 10.3389/fgene.2011.00061
Received: 26 May 2011; Accepted: 18 August 2011;
Published online: 05 September 2011 2011.
Edited by:
Ellen W. Demerath, University of Minnesota School of Public Health, USAReviewed by:
Ellen W. Demerath, University of Minnesota School of Public Health, USAAudrey C. Choh, Wright State University, USA
David Fardo, University of Kentucky, USA
Copyright: © 2011 Martin, Hinton, Zhang, Cripe and Benson. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: D. Woodrow Benson, Division of Cardiology, Cincinnati Children’s Hospital, ML 7042, 3333 Burnet Avenue, Cincinnati, OH 45229, USA. e-mail: woody.benson@cchmc.org