 Dominic Rose1* Peter F. Stadler2,3,4,5,6,7
Dominic Rose1* Peter F. Stadler2,3,4,5,6,7
- 1 Bioinformatics Group, Department of Computer Science, University of Freiburg, Freiburg, Germany
- 2 Bioinformatics Group, Department of Computer Science, Interdisciplinary Center for Bioinformatics, University of Leipzig, Leipzig, Germany
- 3 Max Planck Institute for Mathematics in the Sciences, Leipzig, Germany
- 4 Fraunhofer Institut für Zelltherapie und Immunologie, Leipzig, Germany
- 5 Department of Theoretical Chemistry, University of Vienna, Wien, Austria
- 6 Center for non-coding RNA in Technology and Health, University of Copenhagen, Frederiksberg, Denmark
- 7 Santa Fe Institute, Santa Fe, NM, USA
Eukaryotic genomes are pervasively transcribed. A large fraction of the transcriptional output consists of long, mRNA-like, non-protein-coding transcripts (mlncRNAs). The evolutionary history of mlncRNAs is still largely uncharted territory. In this contribution, we explore in detail the evolutionary traces of the eosinophil granule ontogeny transcript (EGOT), an experimentally confirmed representative of an abundant class of totally intronic non-coding transcripts (TINs). EGOT is located antisense to an intron of the ITPR1 gene. We computationally identify putative EGOT orthologs in the genomes of 32 different amniotes, including orthologs from primates, rodents, ungulates, carnivores, afrotherians, and xenarthrans, as well as putative candidates from basal amniotes, such as opossum or platypus. We investigate the EGOT gene phylogeny, analyze patterns of sequence conservation, and the evolutionary conservation of the EGOT gene structure. We show that EGO-B, the spliced isoform, may be present throughout the placental mammals, but most likely dates back even further. We demonstrate here for the first time that the whole EGOT locus is highly structured, containing several evolutionary conserved, and thermodynamic stable secondary structures. Our analyses allow us to postulate novel functional roles of a hitherto poorly understood region at the intron of EGO-B which is highly conserved at the sequence level. The region contains a novel ITPR1 exon and also conserved RNA secondary structures together with a conserved TATA-like element, which putatively acts as a promoter of an independent regulatory element.
Introduction
Large surveys of transcriptomes, such as ENCODE (ENCODE Project Consortium et al., 2007) and FANTOM (Maeda et al., 2006), demonstrated that eukaryotic genomes are pervasively transcribed (Jacquier, 2009). Long, mRNA-like, non-protein-coding transcripts (mlncRNAs) are an important component of this transcriptional output, often arising from regions unlinked to annotated protein-coding genes (Khalil et al., 2009). Apart from a few exceptions, the detailed function of these transcripts, however, still remains in the dark. The cases that are reasonably well understood, on the other hand, implicate mlncRNAs as key molecules orchestrating essential cellular processes, including gene expression, transcriptional, and post-transcriptional regulation, chromatin-remodeling, differentiation and development (Mercer et al., 2009).
As a group, mlncRNAs show evidence of stabilizing selection (Ponjavic et al., 2007; Marques and Ponting, 2009). Although the evidence for wide-spread evolutionary constraints on the sequence evolution of ncRNAs is the most direct evidence that at least a large fraction of them is in fact functional, we know very little about the evolutionary history of individual transcripts. In contrast to protein-coding genes or short structured ncRNAs, for which comprehensive evolutionary information is available in databases like Pfam (Finn et al., 2010) or Rfam (Gardner et al., 2011), there is no comparable resource for long ncRNAs. The lncRNA database (Amaral et al., 2011) is a first pioneering step in this direction, predominately compiling non-coding transcripts from the model organisms human and mouse.
To-date, only a few detailed case studies are available. Chodroff et al. (2010 recently considered the conservation of a few brain-specific mlncRNAs, reporting weak sequence conservation and major changes in gene structure across amniotes. Even more detailed descriptions of mlncRNA evolution zooming in on the sequences are available only for a few “famous” transcripts. Xist, an eutherian-specific regulatory long ncRNA that plays a central role in inactivation of one female X chromosome by recruiting chromatin-remodeling complexes, reviewed, e.g., by Arthold et al. (2011), is the only long ncRNAs whose evolutionary origin is understood in detail. It arose after the divergence of marsupials and placental mammals from the protein-coding Lnx3 gene upon incorporation of additional, repeat-derived exons (Duret et al., 2006; Elisaphenko et al., 2008; Kolesnikov and Elisafenko, 2010). Xist, along with Kcnq1ot1 (Kanduri, 2011), HOTAIR (Tsai et al., 2010), or HOTTIP (Wang et al., 2011) belongs to a class of chromatin regulatory mlncRNAs. The evolutionary features of HOTAIR were recently studied in some detail by (He et al., 2011). MALAT-1 and its apparent relative MENε/β, on the other hand, are nuclear-retained ncRNAs that are mostly unspliced (Hutchinson et al., 2007), undergo a highly unusual processing of their 3′-ends (Wilusz and Spector, 2010), and function as organizers of nuclear speckle structures (Sasaki et al., 2009). MALAT-1, which exhibits an atypically high level of sequence conservation, dates back at least to the radiation of the gnathostomes (Stadler, 2010).
Besides long intergenic RNAs (lincRNAs), vertebrate genomes also harbor tens of thousands of totally and partially intronic transcripts (TINs and PINs; Nakaya et al., 2007; Louro et al., 2008, 2009). A fraction of these comprises unspliced long antisense intronic RNAs (Rinn et al., 2003; Reis et al., 2004) and other predominately unspliced transcripts (Engelhardt and Stadler, 2011), while another subgroup consists of spliced RNAs. These could potentially be very similar to lincRNAs. In this contribution, we explore in detail the evolution of one particular example of the latter class, the eosinophil granule ontogeny transcript (EGOT).
The eosinophil granule ontogeny transcript is a transcriptional regulator of granule protein expression during eosinophil development (Wagner et al., 2007). Using sucrose density gradients Wagner et al. (2007) demonstrated that EGOT is not associated with ribosomes and thus most likely functions as bona fide non-coding RNA. The same authors proposed that EGOT may act as an siRNA against the eosinophil granule major basic protein (MBP) and eosinophil-derived neurotoxin (EDN). We choose EGOT as an example for a spliced antisense TIN as it is probably the experimentally best-characterized ncRNAs of this type. It is located in an intron of the ITPR1 gene, which codes for the type 1 inositol 1,4,5-triphosphate receptor mediating calcium release from the endoplasmic reticulum upon stimulation by inositol.
Human EGOT has two known isoforms that share the same transcriptional start site. EGO-B consists of two closely spaced exons. Its primary transcript covers about 2.4 kb, of which about 1.4 kb are exonic. In contrast, EGO-A remains unspliced, reaching about 190 nt into the intron. Both transcripts are polyadenylated (Wagner et al., 2007). Overall, EGOT is quite poorly conserved at sequence level. The intron, however, contains a sequence element that was already recognized by Wagner et al. (2007) to be conserved between human and chicken.
Here, we report on an in-depths computational analysis of EGOT, focusing in particular on the spliced and polyadenylated EGO-B transcript, which because of these properties is classified as a mlncRNA.
Materials and Methods
Based on the human EGO-B transcript (acc. no. NR_004428.1), orthologs have been retrieved from the UCSC multiz and the Ensembl EPO alignments but were also manually collected by iterative blat/blast searches against genomes publicly available at the UCSC Genome Browser and the Ensembl database, covering the evolutionary range from human to insects. Finally, a multiple sequence alignment was generated using MUSCLE (Edgar, 2004). Beyond reasonable sequence conservation, we applied additional criteria to collect the putative EGO-B orthologs, i.e., the syntenic conservation of flanking genes or an intact exon/intron gene structure with two conserved splice sites. In order to search for potential homologs outside the eutheria, we first identified the region homologous to the conserved element in the intron of EGO-B, extracted the complete ITPR1 intron plus some flanking sequence and used clustalw to construct separate pairwise alignments of each of the two EGOT exons with the genomic DNA sequence. RNA secondary structures were analyzed using the Vienna RNA package (Hofacker et al., 1994) and RNAz (Washietl et al., 2005). The significance of RNAz-predicted structures was analyzed by a control screen consisting of randomized sequence alignments generated by rnazRandomizeAln.pl, which is part of the RNAz package. This script columnwisely shuffles each sequence alignments such that local alignment characteristics and conservation patterns are preserved while the correlation between columns is destroyed. The UCSC Genome Browser was used for visualization of the EGOT locus. Stabilizing selection was quantified using phastCons (Siepel et al., 2005).
Results
Gene Phylogeny
We identified putative EGO-B orthologs in the genomes of 32 different amniotes, see Table 1; Figure 1. Based on the conservation of DNA sequence, gene structure, splice sites, and synteny, we found 25 strong candidate orthologs in primates, rodents, ungulates, carnivores, afrotherians, and xenarthrans. However, seven of the 32 putative orthologs have to be considered as weak. Their exons exhibit additional insertions, no convincing splice sites, or are extremely diverged in sequence from the members of the strong ortholog set. We could not identify EGO-B in all placental mammals: no homolog was found in pika, alpaca, microbat, and hedgehog genomes. We suspect that this is due to the low coverage and incomplete assembly of these genomes and hence constitutes an artifact rather than true gene loss. No indication for the existence of paralogs of the EGOT locus was found.

Figure 1. Overview of eutherian EGO-B orthologs. Species in which a (nearly) complete EGO-B gene was found are highlighted. EGO-B candidates are labeled with question marks. The figure is based on the Ensembl species tree.
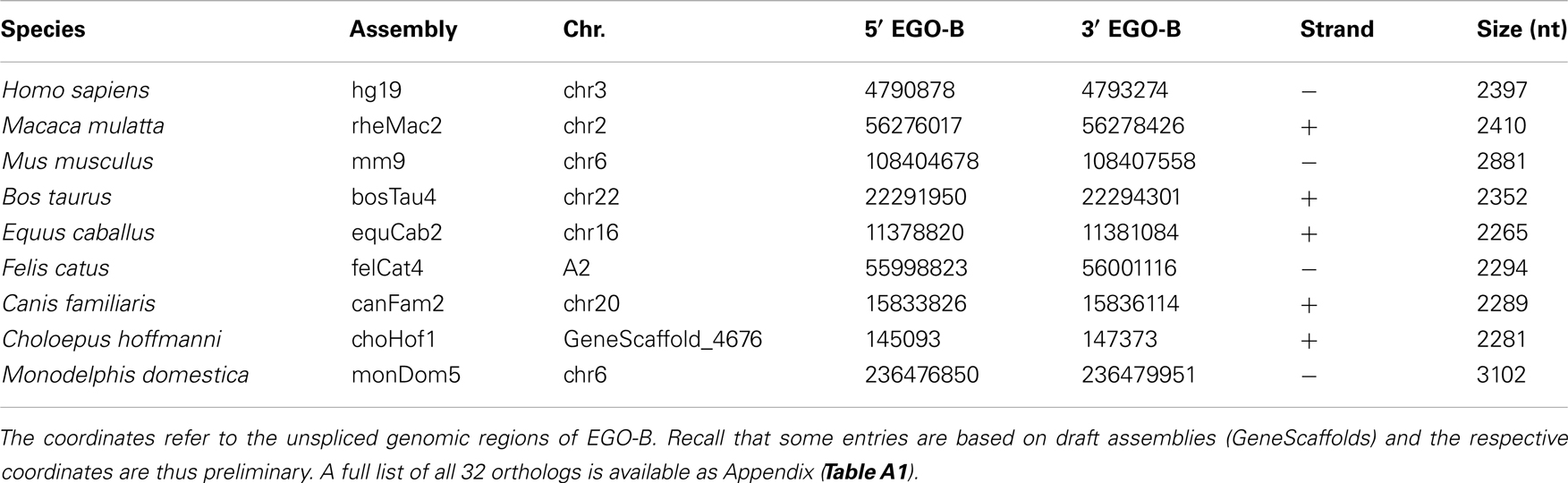
Table 1. Approximate genomic locations of EGO-B orthologs.
Trying to resolve distant homologies, we have also compiled EGO-B candidates for opossum, platypus, and chicken. This search was restricted to the ITPR1 locus to increase sensitivity. The putative ortholog in the opossum genome is most likely a true positive: it shows several compositional and syntenic features conserved throughout the eutherian orthologs, such as comparable exon/intron lengths, putatively functional splice sites, as well as the highly conserved intronic element discussed in detail below. Although the sequence of both exons is highly diverged, and hence the alignment of the opossum candidate to the eutherian sequences is rather poor, we hypothesize that EGOT most likely dates back before the divergence of eutheria and marsupials. In contrast, the candidates in platypus and sauropsids are not well supported.
Sequence Conservation
The two known human EGO-B exons exhibit average phastCons (Siepel et al., 2005) scores close to zero (∼0.04) among mammals (as well as vertebrates) suggesting a remarkably low level of sequence conservation, see Figure 2. In contrast, the two ITPR1 exons flanking EGO-B have phastCons scores of 0.87 and 0.96, respectively. At first glance, this observation conflicts with the initial findings of Wagner et al. (2007) who reported a high level of sequence conservation, which is present nearly exclusively in a highly conserved element (HCE) inside the intron of EGO-B, however. We used phastCons to quantify stabilizing selection. PhastCons uses a hidden Markov model to estimate the probability that each nucleotide of a multiple alignment belongs to a conserved element. Despite differences in detail, the alignment method has surprisingly little influence on the estimates. The average phastCons-score is about 0.09 for the 5′-exon and 0.02 for the 3′-exon, see Figure A1 in Appendix. In fact, major parts of both exons have no measurable conservation signal.
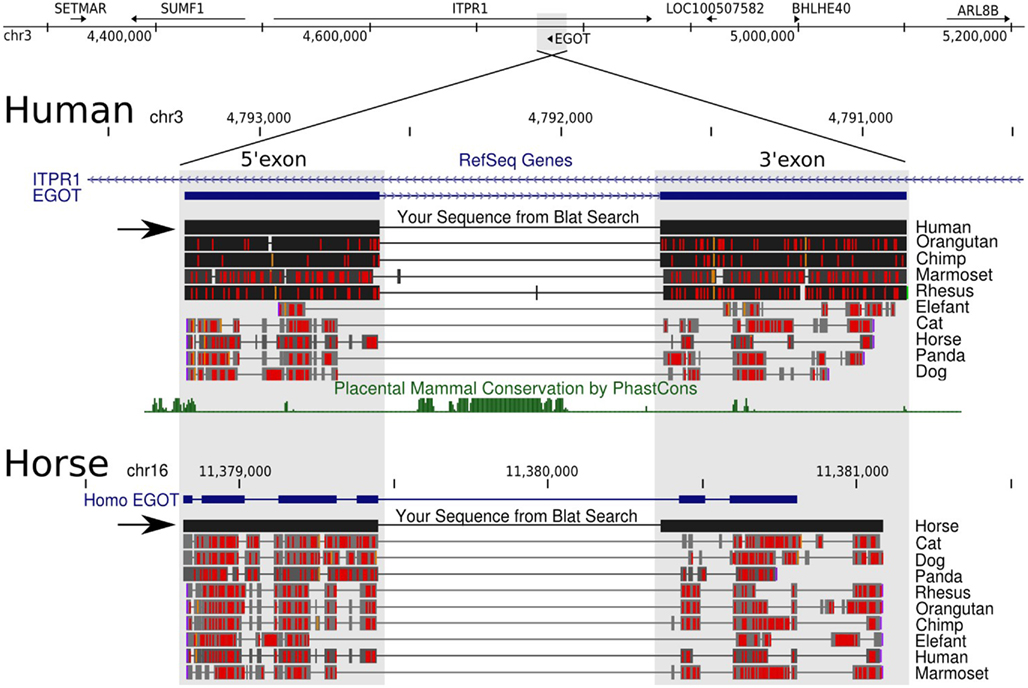
Figure 2. UCSC Genome Browser view. The figure illustrates the difficulties of obtaining orthologs of long ncRNAs due to a lack of sequence conservation. Black horizontal arrows highlight our manually curated EGO-B orthologs of human and horse. Blat searches are basically not sensitive enough, since they only recover fragments of the actual EGO-B exons. In case of horse, for example, even the RefSeq track insufficiently lists only the partial gene structure. Next, sequence conservation, or actually the probability of the EGO-B locus to be under negative selection, is close to zero according to the phastCons program. Nevertheless, we were able to compile a set of at least 25 strong candidates of EGO-B orthologs.
Gene Structures
RefSeq annotated human exons are on average 307 nt long (Pruitt et al., 2009). In contrast, exons of human pseudogenes are substantially longer. For example, the exons of the Yale pseudogene annotation have average lengths of 482 nt (Zhang et al., 2003). This difference can be explained by a lack of selective constraints to preserve the gene structure of pseudogenes. Among others, retrotransposition may lead to the acquirement of repeats and other artifact sequences. We used the two EGO-B exons as anchors for a local alignment approach to collect orthologs. Thus, the loss or inclusion of additional sequence elements at orthologous EGO-B loci can easily be measured. The lengths of orthologous EGO-B genes vary between 1.9 and 3.2 kb, given that we neglect the 9 kb long Procavia capensis or the 12 kb long Ornithorhynchus anatinus loci because of assembly issues. However, the average gene size (2.4 kb) of all collected orthologs is in perfect agreement with the initially reported 2.4 kb of EGO-B in human (Wagner et al., 2007). In particular, the sizes of the EGO-B 5′-exon, the intron, as well the 3′-exon fit fairly well to the human reference transcript for the majority of orthologs, see Figure 3. The deeply conserved gene structure supports our set of EGO-B candidates and suggests selective constraints acting on EGOT to preserve the spliced isoform.
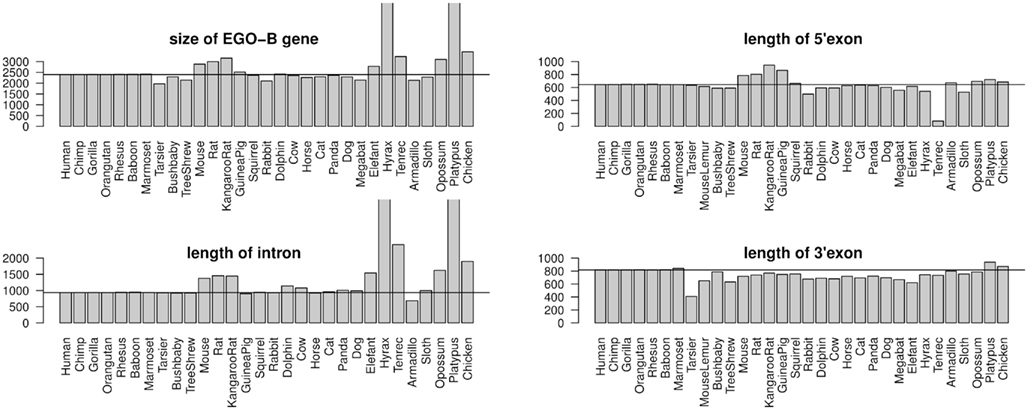
Figure 3. Conservation of the EGO-B gene structure. The gene structure, in particular the exon/intron lengths of the identified EGO-B transcripts are well conserved among orthologs. Horizontal lines mark the lengths of the human reference. In summary, the gene structure of the majority of species fits fairly well to the human reference. Notable exceptions are hyrax and platypus due to incomplete genome assemblies. As expected, especially rodents exhibit additional insertions compared to other higher vertebrates.
Splice Site Conservation
The presence of evolutionary conserved splice sites would further support our set of putative EGO-B orthologs and is usually indicative of a functionally relevant transcript. The majority of the 32 transcript candidates shows canonical splice site sequences at positions homologous to the known splice sites in human: 56% (18/32) have both a standard GT donor and an AG acceptor (59% (19/32) have a GT donor, 88% (28/32) an AG acceptor). Furthermore, we classified the EGO-B splice sites using MaxEntScan (Yeo and Burge, 2004), a maximum entropy modeling approach that discriminates real from false splice sites. As depicted in Figure 4A, 50% (16/32) of all donors and 94% (30/32) of all acceptors yield positive MaxEntScan scores implying that the sequence motifs of these sites are in agreement with known splice sites and therefore likely functional. Scoring the potential splice sites with a novel log-odds scoring scheme that evaluates substitution patterns of vertebrate splice sites and their ancestral sequences along a phylogenetic tree (Rose et al., 2011) yields −16.48 for EGO-B donors and 14.67 for EGO-B acceptors. Again, positive scores are indicative of functional splice sites. The evolutionary traces of substitutions at EGO-B acceptors are summarized in Figure 4B. Interestingly, we observed twice as many (24) substitution events typical for real acceptors compared to 12 atypical substitution events. However, there is a highly conserved TATA box-like motif at the EGO-B donor (see Figure 4C), which might explain the low donor scores as the consequence of an additional selective constraint. Even in human, the MaxEntScan donor score is only half of the corresponding acceptor signal. In summary, our results suggest intact splice sites for at least half (16/32) but likely even more of the analyzed species.
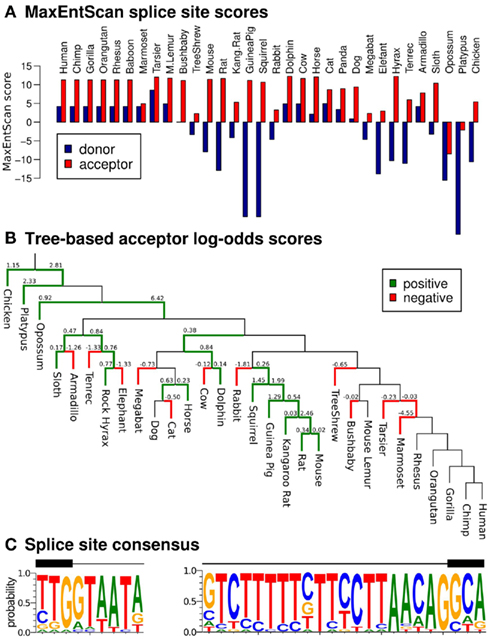
Figure 4. Splice site conservation. We evaluated the similarity of our splice site candidates to real splice sites using MaxEntScan (Yeo and Burge, 2004) and a novel log-odds scoring scheme that also takes phylogenetic information into account (Rose et al., 2011). (A) Draft genomes like hyrax or tenrec as well as genomes with additional insertions compared to human, such as mouse or rat, exhibit a weak MaxEntScan donor signal. However, the majority (75%) of tested splice sites (46/64) are likely real according to MaxEntScan. (B) Evolutionary traces of substitutions at EGO-B acceptors. Green edges indicate substitution events that are in agreement to real splice sites, red edges indicate unusual substitution patterns. (C) Sequence logos for the putative donor (left) and acceptor sites (right).
Syntenic Conservation
Wagner et al. (2007) have previously reported that EGO-B is transcribed antisense to an intron of the ITPR1 gene inositol triphosphate receptor type 1. However, ITPR1 is strictly syntenically linked to SUMF1 and BHLHE40 throughout vertebrates. The ancestral gene order of the ITPR1 locus seems to be SETMAR(+), SUMF1(−), ITPR1(+), BHLHE40(+), ARL8B(+), since this arrangement is present in basically all species in which we have detected EGO-B. Figure 2 (top) gives a compact overview of the gene synteny in human. The fact that synteny is intact and deeply conserved among a variety of vertebrate species supports our collection of EGO-B orthologs. The ITPR1 gene is conserved throughout vertebrates and the HCE in the intron of eutherian EGO-B is detectable throughout amniotes, with a plausible candidate also visible in Xenopus. Nevertheless, no convincing EGO-B orthologs were found outside placental mammals and marsupials.
Promoters
Not much is known about the transcriptional regulation of EGOT. ENCODE data suggest four possible promoter regions for EGOT, see Figure A3 in Appendix. On the one hand digital DNase1 hypersensitivity clusters obtained via tiling array experiments (Sabo et al., 2006) indicate three possible promoter regions upstream of EGOT. On the other hand, ChipSeq histone marks (Ernst et al., 2011) suggest an internal promoter located at the 5′-exon of EGO-B. However, the putative promoter regions are only moderately conserved at the sequence level. Among the four candidates, the external one, which is directly located upstream of EGO-B, exhibits the highest sequence conservation, better phastCons scores (0.21) than EGO-B, and can be traced back until zebrafish.
Mysterious Highly Conserved Elements
The EGOT locus contains three elements of unknown function that are highly conserved at the sequence level, see Figure 5. Two of these HCEs flank EGOT and another is located within the intron of EGO-B. As suggested above, the upstream HCE may function as a promoter. Using Q-RT-PCR Wagner et al. (2007) already confirmed abundant expression at the intronic HCE. Next, there is transcriptional evidence from EST data (FN099218) derived from 454 deep sequencing of primary human breast cancer (Guffanti et al., 2009) and an RNA-seq library of healthy breast tissue (Wang et al., 2008). In the recent release of the Rfam database (10.1, June 2011) the intronic HCE is already listed as EGOT (RF01958; Gardner et al., 2011). However, it is still not satisfactorily resolved whether these HCEs are part of novel EGOT isoforms, belong to independent, yet undiscovered, transcripts, or other functionally relevant regions.

Figure 5. Experimental evidence for transcription. The figure depicts human EST profiles of EGOT. Interestingly, transcription intensity as indicated by the EST profile does not correlate with sequence conservation as measured by phastCons. For example, there is only a single EST (FN099218) at the intronic HCE. Therefore, we speculate that for the depicted locus selective constraints are rather placed on the secondary than on primary structure. However, we have previously predicted a novel exon with conserved splice sites at the HCE (Rose et al., 2011). The predicted exon only partially covers the HCE, but larger alternative exons are conceivable from the predicted splice sites. ENCODE Caltech RNA-seq data reveals that the predicted exon is part of a novel ITPR1 isoform (the upstream exon, not depicted here because of space restrictions, is already part of available RefSeq annotation). The figure depicts only a subset of the spliced Caltech reads confirming the predicted exon.
Wagner et al. (2007) considered the intronic HCE to be independent of EGOT, since it, contrary to EGO-B, was not inducible with IL-5. This assumption is further backed by our bioinformatic analyses predicting a putative novel exon with conserved splice sites at the intronic HCE (Rose et al., 2011), see Figure 5. The putative exon cannot be part of another EGOT isoform, since it is in opposite reading direction. Spliced short reads from the ENCODE Caltech RNA-seq track (Mortazavi et al., 2008) verify the predicted splice site and reveal that the predicted exon is part of a novel ITPR1 isoform.
Moreover, the consensus sequence of the TATA box-like motif at the EGO-B donor (see Figure 4) is TAATA. This element might act as a promoter for an individually transcribed element. It has previously been shown that the TAATA motif can enhance transcription, i.e., it is part of the promoter of the human glucocorticoid receptor gene (Govindan et al., 1991).
Experimental Evidence for Transcription
In addition to sequence homology, EST data are typically used to determine the approximate evolutionary extent of a long ncRNA. There are several cDNAs available experimentally confirming EGO-A and EGO-B, see Figure 5. Analyzing the UniGene EST profiles reveals approximate gene expression patterns. EGOT has been detected in various adult human body sites, predominately adipose tissue, bone marrow, and kidney. Beyond healthy cell lines it is also expressed in various tumor tissues, such as liposarcoma or breast cancer.
However, the available EST data for EGOT mainly derives from human tissues, cDNAs from other species are rare. Beyond human cDNAs, there are only ESTs from Macaca fascicularis (BB876778, adult liver; Osada et al., 2008) and Bos taurus (AJ812842, bovine monocytes; McGuire and Glass, 2005). Both sequences strongly support the expression of the EGO-B 3′-exon, but do not provide a complete proof, since they are unspliced and their reading direction is not known. However, many of the human ESTs can successfully be mapped to several non-human EGOT orthologs recovering the established human gene structure.
Non-coding RNA profiling by high throughput sequencing of nuclear RNA in bone marrow-derived macrophages (De Santa et al., 2010) reveals extragenic Pol-II transcription sites at the mouse EGOT ortholog. As depicted in Figure A5 in Appendix, deep sequencing confirms transcription of the intronic HCE and parts of the 3′-end of the mouse EGOT ortholog. Although the data do not validate the full mouse ortholog, their experiments are still in line with our results. On the one hand, the two independently transcribed regions at the intronic HCE support our hypothesis that the HCE consists of two independent domains, a non-coding and a protein-coding one. Next, since it was previously postulated that EGOT may act via siRNAs to repress its targets MBP and EDN (Wagner et al., 2007), the signals at the 3′-end on the other hand might indeed indicate small RNAs that are hosted by EGOT. In summary, the experimental data of (De Santa et al., 2010) from mouse tally well with what is known from human EGOT.
Secondary Structures
We found that EGOT is highly structured. Using RNAz (Washietl et al., 2005), we identified five regions that exhibit thermodynamically stable and evolutionary conserved secondary structure motifs, see Figure 6. EGO-A contains a distinctive secondary structure at its 3′-end, which therefore might act as a termination signal. Remarkably, one of the EGO-B elements is located at the splice junction and thus can only be formed by the mature (spliced) transcript. In total, 43% (635/1462 nt) of the mature EGO-B transcript exhibit such prominent secondary structure motifs. In line with EGOT, the intronic HCE also shows RNAz-predicted signatures of preserved secondary structures. Figure A4 in Appendix depicts the predicted minimum free energy structures for several species and illustrates their evolutionary conservation in more detail. As expected, a sequence/structure-based clustering using LocARNA (Will et al., 2007) of the corresponding orthologs nearly perfectly recovers the six structural groups.
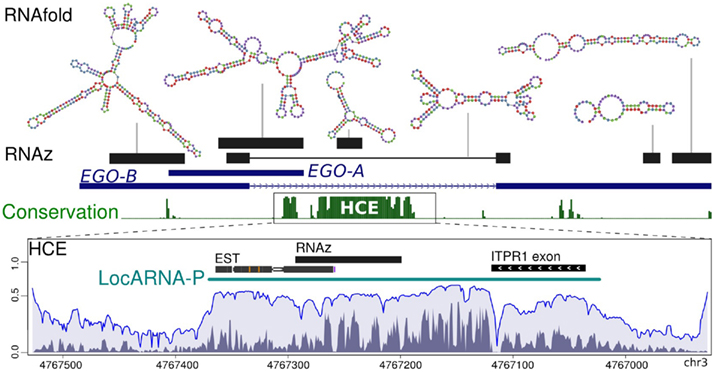
Figure 6. Secondary structural motifs. The figure illustrates RNAz-predicted secondary structures of EGOT and the adjacent intronic HCE. Both loci exhibit signals of thermodynamically stable and evolutionary conserved secondary structures. We provide the approximate genomic position and the predicted minimum free energy structure (RNAfold) for each RNAz hit. Computing the potential gene boundaries of a putative regulatory element located at the HCE using LocARNA-P reveals additional signals of conserved secondary structures. Peaks in the reliability profile are indicative of constrained and therefore most likely functionally relevant regions. Structural contributions to the reliability profile are depicted in dark gray, sequential in light gray.
RNAz is a window based approach. To demonstrate that all six structured regions found at the EGOT locus can indeed be attributed to constraints on EGOT orthologs, we set up a control screen consisting of shuffled alignment windows. The standard screen consisted of 351 input alignment windows, which partially overlap, not only because EGO-B and EGO-A already overlap, but also because several window sizes and various step-widths were tested. Overall, 45 of 351 windows were classified as structured RNA in the standard screen. However, only a single window was classified as structured in the control screen. This significant enrichment of structured windows in real versus control screen supports the significance of these RNAz predictions. We note that genome-wide RNAz-based studies have estimated their false discovery rates (FDR) at ∼20–60% (Missal et al., 2005, 2006; Rose et al., 2007, 2008). Here, we consider only a small locus with a highly significant signal for conserved structure.
Next, we applied LocARNA-P (Will et al., submitted), a novel approach estimating the precise boundaries of non-coding RNAs. It combines sequential and structural reliability information to a profile that depicts constrained and therefore likely functional regions. As illustrated in Figure 6, RNAz considers only a sub-region of the HCE to be structured. It is at least partially confirmed by EST data. However, the LocARNA-P reliability profile reveals additional signals of viable secondary structures next to the RNAz hit and suggests a larger non-coding gene. In summary, the HCE is not only conserved at the sequence level, it also harbors distinct secondary structures possibly associated with relevant biological functions.
We propose that the intronic HCE has ambiguous functions (at least dual), since we could show that it contains both protein-coding domains as well as non-coding elements. Most strikingly, the LocARNA-P-derived reliability profile apparently visualizes this dual character of the HCE. The sharp decrease of reliability signal clearly separates the patterns of putative non-coding RNAs in form of conserved secondary structures from the novel protein-coding ITPR1 exon.
Discussion
We have traced here the evolutionary history of EGOT, one of the first totally intronic long ncRNA that has been studied in detail. The spliced isoform, EGO-B, may be present throughout the placental mammals, and most likely dates back even further. Although both the genomic location in an intron of ITPR1 and the gene structure (i.e., both splice sites) is conserved at least throughout the placental mammals, the putative transcript is quite poorly conserved at the sequence level. In contrast to protein-coding genes and short, structured ncRNAs, this is a rather common feature of long ncRNAs in general (Marques and Ponting, 2009; Chodroff et al., 2010). Hence EGOT appears to be a rather typical representative of the mRNA-like ncRNAs.
Superimposed on the overall low level of sequence conservation, the EGOT locus contains also highly conserved regions. In particular, we have characterized the intronic HCE and untangled its complex nature. The 3′ part of the HCE can be recognized as an undescribed exon of ITPR1. Thus, it might even be that EGOT expression affects the (alternative) splicing of ITPR1 as it is known from the Saf/Fas locus (Yan et al., 2005). Its 5′ side shows evidence for expression unrelated to both ITPR1 and EGOT, exhibits a well conserved secondary structure element and features a conserved TATA-like element potentially acting as a promoter. Our results could furthermore be used to extend and refine the EGOT Rfam entry (RF01958), which at the moment just covers the intronic HCE but not the actual EGOT transcript.
In order to assess EGOT orthologs computationally, we have analyzed apparent indexes of conservation like synteny or the presence of functional splice sites at the EGOT locus. Although we have collected computational indication for a deep evolutionary conservation of EGOT, it is still theoretically possible that some of our signals might not be due to the putative EGO-B transcript orthologs, but to other yet unidentified functional elements in the region.
Surprisingly, a large part of EGO-B is folded into evolutionary conserved secondary structures. This sets it apart from the few other well-studied long ncRNAs. HOTAIR, for instance, has been reported to contain functional secondary structure elements whose evolutionary conservation appears to be weak (Tsai et al., 2010; He et al., 2011; Schorderet and Duboule, 2011) and requires further analysis. MALAT-1, on the other hand, exhibits only a few small conserved structured elements despite its overall high level of sequence conservation (Stadler, 2010). Furthermore, Marques and Ponting 2009) reported a moderate enrichment of conserved structural elements in some but not all types of long ncRNAs. This calls for a more systematic analysis of RNA secondary structures in long ncRNAs. The difference in structure content suggests, in particular, that this could be an important means of distinguishing functional classes of long ncRNAs.
The overall low level of sequence conservation is a serious obstacle for comparative genomics approaches. It limits first the sensitivity of homology search and then the accuracy of multiple sequence alignments. The large size of the molecules and the often complex and variable exon/intron structures, on the other hand, makes it extremely tedious to resort to manual improvements of alignments, in particular since currently available alignment editors are unable to accommodate complex annotation data. Recently developed tools (Rose et al., 2011) for the systematic assessment of splice site conservation were instrumental both in recognizing the additional exon in the HCE and in providing computational evidence for the conservation of the EGO-B orthologs.
The comparison of original genome-wide alignments and manually curated alignments of the EGOT locus demonstrates several drawbacks of pre-computed alignments (see also Figure A2 in Appendix). Pre-computed genome-wide alignments require substantial post-processing. Separated into alignment blocks, reference-based alignments often contain only partial sequences for some species since the orthologous sequence is not included in some alignment blocks, while on the other hand insertions not included in the reference are not represented at all. A third type of artifact consists in misaligned sequences that violate synteny. Of course, all these issues in principle also pertain to protein-coding regions. High levels of sequence conservation of coding regions and comparably little variability of intron/exon structure in coding regions, however, makes coding regions the most high-quality parts of genome-wide alignments. In-depth case studies such as the present one are thus instrumental in determining the types of problems that need to be considered in constructing analysis pipelines that deal with long non-coding RNAs at genome-wide levels.
Eosinophil granule ontogeny transcript has previously been proposed to affect myeloid development by regulating eosinophil gene expression in human. Eosinophils are generally responsible for an immune response to multicellular parasites and certain infections, not only in human, but in all vertebrates. Therefore, it would be conclusive that EGOT is also present in vertebrates fulfilling similar regulatory roles as in human. In turn, the functional assessment of a putative human-specific EGOT gene bears also great potential for evolutionary as well as clinical bioinformatics. However, further experimental evidence validating the expression of the proposed EGOT orthologs is required to ultimately assess the depth of evolutionary conservation of EGO-B.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful to Sebastian Will and co-authors for permission to use LocARNA-P prior to its publication. We gratefully acknowledge the contributions of Manja Marz and Annegret Wilde.
References
Amaral, P., Clark, M., Gascoigne, D., Dinger, M., and Mattick, J. (2011). lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 39, D146–D151.
Arthold, S., Kurowski, A., and Wutz, A. (2011). Mechanistic insights into chromosome-wide silencing in X inactivation. Hum. Genet. 130, 295–305.
Chodroff, R., Goodstadt, L., Sirey, T., Oliver, P., Davies, K., Green, E., Molnár, Z., and Ponting, C. (2010). Long noncoding RNA genes: conservation of sequence and brain expression among diverse amniotes. Genome Biol. 11, R72.
De Santa, F., Barozzi, I., Mietton, F., Ghisletti, S., Polletti, S., Tusi, B., Muller, H., Ragoussis, J., Wei, C., and Natoli, G. (2010). A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 8, e1000384. doi: 10.1371/journal.pbio.1000384
Duret, L., Chureau, C., Samain, S., Weissenbach, J., and Avner, P. (2006). The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 312, 1653–1655.
Edgar, R. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797.
Elisaphenko, E. A., Kolesnikov, N. N., Shevchenko, A. I., Rogozin, I. B., Nesterova, T. B., Brockdorff, N., and Zakian, S. M. (2008). A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS ONE 3, e2521. doi: 10.1371/journal.pone.0002521
ENCODE Project ConsortiumBirney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigó, R., Gingeras, T. R., Margulies, E. H., Weng, Z., Snyder, M., Dermitzakis, E. T., Thurman, R. E., Kuehn, M. S., Taylor, C. M., Neph, S., Koch, C. M., Asthana, S., Malhotra, A., Adzhubei, I., Greenbaum, J. A., Andrews, R. M., Flicek, P., Boyle, P. J., Cao, H., Carter, N. P., Clelland, G. K., Davis, S., Day, N., Dhami, P., Dillon, S. C., Dorschner, M. O., Fiegler, H., Giresi, P. G., Goldy, J., Hawrylycz, M., Haydock, A., Humbert, R., James, K. D., Johnson, B. E., Johnson, E. M., Frum, T. T., Rosenzweig, E. R., Karnani, N., Lee, K., Lefebvre, G. C., Navas, P. A., Neri, F., Parker, S. C., Sabo, P. J., Sandstrom, R., Shafer, A., Vetrie, D., Weaver, M., Wilcox, S., Yu, M., Collins, F. S., Dekker, J., Lieb, J. D., Tullius, T. D., Crawford, G. E., Sunyaev, S., Noble, W. S., Dunham, I., Denoeud, F., Reymond, A., Kapranov, P., Rozowsky, J., Zheng, D., Castelo, R., Frankish, A., Harrow, J., Ghosh, S., Sandelin, A., Hofacker, I. L., Baertsch, R., Keefe, D., Dike, S., Cheng, J., Hirsch, H. A., Sekinger, E. A., Lagarde, J., Abril, J. F., Shahab, A., Flamm, C., Fried, C., Hackermüller, J., Hertel, J., Lindemeyer, M., Missal, K., Tanzer, A., Washietl, S., Korbel, J., Emanuelsson, O., Pedersen, J. S., Holroyd, N., Taylor, R., Swarbreck, D., Matthews, N., Dickson, M. C., Thomas, D. J., Weirauch, M. T., Gilbert, J., Drenkow, J., Bell, I., Zhao, X., Srinivasan, K. G., Sung, W. K., Ooi, H. S., Chiu, K. P., Foissac, S., Alioto, T., Brent, M., Pachter, L., Tress, M. L., Valencia, A., Choo, S. W., Choo, C. Y., Ucla, C., Manzano, C., Wyss, C., Cheung, E., Clark, T. G., Brown, J. B., Ganesh, M., Patel, S., Tammana, H., Chrast, J., Henrichsen, C. N., Kai, C., Kawai, J., Nagalakshmi, U., Wu, J., Lian, Z., Lian, J., Newburger, P., Zhang, X., Bickel, P., Mattick, J. S., Carninci, P., Hayashizaki, Y., Weissman, S., Hubbard, T., Myers, R. M., Rogers, J., Stadler, P. F., Lowe, T. M., Wei, C. L., Ruan, Y., Struhl, K., Gerstein, M., Antonarakis, S. E., Fu, Y., Green, E. D., Karaöz, U., Siepel, A., Taylor, J., Liefer, L. A., Wetterstrand, K. A., Good, P. J., Feingold, E. A., Guyer, M. S., Cooper, G. M., Asimenos, G., Dewey, C. N., Hou, M., Nikolaev, S., Montoya-Burgos, J. I., Löytynoja, A., Whelan, S., Pardi, F., Massingham, T., Huang, H., Zhang, N. R., Holmes, I., Mullikin, J. C., Ureta-Vidal, A., Paten, B., Seringhaus, M., Church, D., Rosenbloom, K., Kent, W. J., Stone, E. A., NISC Comparative Sequencing Program Baylor College of Medicine Human Genome Sequencing Center, Washington University Genome Sequencing Center, Broad Institute, Children’s Hospital Oakland Research Institute, Batzoglou, S., Goldman, N., Hardison, R. C., Haussler, D., Miller, W., Sidow, A., Trinklein, N. D., Zhang, Z. D., Barrera, L., Stuart, R., King, D. C., Ameur, A., Enroth, S., Bieda, M. C., Kim, J., Bhinge, A. A., Jiang, N., Liu, J., Yao, F., Vega, V. B., Lee, C. W., Ng, P., Shahab, A., Yang, A., Moqtaderi, Z., Zhu, Z., Xu, X., Squazzo, S., Oberley, M. J., Inman, D., Singer, M. A., Richmond, T. A., Munn, K. J., Rada-Iglesias, A., Wallerman, O., Komorowski, J., Fowler, J. C., Couttet, P., Bruce, A. W., Dovey, O. M., Ellis, P. D., Langford, C. F., Nix, D. A., Euskirchen, G., Hartman, S., Urban, A. E., Kraus, P., Van Calcar, S., Heintzman, N., Kim, T. H., Wang, K., Qu, C., Hon, G., Luna, R., Glass, C. K., Rosenfeld, M. G., Aldred, S. F., Cooper, S. J., Halees, A., Lin, J. M., Shulha, H. P., Zhang, X., Xu, M., Haidar, J. N., Yu, Y., Ruan, Y., Iyer, V. R., Green, R. D., Wadelius, C., Farnham, P. J., Ren, B., Harte, R. A., Hinrichs, A. S., Trumbower, H., Clawson, H., Hillman-Jackson, J., Zweig, A. S., Smith, K., Thakkapallayil, A., Barber, G., Kuhn, R. M., Karolchik, D., Armengol, L., Bird, C. P., de Bakker, P. I., Kern, A. D., Lopez-Bigas, N., Martin, J. D., Stranger, B. E., Woodroffe, A., Davydov, E., Dimas, A., Eyras, E., Hallgrímsdóttir, I. B., Huppert, J., Zody, M. C., Abecasis, G. R., Estivill, X., Bouffard, G. G., Guan, X., Hansen, N. F., Idol, J. R., Maduro, V. V., Maskeri, B., McDowell, J. C., Park, M., Thomas, P. J., Young, A. C., Blakesley, R. W., Muzny, D. M., Sodergren, E., Wheeler, D. A., Worley, K. C., Jiang, H., Weinstock, G. M., Gibbs, R. A., Graves, T., Fulton, R., Mardis, E. R., Wilson, R. K., Clamp, M., Cuff, J., Gnerre, S., Jaffe, D. B., Chang, J. L., Lindblad-Toh, K., Lander, E. S., Koriabine, M., Nefedov, M., Osoegawa, K., Yoshinaga, Y., Zhu, B., and de Jong, P. J. (2007). Identification and analysis of functional elements in 1 genome by the ENCODE pilot project. Nature 447, 799–816.
Engelhardt, J., and Stadler, P. F. (2011). “Hidden treasures in unspliced EST data,” in Proceedings of the 6th International Symposium on Heath Informatics and Bioinformatics (HIBIT), Izmir, Turkey.
Ernst, J., Kheradpour, P., Mikkelsen, T. S., Shoresh, N., Ward, L. D., Epstein, C. B., Zhang, X., Wang, L., Issner, R., Coyne, M., Ku, M., Durham, T., Kellis, M., and Bernstein, B. E. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49.
Finn, R., Mistry, J., Tate, J., Coggill, P., Heger, A., Ponton, J. E., Gavin, O. L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L., Eddy, S. R., and Bateman, A. (2010). The pfam protein families database. Nucleic Acids Res. 38, D211–D222.
Gardner, P., Daub, J., Tate, J., Moore, B. L., Osuch, I. H., Griffiths-Jones, S., Finn, R. D., Nawrocki, E. P., Kolbe, D. L., Eddy, S. R., and Bateman, A. (2011). Rfam: wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39, D141–D145.
Govindan, M., Pothier, F., Leclerc, S., Palaniswami, R., and Xie, B. (1991). Human glucocorticoid receptor gene promoter-homologous down regulation. J. Steroid Biochem. Mol. Biol. 40, 317–323.
Guffanti, A., Iacono, M., Pelucchi, P., Kim, N., Soldà, G., Croft, L. J., Taft, R. J., Rizzi, E., Askarian-Amiri, M., Bonnal, R. J., Callari, M., Mignone, F., Pesole, G., Bertalot, G., Bernardi, L. R., Albertini, A., Lee, C., Mattick, J. S., Zucchi, I., and De Bellis, G. (2009). A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics 10, 163. doi: 10.1186/1471-2164-10-163
He, S., Liu, S., and Zhu, H. (2011). The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evol. Biol. 11, 102. doi: 10.1186/1471-2148-11-102
Hofacker, I. L., Fontana, W., Stadler, P. F., Bonhoeffer, L. S., Tacker, M., and Schuster, P. (1994). Fast folding and comparison of RNA secondary structures. Monatshefte Chem. Chem. Mon. 125, 167–188.
Hutchinson, J., Ensminger, A., Clemson, C., Lynch, C., Lawrence, J., and Chess, A. (2007). A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8, 39. doi: 10.1186/1471-2164-8-39
Jacquier, A. (2009). The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat. Rev. Genet. 10, 833–844.
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D., Thomas, K., Presser, A., Bernstein, B. E., van Oudenaarden, A., Regev, A., Lander, E. S., and Rinn, J. L. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U.S.A. 106, 11675–11680.
Kolesnikov, N., and Elisafenko, E. (2010). Comparative organization and the origin of noncoding regulatory RNA genes from x-chromosome inactivation center of human and mouse. Genetika 46, 1386–1391.
Louro, R., El-Jundi, T., Nakaya, H. I., Reis, E. M., and Verjovski-Almeida, S. (2008). Conserved tissue expression signatures of intronic noncoding RNAs transcribed from human and mouse loci. Genomics 92, 18–25.
Louro, R., Smirnova, A. S., and Verjovski-Almeida, S. (2009). Long intronic noncoding RNA transcription: expression noise or expression choice? Genomics 93, 291–298.
Maeda, N., Kasukawa, T., Oyama, R., Gough, J., Frith, M., Engström, P. G., Lenhard, B., Aturaliya, R. N., Batalov, S., Beisel, K. W., Bult, C. J., Fletcher, C. F., Forrest, A. R., Furuno, M., Hill, D., Itoh, M., Kanamori-Katayama, M., Katayama, S., Katoh, M., Kawashima, T., Quackenbush, J., Ravasi, T., Ring, B. Z., Shibata, K., Sugiura, K., Takenaka, Y., Teasdale, R. D., Wells, C. A., Zhu, Y., Kai, C., Kawai, J., Hume, D. A., Carninci, P., and Hayashizaki, Y. (2006). Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genet. 2, e62. doi: 10.1371/journal.pgen.0020062
Marques, A., and Ponting, C. (2009). Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome Biol. 10, R124.
McGuire, K., and Glass, E. (2005). The expanding role of microarrays in the investigation of macrophage responses to pathogens. Vet. Immunol. Immunopathol. 105, 259–275.
Mercer, T., Dinger, M., and Mattick, J. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159.
Missal, K., Rose, D., and Stadler, P. (2005). Non-coding RNAs in ciona intestinalis. Bioinformatics 21(Suppl. 2), ii77–ii78.
Missal, K., Zhu, X., Rose, D., Deng, W., Skogerbø, G., Chen, R., and Stadler, P. (2006). Prediction of structured non-coding RNAs in the genomes of the nematodes caenorhabditis elegans and caenorhabditis briggsae. J. Exp. Zool. B Mol. Dev. Evol. 306, 379–392.
Mortazavi, A., Williams, B., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-S. Nat. Methods 5, 621–628.
Nakaya, H. I., Amaral, P. P., Louro, R., Lopes, A., Fachel, A. A., Moreira, Y. B., El-Jundi, T. A., da Silva, A. M., Reis, E. M., and Verjovski-Almeida, S. (2007). Genome mapping and expression analyses of human intronic noncoding RNAs reveal tissue-specific patterns and enrichment in genes related to regulation of transcription. Genome Biol. 8, R43.
Osada, N., Hashimoto, K., Kameoka, Y., Hirata, M., Tanuma, R., Uno, Y., Inoue, I., Hida, M., Suzuki, Y., Sugano, S., Terao, K., Kusuda, J., and Takahashi, I. (2008). Large-scale analysis of macaca fascicularis transcripts and inference of genetic divergence between M. fascicularis and M. mulatta. BMC Genomics 9, 90. doi: 10.1186/1471-2164-9-90
Ponjavic, J., Ponting, C. P., and Lunter, G. (2007). Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 17, 556–565.
Pruitt, K., Tatusova, T., Klimke, W., and Maglott, D. (2009). NCBI reference sequences: current status, policy and new initiatives. Nucleic Acids Res. 37, D32–D36.
Reis, E. M., Nakaya, H. I., Louro, R., Canavez, F. C., Flatschart, A. V., Almeida, G. T., Egidio, C. M., Paquola, A. C., Machado, A. A., Festa, F., Yamamoto, D., Alvarenga, R., da Silva, C. C., Brito, G. C., Simon, S. D., Moreira-Filho, C. A., Leite, K. R., Camara-Lopes, L. H., Campos, F. S., Gimba, E., Vignal, G. M., El-Dorry, H., Sogayar, M. C., Barcinski, M. A., da Silva, A. M., and Verjovski-Almeida, S. (2004). Antisense intronic non-coding RNA levels correlate to the degree of tumor differentiation in prostate cancer. Oncogene 23, 6684–6692.
Rinn, J. L., Euskirchen, G., Bertone, P., Martone, R., Luscombe, N. M., Hartman, S., Harrison, P. M., Nelson, F. K., Miller, P., Gerstein, M., Weissman, S., and Snyder, M. (2003). The transcriptional activity of human chromosome 22. Genes Dev. 17, 529–540.
Rose, D., Hackermüller, J., Washietl, S., Reiche, K., Hertel, J., Findeiss, S., Stadler, P., and Prohaska, S. (2007). Computational RNomics of drosophilids. BMC Genomics 8, 406. doi: 10.1186/1471-2164-8-406
Rose, D., Hiller, M., Schutt, K., Hackermüller, J., Backofen, R., and Stadler, P. (2011). Computational discovery of human coding and non-coding transcripts with conserved splice sites. Bioinformatics 27, 1894–1900.
Rose, D., Jöris, J., Hackermüller, J., Reiche, K., Li, Q., and Stadler, P. (2008). Duplicated RNA genes in teleost fish genomes. J. Bioinform. Comput. Biol. 6, 1157–1175.
Sabo, P., Kuehn, M. S., Thurman, R., Johnson, B. E., Johnson, E. M., Cao, H., Yu, M., Rosenzweig, E., Goldy, J., Haydock, A., Weaver, M., Shafer, A., Lee, K., Neri, F., Humbert, R., Singer, M. A., Richmond, T. A., Dorschner, M. O., McArthur, M., Hawrylycz, M., Green, R. D., Navas, P. A., Noble, W. S., and Stamatoyannopoulos, J. A. (2006). Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat. Methods 3, 511–518.
Sasaki, Y. T. F., Ideue, T., Sano, M., Mituyama, T., and Hirose, T. (2009). MEN ε/β noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl. Acad. Sci. U.S.A. 106, 2525–2530.
Schorderet, P., and Duboule, D. (2011). Structural and functional differences in the long non-coding RNA Hotair in mouse and human. PLoS Genet. 7, e1002071. doi: 10.1371/journal.pgen.1002071
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K., Clawson, H., Spieth, J., Hillier, L. W., Richards, S., Weinstock, G. M., Wilson, R. K., Gibbs, R. A., Kent, W. J., Miller, W., and Haussler, D. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050.
Stadler, P. F. (2010). “Evolution of the long non-coding RNAs MALAT1 and MEN ε/β,” in Advances in Bioinformatics and Computational Biology, Vol. 6268, eds C. Ferreira, S. Miyano, and P. Stadler (Heidelberg: Springer), 1–12.
Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., Shi, Y., Segal, E., and Chang, H. Y. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693.
Wagner, L., Christensen, C., Dunn, D., Spangrude, G., Georgelas, A., Kelley, L., Esplin, M., Weiss, R., and Gleich, G. (2007). EGO, a novel, noncoding RNA gene, regulates eosinophil granule protein transcript expression. Blood 109, 5191–5198.
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S. F., Schroth, G. P., and Burge, C. B. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476.
Wang, K. C., Yang, Y. W., Liu, B., Sanyal, A., Corces-Zimmerman, R., Chen, Y., Lajoie, B. R., Protacio, A., Flynn, R. A., Gupta, R. A., Wysocka, J., Lei, M., Dekker, J., Helms, J. A., and Chang, H. Y. (2011). A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124.
Washietl, S., Hofacker, I., and Stadler, P. (2005). Fast and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. U.S.A. 102, 2454–2459.
Will, S., Reiche, K., Hofacker, I., Stadler, P., and Backofen, R. (2007). Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 3, e65. doi: 10.1371/journal.pcbi.0030065
Wilusz, J. E., and Spector, D. L. (2010). An unexpected ending: noncanonical 3’ end processing mechanisms. RNA 16, 259–266.
Yan, M., Hong, C., Lai, G., Cheng, A., Lin, Y., and Chuang, S. (2005). Identification and characterization of a novel gene saf transcribed from the opposite strand of fas. Hum. Mol. Genet. 14, 1465–1474.
Yeo, G., and Burge, C. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394.
Zhang, Z., Harrison, P., Liu, Y., and Gerstein, M. (2003). Millions of years of evolution preserved: a comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 13, 2541–2558.
Appendix
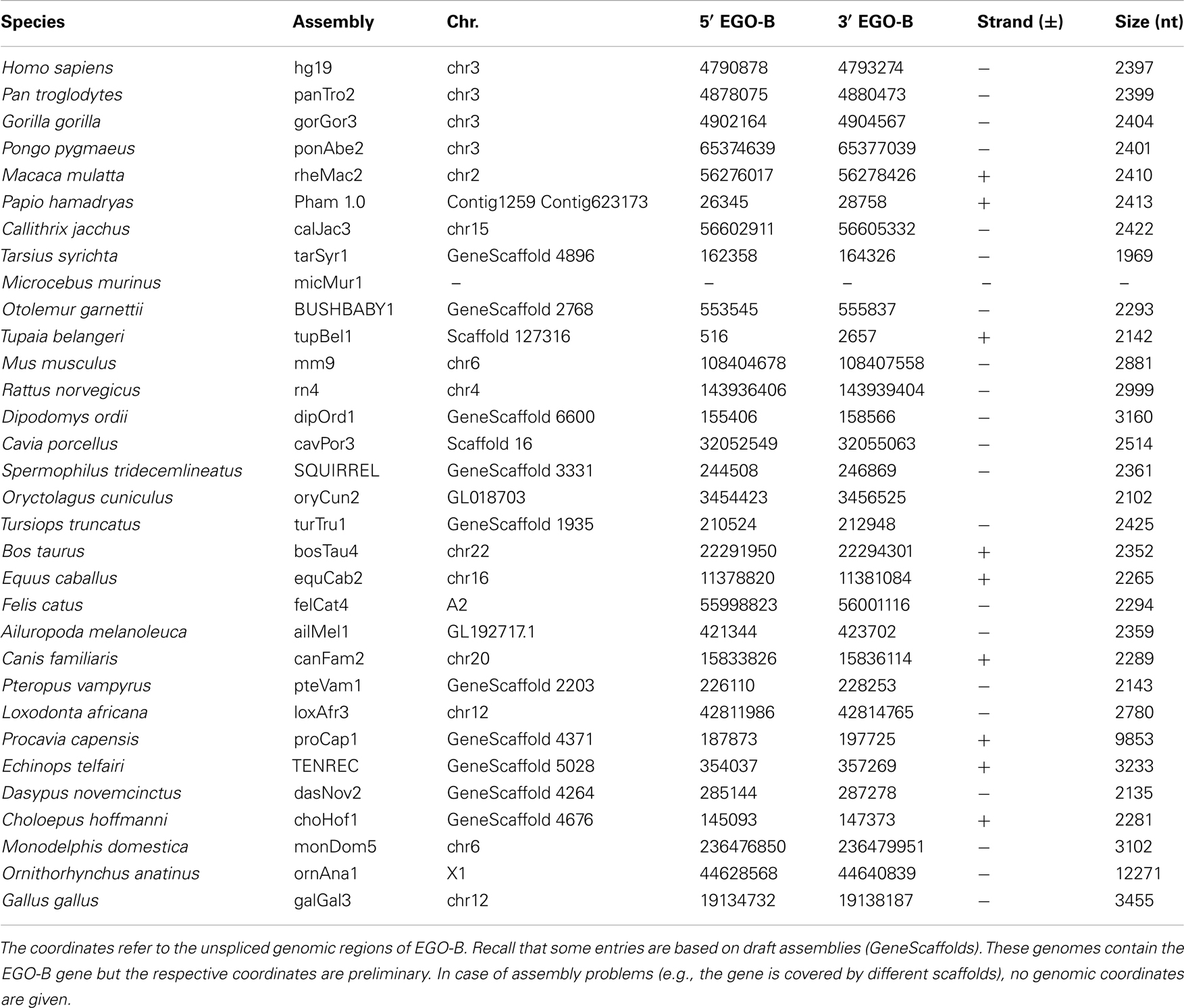
Table A1. Approximate genomic locations of EGO-B orthologs.
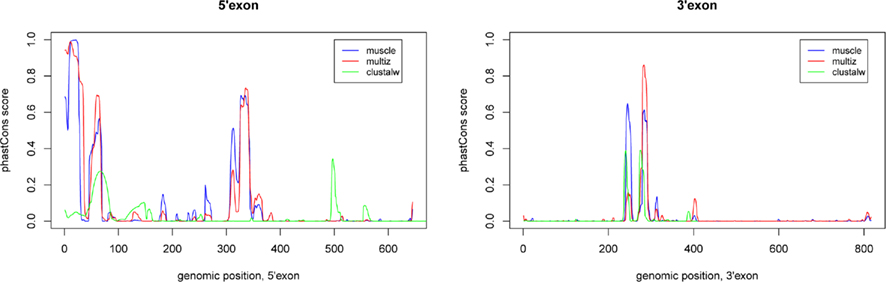
Figure A1. Sequence conservation of EGO-B as indicated by the phastCons program. We have computed the phastCons scores for three different alignment approaches using the vertebrate model available at the UCSC Genome Browser: (1) our own muscle alignment, (2) the pre-computed 46-way vertebrate multiz alignment from the UCSC browser, and (3) a clustalw alignment based on our set of orthologs. The peaks are fairly similar, only clustalw slightly differs due to a higher alignment error rate resulting from the lack of consistency transformation or similar alignment refinement/improvement steps. Applied phastCons parameters: transitions 0.01, 0.01; rho 0.4.
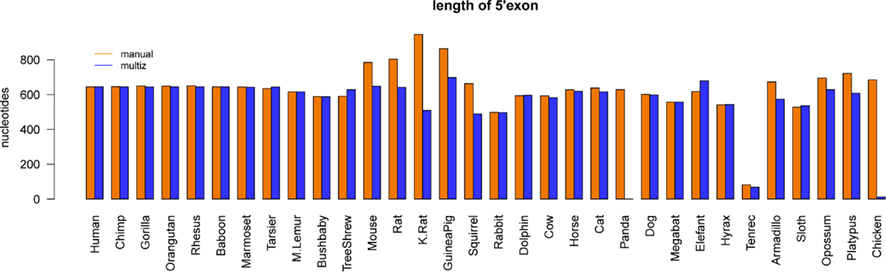
Figure A2. Differences in aligned sequence data between our approach and pre-computed UCSC multiz alignments. We plot the sequence portion of the 5′-exon of EGO-B for different species and alignment approaches. There is a large overlap but also substantial differences between the two alignment approaches. An obvious drawback of pre-computed alignments is missing data. For example, the UCSC alignments do not contain the ortholog of panda because the assembly was not ready at the time the alignments were generated. More strikingly, non-human orthologs are only partially included in the UCSC alignments, since it is a reference (human) based approach. Regions that would cause larger gaps in human, such as mouse or rat which exhibit various exonic insertions, are only partially included in the final alignment. However, partial sequences are crucial for any subsequent analysis relying on valid alignments, i.e., RNA secondary structure prediction.
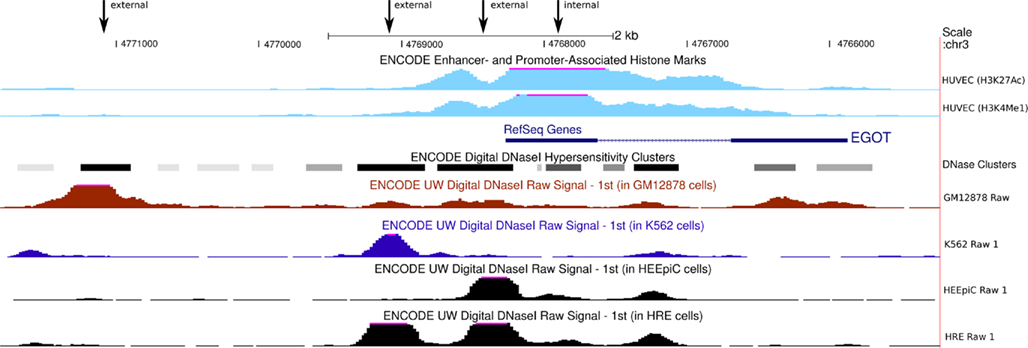
Figure A3. Eosinophil granule ontogeny transcript promoter regions. ENCODE data suggest four possible promoter regions for EGOT (marked by the four arrows). Digital DNase1 hypersensitivity clusters indicate three promoter candidate regions upstream of EGOT. On the other hand, histone marks suggest an internal promoter at the 5′-exon of EGO-B. The figure contains only exemplary cell lines. However, the depicted signal peaks, especially the DNase1 hypersensitivity peaks, are consistently present in numerous cell lines.
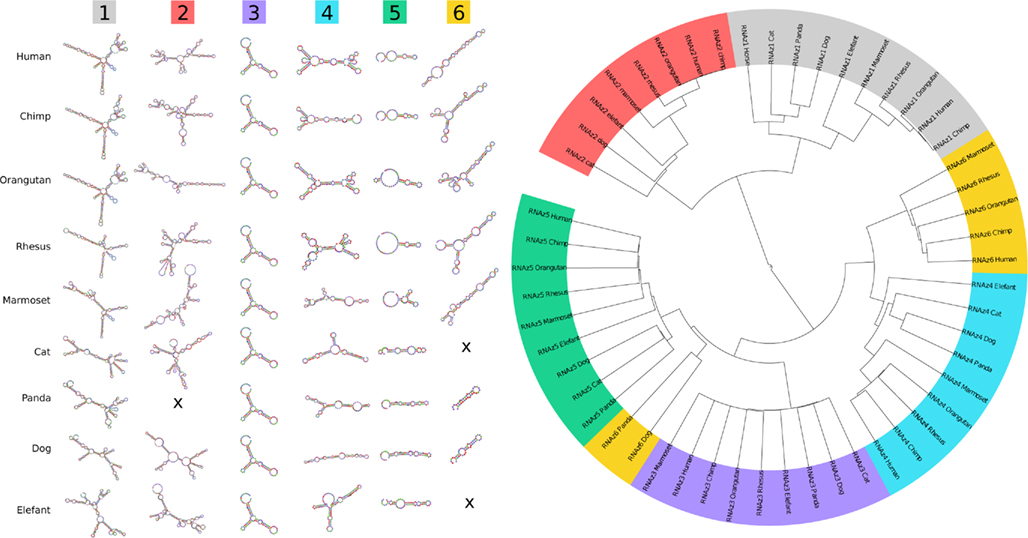
Figure A4. Traces of evolutionary conserved secondary structures. The minimum free energy structures of the six RNAz-predicted regions are at least partially conserved throughout higher eukaryotes. A sequence/structure-based clustering using LocARNA (Will et al., 2007) visualizes the similarities between the predicted structures in more detail. As expected, the structures nearly perfectly cluster into the six groups.
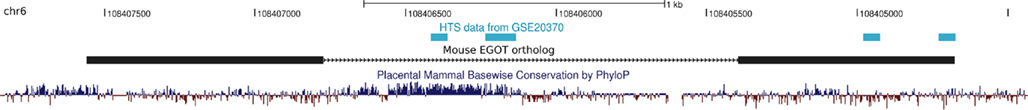
Figure A5. Non-coding RNA profiling by high throughput sequencing reveals extragenic Pol-II transcription sites at the mouse EGOT ortholog. The deep sequencing data from (De Santa et al., 2010) available in the Gene Expression Omnibus (under GEO accession number GSE20370) confirm transcription of the intronic highly conserved element (HCE) and parts of the 3′-end of the mouse EGOT ortholog. Although the data do not validate the full mouse ortholog, we benefit twice from the depicted transcribed regions. On the one hand the two independently transcribed regions at the intronic HCE support our findings that the HCE consists of two independent non-coding as well as protein-coding domains. Next, since it was previously postulated that EGOT may act via siRNAs to repress its targets MBP and EDN (Wagner et al., 2007), the signals at the 3′-end on the other hand might indeed indicate small RNAs that are hosted by EGOT.
Keywords: EGO, EGO-A, EGO-B, EGOT, long non-coding RNA, lncRNA, mlncRNA, evolution
Citation: Rose D and Stadler PF (2011) Molecular evolution of the non-coding eosinophil granule ontogeny transcript. Front. Gene. 2:69. doi: 10.3389/fgene.2011.00069
Received: 01 June 2011;
Accepted: 16 September 2011;
Published online: 05 October 2011.
Edited by:
Helder I. Nakaya, Emory University, USAReviewed by:
Helder I. Nakaya, Emory University, USAFei Li, Nanjing Agricultural University, China
Paulo De Paiva Rosa Amaral, The University of Queensland, Australia
Copyright: © 2011 Rose and Stadler. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Dominic Rose, Bioinformatics Group, Department of Computer Science, University of Freiburg, Georges-Köhler-Allee 106, D-79110 Freiburg, Germany. e-mail: dominic@informatik.uni-freiburg.de