
- Modifiable Risk Factors Branch, Division of Cancer Control and Population Sciences, National Cancer Institute, Bethesda, MD, USA
Epigenetics is the study of heritable changes in gene expression that occur without a change in DNA sequence. Cancer is a multistep process derived from combinational crosstalk between genetic alterations and epigenetic influences through various environmental factors. The observation that epigenetic changes are reversible makes them an attractive target for cancer prevention. Until recently, there have been difficulties studying epigenetic mechanisms in interactions between dietary factors and environmental toxicants. The development of the field of cancer epigenetics during the past decade has been advanced rapidly by genome-wide technologies – which initially employed microarrays but increasingly are using high-throughput sequencing – which helped to improve the quality of the analysis, increase the capacity of sample throughput, and reduce the cost of assays. It is particularly true for applications of cancer epigenetics in epidemiologic studies that examine the relationship among diet, epigenetics, and cancer because of the issues of tissue heterogeneity, the often limiting amount of DNA samples, and the significant cost of the analyses. This review offers an overview of the state of the science in nutrition, environmental toxicants, epigenetics, and cancer to stimulate further exploration of this important and developing area of science. Additional epidemiologic research is needed to clarify the relationship between these complex epigenetic mechanisms and cancer.
Introduction
Cancer represents a group of diseases initiated and driven largely by heritable genetic aberrations resulting from exposure to endogenous and environmental agents (Loeb and Harris, 2008; Pogribny et al., 2009). Therefore, the general approach for elucidating key events in cancer development has focused on identifying genetic aberrations associated with carcinogenesis. However, the presence of genetic lesions alone is not sufficient for tumor formation. Tumor formation results mainly from the inability of cells to maintain and control accurate expression of genetic information. Additionally, genetic alterations alone cannot explain the extremely diverse phenotypic changes observed in preneoplastic and malignant cells. These findings have led to the hypothesis that the transition from promotion – a stage that may be reversible in carcinogenesis – to progression – a stage that may be irreversible – could be driven primarily by epigenetic abnormalities (Pitot, 2007). The term “epigenetics” refers to mechanisms outside of the scope of conventional genetics. Epigenetics is defined as processes that regulate heritable changes in gene expression that are transmitted through meiosis and mitosis, without any modification of the primary DNA sequence. These epigenetic mechanisms include DNA methylation and a complex set of histone modifications, including acetylation, methylation, phosphorylation, ADP-ribosylation, and ubiquitination, leading to chromatin remodeling.
Diet and lifestyle play a crucial role in cancer etiology. It has been estimated that more than one-third of cancer deaths in the United States potentially can be avoided through dietary modification (Doll and Peto, 1981). Different mechanisms, including antioxidant, anti-inflammatory, and anti-estrogenic processes, have been proposed to explain the protective nature of certain dietary components. However, the exact mechanistic pathways in which these effects are exerted on cells to avoid, delay, or reverse carcinogenesis are not as clear. Epigenetic marks are influenced by aging and environmental exposures, including dietary intakes (Issa et al., 1994; Issa, 2003). Emerging evidence indicates that nutrition and environmental factors affect epigenetic changes. The observation that epigenetic changes are reversible makes them an attractive target for cancer prevention. The intent of this review is to provide an overview of the state of the science in nutrition, environmental toxicants, epigenetics, and cancer to stimulate the continuous exploration of how nutritional factors and environmental toxicants in foods affect the functioning of genes via molecular mechanisms other than those observed in conventional genetics, such as mutations and polymorphisms.
DNA Methylation
The most well-known epigenetic mechanism is methylation. DNA methylation is a major epigenetic phenomenon that predominantly involves the covalent addition of a methyl group (CH3) to the 5′-position of cytosine that precedes a guanosine in the DNA sequence (the CpG dinucleotides), thereby regulating genetic expression and integrity in various biological processes, such as differentiation, genomic imprinting, DNA mutation, and DNA repair (Fang et al., 2007). This is referred to as an epigenetic modification because it does not change the coding sequence of the DNA. The CpG dinucleotides are not distributed evenly in the whole genome, but tend to be clustered in small stretches of DNA termed “CpG islands” (Robertson and Wolffe, 2000). These regions often are associated with the promoter regions of genes. Approximately one-half of the genes in the human genome have such CpG-rich promoter regions (Herman and Baylin, 2003). A majority of the CpG dinucleotides in the human genome that are not associated with CpG islands are heavily methylated (Herman and Baylin, 2003). On the other hand, the dinucleotides in CpG islands, in particular those associated with gene promoters, usually are unmethylated (Bird, 2002). Patterns of DNA methylation are generated during development involving de novo methylation and demethylation activities. DNA methylation is an enzymatic process that is mediated by DNA methyltransferases (DNMTs). DNMT3 regulates de novo methylation during development, whereas DNMT1 maintains DNA methylation patterns during cell replication. The genome methylation pattern is precisely inherited during mitosis and is highly tissue specific. Cytosine methylation changes the structure of the major groove in the DNA molecule and disrupts the attachment of DNA-binding proteins and transcription factors. Genes methylated at specific sites, such as upstream of a promoter region, are either not transcribed into messenger RNA (mRNA) or are transcribed at a reduced rate, thus reducing translation of the gene into the encoded protein. As a result, epigenetic DNA methylation contributes to the control of gene and ultimately protein expression (Costello and Plass, 2001; Duthie, 2011). It can induce the gene to turn on or off, and to up- or down-regulate.
Aberrant DNA methylation patterns in cancer cells have been recognized for decades (Boehm and Drahovsky, 1983). However, the interpretation of observed DNA methylation and cancer risk remains a challenge because virtually all types of cancer that have been examined have shown both global hypomethylation and gene-specific hypermethylation in gene promoter regions (Baylin et al., 1998). Hypermethylation of promoter regions, which is associated with transcription silencing, similar to DNA mutation, has been proposed as a mechanism for tumor suppressor gene inactivation in human cancers (Jones and Baylin, 2007). In addition, a number of candidate tumor suppressor genes, such as HIC1, INK4b (p15), TIMP3, and others that are not commonly inactivated by mutation, are transcriptionally silenced by methylation. The aberrant methylation of genes that suppress tumorigenesis appears to occur early in tumor development, accumulates progressively, and eventually leads to the malignant phenotype (Fearon and Vogelstein, 1990; Davis and Uthus, 2004). Hypermethylation has been found to be associated with the dysregulation of pathways involved in the cancer process, such as DNA repair (hMLH1, BRCA1, MGMT), cell-cycle regulation (p16, p14, p15), carcinogen metabolism (GSTP1), apoptosis (DAPK, APAF-1), hormonal response (RARβ2), and cell adherence (CDH1, CDH3; Costello and Plass, 2001; Momparler, 2003; Davis and Uthus, 2004; Esteller, 2008).
Histone Modifications and the Histone Code
A complex of DNA and globular proteins, known as “histones,” package heritable genetic information in eukaryotic nuclei in the form of nucleosomes that constitute chromatin. Nucleosomes are composed of 147 bp of DNA wrapped around core histone proteins named H2A, H2B, H3, and H4. An additional histone, H1, is located at the outer surface of the nucleosome to anchor DNA to the nucleosome. H3 and H4 have long tails that extend out from the nucleosome and can be modified chemically by acetylation, methylation, ubiquitination, phosphorylation, sumoylation, citrullination, and ADP-ribosylation (Baccarelli and Bollati, 2009). These chemical modifications can result in a closed chromatin conformation that suppresses transcription, or in an open conformation that activates transcription. The most common modifications are acetylation and methylation on histone lysine residues. Increased acetylation results in transcription activation, and methylation of histones can result in either repression or activation of transcription, as determined by the lysine residue position involved (Bollati and Baccarelli, 2010).
Growing lists of histone modifications, histone-modifying complexes, and their interactions have been developed into what is commonly referred to as a “histone code.” Together with methylation, the histone code ensures that chromatin exists in a transcriptionally inactive mode that undergoes activation of specific gene regions when needed. The timely on/off switching of genes requires relevant changes in the epigenetic profile. Given that chromatin transcriptional activity is related to germline development, X-inactivation, stem-cell identity, cell-cycle regulation, and mitosis/meiosis, the decoding of epigenetic mechanisms should hold insights for studying cancer (Delage and Dashwood, 2008). When histone modifications and chromatin remodeling processes become deregulated, they could result in mutations in oncogenes, tumor suppressor genes, or DNA repair genes. Such changes then could result in genomic instability, oncogenic transformation, and the development of cancer. Histone modifications are extremely dynamic and highly regulated. When histone-modifying complexes, including histone acetyltransferases (HATs), deacetylases, or histone methyltransferases, display aberrant activity, cancer development by misregulation of chromatin structure and activity may occur, as happens frequently in human leukemia (Herceg, 2007).
Histone acetylation typically results in an open chromatin configuration that facilitates transcription factor access to DNA and gene transcription. It also can silence tumor suppressor genes in cancer cells if deacetylated (Wade, 2001; Nian et al., 2009). HATs are responsible for acetylation; whereas histone deacetylases (HDACs) are responsible for histone deacetylation. Overexpression and or increased activity of HDACs is found in many malignancies, and the repression of transcription can result in dysregulated cell-cycle kinetics, apoptosis, and differentiation (McLaughlin and La Thangue, 2004; Dokmanovic and Marks, 2005; Mariadason, 2008). Many dietary components, such as sulforaphane, a major component in cruciferous vegetables, have shown promising results in direct or indirect inhibition of HDAC activity as well as other histone modification activities in cancer prevention and therapy (Myzak et al., 2004; Meeran et al., 2010).
RNA Interference
RNA interference plays an important role in controlling the activity level of specific genes. One type of small RNA molecule involved in RNA interference is microRNA (miRNA). miRNAs are single-stranded non-coding RNAs of approximately 21–23 nucleotides in length whose main function is to down-regulate gene expression by interfering with mRNA processes. miRNA suppresses gene expression by affecting mRNA stability, targeting the mRNA for degradation, or both (Mathers et al., 2010). In limited cases, miRNA also may increase gene transcription (Barros and Offenbacher, 2009). Close to 1,000 miRNAs have been identified in human cells with the potential to regulate the expression of about one-third of human mRNAs and influence almost all genetic pathways through their effects on transcription factors, receptors, and transporters (Esquela-Kerscher and Slack, 2006). miRNAs have an established repressing role in cancer initiation, development, maintenance, and proliferation (Esquela-Kerscher and Slack, 2006). Mutation or mis-expression of miRNA occurs in several human cancers, and miRNAs have been reported to repress the expression of cancer-related genes. Taken together, these two observations suggest the potential importance of miRNA in tumorigenesis (Esquela-Kerscher and Slack, 2006). It has been demonstrated that chemopreventive agents, such as n−3 polyunsaturated fatty acid, pectin, folate, retinoids, and curcumin, favorably modulate carcinogen-directed non-coding miRNA signatures in various cancer (Kutay et al., 2006; Sun et al., 2008; Wang et al., 2008; Davidson et al., 2009; Shah et al., 2011). These data suggest a persistent effect of diet in miRNA-mediated oncogenic transformation.
Chromosomal Instability
The stability of the genome in general and chromosomes in particular plays a large role in several diseases including cancer. Mechanisms by which stability is undermined include DNA metabolism and repair gene silencing by aberrant methylation near CpG promoter regions, and accelerated telomere shortening. Nutritional factors may control both mechanisms. For example, zinc and magnesium levels help determine the effectiveness of DNA repair and DNA metabolism, respectively, and telomere length may be associated with folate and nicotinic acid levels (Bull and Fenech, 2008).
Transposons
Transposable elements are sequences of DNA that move to new positions within the genome of a single cell. Transposons are among several types of transposable elements which include plasmids, bacteriophages, and certain introns (Wicker et al., 2007). Transposons can behave as mutagens that cause genomic damage by various pathways. For example a given section of DNA could be cut and pasted into another DNA region potentially resulting in disruption of the functional gene it was pasted into or leaving behind an irreparable gap in the gene it was cut from, both resulting in a mutagenic effect. In other cases multiple sequences of pasted copies result which disturb chromosomal pairing during mitosis and meiosis. Some human blood dyscrasias can be caused by transposons including hemophilia A and B and porphyria (Kazazian, 2004). Eukaryotic cells use RNA interference to silence transposable elements immediately after they are transcribed thereby inhibiting transposon activity levels to protect against genomic damage.
Loss of Imprinting
Imprinting is a process in which genes are silenced, depending on whether they are maternal or paternal in origin. The epigenome cycles through a series of precisely timed methylation changes designed to ensure proper development, both in utero and throughout life, of the organism. In humans, the paternal genome is actively demethylated, and the maternal genome is passively demethylated following fertilization (Bernal and Jirtle, 2010). Genomic imprinting results in parent-of-origin-dependent monoallelic expression of a subset of critical autosomal genes (Murphy and Jirtle, 2003). Loss of methylation can either shut down these critical genes or lead to overexpression of the gene product. Because of their monoallelic expression, imprinted genes are particularly susceptible to dysregulated expression that results from epigenetic aberrations. Environmental exposures that alter the imprinting of these genes will lead to enhanced susceptibility to disease (Das et al., 2009). For example, a study has demonstrated that loss of imprinting at the insulin-like growth factor 2 (igf2) locus could be induced by synthetic methyl-donor-deficient post weaning diet in mice (Waterland et al., 2006). Loss of imprinting is associated specifically with hypomethylation in colon cancer (Cui et al., 2002).
Effects of Nutrition on Epigenetics
Established epigenetic patterns during the fetal period can be changed in adult life by environmental factors, including nutrition. Diet can profoundly alter epigenetic patterns in animals. The Agouti mouse has been used extensively to demonstrate the impact of maternal nutrition on the fetal epigenome and the phenotype of the offspring (Wolff et al., 1998). Coat color is linked to the methylation status of the Agouti gene, which is highly dependent on maternal diet. Most importantly, altered epigenetic patterns are associated with the increased risk of diabetes-like conditions, obesity, and tumorigenesis (Yen et al., 1994; Dolinoy, 2008; Duthie, 2011). Evidence also suggests that specific nutrients in the human diet that are strongly associated with risk of cancer can modulate DNA methylation (Kim et al., 2009). For example, a study found that identical twins possess the same genotype and no distinguishable epigenetic differences in their early life, but remarkable differences were found in genomic methylation and histone acetylation patterns later in their life (Fraga et al., 2005). These epigenetic differences may result in different gene expression and disease susceptibility.
Nutrient intakes have been found to play an important role in regulating one-carbon metabolism (Davis and Uthus, 2004; Lee et al., 2005). Excessive or deficient nutrient status may have an effect on DNA methylation. This is based on the observation that the primary methyl-donor, S-adenosylmethionine (SAM), may be regenerated from S-adenosylhomocysteine (SAH) through a series of chemical reactions that rely on the presence of several nutrients, such as folate, betaine, choline, zinc, vitamin B6, and vitamin B12, as cofactors or intermediates. As shown in Figure 1, the metabolism of folate, choline, betaine, and methionine are interrelated. They interact at the point in the cycle where homocysteine is converted to methionine, and therefore, the metabolites of these nutrients have critical roles in forming SAM, known as the universal methyl-donor. In a set of reactions necessary for the normal functioning of cells, SAM donates its methyl group in methylation reactions, including the methylation of DNA, RNA, and protein. Deficiency of any of these nutrients can result in a depletion of cellular pools of SAM, which may cause aberrant DNA methylation (Gilbert and Liu, 2010). Other dietary factors, including alcohol, arsenic, cadmium, coumestrol, equol, genistein, nickel, selenium, tea polyphenol, vitamin A, and zinc, also are known to influence DNA methylation and cancer susceptibility (Davis and Uthus, 2004).
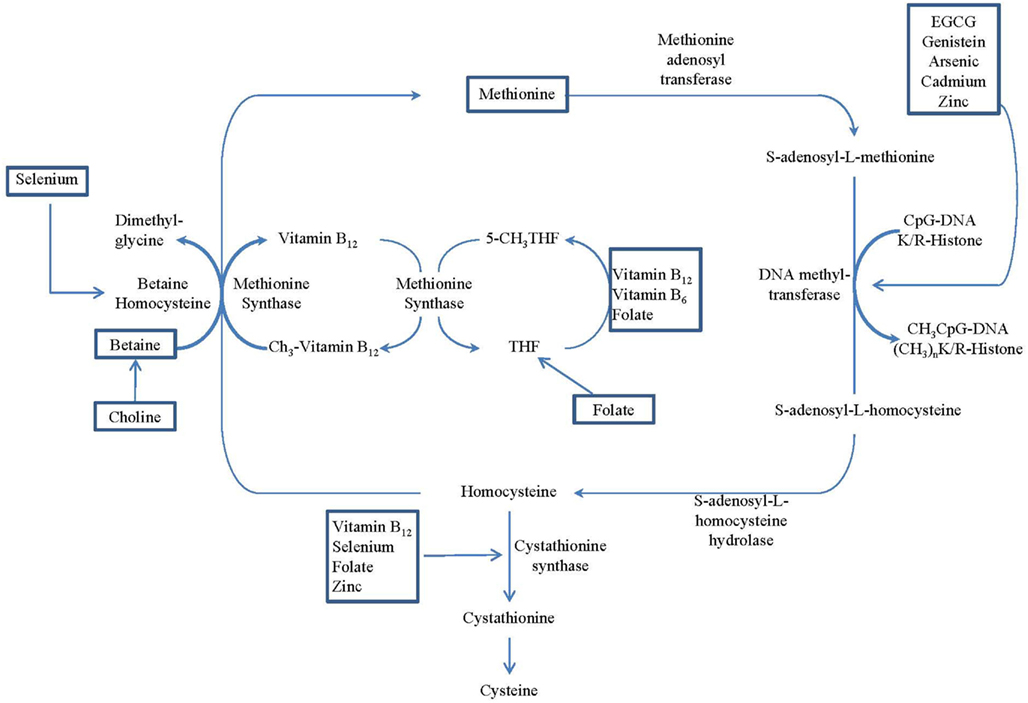
Figure 1. Overview of the methionine–homocysteine–folate-B12 cycle, which provides methyl donors for methyltransferase. Nutritional regulations of the cycle are identified by rectangle frames. (Adopted from Delage and Dashwood, 2008).
Literature that explores possible nutritional effects on histone regulation is scarce (Mathers et al., 2010). Butyrate, a product of carbohydrate fermentation, has been shown to inhibit certain histone deacetylases. The status and function of acetyl transferases and acetylation/deacetylation of histones can be affected by organosulfur compounds from garlic, isothiocyanates from cruciferous vegetables, and compounds such as resveratrol found in grapes (Delage and Dashwood, 2008). These bioactive food components also have been shown to alter epigenetic processes in cell functions, such as control of proliferation, upregulated apoptosis, and reduction in inflammation (Ross et al., 2008). Additionally, changes in miRNA expression in response to dietary exposure may mediate effects on cancer risk. Emerging evidence indicates that a wide range of dietary factors, including both macronutrients (fat, protein, and alcohol) and micronutrients (folate and vitamin E), alter the expression of many miRNAs in rodents and humans (Marsit et al., 2006b). Marsit et al. (2006a) reported that both folate deficiency and arsenic exposure resulted in a global increase in miRNA expression in the human immortalized lymphoblastoid cell line TK-6.
Maternal Nutrition and Epigenetics
A mother’s diet is passed on to her fetus and affects fetal growth and development. The fetal origins of disease hypothesis, also known as Barker’s hypothesis, essentially proposes that under-nutrition during gestation increases the risk of disease later in life (Barker, 2007). Barker et al. (1989) published data showing that men with the lowest weights at birth and at 1 year had the highest death rates from ischemic heart disease, and increasing weight was associated with a graded decrease in the risk. The authors proposed that an environment that induces poor fetal and infant growth will be followed by high risk of ischemic heart disease in adulthood. This idea has been validated in several prospective cohort studies. For example, in Sweden (Leon et al., 1998), Finland (Forsén et al., 1997), Denmark (Baker et al., 2008), Norway (Risnes et al., 2009), England (Syddall et al., 2005), and Scotland (Lawlor et al., 2005), mortality from ischemic heart disease was inversely associated with birth-weights. In the United States (Rich-Edwards et al., 1997, 2005) and India (Stein et al., 1996), a strong inverse association between birth weight and non-fatal coronary heart disease was reported. A recent systematic review of 17 published studies concluded that the findings are consistent with low birth-weights increasing risk for ischemic heart disease (Huxley et al., 2007). If early life exposures indeed affect cardiovascular disease (CVD) risk, by logical extension they also may affect cancer risk because some of the biological risk factors for CVD, such as abnormalities in lipid metabolism and non-insulin-dependent diabetes mellitus, also can affect cancer risk.
Establishing a clear link between maternal diet and epigenetic states of the genome would require a highly sensitive phenotypic outcome. This evidence is not only available but very convincing from the viable yellow agouti (Avy) mouse model. The wild-type murine Agouti gene encodes a paracrine signaling molecule that produces either black eumelanin (a) or yellow pheomelanin (A) (pigment in the hair follicles). Transient A expression in hair follicles during a specific stage of hair growth results in a sub-apical yellow band on each black hair shaft, causing the brown agouti coat color of wild-type mice (Duhl et al., 1994). Wolff et al. (1998) were the first to demonstrate that feeding pregnant black a/a dams a methyl-supplemented diet shifts the coat color distribution of their Avy offspring. As it became evident that Avy/a coat color correlates with Avy methylation status (Morgan et al., 1999), it was argued that maternal methyl-donor diets change the coat color of the offspring via Avy methylation (Cooney et al., 2002). However, Waterland and Jirtle (2003) were able to demonstrate that dietary supplementation of viable yellow Agouti (Avy) mice during pregnancy with methyl donors (folic acid, vitamin B12, choline, and betaine) alters the coat color of their offspring because of the increased DNA methylation of a transposable element upstream of the Agouti gene, rather than mutation of the gene. When a mouse’s agouti gene is completely unmethylated it has a yellow coat color, is obese and prone to diabetes and cancer. When the agouti gene is methylated (as it is in normal mice) the coat color is brown and the mouse has a low disease risk. These observations provide clues that epigenetic modifications resulting from dietary effects may have long-term health consequences. Early life epigenetic alteration could, for example, contribute to carcinogenesis that is known to be affected by dietary intake.
Potential Application to Reverse Epigenetics Changes
Although epigenetic changes are heritable in somatic cells, the notion that these modifications are also potentially reversible as observed through the finding of Agouti mouse model makes them attractive and promising avenues for tailoring cancer preventive and therapeutic strategies (Link et al., 2010). It is thus thought that certain adverse epigenetic modifications as the results of environmental and dietary exposure may be modulated. Many studies, both animal and human, had been exploring the potential reversible characteristics of epigenetic changes since the initial findings from Agouti model. For example, significant increases in both Igf2 and H19 expression were found in the prostate tissues in mice fed with choline–methionine deficient diet prostate compared to controls. These expression changes were reversible with shorter exposure to the choline–methionine deficient diet (Dobosy et al., 2008). A randomized feeding study of low birth weight and normal birth weight young men showed that low birth weight subjects developed peripheral insulin resistance and reduced PPARGC1A and OXPHOS gene expression after 5 days of high-fat overfeeding (Brons et al., 2010). However, PPARGC1A methylation increased in only normal birth weight subjects after overfeeding in a reversible manner. This is the first study demonstrated in human that DNA methylation induced by overfeeding is reversible. Most recently, a primate study reported that exposure to a maternal high-fat diet in utero significantly alters the expression of fetal hepatic Npas2 compared with that in control diet-exposed animals (Suter et al., 2011). It is reversible in fetal offspring from obese dams reversed to a control diet. Although the Npas2 promoter remains largely unmethylated, differential Npas2 promoter occupancy of acetylation of fetal histone H3 at lysine 14 (H3K14ac) occurs in response to maternal high-fat diet exposure compared with control diet-exposed animals. In addition, disruption of Npas2 is found to be consistent with high-fat diet exposure in juvenile animals, regardless of in utero diet exposure. The findings suggest that peripheral Npas2 expression is uniquely vulnerable to diet exposure.
Effects of Environmental Toxicants on Epigenetics
The effect of several nutrients, such as folate, vitamin B12, vitamin B1, polyphenols, flavonoids, phytoestrogens, sulforaphane/isothiocyanates, vitamin A, fat, and selenium, and dietary components, such as butyrate, biotin, lipoic acid, garlic organosulfur compounds, and vitamin E metabolites, on epigenetics have been reviewed to some extents (Dashwood et al., 2006; Barnett et al., 2010; Chen and Xu, 2010; Mathers et al., 2010; vel Szic et al., 2010; Duthie, 2011; McKay and Mathers, 2011). However, the epigenetic effect of environmental toxicants consumed along with foods on diseases, such as cancer has not been systematically examined. Therefore, this review will focus on the environmental toxicant exposures through dietary intake.
Food as a Vehicle for Environmental Toxicants
Environmental toxicants are found naturally in foods or may be introduced by way of cooking practices, production processes, or pesticide exposure. Many environmental toxicants introduced into food either naturally or anthropogenically are heavy metals and endocrine disruptors. These ingested toxicants have been linked to modulations in epigenetic pathways and increased disease susceptibility.
Environmental Toxicants and Epigenetics
Epidemiological and experimental studies link epigenetic modulations with both nutritional factors and environmental toxicants (Feil, 2006; Baccarelli and Bollati, 2009). Nutrition and environmental toxicants affect epigenetic pathways mainly through DNA methylation and complex histone modifications. Evidence linking nutritional and environmental factors with DNA hyper or hypomethylation provides the most compelling support for such an association (Mathers et al., 2010). Further, nutrition may modulate the toxicity of environmental pollutants, through either synergistic or antagonistic effects (Hennig et al., 2007). Environmental toxicants also may cause aberrant modifications in the metabolic activity of nutritional factors (Hennig et al., 2004). The relationship between nutrition and environmental toxicants and epigenetic modulations is complex, however, and an exact causal relationship has yet to be identified. In addition, the complexity of the relationship is compounded by the interaction of multiple toxicants and dietary factors. Nonetheless, nutritional factors and environmental toxicants may affect similar biological and epigenetic pathways and thus should be examined simultaneously.
Arsenic
Arsenic occurs naturally in the earth’s crust with higher concentrations in some geographic areas. Arsenic is usually found combined with other elements. When combined with elements other than carbon, it is called “inorganic arsenic.” Arsenic and inorganic arsenic compounds can be emitted into air and then deposited into water and soil during industrial operations such as ore mining and smelting, and during volcanic eruptions and forest fires (U.S. Environmental Protection Agency, 2007). Arsenic (As) and inorganic arsenic compounds are established carcinogens that have been linked to cancers of the skin, bladder, liver, lung, and prostate (IARC, 2004). Arsenic is ubiquitous in the environment and can contaminate air, food, and water, with more than 137 million people in more than 70 countries having been exposed (Bagchi, 2007). In fact, the major source of human exposure is through drinking water from contaminated well sources. Exposure usually occurs in the form of arsenite or arsenate, with exposure to inorganic arsenite being more toxic (Salnikow and Zhitkovich, 2008). Those not exposed to arsenic through drinking water may be exposed by consuming rice products (Dwivedi et al., 2010; Tuli et al., 2010). Khan et al. (2010) recently investigated the mechanisms by which arsenic may enter the food chain in Bangladesh and found that arsenic and cadmium were present throughout the food chain, and that cooking practices may influence arsenic levels in food. Their results showed that the presence of arsenic in food decreased the uptake of essential nutrients such as iron, manganese, copper, and zinc (Khan et al., 2010). High levels of arsenic also are found in fish and shellfish, though this exposure usually is in the organic non-toxic form. As with many chemicals, the absorption of arsenic can be modulated by nutritional status (Li et al., 2008). Thus, malnourished individuals, especially children, may be more susceptible to increased arsenic exposure levels.
The mechanisms of arsenic’s carcinogenicity remain unclear. Arsenic is thought to exert its carcinogenic effect by inducing DNA hypomethylation, leading to aberrant gene expression (Zhao et al., 1997; Verma and Srivastava, 2002), or by DNA methylation silencing genes associated with controlling tumorigenesis (Verma and Srivastava, 2002; Vaissière et al., 2008). Consequently, exposure to arsenic has been associated with DNA hypo and hypermethylation (Salnikow and Zhitkovich, 2008). Global DNA hypomethylation has been observed in vitro in animal studies and is thought to result from the depletion of SAM and repressed expression of DNA methyltransferase genes DNMT1 and DNMT3A (Reichard et al., 2007). Zhao et al. (1997) found evidence of low SAM, decreased methyltransferase activity, and global DNA hypomethylation in rat liver epithelial cells that were chronically exposed to arsenic. A reduction in DNA methylation also has been observed in human cell lines. For example, global DNA hypomethylation was observed in HaCaT keratinocytes exposed to low-level arsenic (Reichard et al., 2007). Normal human prostate cells were malignantly transformed after chronic low-level arsenic exposure, and the transformed cells showed evidence of genomic DNA hypomethylation (Coppin et al., 2008) and K-ras overexpression (Benbrahim-Tallaa et al., 2005). Although arsenic exposure led to the overexpression of K-ras, there was no evidence of hypomethylation in its promoter region.
Relatively few studies that utilize human subjects have investigated the association of arsenic with epigenetic changes. Arsenic in drinking water was associated with genomic hypomethylation of peripheral blood leukocytes (PBL), and the association was stronger among individuals with higher folate concentrations (Pilsner et al., 2007). These researchers later observed that individuals with hypomethylated PBL DNA had an increased risk of developing skin lesions (Pilsner et al., 2009). Also, hypermethylation of the promoter region of the p53 gene in a dose–response fashion was observed in DNA arsenic-exposed individuals and individuals with arsenic-induced skin cancer (Chanda et al., 2006). In a population-based case study of incident bladder cancer, arsenic exposure was associated with hypermethylation of tumor suppressor genes RASSF1A and PRSS3 (Marsit et al., 2006a). In addition, hypermethylation in the promoter region of DAPK was found in bladder cancers among individuals in arsenic-contaminated areas (Chen et al., 2007). Although most studies that have examined the epigenetic effects of arsenic have focused on DNA methylation, investigations into altered histone modification associated with arsenic-induced gene expression and carcinogenesis are under way (Vaissière et al., 2008; Zhou et al., 2008).
Cadmium
Cadmium (Cd) occurs naturally in the earth’s crust and also can be introduced into the environment by human activities such as coal burning and manufacturing. Cadmium and cadmium compounds are carcinogenic to animals and humans (IARC, 1993; National Toxicology Program, 2005). Air, water, and soil are sources of cadmium exposure, and exposure routes for humans include ingestion, dermal contact, and inhalation. The primary source of cadmium exposure in the general population is through contaminated food (ATSDR, 2008). Fish and shellfish may accumulate high levels of cadmium from aquatic environments and often are thought to be the biggest contributors to cadmium toxicity (Perello et al., 2008). Plants may absorb high levels of cadmium from contaminated soils. Cadmium-contaminated plants first were discovered to cause human toxicity in the 1950s (Kaneta et al., 1986).
Cadmium appears to mimic zinc and calcium, is rapidly cleared from the blood, and concentrates in several tissues, including the liver and kidney (Waalkes, 2003). The main source of epidemiologic evidence of the association between cadmium exposure and human cancer comes from occupational studies of workers involved in the manufacture of nickel–cadmium batteries and electroplating processes. Cadmium exposure has been associated with lung cancer (Beveridge et al., 2010) in a dose–response manner (Stayner et al., 1992). Cadmium also has been associated with prostate, pancreas, renal, liver, stomach, and bladder cancers (Waalkes, 2003). Mechanisms of cadmium carcinogenicity involve the production of oxidative stress, aberrant gene activation and signal transduction, disruption of DNA repair, and suppressed apoptosis (Waisberg et al., 2003).
Cadmium is weakly mutagenic, leading some to believe that its mechanism for carcinogenesis has non-genotoxic or epigenetic components (Takiguchi et al., 2003; Waalkes, 2003; Waisberg et al., 2003; Jiang et al., 2008). Evidence has indicated that acute exposure (1 week) of TRL1215 rat liver cells in vitro to cadmium increased DNA hypomethylation and reduced DNMT activity, whereas more prolonged exposure (10 weeks) led to increased global hypermethylation and enhanced DNMT activity (Takiguchi et al., 2003). This result was replicated in human embryo lung fibroblast cells in which long-term, low-dose exposure increased DNA methylation and DNA methyltransferase activity; but also upregulated mRNA levels of DNMT1, DNMT3a, and DNMT3b, but at higher concentrations (Jiang et al., 2008). Cadmium exposure induced the malignant transformation of prostate epithelial cells and led to overexpression of DNMT3b without changes in DNMT1expression. Importantly, DNMT3b activity was an early event and occurred before the cells were malignantly transformed (Benbrahim-Tallaa et al., 2007). Promoter regions of tumor suppressor genes RASSF1A and p16 were heavily methylated and downregulated at about the same time that DNMT3b overexpression first occurred.
Nickel
Nickel (Ni) compounds are classified as carcinogenic to humans, and metallic nickel possibly is carcinogenic to humans (IARC, 1990). Nickel is used in industrial processes to produce stainless steel, copper–nickel alloys, electroplating, alkaline batteries, coins, welding products, magnets, electrical contacts, and electrodes, as well as surgical and dental prostheses (National Toxicology Program, 2005). Nickel occurs at very low levels in the environment, although it is ubiquitous in air, water, and food. Environmental exposure occurs through inhalation, ingestion, and dermal contact. The general population takes in the most nickel via food, but exposure also can occur via tobacco smoke and nickel-plated materials such as coins, steel, and jewelry (National Toxicology Program, 2005).
The mechanisms of nickel carcinogenesis remain unclear. Several studies have suggested that chromatin structural alterations and epigenetic changes play a role in nickel carcinogenesis (Salnikow and Zhitkovich, 2008). DNA hypermethylation leading to inactivation of gpt expression was found in Chinese hamster G12 cells (Lee et al., 1995). In addition, hypermethylation in the promoter region of tumor suppressor gene p16 was found in tumors of wild-type C57BL/6 mice heterozygous for tumor suppressor p53 implanted with nickel sulfide (Govindarajan et al., 2002). Nickel also has been shown to exert its effect on gene silencing by mechanisms that involve changes in histone acetylation. For example, nickel-induced gene silencing resulted from H4 and H3 histone acetylation and methylation of lysine 9 in histone H3 in the gpt transgenic cell line of Chinese hamsters (Yan et al., 2003). Nickel has also been shown to lead to the deacetylation of H4 in vitro (Broday et al., 2000). Nickel chloride was found to decrease the histone acetylation of H2A, H2B, H3, and H4 and increase the histone ubiquitination of H2A and H2B (Ke et al., 2006). These data suggest that epigenetic changes contribute to nickel-induced carcinogenic effects. However, population studies are needed to determine if these effects are observed in humans.
Chromium
Chromium has been established to be an essential trace element in mammals in regard to maintenance of normal carbohydrate metabolism. Chromium(III) is found in moderate amounts in a wide range of foods, such as egg yolks, whole grain, high-bran cereals, nuts, green beans, broccoli, meat, brewer’s yeast, wine, and beer; Anderson and Kozlovsky, 1985). As a dietary supplement, chromium is available in many multivitamin and mineral products. However, the role of chromium supplementation among subjects with normal diet is still controversial (Wang and Cefalu, 2010). People should also be aware of the form of chromium products presented. Hexavalent chromium [Cr(VI)] compounds are carcinogenic to humans (IARC, 1990; National Toxicology Program, 2005). Cr(VI) is used in the production of stainless steel and pressure-treated wood. The general population may be exposed to Cr(VI) compounds by inhalation, ingestion of water, or dermal contact with products that contain Cr(VI) (National Toxicology Program, 2005). Epidemiologic studies investigating the risk of lung cancer in occupational settings that manufacture chromate products have been consistently demonstrated the relationship between Cr(VI) exposure and lung cancer (Gibb et al., 2000; National Toxicology Program, 2005). Mechanisms of Cr(VI) carcinogenesis are poorly understood. It is believed that Cr(VI) can exert its carcinogenic effect via oxidative stress and DNA damage associated with Cr-DNA adducts, protein–DNA crosslinks, and DNA single- and double-strand breaks (Sugden and Stearns, 2000; Bagchi et al., 2001; Shi et al., 2004; Zhitkovich, 2005). Interest is increasing in Cr(VI)-induced epigenetic changes as a potential carcinogenic mechanism. Indeed, both animal and human studies have shown some evidence of changes in epigenetic marks associated with Cr(VI) exposure. Potassium chromate salts induced aberrant DNA methylation silencing the G12 reporter transgene in Chinese hamster lung cells (Klein et al., 2002). Chromate also was shown to be associated with histone modification. Chromate significantly increased tri-methyl H3K4 after exposure in human lung carcinoma A549 cells (Zhou et al., 2009). In human lung tumors, hypermethylation of the p16 promoter region was found in Japanese workers exposed to Cr(VI); the effect was time- and dose-specific (Kondo et al., 2006). Also among these workers, the DNA mismatch repair gene hMLH1 was hypermethylated, leading to the repression of the hMLH1 protein (Sun et al., 2009).
Mercury
Mercury (Hg) is found naturally in the environment or is emitted into the atmosphere via industrial processes and fuel combustion. Mercury is released into the atmosphere either as elemental mercury, particle-bound mercury, or oxidized mercury (EPA, 2010). Mercury in the atmosphere can reach aquatic environments and be converted into methylmercury (MeHg) by sediment bacteria (Borchers et al., 2010). Once MeHg enters the food chain, it is absorbed by fish and shellfish at toxic levels. MeHg bioaccumulates progressively with each level of increase in the aquatic food chain, from phytoplankton to predatory fish. Thus, mercury levels in fish vary, with fish on the top trophic levels containing the highest levels of MeHg (EPA, 2010). Shark, swordfish, king mackerel, and tilefish contain the highest levels of MeHg, and shrimp, salmon, pollock, and catfish have the lowest levels (FDA U.S. Food and Drug Administration, 2009). MeHg exposure in humans occurs when seafood containing it is consumed. Controversy has arisen because pregnant women have been advised to consume fish for omega-3 fatty acids and other nutrients that are beneficial to the developing fetus, but also are being warned to limit their consumption of mercury because of possible damage to the fetal nervous system. There were speculation whether mercury causes epigenetic modification (Callaghan et al., 2011; Rooney, 2011). However, little evidence to support the notion until a recent publication by Arai et al. (2011). The investigators studied the effects of mercury and a few other chemicals on morphological changes in heterochromatin and DNA methylation status in mouse ES cells. This study found that mercury affected heterochromatin conditions and caused aberrant DNA methylation at gene loci. Additionally, mercury-exposed cells also exhibited impaired formation of the embryoid body, suggesting mercury as a potential epigenetic mutagen. Additional studies may be needed to further confirm the findings.
Endocrine Disruptors
Endocrine disruptors are compounds found in the environment that mimic the actions of hormones. They are ubiquitous in the environment and have been associated with changes in epigenetic marks following in utero and adult exposure, and these changes may be transmitted to subsequent generations through the germline (Li et al., 2003). Diethylstilbestrol (DES) is a synthetic non-steroidal estrogen that is a known human carcinogen. It was prescribed to pregnant women between 1947 and 1971 to reduce the risk of miscarriage (National Toxicology Program, 2005). In a finding that usually has been observed among older women, female offspring of mothers exposed to DES experienced a higher risk of clear-cell adenocarcinoma of the vagina or cervix at younger ages (Herbst et al., 1977). DES has been shown to increase expression of the c-fos proto-oncogene and significantly reduce methylation in mice exposed in utero (Li et al., 2003). Recent studies have shown that in utero exposure to DES caused hypermethylation of HOXA10, which is expressed in uterine epithelial and stromal cells (Bromer et al., 2010) and directs embryonic uterine development; DNMT1 and DNMT3b also were overexpressed (Bromer et al., 2009). DES increased histone H3 trimethylation in human cells and in mice exposed in utero (Doherty et al., 2010).
Food is a major source of exposure to endocrine-disrupting chemicals such as phthalates and bisphenol A (BPA). BPA is a known endocrine disruptor; however, its method of carcinogenicity in humans is not classified. BPA is used in the production of polycarbonate plastics and epoxy resins. Because of its pervasive use in plastics, it is ubiquitous in the environment. Although the available evidence does not suggest that BPA is carcinogenic on its own, evidence indicates that it increases cancer susceptibility through developmental reprogramming as a result of epigenetic changes (Keri et al., 2007). Similar to DES, BPA has been associated with epigenetic alterations following exposure in utero (Ho et al., 2006). For example, exposing pregnant mice to BPA produced alterations in HOXA10 expression associated with significantly reduced methylation in offspring, but this reduced methylation was not due to repression of DNMTs (Bromer et al., 2010). BPA exposure in utero also has been shown to alter expression of EZH2 in mammary tissue. Overexpression of EZH2 is associated with human breast cancer and reduced expression of DNA repair mediators (Doherty et al., 2010). Also similar to DES, BPA increased H3 trimethylation in human MCF-7 cells and in mice exposed in utero. These epigenetic changes persisted into adulthood (Doherty et al., 2010). In the male rat, hypomethylation of PDE4D4 in the prostate tumors of male rats was observed after neonatal BPA exposure; these epigenetic changes appeared to be permanent (Ho et al., 2006). BPA is present at low but detectable levels among pre-adolescent and adolescent males and females (Wolff et al., 2006; Calafat et al., 2007). Although such epigenetic changes have not yet been studied in humans, these data provide compelling evidence to suggest that exposure to BPA early in life may predispose the mammary and prostate glands to preneoplastic lesions in adult life.
Conclusion
Epigenetic mechanisms have been implicated in critical processes, such as gametogenesis, fetal development, and maintenance of early established patterns of gene expression throughout life. The epigenetic information carried by various epigenetic events reviewed earlier, such as DNA methylation and histone modifications, appears to be influenced significantly by dietary and environmental factors. The phenomenon may explain how maternal diets and exposure to exogenous agents, such as diethylstilbestrol (Newbold et al., 2006), can have long consequences on the health and disease risk of the offspring. Both epidemiological data and animal experiments support the hypothesis that diet and environmental toxicants may exert their oncogenic effects through epigenetic as well as genetic mechanisms. The relationship between cancer and both nutritional factors and environmental toxicants also can be modulated either synergistically or antagonistically by epigenetic mechanisms. A synergistic mechanism may exist between deficiencies of essential nutrients and exposure to environmental toxicants. There may also be interactions between food and environmental toxicants that may increase the harmful effects of the toxicants (Mahaffey and Vanderveen, 1979). Many children and adults are experiencing a double burden of nutrient deficiencies and exposure to environmental toxicants such as heavy metals (Grandjean and Landrigan, 2006; Kordas et al., 2007; Wasserman et al., 2008). Nutrients could also play a role in attenuating the toxicity of environmental toxicants such as polychlorinated biphenyls (PCBs), BPA, lead, and arsenic.
The role epigenetics remain to be fully characterized in normal tissue and cancer. One of the major developments in the field of cancer epigenetics during the past decade has been the rapid advance in genome-wide technologies that initially employed microarrays but increasingly are using high-throughput sequencing. In the foreseeable future, cancer epigenomic methods will be able to sufficiently address issues of tissue heterogeneity and the often limiting amount of DNA present in epidemiologic study samples. Nevertheless, methylation profiling of human cancer has revealed tissue specific epigenetic signatures, as well as tumor specific signatures, reflecting the presence of epigenetic instability of a subset of cancers affected by the CpG island methylator phenotype (Toyota and Issa, 2005). It has been suggested that methylation patterns can be traced to a tissue specific, proliferation-dependent accumulation of aberrant promotor methylation in aging tissues, which can be modulated by chronic inflammation and less well-defined mechanism such as diet, environment, and genetic predisposition. Therefore, challenges remained in applying appropriate epigenetic techniques on appropriate human specimens to interpret the findings. Additionally, despite significant amounts of data available on the effect of diet on DNA methylation, less knowledge is available on the involvement of histone modifications in the regulation of chromatin structure, gene expression, and identification of the nature of metabolic pathways that are controlled by nutrition through epigenetic mechanisms, both in early life and adulthood. The relationship between epigenetic events and the latent nature of cancer in humans may also need to be more carefully explored through prospective cohort study designs with multiple measurements of diet and biomarkers during critical time points in life. A paradigm shift with a focus on improving knowledge of the interplay between diet and environmental toxicant exposures and epigenetics could move the field of modifiable risk factors and cancer into an era of targeted lifestyle modification for cancer prevention.
In addition to the technological challenges, until recently, there have been difficulties in studying the interactions between dietary factors and environmental toxicants to understand epigenetic mechanisms. BPA exposure levels during early development was fund in animal study to be able to modify epigenome and increase disease susceptibility throughout life (Dolinoy et al., 2007). It is also found that certain nutrients such as folic acid and genistein may counteract the effects of BPA. Maternal supplementation with folic acid or the phytoestrogen genistein can decrease the hypomethylating effect of BPA (Dolinoy et al., 2007). The findings that that BPA can alter the epigenomes of offspring but maternal dietary supplements may counteract these effects offer a renewed hope affirming the hypotheses by Doll and Peto (1981) that carcinogenesis may be reversible through dietary modification. To date, few epidemiologic studies have examined the effect of nutrients or bioactive food component intakes on epigenetics and their modulating effects on cancer, even fewer examined environmental toxicants in food and their effects Out of more than 450 grants currently funded or co-funded by the Epidemiology and Genetics Research Program, Division of Cancer Control and Population Sciences, National Cancer Institute, 23 research grants (14 Research Project Grants (R01s), four Exploratory/Research Development Grant (R21), and five Small Grant (R03) are directly or indirectly examining the link between epigenetic effects on cancer in humans. Among these funded applications, only four of them focus on the effect of dietary and environmental toxicants. It is obviously an under-representative area in terms of the Epidemiology and Genetic Research Program funding. Additional epidemiologic research is needed to clarify the relationship between these complex epigenetic mechanisms and cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, R. A., and Kozlovsky, A. S. (1985). Chromium intake, absorption and excretion of subjects consuming self-selected diets. Am. J. Clin. Nutr. 41, 1177–1183.
Arai, Y., Ohgane, J., Yagi, S., Ito, R., Iwasaki, Y., Saito, K., Akutsu, K., Takatori, S., Ishii, R., Hayashi, R., Izumi, S., Sugino, N., Kondo, F., Horie, M., Nakazawa, H., Makino, T., and Shiota, K. (2011). Epigenetic assessment of environmental chemicals detected in maternal peripheral and cord blood samples. J. Reprod. Dev. 57, 507–517.
ATSDR. (2008). Toxicological profile for Cadmium (Draft for Public Comment). Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service.
Baccarelli, A., and Bollati, V. (2009). Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 21, 243–251.
Bagchi, D., Bagchi, M., and Stohs, S. J. (2001). Chromium (VI)-induced oxidative stress, apoptotic cell death and modulation of p53 tumor suppressor gene. Mol. Cell. Biochem. 222, 149–158.
Baker, J., Olsen, L., and Sørensen, T. (2008). Weight at birth and all-cause mortality in adulthood. Epidemiology 19, 197–203.
Barker, D., Winter, P., Osmond, C., Margetts, B., and Simmonds, S. (1989). Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580.
Barnett, M., Bermingham, E., Mcnabb, W., Bassett, S., Armstrong, K., Rounce, J., and Roy, N. (2010). Investigating micronutrients and epigenetic mechanisms in relation to inflammatory bowel disease. Mutat. Res. 690, 71–80.
Barros, S. P., and Offenbacher, S. (2009). Epigenetics: connecting environment and genotype to phenotype and disease. J. Dent. Res. 88, 400–408.
Baylin, S. B., Herman, J. G., Graff, J. R., Vertino, P. M., and Issa, J. P. (1998). Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv. Cancer Res. 72, 141–196.
Benbrahim-Tallaa, L., Waterland, R. A., Dill, A. L., Webber, M. M., and Waalkes, M. P. (2007). Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase. Environ. Health Perspect. 115, 1454–1459.
Benbrahim-Tallaa, L., Waterland, R. A., Styblo, M., Achanzar, W. E., Webber, M. M., and Waalkes, M. P. (2005). Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: aberrant genomic DNA methylation and K-ras oncogene activation. Toxicol. Appl. Pharmacol. 206, 288–298.
Bernal, A. J., and Jirtle, R. L. (2010). Epigenomic disruption: the effects of early developmental exposures. Birth Defects Res. A Clin. Mol. Teratol. 88, 938–944.
Beveridge, R., Pintos, J., Parent, M.-É., Asselin, J., and Siemiatycki, J. (2010). Lung cancer risk associated with occupational exposure to nickel, chromium VI, and cadmium in two population-based case–control studies in Montreal. Am. J. Ind. Med. 53, 476–485.
Boehm, T. L., and Drahovsky, D. (1983). Alteration of enzymatic methylation of DNA cytosines by chemical carcinogens: a mechanism involved in the initiation of carcinogenesis. J. Natl. Cancer Inst. 71, 429–433.
Borchers, A., Teuber, S. S., Keen, C. L., and Gershwin, M. E. (2010). Food safety. Clin. Rev. Allergy Immunol. 39, 95–141.
Broday, L., Peng, W., Kuo, M. H., Salnikow, K., Zoroddu, M., and Costa, M. (2000). Nickel compounds are novel inhibitors of histone H4 acetylation. Cancer Res. 60, 238–241.
Bromer, J. G., Wu, J., Zhou, Y., and Taylor, H. S. (2009). Hypermethylation of homeobox A10 by in utero diethylstilbestrol exposure: an epigenetic mechanism for altered developmental programming. Endocrinology 150, 3376–3382.
Bromer, J. G., Zhou, Y., Taylor, M. B., Doherty, L., and Taylor, H. S. (2010). Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J. 24, 2273–2280.
Brons, C., Jacobsen, S., Nilsson, E., Ronn, T., Jensen, C. B., Storgaard, H., Poulsen, P., Groop, L., Ling, C., Astrup, A., and Vaag, A. (2010). Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J. Clin. Endocrinol. Metab. 95, 3048–3056.
Bull, C., and Fenech, M. (2008). Genome-health nutrigenomics and nutrigenetics: nutritional requirements or ‘nutriomes’ for chromosomal stability and telomere maintenance at the individual level. Proc. Nutr. Soc. 67, 146–156.
Calafat, A. M., Ye, X., Wong, L.-Y., Reidy, J. A., and Needham, L. L. (2007). Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 116, 39–44.
Callaghan, B., Feldman, D., Gruis, K., and Feldman, E. (2011). The association of exposure to lead, mercury, and selenium and the development of amyotrophic lateral sclerosis and the epigenetic implications. Neurodegener. Dis. 8, 1–8.
Chanda, S., Dasgupta, U. B., Guhamazumder, D., Gupta, M., Chaudhuri, U., Lahiri, S., Das, S., Ghosh, N., and Chatterjee, D. (2006). DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol. Sci. 89, 431–437.
Chen, W. T., Hung, W. C., Kang, W. Y., Huang, Y. C., and Chai, C. Y. (2007). Urothelial carcinomas arising in arsenic-contaminated areas are associated with hypermethylation of the gene promoter of the death-associated protein kinase. Histopathology 51, 785–792.
Cooney, G., Dave, A., and Wolff, G. (2002). Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J. Nutr. 132, 2393s–2400s.
Coppin, J.-F., Qu, W., and Waalkes, M. P. (2008). Interplay between cellular methyl metabolism and adaptive efflux during oncogenic transformation from chronic arsenic exposure in human cells. J. Biol. Chem. 283, 19342–19350.
Cui, H., Onyango, P., Brandenburg, S., Wu, Y., Hsieh, C. L., and Feinberg, A. P. (2002). Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 62, 6442–6446.
Das, R., Hampton, D. D., and Jirtle, R. L. (2009). Imprinting evolution and human health. Mamm. Genome 20, 563–572.
Dashwood, R. H., Myzak, M. C., and Ho, E. (2006). Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis 27, 344–349.
Davidson, L. A., Wang, N., Shah, M. S., Lupton, J. R., Ivanov, I., and Chapkin, R. S. (2009). n-3 Polyunsaturated fatty acids modulate carcinogen-directed non-coding microRNA signatures in rat colon. Carcinogenesis 30, 2077–2084.
Davis, C. D., and Uthus, E. O. (2004). DNA methylation, cancer susceptibility, and nutrient interactions. Exp. Biol. Med. (Maywood) 229, 988–995.
Delage, B., and Dashwood, R. H. (2008). Dietary manipulation of histone structure and function. Annu. Rev. Nutr. 28, 347–366.
Dobosy, J. R., Fu, V. X., Desotelle, J. A., Srinivasan, R., Kenowski, M. L., Almassi, N., Weindruch, R., Svaren, J., and Jarrard, D. F. (2008). A methyl-deficient diet modifies histone methylation and alters Igf2 and H19 repression in the prostate. Prostate 68, 1187–1195.
Doherty, L., Bromer, J., Zhou, Y., Aldad, T., and Taylor, H. (2010). In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: an epigenetic mechanism linking endocrine disruptors to breast cancer. Horm Cancer 1, 146–155.
Dokmanovic, M., and Marks, P. A. (2005). Prospects: histone deacetylase inhibitors. J. Cell. Biochem. 96, 293–304.
Dolinoy, D. C. (2008). The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr. Rev. 66(Suppl. 1), S7–S11.
Dolinoy, D. C., Huang, D., and Jirtle, R. L. (2007). Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. U.S.A. 104, 13056–13061.
Doll, R., and Peto, R. (1981). The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 66, 1191–1308.
Duhl, D. M., Vrieling, H., Miller, K. A., Wolff, G. L., and Barsh, G. S. (1994). Neomorphic agouti mutations in obese yellow mice. Nat. Genet. 8, 59–65.
Duthie, S. J. (2011). Epigenetic modifications and human pathologies: cancer and CVD. Proc. Nutr. Soc. 70, 47–56.
Dwivedi, S., Tripathi, R. D., Tripathi, P., Kumar, A., Dave, R., Mishra, S., Singh, R., Sharma, D., Rai, U. N., Chakrabarty, D., Trivedi, P. K., Adhikari, B., Bag, M. K., Dhankher, O. P., and Tuli, R. (2010). Arsenate exposure affects amino acids, mineral nutrient status and antioxidants in rice (Oryza sativa L.) genotypes. Environ. Sci. Technol. 44, 9542–9549.
EPA. (2010). Human Exposure to Mercury. Available at: http://www.epa.gov/hg/exposure.htm#1 [accessed 2011].
Esquela-Kerscher, A., and Slack, F. J. (2006). Oncomirs – microRNAs with a role in cancer. Nat. Rev. Cancer 6, 259–269.
Fang, M., Chen, D., and Yang, C. S. (2007). Dietary polyphenols may affect DNA methylation. J. Nutr. 137, 223S–228S.
FDA U.S. Food and Drug Administration. (2009). Mercury Levels in Commercial Fish and Shellfish. Available at: http://www.fda.gov/Food/FoodSafety/Product-SpecificInformation/Seafood/FoodbornePathogensContaminants/Methylmercury/ucm115644.htm [accessed 2011].
Fearon, E. R., and Vogelstein, B. (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767.
Feil, R. (2006). Environmental and nutritional effects on the epigenetic regulation of genes. Mutat. Res. 600, 46–57.
Forsén, T., Eriksson, J., Tuomilehto, J., Teramo, K., Osmond, C., and Barker, D. (1997). Mother’s weight in pregnancy and coronary heart disease in a cohort of Finnish men: follow up study. BMJ 315, 837–840.
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., Heine-Suner, D., Cigudosa, J. C., Urioste, M., Benitez, J., Boix-Chornet, M., Sanchez-Aguilera, A., Ling, C., Carlsson, E., Poulsen, P., Vaag, A., Stephan, Z., Spector, T. D., Wu, Y. Z., Plass, C., and Esteller, M. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609.
Gibb, H. J., Lees, P. S., Pinsky, P. F., and Rooney, B. C. (2000). Lung cancer among workers in chromium chemical production. Am. J. Ind. Med. 38, 115–126.
Gilbert, E. R., and Liu, D. (2010). Flavonoids influence epigenetic-modifying enzyme activity: structure-function relationships and the therapeutic potential for cancer. Curr. Med. Chem. 17, 1756–1768.
Govindarajan, B., Klafter, R., Miller, M. S., Mansur, C., Mizesko, M., Bai, X., Lamontagne, K. Jr., and Arbiser, J. L. (2002). Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase. Mol. Med. 8, 1–8.
Grandjean, P., and Landrigan, P. J. (2006). Developmental neurotoxicity of industrial chemicals. Lancet 368, 2167–2178.
Hennig, B., Ettinger, A. S., Jandacek, R. J., Koo, S., Mcclain, C., Seifried, H., Silverstone, A., Watkins, B., and Suk, W. A. (2007). Using nutrition for intervention and prevention against environmental chemical toxicity and associated diseases. Environ. Health Perspect. 115, 493–495.
Hennig, B., Toborek, M., Bachas, L. G., and Suk, W. A. (2004). Emerging issues: nutritional awareness in environmental toxicology. J. Nutr. Biochem. 15, 194–195.
Herbst, A. L., Cole, P., Colton, T., Robboy, S. J., and Scully, R. E. (1977). Age-incidence and risk of diethylstilbestrol-related clear cell adenocarcinoma of the vagina and cervix. Am. J. Obstet. Gynecol. 128, 43–50.
Herceg, Z. (2007). Epigenetics and cancer: towards an evaluation of the impact of environmental and dietary factors. Mutagenesis 22, 91–103.
Herman, J. G., and Baylin, S. B. (2003). Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 349, 2042–2054.
Ho, S.-M., Tang, W.-Y., Belmonte De Frausto, J., and Prins, G. S. (2006). Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 66, 5624–5632.
Huxley, R., Owen, C., Whincup, P., Cook, D., Rich-Edwards, J., Smith, G., and Collins, R. (2007). Is birth weight a risk factor for ischemic heart disease in later life? Am. J. Clin. Nutr. 85, 1244–1250.
IARC. (1990). “Chromium, nickel and welding,” in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, (ed.) I. S. Publications (Lyon: International Agency for Research on Cancer).
IARC. (1993). “Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry,” in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, (ed.) I. S. Publications (Lyon: International Agency for Research on Cancer).
IARC. (2004). “Some drinking-water disinfectants and contaminants, including arsenic,” in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, (ed.) I. S. Publications (Lyon: International Agency for Research on Cancer).
Issa, J. P. (2003). Age-related epigenetic changes and the immune system. Clin. Immunol. 109, 103–108.
Issa, J. P., Ottaviano, Y. L., Celano, P., Hamilton, S. R., Davidson, N. E., and Baylin, S. B. (1994). Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat. Genet. 7, 536–540.
Jiang, G., Xu, L., Song, S., Zhu, C., Wu, Q., Zhang, L., and Wu, L. (2008). Effects of long-term low-dose cadmium exposure on genomic DNA methylation in human embryo lung fibroblast cells. Toxicology 244, 49–55.
Kaneta, M., Hikichi, H., Endo, S., and Sugiyama, N. (1986). Chemical form of cadmium (and other heavy metals) in rice and wheat plants. Environ. Health Perspect. 65, 33–37.
Ke, Q., Davidson, T., Chen, H., Kluz, T., and Costa, M. (2006). Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis 27, 1481–1488.
Keri, R. A., Ho, S.-M., Hunt, P. A., Knudsen, K. E., Soto, A. M., and Prins, G. S. (2007). An evaluation of evidence for the carcinogenic activity of bisphenol A. Reprod. Toxicol. 24, 240–252.
Khan, S. I., Ahmed, A. K., Yunus, M., Rahman, M., Hore, S. K., Vahter, M., and Wahed, M. A. (2010). Arsenic and cadmium in food-chain in Bangladesh – an exploratory study. J. Health Popul. Nutr. 28, 578–584.
Kim, K. C., Friso, S., and Choi, S. W. (2009). DNA methylation, an epigenetic mechanism connecting folate to healthy embryonic development and aging. J. Nutr. Biochem. 20, 917–926.
Klein, C. B., Su, L., Bowser, D., and Leszczynska, J. (2002). Chromate-induced epimutations in mammalian cells. Environ. Health Perspect. 110(Suppl. 5), 739–743.
Kondo, K., Takahashi, Y., Hirose, Y., Nagao, T., Tsuyuguchi, M., Hashimoto, M., Ochiai, A., Monden, Y., and Tangoku, A. (2006). The reduced expression and aberrant methylation of p16INK4a in chromate workers with lung cancer. Lung Cancer 53, 295–302.
Kordas, K., Lonnerdal, B., and Stoltzfus, R. J. (2007). Interactions between nutrition and environmental exposures: effects on health outcomes in women and children. J. Nutr. 137, 2794–2797.
Kutay, H., Bai, S., Datta, J., Motiwala, T., Pogribny, I., Frankel, W., Jacob, S. T., and Ghoshal, K. (2006). Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J. Cell. Biochem. 99, 671–678.
Lawlor, D., Ronalds, G., Clark, H., Smith, G., and Leon, D. (2005). Birth weight is inversely associated with incident coronary heart disease and stroke among individuals born in the 1950s: findings from the Aberdeen children of the 1950s prospective cohort study. Circulation 112, 1414–1418.
Lee, W. J., Shim, J. Y., and Zhu, B. T. (2005). Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 68, 1018–1030.
Lee, Y., Klein, C., Kargacin, B., Salnikow, K., Kitahara, J., Dowjat, K., Zhitkovich, A., Christie, N., and Costa, M. (1995). Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: a new model for epigenetic carcinogens. Mol. Cell. Biol. 15, 2547–2557.
Leon, D., Lithell, H., Vâgerö, D., Koupilová, I., Mohsen, R., Berglund, L., Lithell, U., and Mckeigue, P. (1998). Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15 000 Swedish men and women born 1915-29. BMJ 317, 241–245.
Li, L., Ekstrom, E. C., Goessler, W., Lonnerdal, B., Nermell, B., Yunus, M., Rahman, A., El Arifeen, S., Persson, L. A., and Vahter, M. (2008). Nutritional status has marginal influence on the metabolism of inorganic arsenic in pregnant Bangladeshi women. Environ. Health Perspect. 116, 315–321.
Li, S., Hursting, S. D., Davis, B. J., Mclachlan, J. A., and Barrett, J. C. (2003). Environmental exposure, DNA methylation, and gene regulation: lessons from diethylstilbestrol-induced cancers. Ann. N. Y. Acad. Sci. 983, 161–169.
Link, A., Balaguer, F., and Goel, A. (2010). Cancer chemoprevention by dietary polyphenols: promising role for epigenetics. Biochem. Pharmacol. 80, 1771–1792.
Loeb, L. A., and Harris, C. C. (2008). Advances in chemical carcinogenesis: a historical review and prospective. Cancer Res. 68, 6863–6872.
Mahaffey, K. R., and Vanderveen, J. E. (1979). Nutrient-toxicant interactions: susceptible populations. Environ. Health Perspect. 29, 81–87.
Marsit, C. J., Karagas, M. R., Danaee, H., Liu, M., Andrew, A., Schned, A., Nelson, H. H., and Kelsey, K. T. (2006a). Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis 27, 112–116.
Marsit, C. J., Karagas, M. R., Schned, A., and Kelsey, K. T. (2006b). Carcinogen exposure and epigenetic silencing in bladder cancer. Ann. N. Y. Acad. Sci. 1076, 810–821.
Mathers, J. C., Strathdee, G., and Relton, C. L. (2010). Induction of epigenetic alterations by dietary and other environmental factors. Adv. Genet. 71, 3–39.
McKay, J. A., and Mathers, J. C. (2011). Diet induced epigenetic changes and their implications for health. Acta Physiol. (Oxf.) 202, 103–118.
McLaughlin, F., and La Thangue, N. B. (2004). Histone deacetylase inhibitors open new doors in cancer therapy. Biochem. Pharmacol. 68, 1139–1144.
Meeran, S. M., Ahmed, A., and Tollefsbol, T. O. (2010). Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin. Epigenetics 1, 101–116.
Morgan, H., Sutherland, H., Martin, D., and Whitelaw, E. (1999). Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 23, 314–318.
Murphy, S. K., and Jirtle, R. L. (2003). Imprinting evolution and the price of silence. Bioessays 25, 577–588.
Myzak, M. C., Karplus, P. A., Chung, F. L., and Dashwood, R. H. (2004). A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 64, 5767–5774.
National Toxicology Program. (2005). National Toxicology Program, Report on Carcinogens, 11th Edn. Research Triangle Park: U.S. Department of Health and Human Services, Public Health Service.
Newbold, R. R., Padilla-Banks, E., and Jefferson, W. N. (2006). Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology 147, S11–S17.
Nian, H., Delage, B., Ho, E., and Dashwood, R. H. (2009). Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane and garlic organosulfur compounds. Environ. Mol. Mutagen. 50, 213–221.
Perello, G., Marti-Cid, R., Llobet, J. M., and Domingo, J. L. (2008). Effects of various cooking processes on the concentrations of arsenic, cadmium, mercury, and lead in foods. J. Agric. Food Chem. 56, 11262–11269.
Pilsner, J. R., Liu, X., Ahsan, H., Ilievski, V., Slavkovich, V., Levy, D., Factor-Litvak, P., Graziano, J. H., and Gamble, M. V. (2007). Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am. J. Clin. Nutr. 86, 1179–1186.
Pilsner, J. R., Liu, X., Ahsan, H., Ilievski, V., Slavkovich, V., Levy, D., Factor-Litvak, P., Graziano, J. H., and Gamble, M. V. (2009). Folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ. Health Perspect. 117, 254–260.
Pogribny, I. P., Shpyleva, S. I., Muskhelishvili, L., Bagnyukova, T. V., James, S. J., and Beland, F. A. (2009). Role of DNA damage and alterations in cytosine DNA methylation in rat liver carcinogenesis induced by a methyl-deficient diet. Mutat. Res. 669, 56–62.
Reichard, J. F., Schnekenburger, M., and Puga, A. (2007). Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem. Biophys. Res. Commun. 352, 188–192.
Rich-Edwards, J., Kleinman, K., Michels, K., Stampfer, M., Manson, J., Rexrode, K., Hibert, E., and Willett, W. (2005). Longitudinal study of birth weight and adult body mass index in predicting risk of coronary heart disease and stroke in women. BMJ 330, 1115–1121.
Rich-Edwards, J., Stampfer, M., Manson, J., Rosner, B., Hankinson, S., Colditz, G., Willett, W., and Hennekens, C. (1997). Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ 315, 396–400.
Risnes, K., Romundstad, P., Nilsen, T., Eskild, A., and Vatten, L. (2009). Placental weight relative to birth weight and long-term cardiovascular mortality: findings from a cohort of 31,307 men and women. Am. J. Epidemiol. 170, 622–632.
Robertson, K. D., and Wolffe, A. P. (2000). DNA methylation in health and disease. Nat. Rev. Genet. 1, 11–19.
Rooney, J. (2011). Further thoughts on mercury, epigenetics, genetics, and amyotrophic sclerosis. Neurodegener. Dis. 8, 523–524.
Ross, S. A., Dwyer, J., Umar, A., Kagan, J., Verma, M., Van Bemmel, D. M., and Dunn, B. K. (2008). Introduction: diet, epigenetic events and cancer prevention. Nutr. Rev. 66(Suppl. 1), S1–S6.
Salnikow, K., and Zhitkovich, A. (2008). Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem. Res. Toxicol. 21, 28–44.
Shah, M. S., Schwartz, S. L., Zhao, C., Davidson, L. A., Zhou, B., Lupton, J. R., Ivanov, I., and Chapkin, R. S. (2011). Integrated microRNA and mRNA expression profiling in a rat colon carcinogenesis model: effect of a chemo-protective diet. Physiol. Genomics 43, 640–654.
Shi, H., Hudson, L. G., and Liu, K. J. (2004). Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radic. Biol. Med. 37, 582–593.
Stayner, L., Smith, R., Thun, M., Schnorr, T., and Lemen, R. (1992). A dose-response analysis and quantitative assessment of lung cancer risk and occupational cadmium exposure. Ann Epidemiol 2, 177–194.
Stein, C., Fall, C., Kumaran, K., Osmond, C., Cox, V., and Barker, D. (1996). FEtal growth and coronary heart disease in South India. Lancet 348, 1269–1273.
Sugden, K. D., and Stearns, D. M. (2000). The role of chromium(V) in the mechanism of chromate-induced oxidative DNA damage and cancer. J. Environ. Pathol. Toxicol. Oncol. 19, 215–230.
Sun, H., Zhou, X., Chen, H., Li, Q., and Costa, M. (2009). Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol. Appl. Pharmacol. 237, 258–266.
Sun, M., Estrov, Z., Ji, Y., Coombes, K. R., Harris, D. H., and Kurzrock, R. (2008). Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol. Cancer Ther. 7, 464–473.
Suter, M., Bocock, P., Showalter, L., Hu, M., Shope, C., Mcknight, R., Grove, K., Lane, R., and Aagaard-Tillery, K. (2011). Epigenomics: maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates. FASEB J. 25, 714–726.
Syddall, H., Sayer, A., Simmonds, S., Osmond, C., Cox, V., Dennison, E., Barker, D., and Cooper, C. (2005). Birth weight, infant weight gain, and cause-specific mortality: the Hertfordshire Cohort Study. Am. J. Epidemiol. 161, 1074–1080.
Takiguchi, M., Achanzar, W. E., Qu, W., Li, G., and Waalkes, M. P. (2003). Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp. Cell Res. 286, 355–365.
Toyota, M., and Issa, J. P. (2005). Epigenetic changes in solid and hematopoietic tumors. Semin. Oncol. 32, 521–530.
Tuli, R., Chakrabarty, A., Trivedi, P. K., and Tripathi, R. D. (2010). Recent advances in arsenic accumulation and metabolism in rice. Mol. Breed. 26, 307–323.
U.S. Environmental Protection Agency. (2007). Inorganic Arsenic: Toxicity and Exposure Assessment for Children’s Health Chemical Summary. Research Triangle Park, NC: U.S. Environmental Protection Agency.
Vaissière, T., Sawan, C., and Herceg, Z. (2008). Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 659, 40–48.
vel Szic, K. S., Ndlovu, M. N., Haegeman, G., and Vanden Berghe, W. (2010). Nature or nurture: let food be your epigenetic medicine in chronic inflammatory disorders. Biochem. Pharmacol. 80, 1816–1832.
Verma, M., and Srivastava, S. (2002). Epigenetics in cancer: implications for early detection and prevention. Lancet Oncol. 3, 755–763.
Wade, P. A. (2001). Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum. Mol. Genet. 10, 693–698.
Waisberg, M., Joseph, P., Hale, B., and Beyersmann, D. (2003). Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 192, 95–117.
Wang, Y., Lee, A. T., Ma, J. Z., Wang, J., Ren, J., Yang, Y., Tantoso, E., Li, K. B., Ooi, L. L., Tan, P., and Lee, C. G. (2008). Profiling microRNA expression in hepatocellular carcinoma reveals microRNA-224 up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific target. J. Biol. Chem. 283, 13205–13215.
Wang, Z. Q., and Cefalu, W. T. (2010). Current concepts about chromium supplementation in type 2 diabetes and insulin resistance. Curr. Diab. Rep. 10, 145–151.
Wasserman, G. A., Liu, X., Factor-Litvak, P., Gardner, J. M., and Graziano, J. H. (2008). Developmental impacts of heavy metals and undernutrition. Basic Clin. Pharmacol. Toxicol. 102, 212–217.
Waterland, R. A., Lin, J. R., Smith, C. A., and Jirtle, R. L. (2006). Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum. Mol. Genet. 15, 705–716.
Waterland, R., and Jirtle, R. (2003). Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell Biol. 23, 5293–5300.
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., Flavell, A., Leroy, P., Morgante, M., Panaud, O., Paux, E., SanMiguel, P., and Schulman, A. H. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982.
Wolff, G. L., Kodell, R. L., Moore, S. R., and Cooney, C. A. (1998). Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 12, 949–957.
Wolff, M. S., Teitelbaum, S. L., Windham, G., Pinney, S. M., Britton, J. A., Chelimo, C., Godbold, J., Biro, F., Kushi, L. H., Pfeiffer, C. M., and Calafat, A. M. (2006). Pilot study of urinary biomarkers of phytoestrogens, phthalates, and phenols in girls. Environ. Health Perspect. 115, 116–121.
Yan, Y., Kluz, T., Zhang, P., Chen, H.-B., and Costa, M. (2003). Analysis of specific lysine histone H3 and H4 acetylation and methylation status in clones of cells with a gene silenced by nickel exposure. Toxicol. Appl. Pharmacol. 190, 272–277.
Yen, T. T., Gill, A. M., Frigeri, L. G., Barsh, G. S., and Wolff, G. L. (1994). Obesity, diabetes, and neoplasia in yellow A(vy)/- mice: ectopic expression of the agouti gene. FASEB J. 8, 479–488.
Zhao, C. Q., Young, M. R., Diwan, B. A., Coogan, T. P., and Waalkes, M. P. (1997). Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. U.S.A. 94, 10907–10912.
Zhitkovich, A. (2005). Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI). Chem. Res. Toxicol. 18, 3–11.
Zhou, X., Li, Q., Arita, A., Sun, H., and Costa, M. (2009). Effects of nickel, chromate, and arsenite on histone 3 lysine methylation. Toxicol. Appl. Pharmacol. 236, 78–84.
Keywords: cancer, epigenetics, diet, nutrient, toxicants
Citation: Su LJ, Mahabir S, Ellison GL, McGuinn LA and Reid BC (2012) Epigenetic contributions to the relationship between cancer and dietary intake of nutrients, bioactive food components, and environmental toxicants. Front. Gene. 2:91. doi: 10.3389/fgene.2011.00091
Received: 31 May 2011; Accepted: 06 December 2011;
Published online: 09 January 2012.
Edited by:
Jamie L. Barger, LifeGen Technologies, USAReviewed by:
Dhanansayan Shanmuganayagam, University of Wisconsin–Madison, USAAngela Mastaloudis, NuSkin Enterprises, USA
Copyright: © 2012 Su, Mahabir, Ellison, McGuinn and Reid. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: L. Joseph Su, Modifiable Risk Factors Branch, Division of Cancer Control and Population Sciences, National Cancer Institute, 6130 Executive Blvd, MSC 7395, Bethesda, MD 20892-7395, USA. e-mail: sulj@mail.nih.gov