
- 1 Bioinformatics and Genomics, Centre for Genomic Regulation, Universitat Pompeu Fabra, Barcelona, Spain
- 2 Departament de Ciències Experimentals i de la Salut, Universitat Pompeu Fabra, Barcelona, Spain
The transcriptome of a cell is represented by a myriad of different RNA molecules with and without protein-coding capacities. In recent years, advances in sequencing technologies have allowed researchers to more fully appreciate the complexity of whole transcriptomes, showing that the vast majority of the genome is transcribed, producing a diverse population of non-protein coding RNAs (ncRNAs). Thus, the biological significance of non-coding RNAs (ncRNAs) have been largely underestimated. Amongst these multiple classes of ncRNAs, the long non-coding RNAs (lncRNAs) are apparently the most numerous and functionally diverse. A small but growing number of lncRNAs have been experimentally studied, and a view is emerging that these are key regulators of epigenetic gene regulation in mammalian cells. LncRNAs have already been implicated in human diseases such as cancer and neurodegeneration, highlighting the importance of this emergent field. In this article, we review the catalogs of annotated lncRNAs and the latest advances in our understanding of lncRNAs.
The Cell, an RNA-Dependent Machinery
Some of the most fundamental cellular processes rely on anciently conserved non-coding RNAs (ncRNAs). These include, for instance, the ribosomal RNAs which are assembled together to constitute ribosomes, the factories for translation of messenger RNAs (mRNAs) into proteins. Other ancient roles of ncRNAs include the transport of amino acids through ribosomes via the transfer RNAs (tRNAs) or the splicing of introns of pre-mRNA which is mediated in part by the snRNAs (small nuclear RNAs). More recently, the crucial role of ncRNA in post-transcriptional gene regulation has been highlighted by the discovery of microRNAs (miRNAs), which repress gene expression by targeting semi-complementary motifs in target mRNAs (Lee et al., 1993). Many additional classes of ncRNAs have been discovered in the last decade reinforcing the view that they are of central importance in the functioning of cells from all the branches of life (Amaral et al., 2008).
Amongst the various ncRNA classes, we know probably least about the long non-coding RNAs (lncRNAs). In particular, what is the total number of lncRNAs in mammalian genomes? Where are they localized? What is their significance in the context of evolution, and particularly in the evolution of complex processing in primate brains? Now that good catalogs of lncRNAs have become available, the most critical question is to address the functionality of these transcripts. This question is particularly acute given that we have no a priori methods for the prediction of lncRNA function based on sequence alone, in contrast to proteins where confident inferences on protein function can be made by simply analysis of the amino acid sequence. Given the sheer number of new unexplored lncRNA transcripts (∼15,000 at last count; Derrien et al., submitted), the field must move forward to address this question of function by using large-scale functional screens. Such moves are already underway, with groups such as Eric Lander’s carrying out siRNA screens (Guttman et al., 2011). Large-scale analysis of protein-binding partners will also add another layer of valuable information to such annotation of lncRNA catalogs. Hopefully, advances in bioinformatic annotation of RNA structures (Torarinsson et al., 2006; Parker et al., 2011), and methods to predict functions based on this, will be developed. In this way, we might build up a richly annotated catalog of lncRNAs with functional predictions, that will enable us to integrate them into existing knowledge of the cell, and infer possible roles in human diseases.
Cis and trans Functions for lncRNAs
Until recently, only a handful of lncRNAs have been described in the literature. One of the earliest examples was XIST, a 19 kb non-protein-coding transcript which is responsible for the inactivation of one of the two X chromosome in placental females through DNA methylation (Brockdorff et al., 1992). Others examples of lncRNAs located in imprinted regions, such as Airn (Sleutels et al., 2002; Nagano et al., 2008), H19 (Gabory et al., 2009), NESPAS (Wroe et al., 2000), or Kcnq1ot1 (Mancini-Dinardo et al., 2006; Mohammad et al., 2010) are involved in the inactivation of gene expression via specific associations with chromatin-modifying complexes. More recently, the HOTAIR lncRNA was shown to epigenetically repress the HOXD locus via the recruitment of the PRC2 complex (Rinn et al., 2007). Strikingly, this study described a trans mechanism of action of a lncRNA located on human Chromosome 5 which modulates expression of multiple genes clustered on human Chromosome 4 (HOXD locus; Rinn et al., 2007). Supporting this hypothesis, two recent papers (Cabili et al., 2011; Guttman et al., 2011) showed that lncRNAs primarily affect gene expression in trans. The latter work used loss-of-function protocols to demonstrate that large intergenic ncRNAs (lincRNAs) both up- and down-regulate hundreds of genes expression in trans which support a primary role of lincRNAs in the circuitry controlling embryonic stem (ES) cell states (Guttman et al., 2011).
On the other hand, previous studies showed that some lncRNAs could also activate expression of protein-coding genes in their immediate genomic neighborhood. This cis-mechanism of action was demonstrated by Ørom and colleagues who used interference RNAs (siRNAs) to knock down candidate lncRNAs annotated as part of the GENCODE project (Harrow et al., 2006). The inactivation of some of these lncRNAs further triggers a down-regulation of protein-coding genes transcription located either in the same or opposite strand within 1 Mb from the lncRNA (Ørom et al., 2010) suggesting the latter functions as a transcriptional activator. Further supporting the cis-mechanism, a lincRNA called HOTTIP transcribed from the HOX A locus coordinates the transcription of several genes localized in cis at the 5′ of the HOXA locus (Wang et al., 2008). HOTTIP was shown to activate gene expression by recruiting the WDR5/MLL complex and thus depositing the activating histone modification H3K4me3. Finally, the distinction between activating lncRNAs and enhancers remains unclear. For instance, about 12,000 actively regulated enhancer were identified based on their bindings to the transcriptional co-activator p300/CBP in mouse neurons (Kim et al., 2010). Using ChipSeq analysis to define RNA polymerase II binding sites, the authors also reported that 25% of the enhancers co-localize with RNAPII sites suggesting that some enhancers are transcribed; they termed these transcripts eRNAs for enhancer RNAs (Kim et al., 2010). It will be important to functionally define whether such eRNAs are all required for enhancer function, or are simply a by-product of some non-functional transcription of enhancers by RNA PolII.
Similarly it will be important to define whether the activating lncRNAs (Ørom et al., 2010) are in fact a subset of eRNAs, or not.
While it is more likely that an lncRNA regulates the co-expression of nearby protein coding genes (as for tandemly duplicated genes, imprinted genes, or ubiquitously expressed genes), an interesting study demonstrate that modulating the expression of a particular locus will also trigger the modification of the expression of nearby transcripts by a mechanism known as « ripple of transcription» (Ebisuya et al., 2008). Taken together and similar to the behavior of protein-coding genes, lncRNAs seem to act both in cis and trans and are a key player of the regulation of gene expression.
LncRNAs in Human Disease
There is growing evidence that lncRNAs are involved in disease progression and especially cancers. For instance, recent work implies a non-coding RNA, lincRNA-p21, in the p53 response though the modulation of multiple p53 dependent gene expression in trans (Huarte et al., 2010). Another example is MEG3, which is thought to directly activate the tumor suppressor gene p53, although the mechanism has yet to be elucidated (Zhou et al., 2007). Finally, another long non-coding RNA, called ANRIL, located in the p15/CDKN2B–p16/CDKN2A–p14/ARF is genetically associated with diverse diseases such as diabetes, gliomas, coronary diseases, and basal cell carcinomas via genome-wide association studies (GWAS; Pasmant et al., 2010; Wapinski and Chang, 2011). More generally, given the lack of annotation of human lncRNAs, one could speculate on the impact of non-coding regions of the human genome in an answer to the “missing heritability” in GWAS studies (Manolio et al., 2009). Indeed, given that at least a half of the human genome is transcribed into RNA molecules (Carninci et al., 2005; ENCODE Project Consortium et al., 2007), it is now exciting to further characterize the 80% of disease-associated variants that are located outside of protein-coding genes (Manolio et al., 2009). Thus lncRNA represent a new frontier in human disease genomics. Presently no drugs against lncRNAs are available. It will be fascinating to observe whether it will be possible to specifically drug lncRNA pathways, perhaps through the use of specific modified small oligonucleotides. It is also worth mentioning that ncRNAs can be detected in human bodily fluids and hold great promise as biomarkers (Gaughwin et al., 2011).
Resources for the Annotation of LncRNAs
Similar to that of protein coding genes, resources for the global annotation of lncRNAs are needed in order to identify, classify and elucidate the roles of these transcripts within the cell machinery.
Particularly relevant is the effort from John Mattick’s group to compile and centralize biologically meaningful information dedicated to lncRNA (Amaral et al., 2011). The lncRNA database (lncRNAdb) provides sequence, structural, and conservation evidence for mutli-species lncRNAs together with a list of lncRNAs that are experimentally known to interact with coding mRNAs.
In mouse in the early 2000s, the FANTOM consortium pioneered the genome-wide discovery of lncRNAs publishing a set of 34,030 lncRNAs based on cDNA sequencing (Maeda et al., 2006). More recently, Guttman and colleagues used chromatin signatures via ChIPSeq (Chromatin Immuno-Precipitation followed by high throughput Sequencing) to reveal ∼1,600 lincRNAs (Guttman et al., 2009). They further showed that some of these lincRNAs are functional and transcriptionally regulated by key transcription factors such Oct4 (Guttman et al., 2009). While expressed in a wide range of tissue, lincRNAs tend to be modestly conserved (Marques and Ponting, 2009) as shown by using a neutral indel model which exploits the patterns of substitutions and insertions or deletions (Lunter et al., 2006). The methodology employed by Guttman and colleagues has been applied to human thus leading to the identification of about ∼3,300 lincRNAs whose functional roles may include guidance of chromatin-modifying complexes to specific regions of the genome (Khalil et al., 2009). Very recently, the growing interest in lincRNAs led to the annotation of more than 8,000 lincRNA genes in human using a combination of computational methods and RNASeq experiments especially from the Human Body Map (HBM) project (Cabili et al., 2011; Table 1).
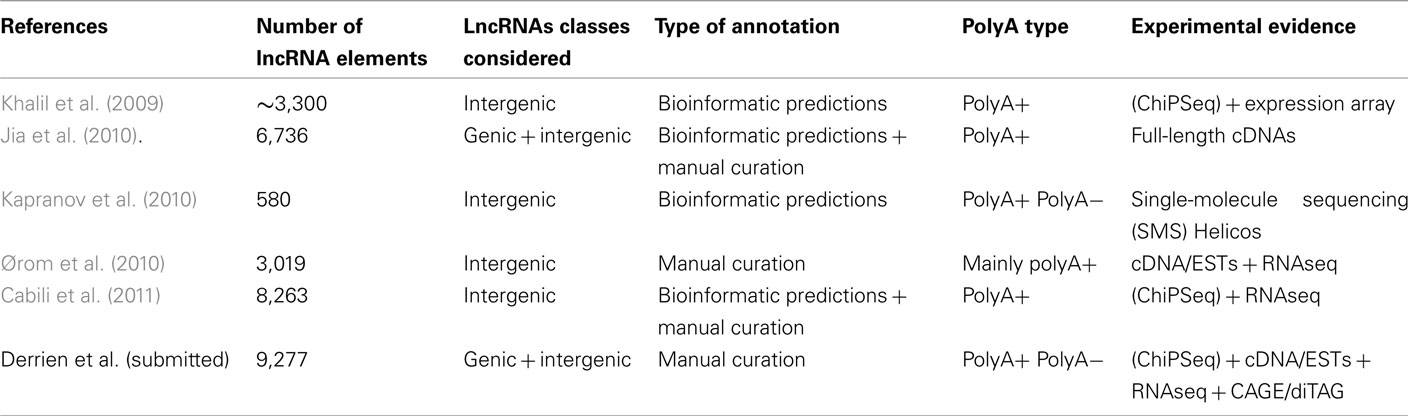
Table 1. Description of human lncRNAs published catalogs.
It is worth mentioning that many of the current RNASeq data (including HBM) mainly select RNA transcripts harboring a polyA tail at their 3′end (polyA+) and therefore offer little information on transcripts lacking polyA (polyA−). To tackle this issue, sequencing technologies such as single-molecule sequencing (SMS; Pushkarev et al., 2009) was used to estimate the abundance of ncRNAs by avoiding amplification and minimizing sample preparation (Kapranov et al., 2010). Interestingly, this studies revealed that “dark matter” transcription may represent the majority of the total (non-ribosomal and non-mitochondrial) RNA of a cell. In addition, it shed light on a new class of very long ncRNAs (min size ∼50 kb), abundantly expressed and localized in intergenic regions of the genome, the so-called vlincRNAs (very long intergenic ncRNAs). Focusing on the total RNA of a cell rather than the highly selected polyA+ transcripts seems to complement the latest catalog of lincRNAs (Cabili et al., 2011) since only 40% of these vlincRNAs overlap the lincRNA genes. We also recently showed that the GENCODE lncRNA set tends to have higher PolyA− representation compared to protein-coding mRNAs (Derrien et al., submitted). Although many studies have concentrated on the intergenic lncRNAs (the lincRNAs), this seriously underestimates the true number of lncRNA transcripts in the genome. Approximately one third (Derrien et al., submitted) to one half (Jia et al., 2010) of lncRNAs overlap protein-coding loci in some way – “genic” lncRNAs. It seems therefore essential to annotate lncRNAs both in intergenic and coding regions since (i) the exact boundaries of protein-coding genes is frequently subject to variations and reannotations (Denoeud et al., 2007; Gingeras, 2007) and thus could lead to the revision of a lincRNAs into a bona-fide lncRNAs, (ii) thousands of protein-coding genes harbor natural antisense transcripts belonging to the lncRNAs class (He et al., 2008; iii) numerous functional genic lncRNAs overlapping protein-coding genes have been experimentally validated, especially in disease states (Faghihi et al., 2008; Pasmant et al., 2011; Wapinski and Chang, 2011). A recent catalog of both genic and intergenic lncRNAs has been released based on genome-wide computational approach combined with intensive manual annotation. This led to the identification oh 6,736 lncRNA genes in human (Jia et al., 2010) among which 63% are localized within or in a close proximity (<10 kb) of known protein coding genes (Jia et al., 2010).
The GENCODE Catalog of Human lncRNAs
Most recently, the GENCODE annotation group has produced the most comprehensive, high-quality human lncRNA annotation to date. In order to identify all evidence-based functional gene features in the human genome, the GENCODE group (Harrow et al., 2006) within the ENCODE framework (ENCyclopedia Of DNA Elements; ENCODE Project Consortium et al., 2007) provides a high-quality collection of lncRNAs. GENCODE annotation involves manual curation, multiple computational analysis, and targeted experimental approaches, all together representing complementary methodologies for the complete identification of all human functional elements (coding and non-coding genes). At present, the GENCODE collection (Version 7) comprises 14,880 lncRNA transcripts arising from 9,277 distinct gene loci (Derrien et al., submitted).
In a recent study, we investigated whether these lncRNAs are under negative evolutionary selection, indicative of functionality (Derrien et al., submitted). Evolutionary scores were computed based both on the phastCons program (Siepel et al., 2005) and custom BLAST alignments within mammals in order to measure the conservation profiles of GENCODE lncRNAs in comparison with protein-coding transcripts and ancestral repeats (ARs), the latter representing a good proxy for measuring neutrally evolving sequences (Ponjavic et al., 2007). Overall, lncRNAs show moderate sequence conservation compared to coding transcripts. This lower sequence conservation may reflect the fact that functional RNA structures are more robust in the face of sequence mutations and insertions–deletions (indels), compared to the higher constraints inherent of protein-coding open reading frames. Nevertheless, lncRNAs and more especially, their promoters, showed statistically significant, non-random conservation, strongly suggesting a functional role for these ncRNAs. Interestingly, about one third of the 15,000 lncRNAs display a primate-specific pattern of conservation (Derrien et al., submitted).
Using whole transcriptome sequencing (RNAseq) of 16 human cell lines produced in the framework of the ENCODE consortium (ENCODE Project Consortium et al., 2007) and 16 tissues from the Human Body Map project (www.illumina.com), we showed that 94% of the GENCODE lncRNAs transcripts are expressed in at least one of these tissue/cell line studied. Strikingly, the level of expression of polyA+ lncRNAs is ∼10–20 times lower than protein-coding transcripts reinforcing the need to use deep sequencing based technologies to identify these low expressed non-coding loci (Figure 1.). We also demonstrated that lncRNAs tend to be enriched in nucleus in comparison with mRNAs; this latter observation being consistent with the idea that many lncRNAs may be devoted to gene regulation in the nucleus. Finally, the question is raised as to whether lincRNAs could encode very small peptides as shown by Ingolia et al. (2011). However, there is still conflicting evidence about this hypothesis since a recent study which used comprehensive mass spectrometry data (MS) produced as part of the ENCODE project only found about a hundred of GENCODE lncRNA to be matched by small peptides (Banfai et al., submitted).
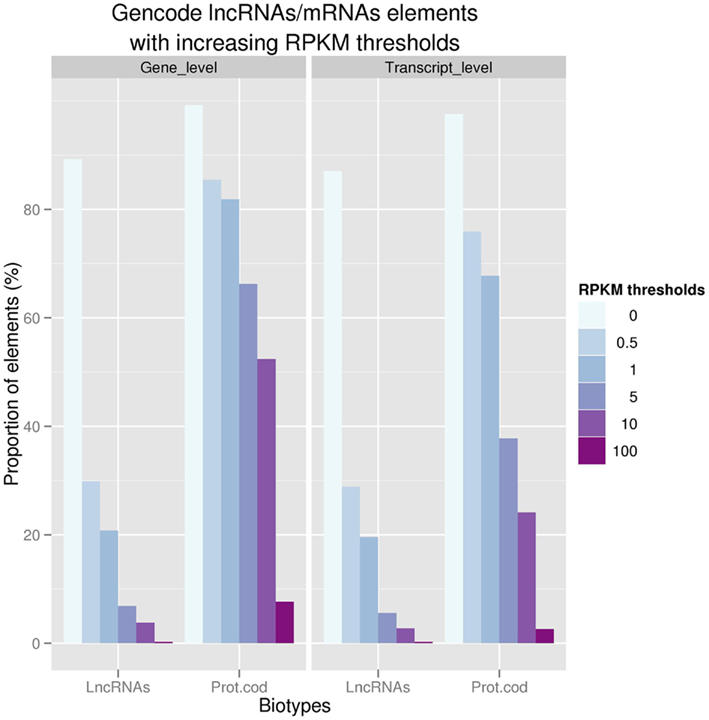
Figure 1. Proportion of GENCODE polyA+ LncRNAs and protein coding at the gene (n = 9,277 and 18,063; respectively) and transcript levels with increasing thresholds of expression values (RPKM) in ENCODE RNASeq experiments.
Conclusion
Over the past decade, the estimation of the proportion of “functional DNA” in the human genome has been constantly revised upward (Ponting and Hardison, 2011).
We now know that the human genome contains thousands of lncRNAs, both genic and intergenic. This new class of non-protein coding RNAs (ncRNAs) lack functional ORFs, are modestly conserved and seem to negatively and positively regulate protein coding gene expression, in cis and trans. Diverse mechanisms of action have been observed (see for reviews Ponting et al., 2009; Nagano and Fraser, 2011) suggesting that lncRNAs are a fundamental regulators of transcription. The classification of lncRNAs remains difficult, and we presently have only a vague idea of what sub-categories exist, and how we might use experimental or sequence information to distinguish between such categories. With the ongoing and increasing number of RNAseq experiments characterizing transcriptomes of multiples cell lines and human tissues (in particular within the ENCODE consortium), it is likely that the number of annotated lncRNAs will increase dramatically in the near future. Future studies will likely focus on identifying functional lncRNAs, and those involved in human disease processes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank reviewers for the helpful comments.
References
Amaral, P. P., Dinger, M. E., Mercer, T. R., and Mattick, J. S. (2008). The Eukaryotic genome as an RNA machine. Science 319, 1787–1789.
Amaral, P. P., Michael, B. C., Dennis, K. G., Marcel, E. D., and John, S. M. (2011). lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res. 39, D146–D151.
Brockdorff, N., Ashworth, A., Kay, G. F., McCabe, V. M., Norris, D. P., Cooper, P. J., Swift, S., and Rastan, S. (1992). The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 71, 515–526.
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., and Rinn, J. L. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. doi:10.1101/gad.17446611
Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., Oyama, R., Ravasi, T., Lenhard, B., Wells, C., Kodzius, R., Shimokawa, K., Bajic, V. B., Brenner, S. E., Batalov, S., Forrest, A. R., Zavolan, M., Davis, M. J., Wilming, L. G., Aidinis, V., Allen, J. E., Ambesi-Impiombato, A., Apweiler, R., Aturaliya, R. N., Bailey, T. L., Bansal, M., Baxter, L., Beisel, K. W., Bersano, T., Bono, H., Chalk, A. M., Chiu, K. P., Choudhary, V., Christoffels, A., Clutterbuck, D. R., Crowe, M. L., Dalla, E., Dalrymple, B. P., de Bono, B., Della Gatta, G., di Bernardo, D., Down, T., Engstrom, P., Fagiolini, M., Faulkner, G., Fletcher, C. F., Fukushima, T., Furuno, M., Futaki, S., Gariboldi, M., Georgii-Hemming, P., Gingeras, T. R., Gojobori, T., Green, R. E., Gustincich, S., Harbers, M., Hayashi, Y., Hensch, T. K., Hirokawa, N., Hill, D., Huminiecki, L., Iacono, M., Ikeo, K., Iwama, A., Ishikawa, T., Jakt, M., Kanapin, A., Katoh, M., Kawasawa, Y., Kelso, J., Kitamura, H., Kitano, H., Kollias, G., Krishnan, S. P., Kruger, A., Kummerfeld, S. K., Kurochkin, I. V., Lareau, L. F., Lazarevic, D., Lipovich, L., Liu, J., Liuni, S., McWilliam, S., Madan Babu, M., Madera, M., Marchionni, L., Matsuda, H., Matsuzawa, S., Miki, H., Mignone, F., Miyake, S., Morris, K., Mottagui-Tabar, S., Mulder, N., Nakano, N., Nakauchi, H., Ng, P., Nilsson, R., Nishiguchi, S., Nishikawa, S., Nori, F., Ohara, O., Okazaki, Y., Orlando, V., Pang, K. C., Pavan, W. J., Pavesi, G., Pesole, G., Petrovsky, N., Piazza, S., Reed, J., Reid, J. F., Ring, B. Z., Ringwald, M., Rost, B., Ruan, Y., Salzberg, S. L., Sandelin, A., Schneider, C., Schönbach, C., Sekiguchi, K., Semple, C. A., Seno, S., Sessa, L., Sheng, Y., Shibata, Y., Shimada, H., Shimada, K., Silva, D., Sinclair, B., Sperling, S., Stupka, E., Sugiura, K., Sultana, R., Takenaka, Y., Taki, K., Tammoja, K., Tan, S. L., Tang, S., Taylor, M. S., Tegner, J., Teichmann, S. A., Ueda, H. R., van Nimwegen, E., Verardo, R., Wei, C. L., Yagi, K., Yamanishi, H., Zabarovsky, E., Zhu, S., Zimmer, A., Hide, W., Bult, C., Grimmond, S. M., Teasdale, R. D., Liu, E. T., Brusic, V., Quackenbush, J., Wahlestedt, C., Mattick, J. S., Hume, D. A., Kai, C., Sasaki, D., Tomaru, Y., Fukuda, S., Kanamori-Katayama, M., Suzuki, M., Aoki, J., Arakawa, T., Iida, J., Imamura, K., Itoh, M., Kato, T., Kawaji, H., Kawagashira, N., Kawashima, T., Kojima, M., Kondo, S., Konno, H., Nakano, K., Ninomiya, N., Nishio, T., Okada, M., Plessy, C., Shibata, K., Shiraki, T., Suzuki, S., Tagami, M., Waki, K., Watahiki, A., Okamura-Oho, Y., Suzuki, H., Kawai, J., Hayashizaki, Y., FANTOM Consortium, RIKEN Genome Exploration Research Group and Genome Science Group (Genome Network Project Core Group). (2005). The transcriptional landscape of the mammalian genome. Science 309, 1559–1563.
Denoeud, F., Kapranov, P., Ucla, C., Frankish, A., Castelo, R., Drenkow, J., Lagarde, J., Alioto, T., Manzano, C., Chrast, J., Dike, S., Wyss, C., Henrichsen, C. N., Holroyd, N., Dickson, M. C., Taylor, R., Hance, Z., Foissac, S., Myers, R. M., Rogers, J., Hubbard, T., Harrow, J., Guigó, R., Gingeras, T. R., Antonarakis, S. E., and Reymond, A. (2007). Prominent use of distal 5′ transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res. 17, 746–759.
Ebisuya, M., Yamamoto, T., Nakajima, M., and Nishida, E. (2008). Ripples from neighbouring transcription. Nat. Cell Biol. 10, 1106–1113.
ENCODE Project Consortium, Birney, E., Stamatoyannopoulos, J. A., Dutta, R., Guigo, A., Gingeras, T. R., Margulies, E. H., Weng, Z., Snyder, M., Dermitzakis, E. T., Thurman, R. E., Kuehn, M. S., Taylor, C. M., Neph, S., Koch, C. M., Asthana, S., Malhotra, A., Adzhubei, I., Greenbaum, J. A., Andrews, R. M., Flicek, P., Boyle, P. J., Cao, H., Carter, N. P., Clelland, G. K., Davis, S., Day, N., Dhami, P., Dillon, S. C., Dorschner, M. O., Fiegler, H., Giresi, P. G., Goldy, J., Hawrylycz, M., Haydock, A., Humbert, R., James, K. D., Johnson, B. E., Johnson, E. M., Frum, T. T., Rosenzweig, E. R., Karnani, N., Lee, K., Lefebvre, G. C., Navas, P. A., Neri, F., Parker, S. C., Sabo, P. J., Sandstrom, R., Shafer, A., Vetrie, D., Weaver, M., Wilcox, S., Yu, M., Collins, F. S., Dekker, J., Lieb, J. D., Tullius, T. D., Crawford, G. E., Sunyaev, S., Noble, W. S., Dunham, I., Denoeud, F., Reymond, A., Kapranov, P., Rozowsky, J., Zheng, D., Castelo, R., Frankish, A., Harrow, J., Ghosh, S., Sandelin, A., Hofacker, I. L., Baertsch, R., Keefe, D., Dike, S., Cheng, J., Hirsch, H. A., Sekinger, E. A., Lagarde, J., Abril, J. F., Shahab, A., Flamm, C., Fried, C., Hackermüller, J., Hertel, J., Lindemeyer, M., Missal, K., Tanzer, A., Washietl, S., Korbel, J., Emanuelsson, O., Pedersen, J. S., Holroyd, N., Taylor, R., Swarbreck, D., Matthews, N., Dickson, M. C., Thomas, D. J., Weirauch, M. T., Gilbert, J., Drenkow, J., Bell, I., Zhao, X., Srinivasan, K. G., Sung, W. K., Ooi, H. S., Chiu, K. P., Foissac, S., Alioto, T., Brent, M., Pachter, L., Tress, M. L., Valencia, A., Choo, S. W., Choo, C. Y., Ucla, C., Manzano, C., Wyss, C., Cheung, E., Clark, T. G., Brown, J. B., Ganesh, M., Patel, S., Tammana, H., Chrast, J., Henrichsen, C. N., Kai, C., Kawai, J., Nagalakshmi, U., Wu, J., Lian, Z., Lian, J., Newburger, P., Zhang, X., Bickel, P., Mattick, J. S., Carninci, P., Hayashizaki, Y., Weissman, S., Hubbard, T., Myers, R. M., Rogers, J., Stadler, P. F., Lowe, T. M., Wei, C. L., Ruan, Y., Struhl, K., Gerstein, M., Antonarakis, S. E., Fu, Y., Green, E. D., Karaöz, U., Siepel, A., Taylor, J., Liefer, L. A., Wetterstrand, K. A., Good, P. J., Feingold, E. A., Guyer, M. S., Cooper, G. M., Asimenos, G., Dewey, C. N., Hou, M., Nikolaev, S., Montoya-Burgos, J. I., Löytynoja, A., Whelan, S., Pardi, F., Massingham, T., Huang, H., Zhang, N. R., Holmes, I., Mullikin, J. C., Ureta-Vidal, A., Paten, B., Seringhaus, M., Church, D., Rosenbloom, K., Kent, W. J., Stone, E. A., NISC Comparative Sequencing Program, Baylor College of Medicine Human Genome Sequencing Center, Washington University Genome Sequencing Center, Broad Institute, Children’s Hospital Oakland Research Institute, Batzoglou, S., Goldman, N., Hardison, R. C., Haussler, D., Miller, W., Sidow, A., Trinklein, N. D., Zhang, Z. D., Barrera, L., Stuart, R., King, D. C., Ameur, A., Enroth, S., Bieda, M. C., Kim, J., Bhinge, A. A., Jiang, N., Liu, J., Yao, F., Vega, V. B., Lee, C. W., Ng, P., Shahab, A., Yang, A., Moqtaderi, Z., Zhu, Z., Xu, X., Squazzo, S., Oberley, M. J., Inman, D., Singer, M. A., Richmond, T. A., Munn, K. J., Rada-Iglesias, A., Wallerman, O., Komorowski, J., Fowler, J. C., Couttet, P., Bruce, A. W., Dovey, O. M., Ellis, P. D., Langford, C. F., Nix, D. A., Euskirchen, G., Hartman, S., Urban, A. E., Kraus, P., Van Calcar, S., Heintzman, N., Kim, T. H., Wang, K., Qu, C., Hon, G., Luna, R., Glass, C. K., Rosenfeld, M. G., Aldred, S. F., Cooper, S. J., Halees, A., Lin, J. M., Shulha, H. P., Zhang, X., Xu, M., Haidar, J. N., Yu, Y., Ruan, Y., Iyer, V. R., Green, R. D., Wadelius, C., Farnham, P. J., Ren, B., Harte, R. A., Hinrichs, A. S., Trumbower, H., Clawson, H., Hillman-Jackson, J., Zweig, A. S., Smith, K., Thakkapallayil, A., Barber, G., Kuhn, R. M., Karolchik, D., Armengol, L., Bird, C. P., de Bakker, P. I., Kern, A. D., Lopez-Bigas, N., Martin, J. D., Stranger, B. E., Woodroffe, A., Davydov, E., Dimas, A., Eyras, E., Hallgrímsdóttir, I. B., Huppert, J., Zody, M. C., Abecasis, G. R., Estivill, X., Bouffard, G. G., Guan, X., Hansen, N. F., Idol, J. R., Maduro, V. V., Maskeri, B., McDowell, J. C., Park, M., Thomas, P. J., Young, A. C., Blakesley, R. W., Muzny, D. M., Sodergren, E., Wheeler, D. A., Worley, K. C., Jiang, H., Weinstock, G. M., Gibbs, R. A., Graves, T., Fulton, R., Mardis, E. R., Wilson, R. K., Clamp, M., Cuff, J., Gnerre, S., Jaffe, D. B., Chang, J. L., Lindblad-Toh, K., Lander, E. S., Koriabine, M., Nefedov, M., Osoegawa, K., Yoshinaga, Y., Zhu, B., and de Jong, P. J. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816.
Faghihi, M. A., Modarresi, F., Khalil, A. M., Wood, D. E., Sahagan, B. G., Morgan, T. E., Finch, C. E., St Laurent, G. III., Kenny, P. J., and Wahlestedt, C. (2008). Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 14, 723–730.
Gabory, A., Ripoche, M.-A., Le Digarcher, A., Watrin, F., Ziyyat, A., Forné, T., Jammes, H., Ainscough, J. F., Surani, M. A., Journot, L., and Dandolo, L. (2009). H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 136, 3413–3421.
Gaughwin, P. M., Ciesla, M., Lahiri, N., Tabrizi, S. J., Brundin, P., and Björkqvist, M. (2011). Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 20, 2225–2237.
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., Huarte, M., Zuk, O., Carey, B. W., Cassady, J. P., Cabili, M. N., Jaenisch, R., Mikkelsen, T. S., Jacks, T., Hacohen, N., Bernstein, B. E., Kellis, M., Regev, A., Rinn, J. L., and Lander, E. S. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227.
Guttman, M., Donaghey, J., Carey, B. W., Garber, M., Grenier, J. K., Munson, G., Young, G., Lucas, A. B., Ach, R., Bruhn, L., Yang, X., Amit, I., Meissner, A., Regev, A., Rinn, J. L., Root, D. E., and Lander, E. S. (2011). lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 1–11.
Harrow, J., Denoeud, F., Frankish, A., Reymond, A., Chen, C. K., Chrast, J., Lagarde, J., Gilbert, J. G., Storey, R., Swarbreck, D., Rossier, C., Ucla, C., Hubbard, T., Antonarakis, S. E., and Guigo, R. (2006). GENCODE: producing a reference annotation for ENCODE. Genome Biol. 7(Suppl. 1), S4.1–S9.
He, Y., Vogelstein, B., Velculescu, V. E., Papadopoulos, N., and Kinzler, K. W. (2008). The antisense transcriptomes of human cells. Science 322, 1855–1857.
Huarte, M., Guttman, M., Feldser, D., Garber, M., Koziol, M. J., Kenzelmann-Broz, D., Khalil, A. M., Zuk, O., Amit, I., Rabani, M., Attardi, L. D., Regev, A., Lander, E. S., Jacks, T., and Rinn, J. L. (2010). A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142, 409–419.
Ingolia, N. T., Lareau, L. F., and Weissman, J. S. (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802.
Jia, H., Osak, M., Bogu, G. K., Stanton, L. W., Johnson, R., and Lipovich, L. (2010). Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 16, 1478–1487.
Kapranov, P., St. Laurent, G., Raz, T., Ozsolak, F., Reynolds, C. P., Sorensen, P. H., Reaman, G., Milos, P., Arceci, R. J., Thompson, J. F., and Triche, T. J. (2010). The majority of total nuclear-encoded non-ribosomal RNA in a human cell is ‘dark matter’ un-annotated RNA. BMC Biol. 8, 149. doi:10.1186/1741-7007-8-149
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D., Thomas, K., Presser, A., Bernstein, B. E., van Oudenaarden, A., Regev, A., Lander, E. S., and Rinn, J. L. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. 106, 11667–11672.
Kim, T. K., Hemberg, M., Gray, J. M., Costa, A. M., Bear, D. M., Wu, J., Harmin, D. A., Laptewicz, M., Barbara-Haley, K., Kuersten, S., Markenscoff-Papadimitriou, E., Kuhl, D., Bito, H., Worley, P. F., Kreiman, G., and Greenberg, M. E. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187.
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854.
Lunter, G., Ponting, C. P., and Hein, J. (2006). Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput. Biol. 2, e5. doi:10.1371/journal.pcbi.0020005
Maeda, N., Kasukawa, T., Oyama, R., Gough, J., Frith, M., Engström, P. G., Lenhard, B., Aturaliya, R. N., Batalov, S., Beisel, K. W., Bult, C. J., Fletcher, C. F., Forrest, A. R., Furuno, M., Hill, D., Itoh, M., Kanamori-Katayama, M., Katayama, S., Katoh, M., Kawashima, T., Quackenbush, J., Ravasi, T., Ring, B. Z., Shibata, K., Sugiura, K., Takenaka, Y., Teasdale, R. D., Wells, C. A., Zhu, Y., Kai, C., Kawai, J., Hume, D. A., Carninci, P., and Hayashizaki, Y. (2006). Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genet. 2, e62. doi:10.1371/journal.pgen.0020062
Mancini-Dinardo, D., Steele, S. J., Levorse, J. M., Ingram, R. S., and Tilghman, S. M. (2006). Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 20, 1268–1282.
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., McCarthy, M. I., Ramos, E. M., Cardon, L. R., Chakravarti, A., Cho, J. H., Guttmacher, A. E., Kong, A., Kruglyak, L., Mardis, E., Rotimi, C. N., Slatkin, M., Valle, D., Whittemore, A. S., Boehnke, M., Clark, A. G., Eichler, E. E., Gibson, G., Haines, J. L., Mackay, T. F., McCarroll, S. A., and Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753.
Marques, A. C., and Ponting, C. P. (2009). Catalogues of mammalian long noncoding RNAs: modest conservation and incompleteness. Genome Biol. 10, R124.
Mohammad, F., Mondal, T., Guseva, N., Pandey, G. K., and Kanduri, C. (2010). Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 137, 2493–2499.
Nagano, T., and Fraser, P. (2011). No-nonsense functions for long noncoding RNAs. Cell 145, 178–181.
Nagano, T., Mitchell, J. A., Sanz, L. A., Pauler, F. M., Ferguson-Smith, A. C., Feil, R., and Fraser, P. (2008). The air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322, 1717–1720.
Ørom, U. A., Derrien, T., Beringer, M., Gumireddy, K., Gardini, A., Bussotti, G., Lai, F., Zytnicki, M., Notredame, C., Huang, Q., Guigo, R., and Shiekhattar, R. (2010). Long noncoding RNAs with enhancer-like function in human cells. Cell 143, 46–58.
Parker, B. J., Moltke, I., Roth, A., Washietl, S., Wen, J., Kellis, M., Breaker, R., and Pedersen, J. S. (2011). New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 21, 1929–1943.
Pasmant, E., Laurendeau, I., Sabbagh, A., Parfait, B., Vidaud, M., Vidaud, D., and Bièche, I. (2010). The amazing story of ANRIL, a long non-coding RNA. Med. Sci. (Paris) 26, 564–566.
Pasmant, E., Sabbagh, A., Vidaud, M., and Bièche, I. (2011). ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 25, 444–448.
Ponjavic, J., Ponting, C. P., and Lunter, G. (2007). Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 17, 556–565.
Ponting, C. P., and Hardison, R. C. (2011). What fraction of the human genome is functional? Genome Res. 21, 1769–1776.
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641.
Pushkarev, D., Neff, N. F., and Quake, S. R. (2009). Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 27, 847–850.
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., Goodnough, L. H., Helms, J. A., Farnham, P. J., Segal, E., and Chang, H. Y. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323.
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K., Clawson, H., Spieth, J., Hillier, L. W., Richards, S., Weinstock, G. M., Wilson, R. K., Gibbs, R. A., Kent, W. J., Miller, W., and Haussler, D. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050.
Sleutels, F., Zwart, R., and Barlow, D. P. (2002). The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415, 810–813.
Torarinsson, E., Sawera, M., Havgaard, J. H., Fredholm, M., and Gorodkin, J. (2006). Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure. Genome Res. 16, 885–889.
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S. F., Schroth, G. P., and Burge, C. B. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476.
Wapinski, O., and Chang, H. Y. (2011). Long noncoding RNAs and human disease. Trends Cell Biol. 21, 354–361.
Wroe, S. F., Kelsey, G., Skinner, J. A., Bodle, D., Ball, S. T., Beechey, C. V., Peters, J., and Williamson, C. M. (2000). An imprinted transcript, antisense to Nesp, adds complexity to the cluster of imprinted genes at the mouse Gnas locus. Proc. Natl. Acad. Sci. U.S.A. 97, 3342–3346.
Keywords: non-coding RNAs, regulation, long non-coding RNA, epigenetics
Citation: Derrien T, Guigó R and Johnson R (2012) The long non-coding RNAs: a new (p)layer in the “dark matter”. Front. Gene. 2:107. doi: 10.3389/fgene.2011.00107
Received: 06 September 2011;
Accepted: 21 December 2011;
Published online: 09 January 2012.
Edited by:
Philipp Kapranov, St. Laurent Institute, USAReviewed by:
Yohei Kirino, Cedars-Sinai Medical Center, USAChris Ponting, MRC Functional Genomics Unit, UK
Copyright: © 2012 Derrien, Guigó and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Thomas Derrien, Bioinformatics and Genomics Group, Centre for Genomic Regulation, Biomedical Research Park of Barcelona, C. Dr. Aiguader 88, Barcelona 08003, Spain. e-mail: toma.derrien@gmail.com