 Tomasz K. Wojdacz1,2,3*
Tomasz K. Wojdacz1,2,3*- 1 The Lundbeck Foundation Centre for International Research in Radiation Oncology, Aarhus C, Denmark
- 2 Department of Experimental Clinical Oncology, Aarhus University Hospital, Aarhus C, Denmark
- 3 Department of Biomedicine, University of Aarhus, Aarhus C, Denmark
The terms methylation quantification and qualification seem self-explanatory however, the results of experiments aiming to quantify or qualify locus specific methylation in clinical material are often difficult to interpret. There are three main reasons for difficulties in understanding methylation status measurement. First, the complexity of locus specific methylation patterns, which oscillate between unmethylated, fully methylated, and heterogeneously methylated. Second the interpretation of methylation-screening results can frequently be problematic due to limitations of the methods used. And finally the specifications of the clinical samples used in laboratory practice frequently hamper the methylation measurement. Thus, the process of quantification and qualification of methylation has to be discussed with consideration of the specific locus analyzed, the methodology used, and the clinical material source used in each specific experiment. The question of the clinical significance of determination of different methylation levels is even more complicated, with substantial evidence for correlation between qualitative methylation changes and clinical features of the disease and at the same time no data showing that different relative levels of methylation alter the disease outcome. The limitations of methylation quantification and qualification are discussed in this mini-review.
Methylation
DNA methylation in humans generally describes a process of addition of a methyl group to carbon 5 of cytosine (5-mC) within a 5′-CpG-3′ dinucleotide. In normal cell physiology DNA methylation modulates gene expression. Deregulation of the normal pattern of methylation leads to aberrant expression of the genes, which in turn can initiate the disease or contribute to the disease phenotype (Esteller, 2008). Hence, measurements of the methylation status of the CpG dinucleotides become very important from both a biological and a medical/clinical point of view.
Complexity of PCR Based Measurement of Methylation
Use of PCR in Methylation Studies
The vast majority of techniques currently used for measurement of methylation are based on PCR amplification. However, the use of PCR amplification in methylation studies is not straightforward. In the cell, methylation marks are added to the replicated DNA strand in a semi conservative fashion by DNMTs (DNA methyltransferase). No DNMTs are present in standard PCR and information on methylation status of CpG sites is lost in the first round of PCR. Therefore, in methylation studies prior to the use of PCR, the methylation marks have to be preserved on the template DNA. Modification of the template DNA with sodium bisulfite is most commonly used to preserve methylation marks before PCR amplification. Sodium bisulfite implements methylation dependent base changes in the DNA strand by converting cytosines into uracil and leaving 5-mC intact. The amplification of bisulfite-modified template results in different nucleotide composition of the amplification product depending on whether it originates from a methylated, unmethylated, or heterogeneously methylated template (Figure 1).
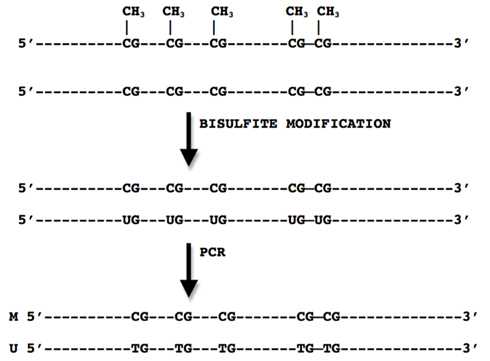
Figure 1. Principles of bisulfite modification where methylated CpG sites remain unchanged while cytosines are converted to uracil.
Design of PCR for Amplification of Bisulfite-Modified Template
When analyzing methylation of a number of CpG sites in a DNA strand of certain length, three scenarios are possible: all CpG sites within the sequence of interest can be devoid of methylation, or can be methylated and the third scenario is referred to as a heterogeneous methylation status where a different number of CpG sites can be methylated at different alleles (Figure 2). The PCR amplification in methylation studies can specifically target fully methylated or unmethylated variant of the locus [methylation-specific PCR (MSP) based amplification, see below] or can amplify the locus of interest regardless of the methylation status for post-PCR methylation analyses [methylation independent primers (MIP) based amplification].
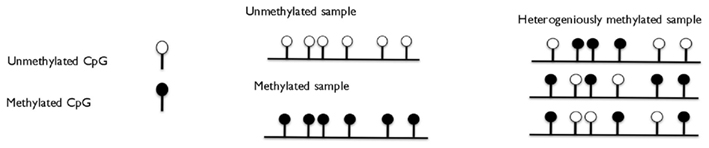
Figure 2. Three stages of methylation of the locus: fully methylated, fully unmethylated, and heterogeneously methylated.
MSP Based Approach
When using the MSP based approach the primers are in principle designed to amplify only fully methylated template (another set can potentially be designed to target unmethylated template; Herman et al., 1996). In order to achieve methylation-specific amplification, the primer set has to, in general, contain as many CpG sites as possible. This will ensure specific binding only to the template where all the CpG sites are methylated and therefore not converted by sodium bisulfite. For this technology no amplification is allowed from partly methylated (heterogeneously methylated) templates and the stringency of PCR conditions have to be optimized to ensure specific amplification. If any amplification from mismatched primers (partly methylated templates) takes place, the assay cannot be considered reliable due to the possibility of generating false positive amplifications (Aggerholm and Hokland, 2000). To obtain quantitative measurement of fully methylated template in the sample, a fluorescent probe can be flanked by MSP primers or DNA intercalating dye can be added to PCR [methods called Q (quantitative)-MSP or MethyLight] to enable real-time monitoring of the amplification (Trinh et al., 2001).
MIP Based Approach
Methylation independent primers based technologies utilize primers that aim to amplify the locus of interest regardless of its methylation status in the same PCR (Clark et al., 1994; Wojdacz et al., 2008c). The PCR product amplified with MIP primers will differ in base content depending whether it originates from methylated, unmethylated, or heterogeneously methylated templates. In this approach a post-PCR technology, e.g., melting or sequencing is required to investigate the methylation status of the sample (Wojdacz and Hansen, 2006). It is important to keep in mind that when using the MIP approach the post-PCR methylation “read out” reflects the average state of methylation of single CpG site for all the PCR products present in the sample. Unless technologies like cloning of PCR products followed by sequencing (pyrosequencing or Sanger sequencing) or second-generation sequencing are used in combination with MIP based amplifications no information on the methylation status of single alleles (PCR products) in the sample can be obtained (Clark et al., 1994; Fazzari and Greally, 2010).
PCR Bias in MIP Based Amplifications
Proportional amplification of methylated and unmethylated templates with one primer set in a single PCR is in most cases compromised by the PCR bias phenomenon (Wojdacz et al., 2009). PCR bias in methylation studies is described as preferential amplification of the unmethylated template (Warnecke et al., 1997). Modification of the PCR chemistry has been shown not to eliminate the PCR bias from MIP based amplifications (Warnecke et al., 1997). However, specific primer design and empirical optimization of the PCR conditions have been shown to resolve the PCR bias problem and reassure proportional amplification of methylated and unmethylated templates (details of the design can be found in Wojdacz et al., 2008c).
Interpretation of PCR Based Methylation-Screening Results
Qualitative Methylation Measurement
To assess methylation in a qualitative manner means to show that the methylated (or unmethylated) sequence of interest is present in the screened sample at detection level that is characteristic for a given methylation detection technology. Both MSP and MIP based methods can be used to qualify methylation. The qualitative result cannot be discussed in the context of methylation levels but only indicate presence or absence of methylation. The only instance when qualitative results are referred to as “methylation levels” in the literature is when reporting a number of methylation positive samples in a total number of screened samples. However, the term “the methylation frequency” is more accurate in this case. Over the years MSP was the gold standard method for qualification of methylation. This method however, only confirms the fully methylated status of the CpG sites within primer binding sites. When MIP amplification is used, not only overall methylation status of the amplicon can be qualified by post-PCR technologies like high resolution melting (HRM), but also the methylation pattern within the amplicon can be investigated if MIP amplification is combined with sequencing based technologies (Wojdacz et al., 2010; Candiloro et al., 2011).
Quantitative Methylation Measurement
To quantify methylation means to “count” all the differently methylated molecules in the sample. Quantification of fully methylated (or fully unmethylated) molecules is relatively easy and can be described as percentage of methylated (or unmethylated) molecules of the overall number of molecules in the sample. Quantification of heterogeneous methylation is challenging. It requires sequencing of every single heterogeneously methylated allele in the screened sample, which currently only can be accomplished using next generation sequencing technologies (see below for detailed description).
The MSP approach can be utilized to investigate the number of fully methylated molecules in the sample. However the MSP primers have to be combined with a fluorescent probe or an intercalating dye to enable real-time monitoring of the increasing amount of the PCR product (Eads et al., 2000; Harden et al., 2003).
The MIP approach can also be used to quantify methylation levels, however this measurement is based on different principles than MSP based methylation level assessment. The MIP based methylation level measurement relies on the assumption that the post-PCR proportion of the PCR product originating from the methylated and unmethylated templates, reflects the pre-PCR proportion of methylated to unmethylated templates (assuming that PCR bias during amplification is controlled; Wojdacz and Dobrovic, 2007; Wojdacz et al., 2008b, 2009; Balic et al., 2009; Stanzer et al., 2010). Different methods can be used to measure the post-PCR proportion of “methylated” to “unmethylated” PCR products. Direct sequencing (or pyrosequencing) of MIP amplification products enables estimation of the average methylation levels at each CpG site (heights of the sequencing peaks at CpG sites). However due to limitations of the sequencing technologies (e.g., relatively low sensitivity) interpretation of the results can often be challenging (Wojdacz et al., 2010). Cloning and sequencing are significantly more precise but cost and labor consuming (Clark et al., 1994). At the same time, the result of single cloning and sequencing experiment is qualitative and reveals the methylation pattern of single allele. However, if the results of sequencing results for given locus from more than one clone are reported, the methylation frequency at each CpG site can be averaged over the number of clones and reported as methylation level. This is a very powerful technology allowing investigation of the methylation pattern of single alleles, but at the same time its precision with regard to methylation levels assessment depends on the number of clones sequenced.
Apart from sequencing technologies two other techniques methylation sensitive high resolution melting (MS-HRM) or combined bisulfite restriction analysis (COBRA) are currently most frequently utilized to measure the proportion of methylated to unmethylated PCR product after MIP amplification (Xiong and Laird, 1997; Wojdacz and Dobrovic, 2007). In MS-HRM methylation levels in screened samples can be calculated by means of comparison of the HRM profile of PCR product characteristic for the screened samples to the HRM profiles of the PCR products obtained by amplification of the mixes of artificially methylated and unmethylated standards with know proportion of methylated to unmethylated template (Wojdacz et al., 2008b). When using COBRA approach, quantification of enzymatic digestion products and comparison to the amount of the digest from control samples are used as surrogate measurement of methylation levels (Xiong and Laird, 1997).
Measurement of Heterogeneous Methylation
Only MIP based primers can be used to amplify heterogeneously methylated loci. The PCR product obtained after amplification of heterogeneously methylated loci derives from a vast number of alleles with different methylation pattern (Wojdacz et al., 2010; Candiloro et al., 2011). Currently, only second-generation sequencing technologies have the capacity to visualize the allelic variations in heterogeneously methylated samples. In the instances where approximate data on methylation status of single CpG sites within each allele in heterogeneously methylated sample is needed, approaches based on cloning and sequencing (as described above) can be applied. Identification of heterogeneous methylation in the sample can be performed by MS-HRM which currently is the only PCR based technique that can robustly identify PCR products derived from heterogeneously methylated samples as they display characteristic HRM profile (Wojdacz et al., 2010). However, MS-HRM results are not quantitative with regards to heterogeneous methylation and do not indicate methylation status of particular CpG sites within the sequence of interest.
In principle heterogeneous methylation can only be described as a qualitative pattern of the methylated CpG sites within the screened amplicon at each allele present is the sample. Consequently heterogeneous methylation should be reported, as a graphical illustration of different patterns of methylation for each specific screened allele. It is relatively easy to do, for the experiments employing cloning and sequencing of bisulfite-modified templates as those experiments normally involve a limited number of alleles in the samples. However it may be challenging for second-generation sequencing. Reporting heterogeneous methylation as average levels of methylation for each CpG site within the sequenced template can be very confusing. That is due to the fact that the raw result of Sanger/pyrosequencing of heterogeneously methylated templates is very similar to the results of sequencing of the sample containing a mix of fully methylated and unmethylated template (Wojdacz et al., 2010). There is however one instance where the averaged percentage of methylation for a number of clones sequenced from heterogeneously methylated sample can be used. It is known that within some loci single CpG sites undergo methylation independently and methylation is “seeded” at particular CpG sites over time, until all CpG sites within a sequence become methylated, e.g., during cancer progression from benign to malignant tumor or aging (Mikeska et al., 2010; Oster et al., 2011). The intermediate stage between fully methylated and unmethylated is in principle a heterogeneously methylated stage of the alleles. Therefore, to illustrate this gradual process of locus methylation, reporting an averaged number of methylated CpG sites over time, can be useful.
Clinical Material as a Limiting Factor for Quantification and Qualification of Methylation
In every day laboratory practice almost all clinical samples screened for methylation contain, apart form the tissue targeted by a given experiment, a fraction of different tissues. The “contaminating” fraction of tissues/cells is a consequence of the sample collection procedure, and may frequently constitute a considerable part of clinical specimen. It is relatively easy to qualify the methylation in such a sample, as the contamination does not interfere with technologies that can identify even a single molecule that undergoing hyper- or hypomethylation as long as the detection limit of the method is sufficiently high. However, when quantifying locus specific methylation in clinical samples the ratio between the abnormal (target) tissue/cells in the sample and the normal tissue/cells (e.g., tissue from which the abnormal tissue was dissected) will significantly influence quantification of methylation. The measurement of methylation levels in a sample with unknown fraction of contaminating tissues reflects only the methylation level in the given sample, and cannot be directly compared with the measurement of methylation in another sample where the fraction of contaminating tissues was not evaluated. In some cases the fraction of abnormal tissue in the clinical specimens can be evaluated prior to methylation analyses (e.g., microdissection or evaluation by pathologist) but normalization for the fraction of “contaminating tissue” in the sample is very difficult to implement in laboratory practice.
The second limitation of methylation quantification in laboratory practice is the heterogeneity of methylation within abnormal tissue, which is a product of different methylation pattern of alleles in abnormal tissue. The above phenomenon is seen in almost all cancer tumors, which develop, by accumulation of genetic/epigenetic abnormalities and clonal expansion. There is no robust method to evaluate tumor heterogeneity and therefore it is virtually impossible to normalize for tumor heterogeneity when investigating methylation levels. Moreover, DNA degradation can occur during sample collection and sample processing procedure involving fixation. And this can significantly influence the results.
Qualification and Quantification of Methylation for Clinical Applications
Potentially any methylation level detected in abnormal tissue can be clinically relevant especially in cancer research when a small part of tumor cells harboring methylated alleles can represent a part of the tumor, resistant to treatment. There is a lot of evidence that qualitative methylation assessment in the clinical sample correlates with clinical outcome. At the same time there is lack of evidence in the literature that two samples of the same malignant tissue (e.g., two breast cancer tumors) with relatively different methylation level at given loci, e.g., 50 and 70% correlate differently with the disease outcomes. Such a correlation has only been shown for imprinted loci where 50% methylation reflects methylation of one allele, 100% methylation of both alleles and 0% loss of methylation (Wojdacz et al., 2008a). However, this does not mean that only qualification of locus specific methylation and qualitative methods are sufficient for diagnostic applications. Recently a lot of attention is paid to the so-called low-levels of methylation that can be found in normal tissues and which do not appear to have pathological consequences (Wallace et al., 2010; Wojdacz et al., 2011a,b). Potential significance of this phenomenon in disease development is largely unknown. However, the fact that for some loci low-level methylation in healthy tissue turns into high levels methylation in abnormal tissue as a consequence of pathological processes, suggests a potential significance of low-level methylation for disease development. The increase of methylation levels in transformed tissue at affected loci can be explained by a stepwise theory of malignant transformation. Where the cell(s) with the acquired aberrant methylation (low-level methylation in normal tissue) only develops into malignant tissue when other initiating events occur and render affected cell capable of clonal proliferation. The clonal proliferation of cells harboring abnormally methylated loci results high levels of methylation of those loci seen in neoplastic tissue. When a qualitative method is used for methylation measurement, non-pathological (low-level) methylation can be mistaken for the pathologically significant methylation level. Therefore, despite the fact that at the current stage of knowledge only qualitative assessment of methylation was shown to have clinical applications the methods used for diagnostic methylation assessment have to be quantitative. Only quantitative methods allow (if needed) a cut-off point for the clinically significant methylation level to be established (Reinert et al., 2011).
Concluding Remarks
The primary issue in evaluation of clinical applicability of disease dependent methylation change at specific loci is correct interpretation of the results and awareness of the limitations of experimental protocols. The vast number of studies in the field has been shown to report discrepant results. One of the most vivid examples of the methodological shortcoming in the filed is a multicenter study by Hegi et al. (2005) which concluded: “The success rate of MSP on paraffin-embedded tumor samples was highly variable and center-dependent” or study by Aggerholm and Hokland (2000). The above may indicate the fact that not all limitations of the experiments are taken in to account when conclusions are drawn. Thus described here basic issues with the results interpretation in methylation studies should be considered in each study aiming to evaluate the clinical applicability of the disease dependent, locus specific methylation changes.
Conflict of Interest Statement
Tomasz K. Wojdacz is listed as an inventor on the patent pending application on the aspects of MS-HRM technology.
Acknowledgments
I would like to thank: Dr. Lise Lotte Hansen, Dr. Emilia Wiechec, and Britta B. Thestrup for the critical review of this manuscript. I would also like to thank: The Danish Cancer Society, CIRRO – The Lundbeck Foundation Center for Interventional Research in Radiation Oncology, and The Danish Council for Strategic Research for the support of my work.
References
Aggerholm, A., and Hokland, P. (2000). DAP-kinase CpG island methylation in acute myeloid leukemia: methodology versus biology? Blood 95, 2997–2999.
Balic, M., Pichler, M., Strutz, J., Heitzer, E., Ausch, C., Samonigg, H., Cote, R. J., and Dandachi, N. (2009). High quality assessment of DNA methylation in archival tissues from colorectal cancer patients using quantitative high-resolution melting analysis. J. Mol. Diagn. 11, 102–108.
Candiloro, I. L., Mikeska, T., and Dobrovic, A. (2011). Assessing combined methylation-sensitive high resolution melting and pyrosequencing for the analysis of heterogeneous DNA methylation. Epigenetics 6, 500–507.
Clark, S. J., Harrison, J., Paul, C. L., and Frommer, M. (1994). High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 22, 2990–2997.
Eads, C. A., Danenberg, K. D., Kawakami, K., Saltz, L. B., Blake, C., Shibata, D., Danenberg, P. V., and Laird, P. W. (2000). MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 28, E32.
Fazzari, M. J., and Greally, J. M. (2010). Introduction to epigenomics and epigenome-wide analysis. Methods Mol. Biol. 620, 243–265.
Harden, S. V., Sanderson, H., Goodman, S. N., Partin, A. A., Walsh, P. C., Epstein, J. I., and Sidransky, D. (2003). Quantitative GSTP1 methylation and the detection of prostate adenocarcinoma in sextant biopsies. J. Natl. Cancer Inst. 95, 1634–1637.
Hegi, M. E., Diserens, A. C., Gorlia, T., Hamou, M. F., De Tribolet, N., Weller, M., Kros, J. M., Hainfellner, J. A., Mason, W., Mariani, L., Bromberg, J. E., Hau, P., Mirimanoff, R. O., Cairncross, J. G., Janzer, R. C., and Stupp, R. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003.
Herman, J. G., Graff, J. R., Myohanen, S., Nelkin, B. D., and Baylin, S. B. (1996). Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U.S.A. 93, 9821–9826.
Mikeska, T., Candiloro, I. L., and Dobrovic, A. (2010). The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2, 561–573.
Oster, B., Thorsen, K., Lamy, P., Wojdacz, T. K., Hansen, L. L., Birkenkamp-Demtroder, K., Sorensen, K. D., Laurberg, S., Orntoft, T. F., and Andersen, C. L. (2011). Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int. J. Cancer 129, 2855–2866.
Reinert, T., Modin, C., Castano, F. M., Lamy, P., Wojdacz, T. K., Hansen, L. L., Wiuf, C., Borre, M., Dyrskjot, L., and Orntoft, T. F. (2011). Comprehensive genome methylation analysis in bladder cancer: identification and validation of novel methylated genes and application of these as urinary tumor markers. Clin. Cancer Res. 17, 5582–5592.
Stanzer, S., Balic, M., Strutz, J., Heitzer, E., Obermair, F., Hauser-Kronberger, C., Samonigg, H., and Dandachi, N. (2010). Rapid and reliable detection of LINE-1 hypomethylation using high-resolution melting analysis. Clin. Biochem. 43, 1443–1448.
Trinh, B. N., Long, T. I., and Laird, P. W. (2001). DNA methylation analysis by MethyLight technology. Methods 25, 456–462.
Wallace, K., Grau, M. V., Levine, A. J., Shen, L., Hamdan, R., Chen, X., Gui, J., Haile, R. W., Barry, E. L., Ahnen, D., Mckeown-Eyssen, G., Baron, J. A., and Issa, J. P. (2010). Association between folate levels and CpG Island hypermethylation in normal colorectal mucosa. Cancer Prev. Res. (Phila.) 3, 1552–1564.
Warnecke, P. M., Stirzaker, C., Melki, J. R., Millar, D. S., Paul, C. L., and Clark, S. J. (1997). Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res. 25, 4422–4426.
Wojdacz, T. K., Borgbo, T., and Hansen, L. L. (2009). Primer design versus PCR bias in methylation independent PCR amplifications. Epigenetics 4, 231–234.
Wojdacz, T. K., and Dobrovic, A. (2007). Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 35, e41.
Wojdacz, T. K., Dobrovic, A., and Algar, E. M. (2008a). Rapid detection of methylation change at H19 in human imprinting disorders using methylation-sensitive high-resolution melting. Hum. Mutat. 29, 1255–1260.
Wojdacz, T. K., Dobrovic, A., and Hansen, L. L. (2008b). Methylation-sensitive high-resolution melting. Nat. Protoc. 3, 1903–1908.
Wojdacz, T. K., Hansen, L. L., and Dobrovic, A. (2008c). A new approach to primer design for the control of PCR bias in methylation studies. BMC Res. Notes 1, 54.
Wojdacz, T. K., and Hansen, L. L. (2006). Techniques used in studies of age-related DNA methylation changes. Ann. N. Y. Acad. Sci. 1067, 479–487.
Wojdacz, T. K., Moller, T. H., Thestrup, B. B., Kristensen, L. S., and Hansen, L. L. (2010). Limitations and advantages of MS-HRM and bisulfite sequencing for single locus methylation studies. Expert Rev. Mol. Diagn. 10, 575–580.
Wojdacz, T. K., Thestrup, B. B., Cold, S., Overgaard, J., and Hansen, L. L. (2011a). No difference in the frequency of locus-specific methylation in the peripheral blood DNA of women diagnosed with breast cancer and age-matched controls. Future Oncol. 7, 1451–1455.
Wojdacz, T. K., Thestrup, B. B., Overgaard, J., and Hansen, L. L. (2011b). Methylation of cancer related genes in tumor and peripheral blood DNA from the same breast cancer patient as two independent events. Diagn. Pathol. 6, 116.
Keywords: methylation, quantification, qualification, MSP, MIP, cancer
Citation: Wojdacz TK (2012) The limitations of locus specific methylation qualification and quantification in clinical material. Front. Gene. 3:21. doi: 10.3389/fgene.2012.00021
Received: 17 October 2011; Accepted: 02 February 2012;
Published online: 28 February 2012.
Edited by:
Richard D. Emes, University of Nottingham, UKReviewed by:
Richard D. Emes, University of Nottingham, UKGuoping Fan, University of California Los Angeles, USA
Copyright: © 2012 Wojdacz. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Tomasz K. Wojdacz, Department of Biomedicine, University of Aarhus, Wilhelm Meyers Alle 4, 8000 Aarhus C, Denmark. e-mail: wojdacz@humgen.au.dk