 Guillermo Gervasini1* Jose M. Vagace2
Guillermo Gervasini1* Jose M. Vagace2- 1Department of Medical and Surgical Therapeutics, Division of Pharmacology, Medical School, University of Extremadura, Badajoz, Spain
- 2Service of Pediatric Hematology, University Hospital Infanta Cristina, Badajoz, Spain
The efficacy of chemotherapy in pediatric acute lymphoblastic leukemia (ALL) patients has significantly increased in the last 20 years; as a result, the focus of research is slowly shifting from trying to increase survival rates to reduce chemotherapy-related toxicity. At the present time, the cornerstone of therapy for ALL is still formed by a reduced number of drugs with a highly toxic profile. In recent years, a number of genetic polymorphisms have been identified that can play a significant role in modifying the pharmacokinetics and pharmacodynamics of these drugs. The best example is that of the TPMT gene, whose genotyping is being incorporated to clinical practice in order to individualize doses of mercaptopurine. However, there are additional genes that are relevant for the metabolism, activity, and/or transport of other chemotherapy drugs that are widely use in ALL, such as methotrexate, cyclophosphamide, vincristine, L-asparaginase, etoposide, cytarabine, or cytotoxic antibiotics. These genes can also be affected by genetic alterations that could therefore have clinical consequences. In this review we will discuss recent data on this field, with special focus on those polymorphisms that could be used in clinical practice to tailor chemotherapy for ALL in order to reduce the occurrence of serious adverse effects.
Introduction
In the last 20 years, the chemotherapy treatment of pediatric acute lymphoblastic leukemia (ALL), the most common malignancy in children, has reached success rates of up to 90%. This achievement has allowed limiting the administration of damaging cranial irradiation to rare cases with central nervous system (CNS) infiltration (Laningham et al., 2007). However, the clinical routine of chemotherapy treatments in these patients includes intrathecal and high intravenous doses of various drugs with a highly toxic profile. This is therefore a source of a wide variety of complications that add up to those caused by peripheral blood cell depression or the disease itself (Vagace and Gervasini, 2011). In fact, the toxicity of chemotherapy is a common cause of morbidity and mortality in children with ALL, as well as a frequent source of sequelae at mid-long term. These adverse effects are often the consequence of direct toxicity in healthy tissue, as a result of the low specificity displayed by these drugs and become more frequent as the treatment is intensified. In particular, central neurotoxicity is a major clinical concern in pediatric patients (Vagace et al., 2012).
In this scenario, it is obvious that any factor able to modify either the pharmacokinetics or pharmacodynamics of chemotherapy drugs holds the potential to be critical for the occurrence of serious adverse effects in ALL patients. One such factor is the presence of genetic polymorphisms in genes coding for drug-metabolizing enzymes, transporters, or drug targets. Genetic association studies in this field have traditionally been focused on efficacy parameters in general and survival in particular and therefore the body of work on the association with toxicity is still scarce.
In this review we will summarize what is currently known about how genetic variability can affect the toxicity induced by the main drugs used in the chemotherapy of ALL (see summary in Table 1). A special focus will be put on the discussion on those genetic analyses with the potential to tailor chemotherapy regimes in the ALL setting.
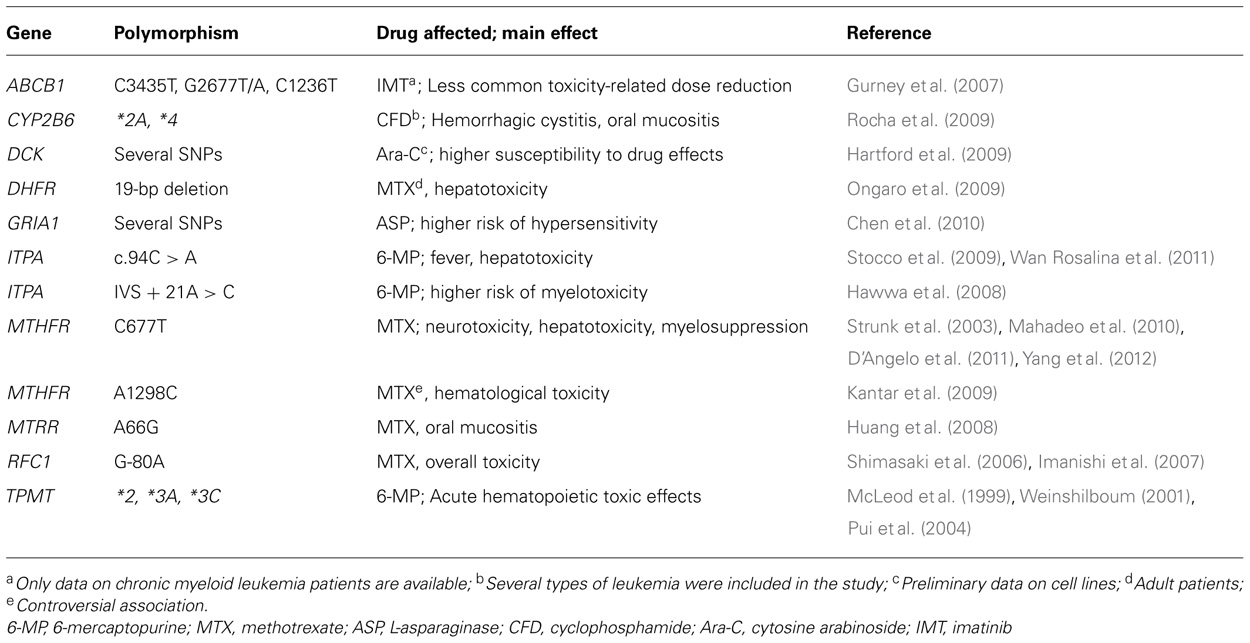
TABLE 1. Summary of genes and polymorphisms with a putative relevant role in chemotherapy-induced toxicity in acute lymphoblastic leukemia
L-Asparaginase
L-asparaginase is a standard component in the initial treatment of childhood ALL which induces the depletion of the essential amino acid L-asparagine in the tumor cells resulting in inhibition of protein synthesis.
This drug has been related to serious adverse effects in ALL patients, such as acute pancreatitis (Flores-Calderon et al., 2009) or cerebrovascular accidents (Gugliotta et al., 1992). In addition, hypersensitivity reactions occur in up to 25% of the patients and 10% of those may experience life-threatening anaphylactic reactions (Cortijo-Cascajares et al., 2012). Studies on the pharmacogenetics of asparaginase in ALL are in their early stages. A large study recently carried out at St Jude Children’s Hospital by Chen et al. (2010) interrogated more than 500 000 single nucleotide polymorphisms (SNPs) in 485 children with ALL and found that five mutations (rs4958351, rs10070447, rs6890057, rs4958676, and rs6889909) in the GRIA1 gene were associated with the occurrence of hypersensitivity to the drug. GRIA1 encodes a subunit of the AMPA receptor, a tetrameric ligand-gated ion channel that transmits glutamatergic signals in the brain. Glutamate not only has a role as a neurotransmitter, but also as an immunomodulator (Pacheco et al., 2007) and the study by Chen et al. (2011) provides the first link between GRIA1 polymorphisms and an immune-related phenotype such as the occurrence of hypersensitivity to L-asparaginase. A later genome-wide study by the same group using the HapMap lymphoblastoid cell lines tested more than 2 million SNPs and identified the aspartate metabolic routes as the most likely candidate pathway for asparaginase sensitivity.
Finally, polymorphisms in other genes that mediate the antileukemic effect of asparaginase, such as the asparaginase synthetase gene (ASNS), the basic region leucine zipper activating transcription factor 5 (ATF5), or the argininosuccinate synthase 1 (ASS1), have been associated to lower event-free survival of ALL patients, albeit the authors reported no associations with adverse effects (Rousseau et al., 2011).
Cyclophosphamide
Cyclophosphamide, an oxazophosphorine, bifunctional DNA alkylating agent, is crucial in the treatment of most pediatric and adult malignancies, including ALL. Cyclophosphamide is bioactivated in the liver by various enzymes of the hepatic P450 system including CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A4, and CYP3A5 (Cox, 1979; Parekh and Sladek, 1993; Dirven et al., 1994; Hayes and Pulford, 1995; Ludeman, 1999; Huang et al., 2000), whilst detoxification of active metabolites is mainly mediated by aldehyde dehydrogenases (ALDH1A1 and ALDH3A1; Parekh and Sladek, 1993) and glutathione S-transferases (GSTA1, GSTM1, GSTP1, and GSTT1; Hayes and Pulford, 1995). These enzymes are known to have functional polymorphisms, some of which have been found to impact toxicity associated with cyclophosphamide-based therapies in several pathologies (Zhong et al., 2006; Cho et al., 2010).
A study conducted in patients with leukemia who underwent hematopoietic stem cell transplantation showed that carriers of CYP2B6*2A or CYP2B6*4 variant alleles treated with cyclophosphamide were at higher risk of developing hemorrhagic cystitis and oral mucositis, respectively (Rocha et al., 2009). Interestingly, at least the CYP2B6*4 variant has been related to increased enzyme activity (Lang et al., 2001; Kirchheiner et al., 2003), and presumably this could lead to increased bioactivation of the drug and therefore could explain the observed higher incidence of toxicity in the patients. Numerous other CYP2B6 alleles that have been associated with changes in the enzyme activity/expression (www.cypalleles.ki.se/cyp2b6.htm) and therefore they could be relevant with regard to cyclophosphamide-induced side effects. It should also be remarked that the CYP2B6 gene exhibits a large intraethnic variability. Indeed, novel allelic variants and different linkage disequilibrium values have been described in certain populations (Restrepo et al., 2011), which should be taking into account when implementing tailored genotyping protocols aimed to determine the potential of CYP2B6 as a biomarker of drug response.
Allan et al. (2001) have reported that the 105Val allele of the GSTP1 Ile105Val polymorphism was overrepresented in a group of patients with therapy-related acute myeloid leukemia. Interestingly, this association was only observed in therapies including GSTP1 substrates such as cyclophosphamide. The authors hypothesized that an overexposure to the drug due to reduced detoxification capabilities could have played a role in leukemogenesis. Moreover, the same variation of the GSTP1 gene has been suggested to be one of the factors determining a higher neurotoxicity of ifosfamide, a closely related agent (Zielinska et al., 2005). A later report showed that the presence of the 677C-1298C haplotype in the methylenetetrahydrofolate reductase (MTHFR) gene was also associated to secondary acute myeloid leukemia after cyclophosphamide treatment of hematologic malignancies (Guillem et al., 2007).
Other polymorphisms reportedly associated with increased cyclophosphamide-induced toxicity are CYP2C19*2 (Takada et al., 2004; Ngamjanyaporn et al., 2011), CYP3A4*1B (Su et al., 2010), GSTM1/T1 null (Cho et al., 2010), ALDH3A1*2 and ALDH1A1*2 (Ekhart et al., 2008), ABCC4 rs9561778 (Low et al., 2009), or ABCG2 Q141K (rs2231142; Kim et al., 2008). It should be stated that some of these studies were conducted in patients with hematological malignancies but there are as yet no available data on ALL.
Cyclophosphamide is a chemotherapeutic agent used in a broad array of malignancies, which somewhat hampers the reproducibility of these findings. Therefore, larger studies and more consistent populations are needed in order to unequivocally establish the impact of the aforementioned SNPs and to identify other variants that could account for increased toxicity in cyclophosphamide-based therapies in ALL.
Cytosine Arabinoside
Cytosine arabinoside (Cytarabine, Ara-C) is an antimetabolite widely used in acute leukemia, which is associated with several adverse side effects, including myelosuppression, infections, mucositis, neurotoxicity, and acute pulmonary syndrome (Hartford et al., 2009). Candidate gene approaches have been used to identify genetic variables that are important in susceptibility to Ara-C. These studies have mainly focused on genes in the pharmacokinetic pathway of the drug. For instance, a common polymorphism, A79C (rs2072671), in the cytidine deaminase (CDA) gene, which catalyzes the rapid deamination of Ara-C, results in lower enzyme activity and hence a decreased rate of Ara-C metabolism (Kirch et al., 1998). Interestingly, Bhatla et al. (2009) have shown that Ara-C-related mortality was significantly elevated in carriers of the 79CC genotype in children with acute leukemia. Moreover, Ciccolini et al. (2012) have related this SNP to life-threatening toxicities induced in a girl with lymphoma treated with Ara-C.
Hartford et al. (2009) utilized an unbiased whole-genome approach to find polymorphisms that might predict the susceptibility to the cytotoxic effects of Ara-C in cell lines derived from persons of European (CEU) and African (YRI) ancestry. The authors identified a unique pharmacogenetic signature consisting of four SNPs explaining 51% of the variability in sensitivity to ara-C among the CEU and five SNPs explaining 58% of the variation among the YRI. These unique genetic signatures comprised novel target genes, most importantly GIT1, RAD51AP1, and SLC25A37, which can be studied further in functional studies. Furthermore, the authors examined 64 SNPs in the deoxycytidine kinase gene (DCK), which catalyzes the most essential step in the Ara-C activation pathway. Their conclusions show that cells that carried the DCK A70G (Ile24Val) polymorphism, which affects protein function, had an increased sensitivity to Ara-C (Hartford et al., 2009). In this regard, a later study has suggested that another DCK SNP, C-360G, is associated with the occurrence of mucositis after low-dose Ara-C in pediatric ALL patients (Banklau et al., 2010).
Recently, a study by Xu et al. (2012) seems to confirm the importance of both CDA and DCK genes as important loci that should be further investigated regarding the outcome of Ara-C-based chemotherapy in leukemia patients.
Cytotoxic Antibiotics
Doxorubicin (adriamycin) and daunorubicin (daunomycin) are anthracycline antibiotics commonly used in the treatment of leukemias whose main concern is their well-known dose-related cardiotoxicity (Gilladoga et al., 1976).
The contribution of pharmacogenetic factors across the doxorubicin biochemical pathway is not well established, but the drug is characterized by inter-individual variation in pharmacokinetic and pharmacodynamic parameters, and genetic variation has been suggested to account for at least part of this variability (Jamieson and Boddy, 2010; Lal et al., 2010). For instance, SNPs in the ABCB1 and SLC22A16 transporter genes have been shown to increase exposure levels that could result in a higher incidence of adverse effects (Lal et al., 2007, 2008). No studies on the impact of these mutations on the incidence of adverse effects in ALL patients have as yet been conducted. In patients of breast cancer treated with doxorubicin, Bray et al. (2010) have shown that several SNPs in the SLC22A16 influx transporter gene (A146G, T312C, and T755C) are related to lower incidence of dose delay, indicative of less toxicity; although it should be noted that the authors considered their own findings as preliminary.
In addition, in vitro studies have shown that the wild-type allele of the Val88Ile (262G > A, rs1143663) SNP in the carbonyl reductase 1 (CBR1) gene, involved in doxorubicin metabolism, exhibits a higher rate of synthesis of cardiotoxic metabolites (Gonzalez-Covarrubias et al., 2007). The conclusions of this work seem to indicate that the small percentage of individuals of African ancestry (to which this SNP is confined) who are homozygous for the low-activity Ile88 allele would therefore be at lower risk of cardiotoxicity. Paradoxically, this would imply that a majority of the African population is at-risk for doxorubicin-induced toxicity. Interestingly enough, a study by Hasan et al. (2004) evaluated 100 African American patients who underwent doxorubicin-based combination therapy and found that they appeared to suffer cardiotoxicity from doxorubicin three times more frequently than previously studied populations. Because of the low sample size analyzed and the lack of head-to-head comparison, larger studies in a multiracial setting seem necessary to clarify this finding.
With regard to daunorubicin, a recent work has shown a trend toward significant increase in the drug systemic exposure in patients carrying the C-allele of the CBR1 G312C (Leu73Leu, rs25678) polymorphism, which could elevate the risk of drug-induced toxicity (Varatharajan et al., 2012). In any case, it should be stated that the genetic associations reported for these two anthracyclines are still relatively recent and have not been consistently observed yet.
Mitoxantrone is an anthracycline analog which is a known substrate for ABC efflux transporters such as those encoded by the ABCB1 and ABCG2 genes (Kodaira et al., 2010). To date, there are no studies in the ALL setting testing the plausible hypothesis that genetic polymorphisms in these genes could affect mitoxantrone disposition and hence modulate the response and side effects to the drug. However, Cotte et al. (2009) have determined the frequencies of seven ABCB1 and ABCG2 SNPs in multiple sclerosis patients treated with mitoxantrone. Several associations were reported regarding the clinical response rate, but no relevant differences in genotype frequencies were observed in a subset of patients with severe hematological or cardiac side effects. However, it was intriguing that one patient presenting cardiomyopathy after a low dose of mitoxantrone was found to carry an uncommon genotype with homozygous variant alleles in two ABCB1 (G2677T/A, rs2032582 and C3435T, rs1045642) and one ABCG2 locus (Gln141Lys, rs2231142), in addition to a variant allele for the ABCC2 C-24T (rs717620) SNP (Cotte et al., 2009).
Etoposide
Etoposide is a topoisomerase II inhibitor used in a variety of malignancies. This drug is a substrate for the P-glycoprotein transporter, CYP3A4 and CYP3A5 isoforms (Relling et al., 1994), whose expression is partly regulated by the vitamin D receptor (VDR; Drocourt et al., 2002), and a number of phase II metabolizing enzymes, including GSTs and UGT1A1 (Watanabe et al., 2003). Therefore, alterations in these genes hold the potential to be relevant for the drug concentrations and clinical effects. However, to our knowledge there are as yet no studies investigating the association of polymorphisms in the etoposide pathways with the occurrence of adverse effects, mainly dose-limiting myelosuppression, in patients treated with the drug. Currently, there are only indications based on the impact of these genetic variations on the level of exposure to the drug. For instance, a study in 109 children diagnosed with ALL showed that carriers of both the CYP3A5*3/*3 and GSTP1 Ile/Ile genotype displayed a lower drug clearance, although this association was only observed in African Americans one month after treatment (Kishi et al., 2004). Moreover, the ABCB1 C3435T SNP was found to be an independent predictor of etoposide clearance disregarding ethnicity. In contrast, one year after treatment, the UGT1A1 6/6 (*1/*1), VDR intron 8 GG and VDR Fok 1 CC genotypes predicted higher clearance in African Americans (Kishi et al., 2004). Despite that the drug pharmacokinetics were shown to correlate with the incidence of adverse effects, no analyses of the association between SNPs and toxicity were conducted by the authors (Kishi et al., 2004).
Two genome-wide studies by an American research group have identified both genomic regions and SNPs associated with cellular sensitivity to etoposide. Huang et al. (2007) identified 63 genetic variants that contributed to etoposide-induced cytotoxicity through the evaluation of cell growth inhibition in cell lines from multi-generational pedigrees. The variants were present in genes whose expression had previously been related to altered cell sensitivity to etoposide, such as AGPAT2, IL1B, and WNT5B, but also in other genes not yet known to be associated with sensitivity to this agent. A limitation of this study was that candidate genes known to contribute to the pharmacokinetics of etoposide, e.g., CYP3A, UGT1A1, and ABCB1, are not expressed or are expressed at very low levels in the lymphoblastoid cell lines utilized.
Using the same cell models, Bleibel et al. (2009) later identified 22 unique SNPs in four genes among three chromosomes significantly associated with cytotoxic phenotypes at one or more treatment conditions. Genes implicated were UVRAG, a DNA repair gene, SEMA5A, which encodes semaphoring-5A protein involved in axonal guidance during development, the SLC7A6 transporter gene, which participates in nitric oxide synthesis that ultimately induces apoptosis and PRMT7, encoding the protein arginine methyltransferase that catalyzes an irreversible protein modification. All these processes would be altered in the presence of functional polymorphisms and could presumably lead to the observed increased cytotoxicity in etoposide-treated cells.
Unfortunately, and despite the interesting background provided by these two genome-wide reports, there are as yet no clinical studies that have investigated the role of any of the aforementioned genetic variants in patients treated with etoposide.
Imatinib
Imatinib is a Bcr-Abl tyrosine kinase inhibitor that is specifically used in the treatment of Phy + leukemia. The drug is demethylated to N-desmethyl-imatinib by CYP3A4/5, with other CYP450 isoforms playing a less important role (Peng et al., 2005). Indeed, coadministration of inhibitors and inducers of CYP3A activity results in significant modifications of the drug’s pharmacokinetics (Bolton et al., 2004), which show a wide interindividual variability (Judson et al., 2005). In order to explain this variability, pharmacogenetics studies have been carried out to find genetic determinants that could modify the pharmacological response to the drug. Most of these studies have aimed to find genetic markers of resistance. Thus, a number of polymorphisms in genes coding for transporters (ABCB1, ABCG2, SLC22A1), drug metabolizing enzymes (CYP3A5), proteins with involved in the nucleotide excision repair pathway (EERC) and proteins related to leukemogenesis (SOCS1 and PTPN22) have been associated to the efficacy of the treatment (Dulucq and Krajinovic, 2010; Kong et al., 2012; Vivona et al., 2012). These studies have mainly been conducted in patients with chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST), probably because of the higher incidence of these two diseases.
On the other hand, pharmacogenetic studies on imatinib-related toxicity are scarce and therefore the clinical impact of genetic polymorphisms on the occurrence of adverse effects has to be inferred from their effect on the drug blood levels, as it has been reported that the severity of the side effects seems to correlate with the drug’s pharmacokinetics (Judson et al., 2005). In this regard, two studies have shown a reduced oral clearance of the drug in patients carrying the CYP2D6*4 (Gardner et al., 2006) or ABCG2 421A variant alleles (Petain et al., 2008), which could hypothetically result in increased adverse effects. Interestingly, Gurney et al. (2007) reported a higher drug clearance but also a less common toxicity-related dose reduction in CML patients on imatinib who were carriers of the TT genotype at each of three key positions in the ABCB1 gene (1236, 2677, and 3435).
These data are not yet sufficiently conclusive to translate into individual drug dose adjustments and therefore further studies are still needed to analyze other genetic variants that can help individualize imatinib therapy.
Mercaptopurine
6-Mercaptopurine (6-MP) is an antimetabolite that has been used for 40 years in the treatment of a variety of diseases. In particular, 6-MP in orally daily regimen associated with weekly methotrexate (MTX) is the backbone of maintenance chemotherapy for ALL. The drug is metabolized toward active, and toxic, 6-thioguanine nucleotides (6-TGN) that are responsible for the elevated myelotoxicity of this chemotherapy agent.
The thiopurine methyltransferase (TPMT) gene, codes for a key enzyme in the metabolism of 6-MP (Figure 1) and other related thiopurine drugs such as 6-thioguanine and azathioprine. This enzyme is affected by functional polymorphisms that have been shown to produce a defective enzyme, amongst them, TPMT*2 (G238C), TMPT*3A (G460A, A719G), and TPMT*3C (A719G) account for 90% of the enzymatic deficiency in most populations (Weinshilboum, 2001). Approximately one in 300 hundred individuals are homozygous for these variants and therefore lack TPMT activity, which results in high levels of 6-TGN and acute hematopoietic toxic effects, greater risk for radiation-induced brain tumors and a greater likelihood of chemotherapy-induced acute myeloid leukemia (Pui et al., 2004). Because of this, extreme caution should be exerted in those children with ALL who lack TPMT activity and are scheduled to receive 6-MP. In turn, these subjects are far less prone to experience a relapse (Lennard and Lilleyman, 1996). Dose reductions of up to 90% have been proved useful in these patients (McLeod et al., 2000). For heterozygous carriers the administration of 50% of the standard dose is recommended, although there has been a certain degree of controversy as to whether these patients with intermediate activity may benefit from lower doses (McLeod et al., 1999; Stanulla et al., 2005).
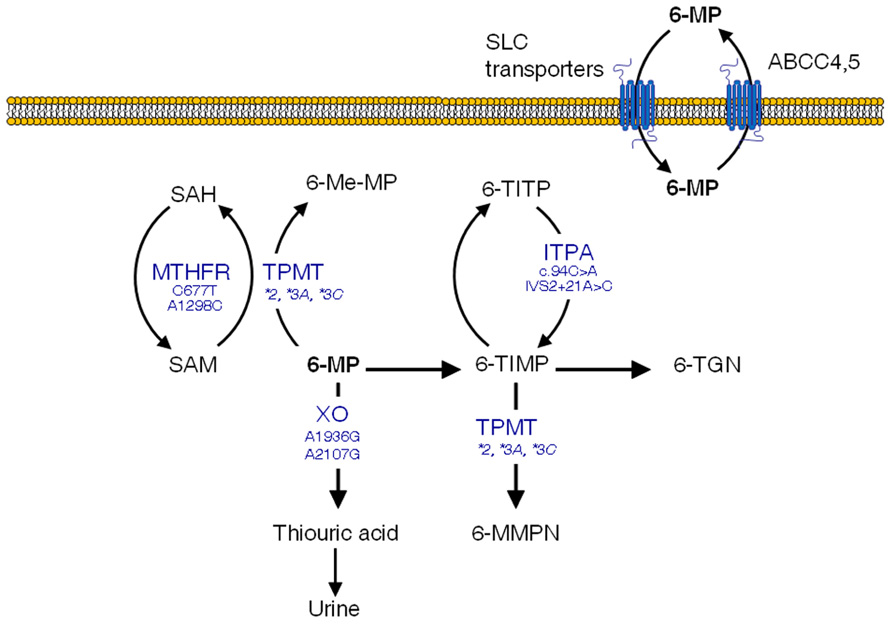
FIGURE 1. 6-mercaptopurine (6-MP) intracellular pathways with polymorphisms suggested to increase the occurrence of adverse effects. SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; TPMT, thiopurine S-methyltransferase; XO, xanthine oxidase; 6-Me-MP, 6-methyl-mercaptopurine; 6-TIMP, 6-thioinosine monophosphate; 6-TITP, 6-thioinosine triphosphate; ITPA, inosine triphosphate pyrophosphatase; 6-MMPN, 6-methyl-mercaptopurine nucleotides; 6-TGN, 6-thioguanine nucleotides
Having said that, the TPMT polymorphism does not explain all 6-MP-induced adverse effects, and some severe toxicities leading to life-threatening conditions remain unexplained (Palmieri et al., 2007). Additional SNPs in genes encoding enzymes involved in 6-MP metabolism and transport might contribute to the drug-induced toxicity. This is the case of the inosine triphosphate pyrophosphatase (ITPA) enzyme, which catalyzes one of the intermediate steps of 6-MP metabolism (Figure 1). Two SNPs in the ITPA gene with a frequency of roughly 10% in Caucasians (Adam de Beaumais and Jacqz-Aigrain, 2012), namely a non-synonymous C94A transition (rs1127354, Pro32Thr) and the intronic IVS2 + 21A > C mutation have been related with defective enzyme activity (Heller et al., 2004) leading to higher risk of myelotoxicity and hepatotoxicity in ALL pediatric patients (Hawwa et al., 2008; Stocco et al., 2009; Wan Rosalina et al., 2011). In addition, MTHFR SNPs have been shown to be more common in subjects with low TPMT activity (Karas-Kuzelicki et al., 2009), probably because of their impact on S-adenosylmethionine, which functions as a cofactor for TPMT. Other polymorphisms in genes involved in 6-MP disposition such as xanthine oxidase or ABCC4 have also been suggested to impact clinical outcomes, although there are as yet no available data in ALL patients (Hawwa et al., 2008; Ban et al., 2010).
The use of the TPMT genotyping for tailoring ALL therapy, and the putative inclusion of other polymorphisms in these genetic tests will be discussed in the final chapter of this review.
Methotrexate
Methotrexate (MTX) is a folate inhibitor widely employed in the chemotherapy of hematologic malignancies and various solid tumors. This drug is the cornerstone for therapy of ALL and has been the focus of a number of pharmacogenetic studies aimed to identify genetic determinants of its toxicity (reviewed elsewhere Gervasini, 2009). Its actions on folate metabolism follow a complex pattern that includes several transporters and metabolizing enzymes whose function and/or expression have been suggested to be altered by genetic polymorphisms.
Briefly, the reduced folate carrier 1 (RFC1, SLC19A1) is responsible for the entry of MTX in the cell (Moscow et al., 1995), whilst the drug is pumped out by a variety of ATP-binding cassette (ABC) efflux transporters (Strand et al., 1999). In the cell, MTX is metabolized to active polyglutamates, which are responsible for the disruption of the folate metabolic pathway by inhibiting enzymes that are essential for the DNA (Chabner et al., 1985). These include thymidylate synthase (TS; Szeto et al., 1979) and dihydrofolate reductase (DHFR; Galivan, 1980). Another key enzyme in the folate pathways is methylenetetrahydrofolate reductase (MTHFR), which produces 5-methyl-tetrahydrofolate (THF) from 5,10-methylene-THF, a major intermediary that is in turn synthesized by serine hydroxymethyltransferase (SHMT1). Finally, the production of 5-methyl-THF is pivotal for biotransformation of homocysteine to methionine, which involves two major enzymes, namely methionine synthase (MS) and methionine synthase reductase (MTRR). Figure 2 depicts an overview of these intracellular pathways pointing out those SNPs that have been related with the occurrence of MTX-induced toxicity.
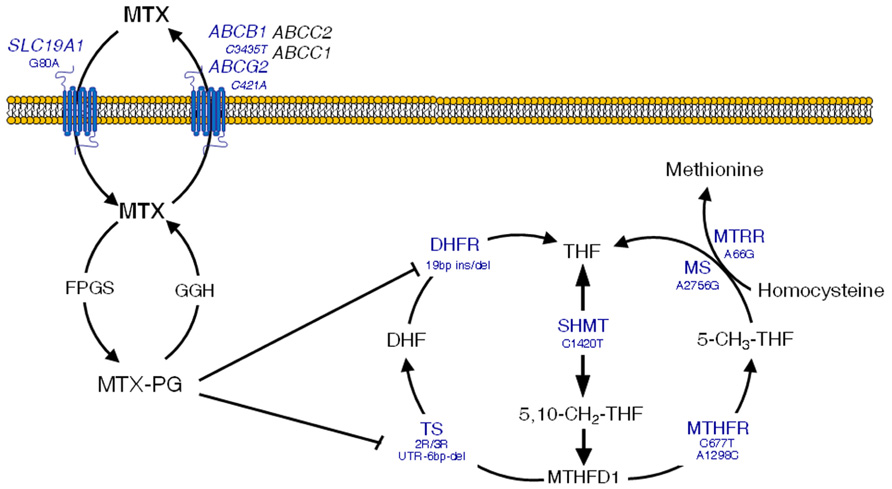
FIGURE 2. Overview of methotrexate (MTX) mechanisms of action in the folate metabolic pathway. Clinically significant polymorphisms are shown for the main genes involved. MTX-PG, methotrexate polyglutamates; SLC19A1, solute carrier 19A1; ABCC1 and 2, ATP-binding cassette C1 and 2 transporters; ABCG2, ATP-binding cassette G2 transporter, breast cancer resistance protein; ABCB1, ATP-binding cassette B1 transporter, multidrug resistance; FPGS, folylpolyglutamyl synthase; GGH, γ-glutamyl hydrolase; TS, thymidylate synthase; UTR, untranslated region; DHFR, dihydrofolate reductase; SHMT, serine hydroxymethyltransferase; MTHFD1, methylenetetrahydrofolate dehydrogenase; CCND1, cyclin D1; ATIC, aminoimidazole 4-carboxamide ribonucleotide (AICAR) transformylase; MS, methionine synthase; MTRR, methionine synthase reductase. Adapted with permission from Gervasini (2009).
MTHFR has by far been the most extensively studied gene in association studies in ALL. Two SNPs, C677T (rs1801133) and A1298C (rs1801131), have been related to increased MTX toxicity (Kantar et al., 2009; D’Angelo et al., 2011). However, a recent meta-analysis (Yang et al., 2012), as well as the general perception in the literature (reviewed by Gervasini, 2009) lead to think that C677T is the only SNP that plays a clinically significant role, albeit contradictory results also exist (Chatzidakis et al., 2006; Kishi et al., 2007; Huang et al., 2008).
Most notably, given the severity of the syndrome, the 677T allele has been suggested to induce MTX-related neurotoxicity (mainly subacute leukoencephalopathy) in young ALL patients (Strunk et al., 2003; Mahadeo et al., 2010; Vagace et al., 2011, 2012). It should be noted that this association has only been observed in clinical case reports. In fact, a study in 53 children with ALL treated with high-dose MTX failed to confirm a relevant role of the 677T allele in the drug-induced neurotoxicity developed by nine of the patients (Kishi et al., 2003). In order to elucidate this controversy, it would be desirable to genotype large populations of pediatric ALL patients who underwent MTX-related neurotoxicity. However, the lack of homogeneity of treatment protocols, which makes multicenter studies hard to conduct, the low number of patients affected, and the more than likely influence of other SNPs, have so far been insurmountable obstacles.
A 19 bp ins/del polymorphism in the DHFR gene has also been associated with increased toxicity in adult leukemia patients treated with MTX (Ongaro et al., 2009). In the same manner, the MTRR A66G SNP has been related to increased risk of developing toxicity in children with ALL treated with high doses of MTX (Huang et al., 2008), although there seems to be no relation with CNS side effects (Krajinovic et al., 2005).
Other genes that could also be involved in the development of MTX-induced toxicity are the ABC efflux transporters. Their location in the blood-brain barrier makes tempting to speculate that polymorphisms in these genes could lead to drug accumulation in the brain and subsequent neurotoxicity. For instance, the combined presence of the C421A (rs2231142, Gln141Lys) SNP in the ABCG2 gene (encoding the Breast Resistance Cancer Protein, BCRP transporter) and the C3435T transition in ABCB1 (coding for P-glycoprotein) has been related to the occurrence of encephalopathy in children with ALL treated with MTX (Erdilyi et al., 2008). Finally, the G-80A polymorphism in the RFC1 influx transporter, which determines intracellular levels of MTX, has been associated with increased overall toxicity in ALL patients (Shimasaki et al., 2006; Imanishi et al., 2007; Kishi et al., 2007).
It should be noted that large studies on the association between MTX toxicity and genetics in childhood ALL are still scarce and mostly focus on MTHFR variants. Studies carried out in other pathologies have found evidences of increased toxicity in the presence of variant alleles in additional ABC transporters (Campalani et al., 2007; Ranganathan et al., 2008), or in the SHMT1 (Weisman et al., 2006) and TS genes (Campalani et al., 2007). Therefore, we should not rule out the implication of these polymorphisms in MXT-related side effects experienced by ALL patients.
Vincristine
The wide pharmacokinetic interindividual variability shown by vincristine, and the occurrence of its dose-limiting neurotoxicity remains largely unpredictable (Moore and Pinkerton, 2009). Renbarger et al. (2008) have showed different grades of neurotoxicity in Caucasians compared to African Americans, suggesting that genetics could influence vincristine-induced toxicity.
The main candidate gene for pharmacogenetic studies is CYP3A5, since it is known to contribute between 55 and 95% to total vincristine metabolism (Dennison et al., 2006). In addition, the drug is a substrate for P-glycoprotein (Song et al., 1999), which makes the encoding ABCB1 gene another suitable candidate for pharmacogenetic studies. Two reports have evaluated the clinical effect of common SNPs (CYP3A5*3 and ABCB1 C3435T and G2677T) in these genes in pediatric ALL patients treated with vincristine. However, the authors failed to find a significant association with increased occurrence of side effects such as impaired motor activity or constipation (Plasschaert et al., 2004; Hartman et al., 2010).These two studies combined added up to only 86 genotyped patients, which is clearly not sufficient to rule out the involvement of these candidate genes in vincristine toxicity.
Perspectives and Conclusion
The utility of pharmacogenetics in clinical routine has turned to be lower than anticipated, as numerous barriers to implementing individualized medicine have appeared over the years (Agundez et al., 2012). Cases in which a genetic test is sufficient to significantly affect a given therapy are discouragingly uncommon (Groenen, 2011) and the leukemia setting is not an exception. Indeed, only 1–2% of marketed drugs have pharmacogenomic-based recommendations (Agundez et al., 2012) and amongst the drugs commonly used in the chemotherapy of pediatric ALL, only 6-MP, thioguanine and azathioprine labels include genetic testing (TPMT) as a recommendation to help individualize therapy. However, steps are being taken in order to revert this situation. The increasing availability of low-cost, high-throughput genetic platforms that allow the simultaneous screening of hundreds of polymorphisms, the education of health practitioners or the implementation of multicenter networks aimed to improve the safety of new drugs, are some of the measures that hopefully will help improve the impact of pharmacogenetics on clinical routine. It should also be pointed out that genetic variability in several of the targets for which genetic testing is recommended by the FDA and other corporations has been shown to modify the risk of acute leukemia (Agundez, 2004), which certainly enhances the utility of these tests.
The body of work on the clinical impact of TPMT and MTHFR polymorphisms on ALL does not mirror the limited current knowledge on the pharmacogenetics of the other drugs that constitute the core of chemotherapy for this disease. Thus, significantly less information is available with regard to vincristine, asparaginase, cyclophosphamide etc. The TPMT gene polymorphism provides the best example of the value of applied pharmacogenetics in ALL and clinical oncology in general. It is now widely acknowledged that the initial dose of 6-MP treatment should be based on the TPMT genotype (Relling et al., 2011), thus allowing the clinician to identify patients at higher risk of toxicity. However, as it was described in the 6-MP chapter, there is still some interindividual variability in the response to this drug that cannot be explained only by the TPMT genotype (Palmieri et al., 2007). Therefore, the implementation of additional genetic analyses to identify polymorphisms in the ITPA, MTHFR, XO and other genes in the 6-MP intracellular pathway, as well as the study of their epistatic interactions seem to be reasonable steps to take in order to better adjust 6-MP doses in ALL patients. Furthermore, pharmacogenomics alone may not be sufficient to explain all the interindividual variability in 6-MP response and efforts should be undertaken in the coming years to create more precise algorithms that can help predict drug response.
With regard to MTHFR, the reasons for the present controversy regarding the influence of its genetic variation on MTX-induced toxicity are diverse. For instance, the wide variety of diseases in which MTX has been proved useful has paradoxically hampered the reproducibility of the results of genetic association studies, because of the heterogeneity of the patients analyzed. Moreover, other unknown variants in the same gene, epistatic interactions with other genes and, most likely, the combination of these factors, may result in genetic backgrounds with different susceptibilities to MTX-induced toxicity. The challenge seems to be to identify which genetic factors and genetic combinations are those in specific populations.
Some ALL chemotherapy regimens contemplate MTX dose reductions for subjects homozygous for the 677T variant or for those carrying both 677CT and 1298AC heterozygous genotypes. One could consider at least premature to make this kind of dose adjustments based on data that are still controversial. In fact, MTX was not included in the list of drugs mentioned in the Table of Pharmacogenomic Biomarkers in Drug Label issued by the Food and Drug Administration (FDA) last year (FDA, 2011). It is probably naive to believe that the determination of SNPs in just one gene (MTHFR) belonging to a highly complex intracellular pathway, such as that of MTX, is enough to accurately anticipate the occurrence of adverse effects.
As a general rule, it would be logical to think that the identification of combinations of mutations in several genes along the pathway of a given drug must be more helpful in terms of identifying subjects at-risk of toxicity than a single-SNP approach (Vagace et al., 2011). This is crucial in the case of drugs with intricate intracellular routes as it is the case of MTX or 6-MP. Genome-wide approaches such as the ones reported by (Huang et al., 2007), Bleibel et al. (2009), and Chen et al. (2011) are able to evaluate whole pathways to identify key routes that can be later studied in detail. In addition, the inclusion of other clinical and demographic factors may increase the predictive value of pharmacogenetic models (Wessels et al., 2007).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been supported in part by grant PRIS11003 from FUNDESALUD, Mérida (Spain) and grant GR10022 from Junta de Extremadura, Consejeria de Economia, Comercio e Innovacion, Merida (Spain).
References
Adam de Beaumais, T., and Jacqz-Aigrain, E. (2012). Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur. J. Clin. Pharmacol. 68, 1233–1242.
Agundez, J. A., Del Barrio, J., Padro, T., Stephens, C., Farre, M., Andrade, R. J., et al. (2012). Trends in qualifying biomarkers in drug safety. Consensus of the 2011 meeting of the spanish society of clinical pharmacology. Front. Pharmacol. 3, 1–6.
Allan, D. S., Kovacs, M. J., and Clark, W. F. (2001). Frequently relapsing thrombotic thrombocytopenic purpura treated with cytotoxic immunosuppressive therapy. Haematologica 86, 844–850.
Ban, H., Andoh, A., Imaeda, H., Kobori, A., Bamba, S., Tsujikawa, T., et al. (2010). The multidrug-resistance protein 4 polymorphism is a new factor accounting for thiopurine sensitivity in Japanese patients with inflammatory bowel disease. J. Gastroenterol. 45, 1014–1021.
Banklau, C., Jindadamrongwech, S., Sawangpanich, R., Apibal, S., Hongeng, S., Paisooksantivatana, K., et al. (2010). Effect of genetic alterations of cytarabine- metabolizing enzymes in childhood acute lymphoblastic leukemia. Hematol. Oncol. Stem Cell Ther. 3, 103–108.
Bhatla, D., Gerbing, R. B., Alonzo, T. A., Conner, H., Ross, J. A., Meshinchi, S., et al. (2009). Cytidine deaminase genotype and toxicity of cytosine arabinoside therapy in children with acute myeloid leukemia. Br. J. Haematol. 144, 388–394.
Bleibel, W. K., Duan, S., Huang, R. S., Kistner, E. O., Shukla, S. J., Wu, X., et al. (2009). Identification of genomic regions contributing to etoposide-induced cytotoxicity. Hum. Genet. 125, 173–180.
Bolton, A. E., Peng, B., Hubert, M., Krebs-Brown, A., Capdeville, R., Keller, U., et al. (2004). Effect of rifampicin on the pharmacokinetics of imatinib mesylate (Gleevec, STI571) in healthy subjects. Cancer Chemother. Pharmacol. 53, 102–106.
Bray, J., Sludden, J., Griffin, M. J., Cole, M., Verrill, M., Jamieson, D., et al. (2010). Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br. J. Cancer 102, 1003–1009.
Campalani, E., Arenas, M., Marinaki, A. M., Lewis, C. M., Barker, J. N., and Smith, C. H. (2007). Polymorphisms in folate, pyrimidine, and purine metabolism are associated with efficacy and toxicity of methotrexate in psoriasis. J. Invest. Dermatol. 127, 1860–1867.
Chabner, B. A., Allegra, C. J., Curt, G. A., Clendeninn, N. J., Baram, J., Koizumi, S., et al. (1985). Polyglutamation of methotrexate. Is methotrexate a prodrug? J. Clin. Invest. 76, 907–912.
Chatzidakis, K., Goulas, A., Athanassiadou-Piperopoulou, F., Fidani, L., Koliouskas, D., and Mirtsou, V. (2006). Methylenetetrahydrofolate reductase C677T polymorphism: association with risk for childhood acute lymphoblastic leukemia and response during the initial phase of chemotherapy in greek patients. Pediatr. Blood Cancer 47, 147–151.
Chen, S. H., Pei, D., Yang, W., Cheng, C., Jeha, S., Cox, N. J., et al. (2010). Genetic variations in GRIA1 on chromosome 5q33 related to asparaginase hypersensitivity. Clin. Pharmacol. Ther. 88, 191–196.
Chen, S. H., Yang, W., Fan, Y., Stocco, G., Crews, K. R., Yang, J. J., et al. (2011). A genome-wide approach identifies that the aspartate metabolism pathway contributes to asparaginase sensitivity. Leukemia 25, 66–74.
Cho, H. J., Eom, H. S., Kim, H. J., Kim, I. S., Lee, G. W., and Kong, S. Y. (2010). Glutathione-S-transferase genotypes influence the risk of chemotherapy-related toxicities and prognosis in Korean patients with diffuse large B-cell lymphoma. Cancer Genet. Cytogenet. 198, 40–46.
Ciccolini, J., Evrard, A., M’Batchi, L., Pourroy, B., Mercier, C., Iliadis, A., et al. (2012). CDA deficiency as a possible culprit for life-threatening toxicities after cytarabine plus 6-mercaptopurine therapy: pharmacogenetic investigations. Pharmacogenomics 13, 393–397.
Cortijo-Cascajares, S., Jimenez-Cerezo, M. J., and Herreros De Tejada, A. (2012). Review of hypersensitivity reactions to antineoplastic agents. Farm. Hosp. 36, 148–158.
Cotte, S., Von Ahsen, N., Kruse, N., Huber, B., Winkelmann, A., Zettl, U. K., et al. (2009). ABC-transporter gene-polymorphisms are potential pharmacogenetic markers for mitoxantrone response in multiple sclerosis. Brain 132, 2517–2530.
Cox, P. J. (1979). Cyclophosphamide cystitis – identification of acrolein as the causative agent. Biochem. Pharmacol. 28, 2045–2049.
D’Angelo, V., Ramaglia, M., Iannotta, A., Crisci, S., Indolfi, P., Francese, M., et al. (2011). Methotrexate toxicity and efficacy during the consolidation phase in paediatric acute lymphoblastic leukaemia and MTHFR polymorphisms as pharmacogenetic determinants. Cancer Chemother. Pharmacol. 68, 1339–1346.
Dennison, J. B., Kulanthaivel, P., Barbuch, R. J., Renbarger, J. L., Ehlhardt, W. J., and Hall, S. D. (2006). Selective metabolism of vincristine in vitro by CYP3A5. Drug Metab. Dispos. 34, 1317–1327.
Dirven, H. A., Venekamp, J. C., Van Ommen, B., and Van Bladeren, P. J. (1994). The interaction of glutathione with 4-hydroxycyclophosphamide and phosphoramide mustard, studied by 31P nuclear magnetic resonance spectroscopy. Chem. Biol. Interact. 93, 185–196.
Drocourt, L., Ourlin, J. C., Pascussi, J. M., Maurel, P., and Vilarem, M. J. (2002). Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J. Biol. Chem. 277, 25125–25132.
Ekhart, C., Rodenhuis, S., Smits, P. H., Beijnen, J. H., and Huitema, A. D. (2008). Relations between polymorphisms in drug-metabolising enzymes and toxicity of chemotherapy with cyclophosphamide, thiotepa and carboplatin. Pharmacogenet. Genomics 18, 1009–1015.
Erdilyi, D. J., Kamory, E., Csokay, B., Andrikovics, H., Tordai, A., Kiss, C., et al. (2008). Synergistic interaction of ABCB1 and ABCG2 polymorphisms predicts the prevalence of toxic encephalopathy during anticancer chemotherapy. Pharmacogenomics J. 8, 321–327.
FDA (2011). Table of Pharmacogenomic Biomarkers in Drug Lables [Online]. Available: http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm [Accessed].
Flores-Calderon, J., Exiga-Gonzalez, E., Moran-Villota, S., Martin-Trejo, J., and Yamamoto-Nagano, A. (2009). Acute pancreatitis in children with acute lymphoblastic leukemia treated with L-asparaginase. J. Pediatr. Hematol. Oncol. 31, 790–793.
Galivan, J. (1980). Evidence for the cytotoxic activity of polyglutamate derivatives of methotrexate. Mol. Pharmacol. 17, 105–110.
Gardner, E. R., Burger, H., Van Schaik, R. H., Van Oosterom, A. T., De Bruijn, E. A., Guetens, G., et al. (2006). Association of enzyme and transporter genotypes with the pharmacokinetics of imatinib. Clin. Pharmacol. Ther. 80, 192–201.
Gervasini, G. (2009). Polymorphisms in methotrexate pathways: what is clinically relevant, what is not, and what is promising. Curr. Drug Metab. 10, 547–566.
Gilladoga, A. C., Manuel, C., Tan, C. T., Wollner, N., Sternberg, S. S., and Murphy, M. L. (1976). The cardiotoxicity of adriamycin and daunomycin in children. Cancer 37, 1070–1078.
Gonzalez-Covarrubias, V., Ghosh, D., Lakhman, S. S., Pendyala, L., and Blanco, J. G. (2007). A functional genetic polymorphism on human carbonyl reductase 1 (CBR1 V88I) impacts on catalytic activity and NADPH binding affinity. Drug Metab. Dispos. 35, 973–980.
Groenen, P. M. (2011). Four decades of modern pharmacogenomics: from promise to clinical utility. World PGX Summit 2011, Boston, USA, 26–27 April, 2011. Pharmacogenomics 12, 1249–1252.
Gugliotta, L., Mazzucconi, M. G., Leone, G., Mattioli-Belmonte, M., Defazio, D., Annino, L., et al. (1992). Incidence of thrombotic complications in adult patients with acute lymphoblastic leukaemia receiving L-asparaginase during induction therapy: a retrospective study. The GIMEMA Group. Eur. J. Haematol. 49, 63–66.
Guillem, V. M., Collado, M., Terol, M. J., Calasanz, M. J., Esteve, J., Gonzalez, M., et al. (2007). Role of MTHFR (677, 1298) haplotype in the risk of developing secondary leukemia after treatment of breast cancer and hematological malignancies. Leukemia 21, 1413–1422.
Gurney, H., Wong, M., Balleine, R. L., Rivory, L. P., Mclachlan, A. J., Hoskins, J. M., et al. (2007). Imatinib disposition and ABCB1 (MDR1, P-glycoprotein) genotype. Clin. Pharmacol. Ther. 82, 33–40.
Hartford, C. M., Duan, S., Delaney, S. M., Mi, S., Kistner, E. O., Lamba, J. K., et al. (2009). Population-specific genetic variants important in susceptibility to cytarabine arabinoside cytotoxicity. Blood 113, 2145–2153.
Hartman, A., Van Schaik, R. H., Van Der Heiden, I. P., Broekhuis, M. J., Meier, M., Den Boer, M. L., et al. (2010). Polymorphisms in genes involved in vincristine pharmacokinetics or pharmacodynamics are not related to impaired motor performance in children with leukemia. Leuk. Res. 34, 154–159.
Hasan, S., Dinh, K., Lombardo, F., and Kark, J. (2004). Doxorubicin cardiotoxicity in African Americans. J. Natl. Med. Assoc. 96, 196–199.
Hawwa, A. F., Millership, J. S., Collier, P. S., Vandenbroeck, K., Mccarthy, A., Dempsey, S., et al. (2008). Pharmacogenomic studies of the anticancer and immunosuppressive thiopurines mercaptopurine and azathioprine. Br. J. Clin. Pharmacol. 66, 517–528.
Hayes, J. D., and Pulford, D. J. (1995). The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 30, 445–600.
Heller, T., Oellerich, M., Armstrong, V. W., and Von Ahsen, N. (2004). Rapid detection of ITPA 94C>A and IVS2+ 21A>C gene mutations by real-time fluorescence PCR and in vitro demonstration of effect of ITPA IVS2+21A>C polymorphism on splicing efficiency. Clin. Chem. 50, 2182–2184.
Huang, L., Tissing, W. J., De Jonge, R., Van Zelst, B. D., and Pieters, R. (2008). Polymorphisms in folate-related genes: association with side effects of high-dose methotrexate in childhood acute lymphoblastic leukemia. Leukemia 22, 1798–1800.
Huang, R. S., Duan, S., Bleibel, W. K., Kistner, E. O., Zhang, W., Clark, T. A., et al. (2007). A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 104, 9758–9763.
Huang, Z., Roy, P., and Waxman, D. J. (2000). Role of human liver microsomal CYP3A4 and CYP2B6 in catalyzing N-dechloroethylation of cyclophosphamide and ifosfamide. Biochem. Pharmacol. 59, 961–972.
Imanishi, H., Okamura, N., Yagi, M., Noro, Y., Moriya, Y., Nakamura, T., et al. (2007). Genetic polymorphisms associated with adverse events and elimination of methotrexate in childhood acute lymphoblastic leukemia and malignant lymphoma. J. Hum. Genet. 52, 166–171.
Jamieson, D., and Boddy, A. V. (2010). Pharmacogenetics of genes across the doxorubicin pathway. Expert Opin. Drug Metab. Toxicol. 7, 1201–1210.
Judson, I., Ma, P., Peng, B., Verweij, J., Racine, A., Di Paola, E. D., et al. (2005). Imatinib pharmacokinetics in patients with gastrointestinal stromal tumour: a retrospective population pharmacokinetic study over time. EORTC Soft Tissue and Bone Sarcoma Group. Cancer Chemother. Pharmacol. 55, 379–386.
Kantar, M., Kosova, B., Cetingul, N., Gumus, S., Toroslu, E., Zafer, N., et al. (2009). Methylenetetrahydrofolate reductase C677T and A1298C gene polymorphisms and therapy-related toxicity in children treated for acute lymphoblastic leukemia and non-Hodgkin lymphoma. Leuk. Lymphoma 50, 912–917.
Karas-Kuzelicki, N., Jazbec, J., Milek, M., and Mlinaric-Rascan, I. (2009). Heterozygosity at the TPMT gene locus, augmented by mutated MTHFR gene, predisposes to 6-MP related toxicities in childhood ALL patients. Leukemia 23, 971–974.
Kim, I. S., Kim, H. G., Kim, D. C., Eom, H. S., Kong, S. Y., Shin, H. J., et al. (2008). ABCG2 Q141K polymorphism is associated with chemotherapy-induced diarrhea in patients with diffuse large B-cell lymphoma who received frontline rituximab plus cyclophosphamide/ doxorubicin/vincristine/prednisone chemotherapy. Cancer Sci. 99, 2496–2501.
Kirch, H. C., Schroder, J., Hoppe, H., Esche, H., Seeber, S., and Schutte, J. (1998). Recombinant gene products of two natural variants of the human cytidine deaminase gene confer different deamination rates of cytarabine in vitro. Exp. Hematol. 26, 421–425.
Kirchheiner, J., Klein, C., Meineke, I., Sasse, J., Zanger, U. M., Murdter, T. E., et al. (2003). Bupropion and 4-OH-bupropion pharmacokinetics in relation to genetic polymorphisms in CYP2B6. Pharmacogenetics 13, 619–626.
Kishi, S., Cheng, C., French, D., Pei, D., Das, S., Cook, E. H., et al. (2007). Ancestry and pharmacogenetics of antileukemic drug toxicity. Blood 109, 4151–4157.
Kishi, S., Griener, J., Cheng, C., Das, S., Cook, E. H., Pei, D., et al. (2003). Homocysteine, pharmacogenetics, and neurotoxicity in children with leukemia. J. Clin. Oncol. 21, 3084–3091.
Kishi, S., Yang, W., Boureau, B., Morand, S., Das, S., Chen, P., et al. (2004). Effects of prednisone and genetic polymorphisms on etoposide disposition in children with acute lymphoblastic leukemia. Blood 103, 67–72.
Kodaira, H., Kusuhara, H., Ushiki, J., Fuse, E., and Sugiyama, Y. (2010). Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J. Pharmacol. Exp. Ther. 333, 788–796.
Kong, J. H., Mun, Y. C., Kim, S., Choi, H. S., Kim, Y. K., Kim, H. J., et al. (2012). Polymorphisms of ERCC1 genotype associated with response to imatinib therapy in chronic phase chronic myeloid leukemia. Int. J. Hematol. 96, 327–333.
Krajinovic, M., Robaey, P., Chiasson, S., Lemieux-Blanchard, E., Rouillard, M., Primeau, M., et al. (2005). Polymorphisms of genes controlling homocysteine levels and IQ score following the treatment for childhood ALL. Pharmacogenomics 6, 293–302.
Lal, S., Mahajan, A., Chen, W. N., and Chowbay, B. (2010). Pharmacogenetics of target genes across doxorubicin disposition pathway: a review. Curr. Drug Metab. 11, 115–128.
Lal, S., Wong, Z. W., Jada, S. R., Xiang, X., Chen Shu, X., Ang, P. C., et al. (2007). Novel SLC22A16 polymorphisms and influence on doxorubicin pharmacokinetics in Asian breast cancer patients. Pharmacogenomics 8, 567–575.
Lal, S., Wong, Z. W., Sandanaraj, E., Xiang, X., Ang, P. C., Lee, E. J., et al. (2008). Influence of ABCB1 and ABCG2 polymorphisms on doxorubicin disposition in Asian breast cancer patients. Cancer Sci. 99, 816–823.
Lang, T., Klein, K., Fischer, J., Nussler, A. K., Neuhaus, P., Hofmann, U., et al. (2001). Extensive genetic polymorphism in the human CYP2B6 gene with impact on expression and function in human liver. Pharmacogenetics 11, 399–415.
Laningham, F. H., Kun, L. E., Reddick, W. E., Ogg, R. J., Morris, E. B., and Pui, C. H. (2007). Childhood central nervous system leukemia: historical perspectives, current therapy, and acute neurological sequelae. Neuroradiology 49, 873–888.
Lennard, L., and Lilleyman, J. S. (1996). Individualizing therapy with 6-mercaptopurine and 6-thioguanine related to the thiopurine methyltransferase genetic polymorphism. Ther. Drug Monit. 18, 328–334.
Low, S. K., Kiyotani, K., Mushiroda, T., Daigo, Y., Nakamura, Y., and Zembutsu, H. (2009). Association study of genetic polymorphism in ABCC4 with cyclophosphamide-induced adverse drug reactions in breast cancer patients. J. Hum. Genet. 54, 564–571.
Ludeman, S. M. (1999). The chemistry of the metabolites of cyclophosphamide. Curr. Pharm. Des. 5, 627–643.
Mahadeo, K. M., Dhall, G., Panigrahy, A., Lastra, C., and Ettinger, L. J. (2010). Subacute methotrexate neurotoxicity and cerebral venous sinus thrombosis in a 12-year-old with acute lymphoblastic leukemia and methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: homocysteine-mediated methotrexate neurotoxicity via direct endothelial injury. Pediatr. Hematol. Oncol. 27, 46–52.
McLeod, H. L., Coulthard, S., Thomas, A. E., Pritchard, S. C., King, D. J., Richards, S. M., et al. (1999). Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 105, 696–700.
McLeod, H. L., Krynetski, E. Y., Relling, M. V., and Evans, W. E. (2000). Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia 14, 567–572.
Moore, A., and Pinkerton, R. (2009). Vincristine: can its therapeutic index be enhanced? Pediatr. Blood Cancer 53, 1180–1187.
Moscow, J. A., Gong, M., He, R., Sgagias, M. K., Dixon, K. H., Anzick, S. L., et al. (1995). Isolation of a gene encoding a human reduced folate carrier (RFC1) and analysis of its expression in transport-deficient, methotrexate-resistant human breast cancer cells. Cancer Res. 55, 3790–3794.
Ngamjanyaporn, P., Thakkinstian, A., Verasertniyom, O., Chatchaipun, P., Vanichapuntu, M., Nantiruj, K., et al. (2011). Pharmacogenetics of cyclophosphamide and CYP2C19 polymorphism in Thai systemic lupus erythematosus. Rheumatol. Int. 31, 1215–1218.
Ongaro, A., De Mattei, M., Della Porta, M. G., Rigolin, G., Ambrosio, C., Di Raimondo, F., et al. (2009). Gene polymorphisms in folate metabolizing enzymes in adult acute lymphoblastic leukemia: effects on methotrexate-related toxicity and survival. Haematologica 94, 1391–1398.
Pacheco, R., Gallart, T., Lluis, C., and Franco, R. (2007). Role of glutamate on T-cell mediated immunity. J. Neuroimmunol. 185, 9–19.
Palmieri, O., Latiano, A., Bossa, F., Vecchi, M., D’Inca, R., Guagnozzi, D., et al. (2007). Sequential evaluation of thiopurine methyltransferase, inosine triphosphate pyrophosphatase, and HPRT1 genes polymorphisms to explain thiopurines’ toxicity and efficacy. Aliment. Pharmacol. Ther. 26, 737–745.
Parekh, H. K., and Sladek, N. E. (1993). NADPH-dependent enzyme-catalyzed reduction of aldophosphamide, the pivotal metabolite of cyclophosphamide. Biochem. Pharmacol. 46, 1043–1052.
Peng, B., Lloyd, P., and Schran, H. (2005). Clinical pharmacokinetics of imatinib. Clin. Pharmacokinet. 44, 879–894.
Petain, A., Kattygnarath, D., Azard, J., Chatelut, E., Delbaldo, C., Geoerger, B., et al. (2008). Population pharmacokinetics and pharmacogenetics of imatinib in children and adults. Clin. Cancer Res. 14, 7102–7109.
Plasschaert, S. L., Groninger, E., Boezen, M., Kema, I., De Vries, E. G., Uges, D., et al. (2004). Influence of functional polymorphisms of the MDR1 gene on vincristine pharmacokinetics in childhood acute lymphoblastic leukemia. Clin. Pharmacol. Ther. 76, 220–229.
Pui, C. H., Relling, M. V., and Downing, J. R. (2004). Acute lymphoblastic leukemia. N. Engl. J. Med. 350, 1535–1548.
Ranganathan, P., Culverhouse, R., Marsh, S., Mody, A., Scott-Horton, T. J., Brasington, R., et al. (2008). Methotrexate (MTX) pathway gene polymorphisms and their effects on MTX toxicity in Caucasian and African American patients with rheumatoid arthritis. J. Rheumatol. 35, 572–579.
Relling, M. V., Gardner, E. E., Sandborn, W. J., Schmiegelow, K., Pui, C. H., Yee, S. W., et al. (2011). Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharmacol. Ther. 89, 387–391.
Relling, M. V., Nemec, J., Schuetz, E. G., Schuetz, J. D., Gonzalez, F. J., and Korzekwa, K. R. (1994). O-demethylation of epipodophyllotoxins is catalyzed by human cytochrome P450 3A4. Mol. Pharmacol. 45, 352–358.
Renbarger, J. L., Mccammack, K. C., Rouse, C. E., and Hall, S. D. (2008). Effect of race on vincristine-associated neurotoxicity in pediatric acute lymphoblastic leukemia patients. Pediatr. Blood Cancer 50, 769–771.
Restrepo, J. G., Martinez, C., Garcia-Agundez, A., Gaviria, E., Laguna, J. J., Garcia-Martin, E., et al. (2011). Cytochrome P450 CYP2B6 genotypes and haplotypes in a Colombian population: identification of novel variant CYP2B6 alleles. Pharmacogenet. Genomics 21, 773–778.
Rocha, V., Porcher, R., Fernandes, J. F., Filion, A., Bittencourt, H., Silva, W. Jr., et al. (2009). Association of drug metabolism gene polymorphisms with toxicities, graft-versus-host disease and survival after HLA-identical sibling hematopoietic stem cell transplantation for patients with leukemia. Leukemia 23, 545–556.
Rousseau, J., Gagne, V., Labuda, M., Beaubois, C., Sinnett, D., Laverdiere, C., et al. (2011). ATF5 polymorphisms influence ATF function and response to treatment in children with childhood acute lymphoblastic leukemia. Blood 118, 5883–5890.
Shimasaki, N., Mori, T., Samejima, H., Sato, R., Shimada, H., Yahagi, N., et al. (2006). Effects of methylenetetrahydrofolate reductase and reduced folate carrier 1 polymorphisms on high-dose methotrexate-induced toxicities in children with acute lymphoblastic leukemia or lymphoma. J. Pediatr. Hematol. Oncol. 28, 64–68.
Song, S., Suzuki, H., Kawai, R., and Sugiyama, Y. (1999). Effect of PSC 833, a P-glycoprotein modulator, on the disposition of vincristine and digoxin in rats. Drug Metab. Dispos. 27, 689–694.
Stanulla, M., Schaeffeler, E., Flohr, T., Cario, G., Schrauder, A., Zimmermann, M., et al. (2005). Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 293, 1485–1489.
Stocco, G., Cheok, M. H., Crews, K. R., Dervieux, T., French, D., Pei, D., et al. (2009). Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin. Pharmacol. Ther. 85, 164–172.
Strand, V., Cohen, S., Schiff, M., Weaver, A., Fleischmann, R., Cannon, G., et al. (1999). Treatment of active rheumatoid arthritis with leflunomide compared with placebo and methotrexate. Leflunomide Rheumatoid Arthritis Investigators Group. Arch. Intern. Med. 159, 2542–2550.
Strunk, T., Gottschalk, S., Goepel, W., Bucsky, P., and Schultz, C. (2003). Subacute leukencephalopathy after low-dose intrathecal methotrexate in an adolescent heterozygous for the MTHFR C677T polymorphism. Med. Pediatr. Oncol. 40, 48–50.
Su, H. I., Sammel, M. D., Velders, L., Horn, M., Stankiewicz, C., Matro, J., et al. (2010). Association of cyclophosphamide drug-metabolizing enzyme polymorphisms and chemotherapy-related ovarian failure in breast cancer survivors. Fertil. Steril. 94, 645–654.
Szeto, D. W., Cheng, Y. C., Rosowsky, A., Yu, C. S., Modest, E. J., Piper, J. R., et al. (1979). Human thymidylate synthetase – III. Effects of methotrexate and folate analogs. Biochem. Pharmacol. 28, 2633–2637.
Takada, K., Arefayene, M., Desta, Z., Yarboro, C. H., Boumpas, D. T., Balow, J. E., et al. (2004). Cytochrome P450 pharmacogenetics as a predictor of toxicity and clinical response to pulse cyclophosphamide in lupus nephritis. Arthritis Rheum. 50, 2202–2210.
Vagace, J. M., Caceres-Marzal, C., Jimenez, M., Casado, M. S., De Murillo, S. G., and Gervasini, G. (2011). Methotrexate-induced subacute neurotoxicity in a child with acute lymphoblastic leukemia carrying genetic polymorphisms related to folate homeostasis. Am. J. Hematol. 86, 98–101.
Vagace, J. M., De La Maya, M. D., Caceres-Marzal, C., Gonzalez De Murillo, S., and Gervasini, G. (2012). Central nervous system chemotoxicity during treatment of pediatric acute lymphoblastic leukemia/lymphoma. Crit. Rev. Oncol. Hematol. 84, 274–286.
Vagace, J.M., and Gervasini, G. (2011). “Chemotherapy toxicity in patients with acute leukemia,” in Acute Leukemia: The Scientist’s Perspective and Challenge, ed. M. Antica (Rijeka, Croatia: INTECH), 391–414.
Varatharajan, S., Abraham, A., Zhang, W., Shaji, R. V., Ahmed, R., Abraham, A., et al. (2012). Carbonyl reductase 1 expression influences daunorubicin metabolism in acute myeloid leukemia. Eur. J. Clin. Pharmacol. doi: 10.1007/s00228-012-1291-9 [Epub ahead of print].
Vivona, D., Bueno, C. T., Lima, L. T., Hirata, R. D., Hirata, M. H., Luchessi, A. D., et al. (2012). ABCB1 haplotype is associated with major molecular response in chronic myeloid leukemia patients treated with standard-dose of imatinib. Blood Cells Mol. Dis. 48, 132–136.
Wan Rosalina, W. R., Teh, L. K., Mohamad, N., Nasir, A., Yusoff, R., Baba, A. A., et al. (2011). Polymorphism of ITPA 94C>A and risk of adverse effects among patients with acute lymphoblastic leukaemia treated with 6-mercaptopurine. J. Clin. Pharm. Ther. 37, 237–241.
Watanabe, Y., Nakajima, M., Ohashi, N., Kume, T., and Yokoi, T. (2003). Glucuronidation of etoposide in human liver microsomes is specifically catalyzed by UDP-glucuronosyltransferase 1A1. Drug Metab. Dispos. 31, 589–595.
Weinshilboum, R. (2001). Thiopurine pharmacogenetics: clinical and molecular studies of thiopurine methyltransferase. Drug Metab. Dispos. 29, 601–605.
Weisman, M. H., Furst, D. E., Park, G. S., Kremer, J. M., Smith, K. M., Wallace, D. J., et al. (2006). Risk genotypes in folate-dependent enzymes and their association with methotrexate-related side effects in rheumatoid arthritis. Arthritis Rheum. 54, 607–612.
Wessels, J. A., Van Der Kooij, S. M., Le Cessie, S., Kievit, W., Barerra, P., Allaart, C. F., et al. (2007). A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum. 56, 1765–1775.
Xu, P. P., Chen, B. A., Feng, J. F., Cheng, L., Xia, G. H., Li, Y. F., et al. (2012). Association of polymorphisms of cytosine arabinoside-metabolizing enzyme gene with therapeutic efficacy for acute myeloid leukemia. Chin. Med. J. (Engl.) 125, 2137–2143.
Yang, L., Hu, X., and Xu, L. (2012). Impact of methylenetetrahydrofolate reductase (MTHFR) polymorphisms on methotrexate-induced toxicities in acute lymphoblastic leukemia: a meta-analysis. Tumour Biol. 33, 1445–1454.
Zhong, S., Huang, M., Yang, X., Liang, L., Wang, Y., Romkes, M., et al. (2006). Relationship of glutathione S-transferase genotypes with side-effects of pulsed cyclophosphamide therapy in patients with systemic lupus erythematosus. Br. J. Clin. Pharmacol. 62, 457–472.
Keywords: acute lymphoblastic leukemia, pharmacogenetics, toxicity, chemotherapy, genetic polymorphisms
Citation: Gervasini G and Vagace JM (2012) Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front. Gene. 3:249. doi: 10.3389/fgene.2012.00249
Received: 05 July 2012; Accepted: 26 October 2012;
Published online: 22 November 2012.
Edited by:
Kathrin Klein, Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, GermanyReviewed by:
Maria J. Prata, Institute of Molecular Pathology and Immunology of the University of Porto, PortugalNatasa Djordjevic, University of Kragujevac, Serbia
Copyright: © 2012 Gervasini and Vagace. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Guillermo Gervasini, Department of Medical and Surgical Therapeutics, Division of Pharmacology, Medical School, University of Extremadura, 06071 Badajoz, Spain. e-mail: ggervasi@unex.es