
- Division of Computational Genetics, Department of Clinical Sciences, Swedish University of Agricultural Sciences, Uppsala, Sweden
Background: Non-synonymous single-nucleotide polymorphisms (nsSNPs) within the coding regions of genes causing amino acid substitutions (AASs) may have a large impact on protein function. The possibilities to identify nsSNPs across genomes have increased notably with the advent of next-generation sequencing technologies. Thus, there is a strong need for efficient bioinformatics tools to predict the functional effect of AASs. Such tools can be used to identify the most promising candidate mutations for further experimental validation.
Results: Here we present prediction of AAS effects (PASE), a novel method that predicts the effect of an AASs based on physicochemical property changes. Evaluation of PASE, using a few AASs of known phenotypic effects and 3338 human AASs, for which functional effects have previously been scored with the widely used SIFT and PolyPhen tools, show that PASE is a useful method for functional prediction of AASs. We also show that the predictions can be further improved by combining PASE with information about evolutionary conservation.
Conclusion: PASE is a novel algorithm for predicting functional effects of AASs, which can be used for pinpointing the most interesting candidate mutations. PASE predictions are based on changes in seven physicochemical properties and can improve predictions from many other available tools, which are based on evolutionary conservation. Using available experimental data and predictions from the already existing tools, we demonstrate that PASE is a useful method for predicting functional effects of AASs, even when a limited number of query sequence homologs/orthologs are available.
Introduction
A non-synonymous single-nucleotide polymorphism (nsSNP) is a single-nucleotide change in a protein-coding region of a gene that causes an amino acid substitution (AAS) in the resulting protein. The importance of nsSNPs has been demonstrated in many studies (Risch and Merikangas, 1996; Collins et al., 1997; Chasman and Adams, 2001), and in databases such as the Online Mendelian Inheritance in Man (OMIM), AASs represent most of the genetic variants known to cause disease in human (Hamosh et al., 2005). An increasing use of next-generation sequencing (NGS) technologies to re-sequence genomes has resulted in an accelerated identification of nsSNPs.
As the functional effects of most AASs are experimentally unexplored, there is a need for development of tools to efficiently predict substitution effects on protein structure and function. Currently, several methods exist that are based on the degree of evolutionary conservation as an indicator of the functional effect of an AAS. Widely used software tools are, for example, SIFT (Ng and Henikoff, 2002, 2003) and PolyPhen (Stitziel et al., 2003, 2004). SIFT is based on sequence homology and position-specific scoring matrices with Dirichlet priors, whereas PolyPhen uses sequence conservation and ternary structure to model AAS sites combined with SWISS-PROT annotation (Ng and Henikoff, 2006). However, when the degree of sequence conservation in distant related species is difficult to assess due to a limited amount of orthologous sequences, alternative information such as physicochemical properties of amino acid (AA) may be applied for functional prediction of AAS. Here we present prediction of AAS effects (PASE), a novel method for efficient computational prediction and visualization of the effect of nsSNPs on the final protein. The prediction from PASE is based on the selected seven physicochemical properties (Table 1). The information obtained from PASE can also be combined with knowledge about sequence conservation to further improve functional predictions. Several examples are provided to illustrate the predictive ability of PASE both independently and combined with conservation information.
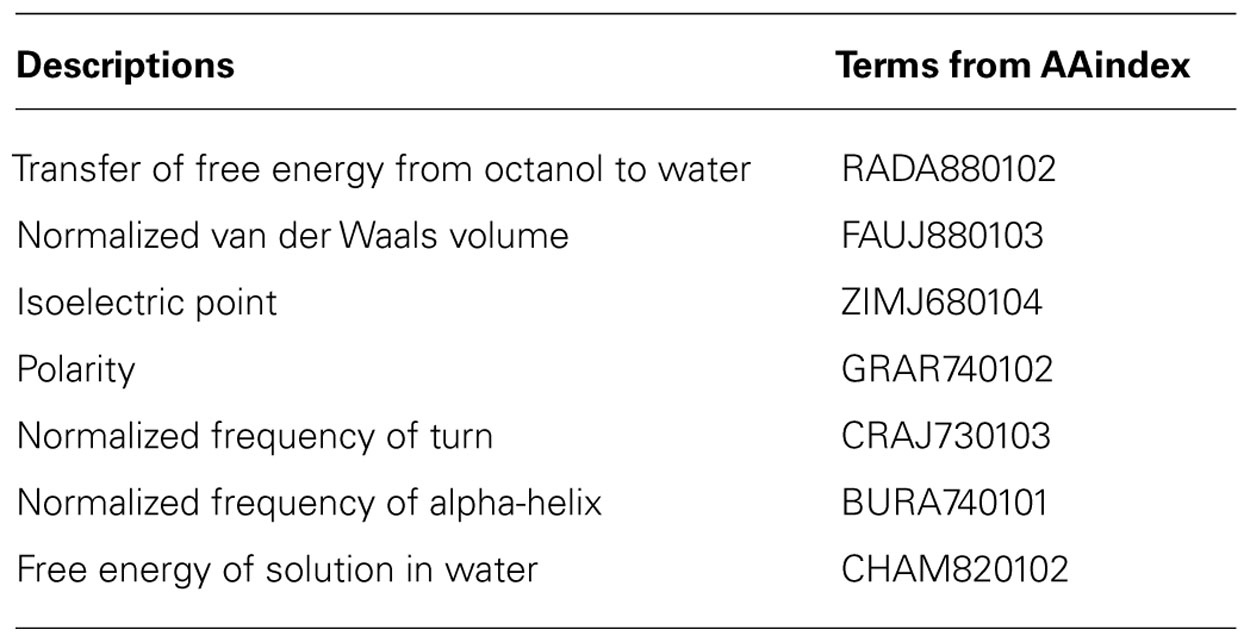
TABLE 1. Physicochemical properties of amino acids as described in the AAindex (Kawashima et al., 1999, 2008).
Implementation
The PASE algorithm uses physicochemical properties of AAs and sequence conservation to estimate the effect of each AAS on the protein (Figure 1). To calculate the AAS-induced change in the physicochemical properties, we imported seven physicochemical properties from the AAindex database previously selected by (Rudnicki and Komorowski (2004); Table 1), which gives every AA a unique profile. To illustrate how PASE can be combined with information about sequence conservation, we calculate a simple conservation score from a given protein sequence based on the alignment of homologous and orthologous sequences.
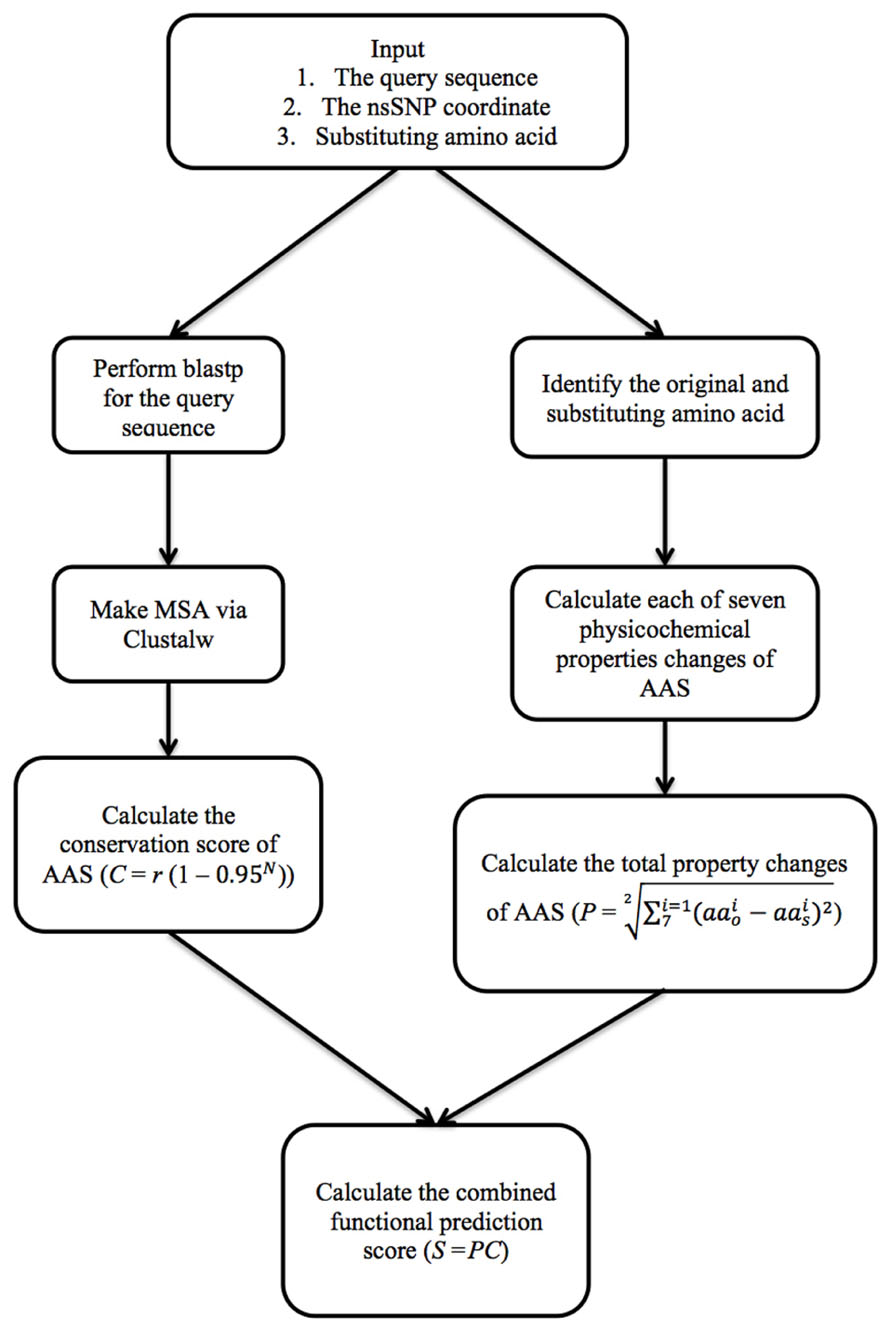
FIGURE 1. The algorithm of PASE.
Physicochemical Property (PASE) Score
Each AA has a characteristic profile of physicochemical properties. Therefore, any AAS may influence the final protein structure by altering its physicochemical properties. PASE applies Euclidean distance formula to compute the physicochemical property changes of AAS between the original and the substituting AA:
where and indicate one of the seven physicochemical properties of original AA and substituted AA, respectively.
The seven physicochemical properties: (1) transfer of free energy from octanol to water, (2) normalized van der Waals volume, (3) isoelectric point, (4) polarity, (5) normalized frequency of turn, (6) normalized frequency of alpha-helix, and (7) free energy of solution in water used by PASE, have been selected from the AAindex database of protein indices (Kawashima et al., 1999, 2008) using an algorithm described by Rudnicki and Komorowski (2004). Briefly, the properties were selected to reflect the major biologically meaningful features of protein sequences: (1) hydrophobicity, (2) polarity, (3) size, (4) tendency to form particular secondary structure, and (5) electrostatic properties of the AA. At least one property has been selected from each of these broad groups and resulted in a seven tuple of properties. Due to the low pairwise correlation within the selected properties, the ability to uniquely identify each AA is preserved (Kierczak et al., 2009; Figure 2). In other words, the selected properties span a nearly orthogonal 7D coordinates space in which every AA is a unique point (Figure 3).
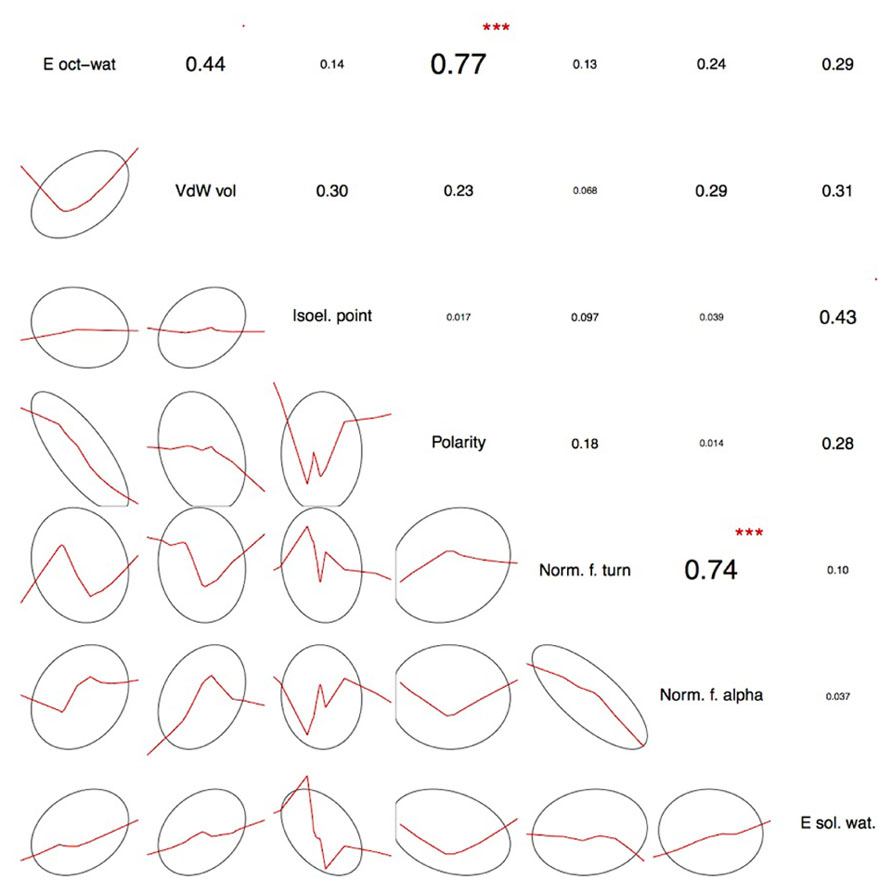
FIGURE 2. Pairwise correlations between the selected physicochemical amino acid properties. The actual correlation coefficients are presented in the upper triangle together with correlation significance symbols: ***p < 0.001, **p < 0.01, *p < 0.05, ’p < 0.1. Lower triangle shows smoothed trend lines together with confidence 1 SD ellipses. The vast majority of the selected properties show low pairwise correlation thus spanning a close-to-orthogonal coordinates frame in the physicochemical property space. As expected, there is a significant correlation between polarity-and transfer of free energy from octanol to water as the latter is to a large degree amino acid polarity-dependent. Similarly, normalized frequency of alpha-helix and normalized frequency of turn, the two secondary structure-related properties show significant pairwise correlation. This partial redundancy is the result of the physicochemical property selection procedure where the initial set of available AAindex descriptors has been narrowed down to only the easy-to-interpret properties. Thus, the selected seven properties are a reasonable trade-off between minimizing the number of dimensions necessary to preserve amino acid discernibility and the ease of interpretation. For a more detailed discussion, see Rudnicki and Komorowski (2004) and Kierczak et al. (2009).
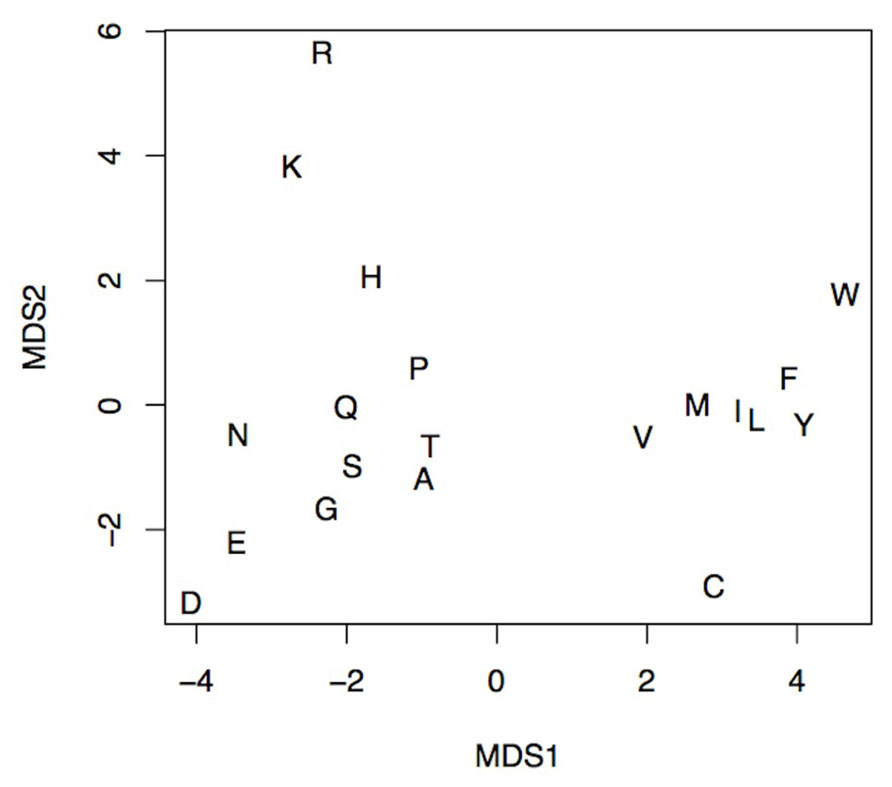
FIGURE 3. An MDS projection of the AA’s property space spanned by the seven selected physicochemical properties. The distance between any pair of letters corresponds to the physicochemical similarity between respective amino acids. The figure shows the ability to discern any pair of amino acids using selected physicochemical properties. The more similar amino acids, the smaller the distance between them, e.g., Ile (I) and Leu (L). Likewise, amino acids with similar physicochemical properties like basic Lys (K), Arg (R), and His (H) form distinct groups.
MSA Conservation Score
Highly conserved sequences often indicate a functional importance of the AA and substitutions tend to be deleterious, whereas those within areas of low conservation are often tolerated. We calculate a simple sequence conservation score (multiple sequence alignment conservation, MSAC), by searching for functionally related protein sequences by using NCBI-blastp (Thompson et al., 1994) and generating an alignment with multiple homologous sequences with an arbitrary threshold of 0.01, where the redundant homologous sequences are filtered out in order to retrieve only one hit per species. ClustalW (Larkin et al., 2007) is used to make a multiple sequence alignment (MSA) to score conservation at each site. To accomplish it, we use the following formula:
where N is the number of assessed sequences of MSA, and r is the proportion of the AAs of interest in MSA. The formula (1 - 0.95N; Pei and Grishin, 2001) indicates the probability of 20 different AAs in a position for N random equal frequent AA sequences. For example, when N = 1, the probability of each AA in a position is 0.05, which is consistent with 20 AAs with frequency 1/20 each.
Combined PASEC Score
The PASE score can be combined with information about sequence conservation by creating a combined score – PASEC. PASEC is computed as S = PC, where the score of physicochemical properties changes is multiplied by the conservation score. The PASEC score ranges from 0 to 1, where 0 is neutral, and higher ratio indicate stronger predicted effects on the protein. We have here for illustration combined PASE with a simple MSAC score, but other and more advanced measures of conservation can be used in the same manner.
Results and Discussion
To explore the predictive ability of the new method, we have studied two protein sequences, with known AASs that have well-characterized phenotypic effects.
nsSNPs in the Human Cx50 Gene
Gap junction proteins, also called connexins, belong to a family of channel-forming structure proteins in contacting plasma membranes (Dbouk et al., 2009). Recently, two consecutive AASs (W45S and G46V) in the human connexin 50 (Cx50) gene have been associated with cataracts. It has been shown that these two mutations cause the disease through different mechanism (Tong et al., 2011).
As shown in Table 2, both W45S and G46V show large properties changes (PASE scores 0.80 and 0.56) as well as high conservation scores (MSAC scores for both of 0.93). The combined PASEC scores are 0.74 and 0.52 for W45S and G46V, respectively, indicating considerable expected effects on protein function. This is consistent with the disease association of both these AASs (Tong et al., 2011) as well as with predictions made using the SIFT and PolyPhen tools, although the PolyPhen prediction were less decisive regarding the effect of the mutations (Table 2).
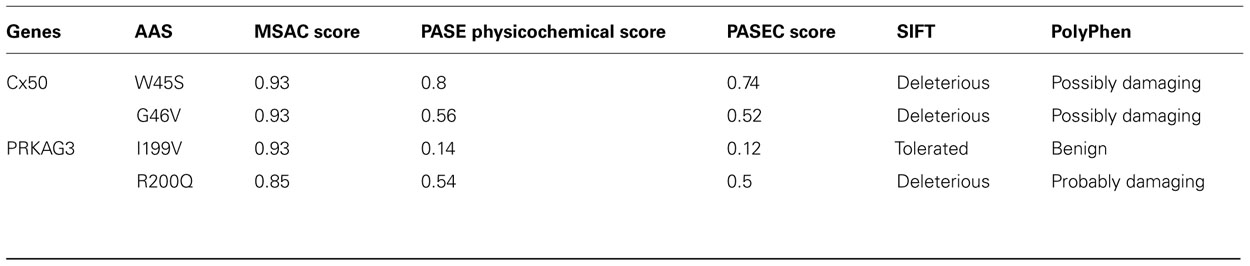
TABLE 2. Functional prediction of AASs in Cx50 and PRKAG3.
nsSNPs in the Porcine PRKAG3 Gene
The protein kinase AMP-activated gamma 3 (PRKAG3) gene encodes the regulatory γ subunit of adenosine monophosphate-activated protein kinase (AMPK), which is prevalently expressed in white skeletal muscle fibers (Mahlapuu et al., 2004). The dominant RN mutation that causes excess glycogen content in pig skeletal muscle is caused by the R200Q AAS in AMPKγ3 of purebred Hampshire pigs (Enfält et al., 1997; Milan et al., 2000). A substitution of the adjacent AA, I199V accounts for smaller increase in the muscle glycogen content than the R200Q substitution, but also co-participates with R200Q in the process (Ciobanu et al., 2001).
Exploration of the conservation and changes in physicochemical property of the AASs in the porcine PRKAG3 revealed large physicochemical property changes for R200Q (PASE score 0.54) and smaller for I119V (PASE score 0.14) as well as high degrees of conservation at both 199th (MSAC score 0.85) and 200th (MSAC score 0.93) sites (see Table 2). The combined PASEC functional prediction scores are 0.50 and 0.12, respectively, thus consistent with the observed phenotypic effects and with the SIFT and PolyPhen predictions for these mutations (see Table 2).
Comparison of Functional Prediction Scores for PASE, PASEC, Sift, and PolyPhen
To further evaluate the PASE and PASEC predictions, we used 3338 AASs in the Ensembl database representing nsSNPs on human chromosome 22 with functional effects previously predicted with the widely used SIFT and PolyPhen tools. For AASs predicted with SIFT as “tolerated” (1987 AASs) or with PolyPhen as “benign” (1637 AASs), PASE/PASEC showed average scores of X/0.18 and Y/0.16, respectively, whereas AASs predicted with SIFT as “deleterious” (1351 AASs) or with PolyPhen as “probably damaging” (1162 AASs) showed average scores of Z/0.3 and A/0.33 (Table 3). The distributions of PASE, MSAC, and PASEC scores are shown in Figures 4A–C, respectively.
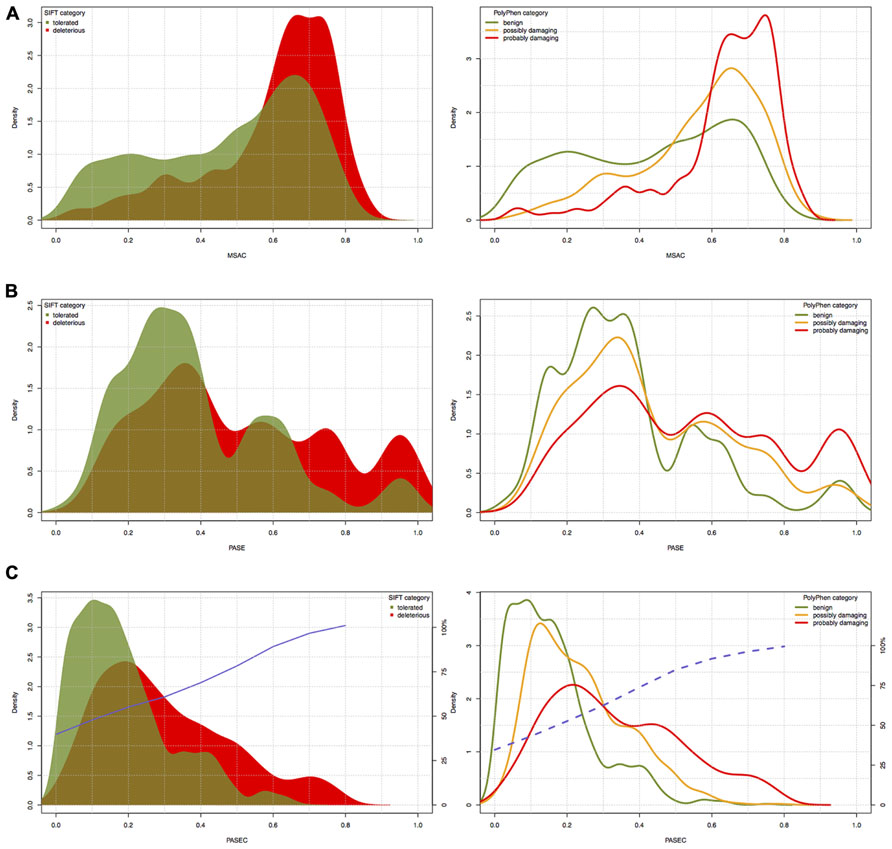
FIGURE 4. The distribution of (A) PASE, (B) MSAC, and (C) PASEC scores within different SIFT and PolyPhen prediction classes. Blue solid and dashed lines in panel (C) correspond to the probability of deleterious/damaging prediction from AAS’s PASEC scores.
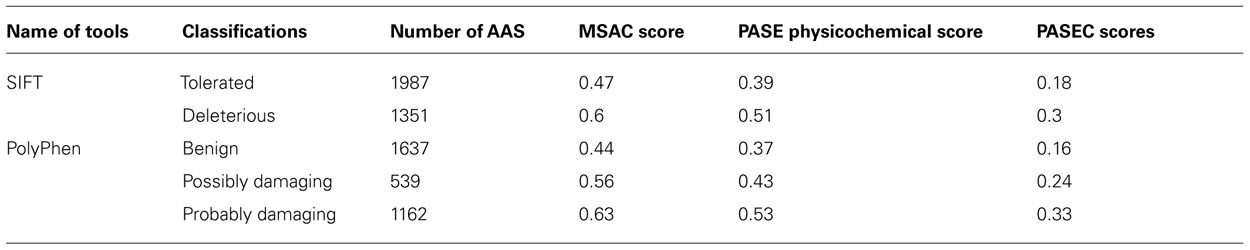
TABLE 3. Functional predictions of AASs in Human Chromosome 22.
The PASE is novel in its approach to utilize seven physicochemical properties of AA’s to predict the effect of an AAS. In Figure 4, we illustrate the overlap between the scores obtained with PASE, MSAC, and PASEC and those obtained using the conservation-based scores from SIFT and PolyPhen. Figure 4A shows that the PASE score provides novel predictions regarding which AAS are likely to have a functional effect in that both AAS that have high and low SIFT and PolyPhen scores have high PASE scores. Interestingly, however, there is an overrepresentation of AASs predicted to be deleterious/damaging among those with high PASE scores, which indicates that combining information on conservation and physicochemical properties might help to further refine the predictions of function of AAS. Figure 4B shows the overlap between the simple conservation score MSAC and SIFT/PolyPhen. The MSAC score overlaps to a large extent with the predictions of deleterious/damaging AAS from SIFT/PolyPhen, in that such AAS have higher MSAC scores, but shows little overlap for the tolerated/benign AAS. Figure 4C shows that the combined PASEC score, combines the information about the physiochemical properties in the PASE score and the conservation information in the MSAC score to further refine the predictions. As shown in Table 4, at PASEC scores above 0.8, all of these are predicted by SIFT/PolyPhen to be damaging/deleterious and at a score of 0.5 this has decreased to 81/87%. Most of the AASs with high PASEC scores are thus defined as deleterious/damaging in SIFT/PolyPhen. There are, however, also a few AAS defined as tolerated/benign with high PASEC scores and a large number of AAS defined as deleterious/damaging with low PASEC scores. This probably reflects that at poorly conserved locations AASs often do not affect the protein function significantly, and even large physicochemical property changes are tolerated and more easily become abundant. Conversely, at highly conserved locations the protein function is very sensitive even for AASs with small physicochemical property changes. Combining conservation and physicochemical property scores thus identifies a smaller number of functional candidates that simultaneously fulfills both criteria and thus are the most interesting candidates for further characterization. We have here used MSAC as a simple score for measuring sequence conservation and compared the overlap to results from other methods based on sequence conservation. It should be noted that MSAC only serves an illustrative purpose in our description of PASEC and could also be exchanged by sequence conservation scores from SIFT or PolyPhen when applied to real data.
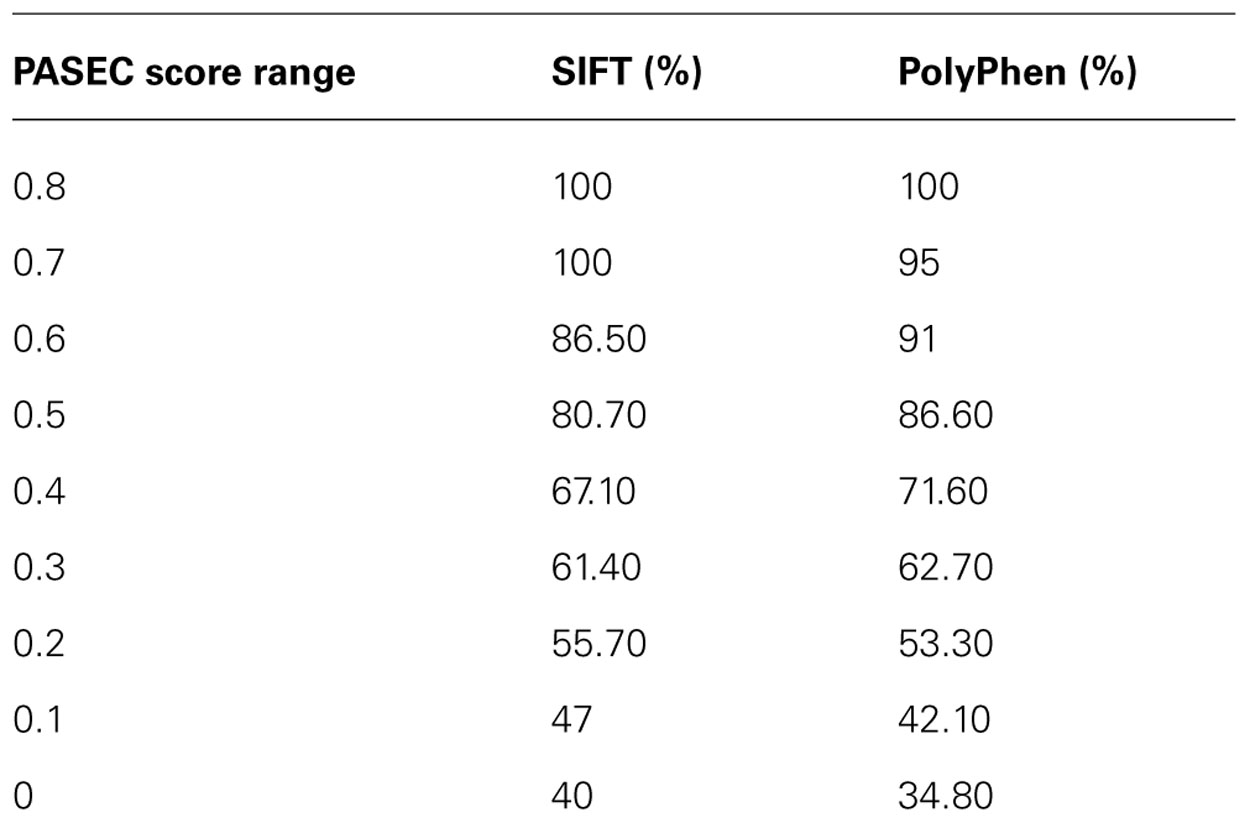
TABLE 4. Proportion of AAS’s with deleterious/damaging predictions from the SIFT/PolyPhen algorithm at different PASEC scores.
Our future research will focus on tailoring PASEC to specific protein classes (e.g., trans-membrane proteins) and protein families. We are planning to use statistical learning methods to take into account the specific microenvironment of the substituted AA and to infer more general, interpretable rules describing the degree of AAS impact on protein function. To this end we need to collect larger datasets of carefully curated examples of different classes of AAS and to integrate information available from several sources, like databases, structure prediction servers, etc.
Conclusion
Functional prediction of AASs is highly important in genetic studies. Functional importance of an AA is often reflected by a high degree of evolutionary conservation. This has earlier been used for functional prediction of AAS in, e.g., the SIFT and PolyPhen algorithm predictions. Here, we propose a new method, PASE, which predicts the functional effect of an AAS based on the physicochemical properties of the alternative AAs. This allows prediction of the potential effect of an AAS in the protein structure and its interaction with other residues. PASE complements already available tools by allowing such functional prediction of an AAS’s effect in situations where information on evolutionary conservation is not available or applicable. As the physiochemical property of an AA and its degree of evolutionary conservation are independent, these two sources of information should if combined be able to better predict the potential functional importance of an AAS. Here, we also show the usefulness of this combined approach and its potential to refine predictions made by the approaches independently.
Availability and Requirements
Project homepage:http://www.computationalgenetics.se/wp-content/uploads/software/PASE/pase.tar
Project name: PASE
Operating system(s): Linux and Unix
Programing language: Python and Biopython
Other requirements: Python 2.6 or higher, Biopython 1.58 or higher and ClustalW (1.82) or higher
License: GNU
Any restrictions to use by non-academics: none
Author Contributions
Marcin Kierczak initiated the study. Marcin Kierczak, Xidan Li, Stefan Marklund, and Örjan Carlborg planned the study. Xidan Li, Marcin Kierczak, Xia Shen, Stefan Marklund, and Örjan Carlborg developed the algorithm that was implemented by Xidan Li and Marcin Kierczak. Xidan Li and Muhammad Ahsan evaluated the algorithm. Xidan Li and Marcin Kierczak drafted the manuscript and all authors contributed to the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by a EURYI award to Örjan Carlborg and a Future research leader grant to Örjan Carlborg from the Swedish Foundation for Strategic Research. The contribution of Muhammad Ahsan was supported by his scholarship from the Higher Education Commission of Pakistan.
Abbreviations
AA, amino acid; AAS, amino acid substitution; MDS, multiple dimensional scaling; MSA, multiple sequence alignment; MSAC, MSA conservation; nsSNP, non-synonymous single-nucleotide polymorphism; PASE, prediction of amino acid substitution effects; PRKAG3, protein kinase AMP-activated gamma 3; SNP, single-nucleotide polymorphism.
References
Chasman, D., and Adams, R. M. (2001). Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: structure-based assessment of amino acid variation. J. Mol. Biol. 307, 683–706.
Ciobanu, D., Bastiaansen, J., Malek, M., Helm, J., Woollard, J., Plastow, G., et al. (2001). Evidence for new alleles in the protein kinase adenosine monophosphate-activated γ3-subunit gene associated with low glycogen content in pig skeletal muscle and improved meat quality. Genetics 159, 1151–1162.
Collins, F. S., Guyer, M. S., and Charkravarti, A. (1997). Variations on a theme: cataloging human DNA sequence variation. Science 278, 1580–1581.
Dbouk, H. A., Mroue, R. M., El-Sabban, M. E., and Talhouk, R. S. (2009). Connexins: a myriad of functions extending beyond assembly of gap junction channels. Cell Commun. Signal. 7, 4.
Enfält, A. C., Lundström, K., Karlsson, A., and Hansson, I. (1997). Estimated frequency of the RN-allele in Swedish Hampshire pigs and comparison of glycolytic potential, carcass composition, and technological meat quality among Swedish Hampshire, Landrace, and Yorkshire pigs. J. Anim. Sci. 75, 2924–2935.
Hamosh, A., Scott, A. F., Amberger, J. S., Bocchini, C. A., and McKusick, V. A. (2005). Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517.
Kawashima, S., Ogata, H., and Kanehisa, M. (1999). AAindex: amino acid index database. Nucleic Acids Res. 27, 368–369.
Kawashima, S., Pokarowski, P., Pokarowska, M., Kolinski, A., Katayama, T., and Kanehisa, M. (2008). AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 36, D202–D205.
Kierczak, M., Ginalski, K., Draminski, M., Koronacki, J., Rudnicki, W. R., and Komorowski, J. (2009). A rough set-based model of HIV-1 reverse transcriptase resistome. Bioinform. Biol. Insights 3, 109–127.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948.
Mahlapuu, M., Johansson, C., Lindgren, K., Hjälm, G., Barnes, B. R., Krook, A., et al. (2004). Expression profiling of the gamma-subunit isoforms of AMP-activated protein kinase suggests a major role for gamma3 in white skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 286, E194–E200.
Milan, D., Jeon, J. T., Looft, C., Amarger, V., Robic, A., Thelander, M., et al. (2000). A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288, 1248–1251.
Ng, P. C., and Henikoff, S. (2002). Accounting for human polymorphisms predicted to affect protein function. Genome Res. 12, 436–446.
Ng, P. C., and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814.
Ng, P. C., and Henikoff, S. (2006). Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 7, 61–80.
Pei, J., and Grishin, N. V. (2001). AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 17, 700–712.
Risch, N., and Merikangas, K. (1996). The future of genetic studies of complex human diseases. Science 273, 1516–1517.
Rudnicki, W., and Komorowski, J. (2004). “Feature synthesis and extraction for the construction of generalized properties of amino acids,” in Proceedings of Rough Sets and Current Trends in Computing: 4th International Conference, Uppsala, Sweden, June 1–5, 786–791.
Stitziel, N. O., Binkowski, T. A., Tseng, Y. Y., Kasif, S., and Liang, J. (2004). topoSNP: a topographic database of non-synonymous single nucleotide polymorphisms with and without known disease association. Nucleic Acids Res. 32, D520–D522.
Stitziel, N. O., Tseng, Y. Y., Pervouchine, D., Goddeau, D., Kasif, S., and Liang, J. (2003). Structural location of disease-associated single-nucleotide polymorphisms. J. Mol. Biol. 327, 1021–1030.
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680.
Keywords: PASE, amino acid substitution, physicochemical properties, functional prediction, mutation effect
Citation: Li X, Kierczak M, Shen X, Ahsan M, Carlborg Ö and Marklund S (2013) PASE: a novel method for functional prediction of amino acid substitutions based on physicochemical properties. Front. Genet. 4:21. doi: 10.3389/fgene.2013.00021
Received: 24 October 2012; Paper pending published: 23 November 2012;
Accepted: 11 February 2013; Published online: 06 March 2013.
Edited by:
Andrzej M. Kierzek, University of Surrey, UKReviewed by:
Franca Fraternali, King’s College London, UKAli Rana Atilgan, Sabanci University, Turkey
Copyright: © 2013 Li, Kierczak, Shen, Ahsan, Carlborg and Marklund. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Xidan Li, Division of Computational Genetics, Department of Clinical Sciences, Swedish University of Agricultural Sciences, Box 7078, SE-750 07 Uppsala, Sweden. e-mail: xidan.li@slu.se
†Xidan Li and Marcin Kierczak have contributed equally to this work.