
- Department of Biochemistry and Molecular Biology, University of Texas MD Anderson Cancer Center, Houston, TX, USA
It is well accepted that non-coding RNAs play a critical role in regulating gene expression. Recent paradigm-setting studies are now revealing that non-coding RNAs, other than microRNAs, also play intriguing roles in the maintenance of chromatin structure, in the DNA damage response, and in adult human stem cell aging. In this review, we will discuss the complex inter-dependent relationships among non-coding RNA transcription, maintenance of genomic stability, chromatin structure, and adult stem cell senescence. DNA damage-induced non-coding RNAs transcribed in the vicinity of the DNA break regulate recruitment of the DNA damage machinery and DNA repair efficiency. We will discuss the correlation between non-coding RNAs and DNA damage repair efficiency and the potential role of changing chromatin structures around double-strand break sites. On the other hand, induction of non-coding RNA transcription from the repetitive Alu elements occurs during human stem cell aging and hinders efficient DNA repair causing entry into senescence. We will discuss how this fine balance between transcription and genomic instability may be regulated by the dramatic changes to chromatin structure that accompany cellular senescence.
Introduction
Cellular senescence is an irreversible cell cycle arrest caused by intrinsic or extrinsic stress, such as shortened telomeres, DNA damage, induced oncogenes, or chromatin perturbation. This phenotype was first observed by Hayflick (1965) using human diploid cells which could undergo a limited number of cell divisions in in vitro culture. Various signaling pathway and regulatory factors of senescence have been reported over the last four decades (Campisi and d’Adda di Fagagna, 2007). Cellular senescence contributes to the aging process (Campisi and d’Adda di Fagagna, 2007; Baker et al., 2011). Aging is highly complex and poorly understood. It is characterized by time-dependent degenerative changes caused by genetic (intrinsic) or environmental (extrinsic) stimuli. Although once thought to be an amalgamation of random detrimental events, the identification of mutations and diets that promote longevity has led to the realization that particular cellular pathways influence the aging process. The challenge now is to understand the mechanistic basis of how these pathways and metabolic states regulate aging.
Although numerous different pathways for aging have been proposed, it is striking that human premature aging syndromes are caused by defects in the maintenance of genomic stability (Coppede, 2012), indicating that genomic integrity plays a key role in determining lifespan. In agreement, the accumulation of DNA damage is a hallmark of aging and senescing cells (Sedelnikova et al., 2004), and the DNA damage signal emanating from shortened telomeres triggers cells to enter a state of senescence (d’Adda di Fagagna et al., 2003; Takai et al., 2003; Herbig et al., 2004). In turn, genomic stability is closely intertwined with the chromatin structure. The chromatin structure not only regulates the accessibility of DNA damaging agents to the genome, but also plays critical roles in the signaling of DNA lesions and their repair (see Papamichos-Chronakis and Peterson, 2013 for a very nice recent review). Given the critical role of chromatin in regulating genomic stability and gene expression, it is tempting to speculate that some of the changes in gene expression and genomic integrity that occur during aging may be caused by the global changes to the chromatin structure that accompany aging. These chromatin changes span from the global loss of heterochromatin in aging human cells (Howard, 1996; Villeponteau, 1997; Kitano and Imai, 1998; Tsurumi and Li, 2012) to the global reduction of histone levels during mitotic aging in yeast (Feser et al., 2010) and human fibroblasts (O’Sullivan et al., 2010). Here we will discuss how non-coding RNAs contribute to the relationships among chromatin structure, genomic integrity, and cellular senescence.
Non-Coding RNA Transcription is Induced by DNA Damage
DNA damage is induced by various genotoxic stresses from inside the cell or from the environment. Arguably the most dangerous type of DNA lesion is a double-strand break (DSB) because a single DSB can lead to the loss of a chromosome arm during mitosis if not repaired, while its inaccurate repair can lead to chromosomal translocations or mutations. DSBs are repaired by either homologous recombination (HR) which copies the identical undamaged DNA information usually from the sister chromatid, or non-homologous end joining (NHEJ) in which the two DNA ends are ligated back together (reviewed in Ciccia and Elledge, 2010). Following the generation of a DSB, the cell mediates a highly orchestrated series of events termed the DNA damage response (DDR; reviewed in Finn et al., 2012; Papamichos-Chronakis and Peterson, 2013). The function of the DDR is at least five fold: (i) to arrest the cell cycle until the DNA damage is repaired (checkpoint activation), (ii) to upregulate gene expression of the repair machinery, and ultimately (iii) to remodel and restore chromatin structure around damage site, (iv) to repair the DNA molecule per se by recruitment of the DNA repair machinery, (v) to trigger apoptosis if there is too much DNA damage to be repaired. At an early stage of the DDR phosphatidylinositol 3-kinase-like protein kinase (PIKKs) family [ATM (ataxia-telangiectasia mutated), ATR (ATM and Rad3-related), and DNA-PKcs (DNA-dependent protein kinase)] and MRN (Mre11–Rad50–Nbs1) complexes are recruited to the DNA breaks. The PIKKs phosphorylate downstream checkpoint proteins, such as Chk1 and Chk2, and the histone variant H2A.X that is present in the chromatin around the breaks. Phosphorylated Chk1 and Chk2 phosphorylate various effector proteins to arrest the cell cycle to allow time for DNA repair. Phosphorylated H2A.X (called γH2A.X) assists to recruit other downstream signaling molecules that facilitate the DDR. The SWI/SNF (switch/sucrose non-fermentable) complex (ATP-dependent chromatin remodeling factor) triggers chromatin relaxation after UV treatment. This chromatin relaxation is involved in late stages of DNA repair because it does not affect the recruitment of early DDR response factors but later DDR response factors, such as xeroderma pigmentosum complementation group G (XPG) and proliferating cell nuclear antigen (PCNA) to UV damage sites (Zhao et al., 2009). The proteins that mediate the DDR are conserved from yeast to human, meaning that our understanding of the DDR can be gained from studies in many different model systems.
Several different types of non-coding RNAs have been implicated in the DDR. For example, many microRNAs (miRNAs) regulate genes that are involved in the DDR (reviewed in Wan et al., 2011; Han et al., 2012). However, these miRNAs are not necessarily encoded by genes in the vicinity of the DNA damage (Figure 1). Moreover, other non-coding RNAs are more directly involved in the DDR. Over 5% of the mammalian genome consists of short interspersed elements (SINEs), typified by the human Alu repeat. These are RNA polymerase III transcribed sequences whose expression is usually silenced. However, upon prolonged exposure to DNA damaging agents that predominantly cause DSBs, transcription from the human Alu elements and murine SINEs is strongly induced (Rudin and Thompson, 2001). The reason for the induction of the SINE/Alu non-coding transcription in response to DSBs is not clear, but it may be relevant that their silencing can be at least partially reversed by DNA demethylating agents in the absence of DNA damaging agents (Liu et al., 1994; Vorce et al., 1994). Pericentromeric DNA damage accumulation also leads to increased Alu transcript levels during adult human stem cell aging (Wang et al., 2011; this will be discussed more below). As such, it seems to be a reasonable prediction that the induction of SINE and Alu transcription in response to DSBs may be a consequence of decompaction of their normally repressive chromatin structure. In agreement with this, the association of histones with DNA is looser globally, as measured by their ability to be salt extracted, following treatment with DNA damaging agents (Xu et al., 2010). Similarly, transcription from the normally silenced yeast Ty retrotransposons is induced by exposure to a variety of DNA damaging agents including UV and γ-radiation (Morawetz, 1987; Bradshaw and McEntee, 1989; Morawetz and Hagen, 1990). Transcription of non-coding RNAs from Ty elements in response to DNA damage is unlikely to be beneficial to the host organism, given that this can lead to retrotransposition and insertion elsewhere into the genome, which itself is mutagenic. Insertion of new retrotransposon elements also increases the opportunity for unequal sister chromatid recombination.
It has been reported that new types of non-coding RNAs (rather than miRNAs or Alu elements) were induced by DNA damage, that are actually beneficial for DNA repair, in the filamentous fungus Neurospora crassa (Lee et al., 2009). However, this is not unique to Neurospora, as this has now been shown to be the case also in Arabidopsis and humans, as discussed below. In Neurospora, hydroxyurea and methyl methanesulfonate (agents that result in DNA breaks during replication) induce expression of a new class of small RNAs, about 20–21 nucleotides long, that originate mostly from the highly repetitive ribosomal DNA (rDNA) locus (Lee et al., 2009). These small RNAs originate from both strands of DNA in the region corresponding to the mature rRNAs but many also derive from the internal spacer regions of the rDNA. The remainder originates from genomic regions that encode transfer RNAs, other intergenic regions, and open reading frames. Production of these short RNAs requires RNA-dependent RNA polymerase, which can transcribe long RNAs that are 500 bp to 2 kb long (Lee et al., 2009). These long RNAs are then processed by Dicer to make the short RNAs, which bind to an active RNA-induced silencing complex (RISC) that includes Argonaute. Consistent with a role for these short RNAs in the DDR, Neurospora mutants lacking RNA-dependent RNA polymerase or Dicer are sensitive to DNA damaging agents. Because DNA damage is well known to result in a decrease in protein synthesis (Morley et al., 1998; Tee and Proud, 2000; Deng et al., 2002; Braunstein et al., 2009; Powley et al., 2009; Spriggs et al., 2010), Lee et al. (2009) hypothesized that production of the short DNA damage-induced RNAs from the rDNA may inhibit rRNA biogenesis and protein synthesis after DNA damage. Indeed, mutants defective in the synthesis of DNA damage-induced short RNAs failed to show a strong DNA damage-induced reduction in protein synthesis (Lee et al., 2009). Taken together, these results indicate that the production of small RNAs in Neurospora in response to DNA damage contributes to the DNA damage checkpoint by down-regulating protein synthesis.
One may ask: Why are small DNA damage-induced RNAs generated from the rDNA locus? The answer to this question may lie in the requirement for the single-strand DNA binding protein replication protein A (RPA) and the Neurospora counterpart of the Werner/Bloom syndrome helicase, QDE-1, for making small DNA damage-induced RNAs (Lee et al., 2010). A likely model is that the repetitive nature of the rDNA predisposes it to either DNA damage or the formation of aberrant structures upon replication stress, the repair or resolution of which requires unwinding of the DNA duplex by DNA helicases and entails processing of the rDNA into single-stranded DNA (ssDNA) coated by RPA. Consistent with this model, the RNA-dependent RNA polymerase uses ssDNA but not dsDNA as a template (Lee et al., 2010). Furthermore, RPA appears to recruit the RNA-dependent RNA polymerase to sites of ssDNA via their physical interaction. RPA also promotes the formation of dsRNA by the RNA-dependent RNA polymerase by disfavoring the formation of DNA/RNA hybrids (Lee et al., 2010). Taken together, the model for how small RNAs are generated from rDNA during replicational stress appears to be fairly sound. How these small DNA damage-induced RNAs result in downregulation of protein synthesis is less clear. The mechanism could be via small RNA-mediated degradation of the rRNAs or heterochromatinization of the rRNA locus, as both are mechanisms that are used in other scenarios in Neurospora (Dang et al., 2011).
A recent study showed that small non-coding RNAs can be induced around DSB sites in Arabidopsis. This work showed that small RNAs, referred to as diRNA (DSB-induced small RNAs), are induced in the vicinity of DSB sites, and revealed that these diRNAs promote repair of the DNA lesion (Wei et al., 2012). Generation of the diRNAs required the RNAi machinery such as Dicer and Argonaute 2 (AGO2). However, unlike the miRNAs that have also been implicated in the DDR (Hu and Gatti, 2011), diRNAs act in a manner distinct from translational inhibition through mRNA degradation. Following induction of a DSB using the endonuclease I-SceI that cuts at a known location in the genome, all the diRNAs mapped specifically to the vicinity of the DSB site. The diRNAs mapped to both strands of the DNA, spanning a few kilobases on either side of the DSB. The generation of the diRNA depended on the PI3 kinase-related kinase ATR (Wei et al., 2012). While the phosphorylation of γH2A.X was not dependent on the induction of the diRNAs, the efficiency of DNA repair was greatly reduced when diRNA induction was blocked. The diRNAs bind to the AGO2 protein, which had previously been shown to be induced upon ionizing radiation (Culligan et al., 2006). Indeed ionizing radiation also resulted in the binding of AGO2 to diRNAs that were induced from 150 different genomic loci, suggesting that this is a general phenomenon in response to DNA lesions generated at many locations throughout the genome (Wei et al., 2012).
Mammalian cells also show a similar local induction of small RNAs upon DNA damage. Wei et al. (2012) demonstrated that diRNAs are induced around a DSB in human cells, with them being generated up to 6000 bp away from the DNA break. In human cells, knocking down Dicer or Ago2 significantly reduced the efficiency of DNA repair. Similarly, knocking down human Dicer or Drosha reduced the number of cells with 53BP1 localizing to repair foci, which is a protein involved in driving repair into the NHEJ pathway and its recruitment efficiency is affected by chromatin changes such as histone modifications including phosphorylation on histone H2AX S139 (γ-H2AX; Celeste et al., 2002) methylation on histone H4 lysine 20 (Botuyan et al., 2006), and histone H3 lysine 79 (Huyen et al., 2004; Wakeman et al., 2012), the histone acetyltransferase (HAT) activity of Tip60 (Murr et al., 2006) and recruitment of both MDC1 (Stewart et al., 2003) and RNF8 (Huen et al., 2007; Mailand et al., 2007). Knocking down human Dicer or Drosha also reduced the number of cells with foci of ATM autophosphorylation and the phosphorylated substrates of ATM, but did not reduce the number of cells with γH2A.X foci (Francia et al., 2012). As such, one can conclude that the DNA damage-induced RNAs act downstream of γH2A.X in the DDR. Interestingly, MDC1 recruitment to chromatin is via binding to γH2A.X (Stucki et al., 2005), yet knockdown of Dicer and Drosha also reduced MDC1 foci formation (Francia et al., 2012). This suggests that γH2A.X is not sufficient for MDC1 recruitment because diRNA are also required. Consistent with this idea, RNase A treatment of cells reduced the accumulation of MDC1 and 53BP1 in repair foci (Pryde et al., 2005; Francia et al., 2012), but had no effect on γH2A.X (Francia et al., 2012). MDC1 in turn is required for the amplification of the DDR signal (Lou et al., 2006), which would explain why ATM autophosphorylation and ATM-mediated phosphorylation of its substrates fails to occur upon the knockdown of Dicer and Drosha (Francia et al., 2012). One possible model that remains to be tested is that the diRNAs, or the process of their transcription per se, may help to generate an open chromatin structure that is permissive for the recognition of γH2A.X by its binding partners such as MDC1 (Figure 1).
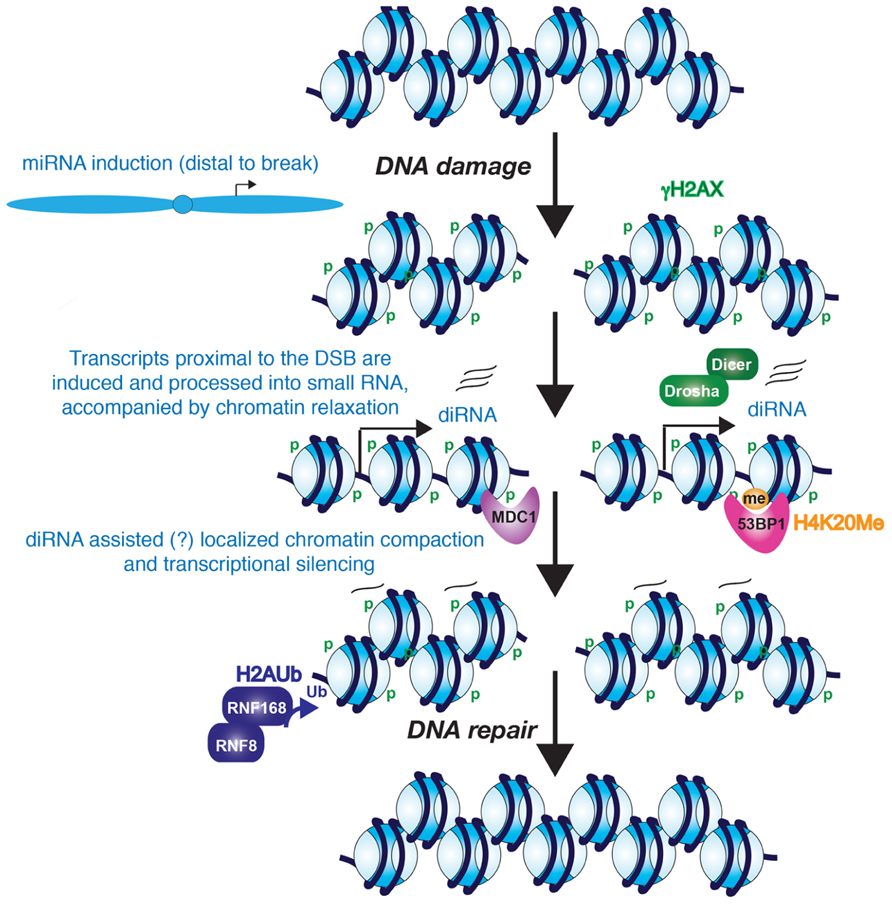
FIGURE 1. Potential role for diRNAs in the DNA damage response. The evidence seems likely that diRNAs are induced from the vicinity of a DNA break. Furthermore, these diRNAs, although not required for phosphorylation of H2AX, they are required for the recruitment of MDC1 and 53BP1 to their modified histone binding partners. Whether the diRNAs remain at the site of chromatin and contribute to heterochromatinization of the region around the DSB is unknown. However, it is clear that the chromatin becomes more highly compacted, perhaps at later stages of DNA repair, and this promotes the process of DNA repair.
RNA-Binding Proteins are Recruited to Double-Strand Break Sites
Perhaps related to the local transcriptional induction of small RNAs in response to DNA damage, several proteins that regulate RNA function have been shown to be transiently recruited to DNA damage sites. Adamson et al. (2012) found that the RNA-binding protein RBMX (RNA-binding motif protein encoded on the X chromosome) is recruited to DNA damage sites by genome-wide siRNA library screening. Furthermore, this appears to be important for DNA repair because silencing of RBMX leads to defects in HR-mediated DNA repair (Adamson et al., 2012). PPM1G, a phosphatase regulating splicing, is also recruited to DNA lesions (Beli et al., 2012). It has been reported that NONO, another RNA-binding protein, is recruited to a DSB site (Salton et al., 2010; Krietsch et al., 2012). Knockdown of NONO leads to a significant decrease in plasmid end joining and knockdown of NONO preferentially compromises NHEJ whereas HR is increased using in vivo NHEJ and HR reporter cell lines. Both RBMX and NONO are recruited to DNA lesions following laser-microirradiation in a poly-ADP-ribose polymerase (PARP)-dependent manner, where PARP mediates one of the earliest chromatin modifications in response to DNA damage (Adamson et al., 2012; Krietsch et al., 2012). Although the RNA targets at DSB sites that are bound by these factors are still unknown, these observations suggest possible active roles for RNA in DNA repair at DSB sites.
Transcriptional Repression Around Double-Strand Break Sites
The findings described above indicate that there is transcription in the immediate vicinity of DSBs in response to DNA damage. On the contrary, there is also evidence for transcriptional silencing induced in cis to a DSB, which may accompany, and facilitate, the later stages of DNA repair (Figure 1). Using a reporter that allows for visualization of the nascent transcript containing the MS2 RNA stem loop by binding to YFP-MS2 protein, silencing of transcription in cis to the DSB site has been observed (Shanbhag et al., 2010). However, DNA damage induced by ionizing radiation and laser-microirradiation does not cause global transcriptional silencing, indicating that the mechanism to induce transcriptional silencing only functions in the immediate proximity to the DSB (Shanbhag et al., 2010). Kruhlak et al. (2007) also reported that RNA polymerase I-mediated transcription of rDNA is inhibited in the vicinity of DSBs. Both forms of transcriptional silencing required ATM, suggesting that both pol I and pol II silencing may occur via a common mechanism around a DSB. Indeed, transcriptional arrest and dissociation of RNA polymerase II is induced around DSBs in active genes in a DNA-PK catalytic subunit-dependent manner (Pankotai et al., 2012). Consistent with the idea of transcriptional arrest, exclusion of the RNA processing factor THRAP3 (thyroid hormone receptor associated protein 3) from the vicinity of DSBs has also been shown, and this is dependent on the E3-ubiquitin ligases RNF8 and RNF168 (Shanbhag et al., 2010; Beli et al., 2012). Shanbhag et al. (2010) also observed ubiquitylated H2A around DSBs. H2A ubiquitylation has been strongly implicated in transcriptional silencing, suggesting that a repressive chromatin structure is formed around DSBs. DSB-induced chromatin condensation is likely to take place in cis not in trans since global chromatin decondensation is induced in response to DNA damage (Ziv et al., 2006).
Small non-coding RNAs are known to be involved in the formation of repressive heterochromatin structures over repetitive DNA elements, such as centromeric repeats and retrotransposons (Reyes-Turcu and Grewal, 2012). Although it has not yet been tested, it is possible that the diRNAs may function similarly to promote a more repressive chromatin environment around DSBs that may facilitate amplification of the DDR and DNA repair (Figure 1).
In previous sections, we discussed the possibility of chromatin decompaction and/or chromatin compaction by diRNA during the DDR. But what is the real function of diRNAs for regulation of chromatin structure around DSB sites? The chromatin structure around DSB site should be opened for protein recruitment (proximal to DSB site) and compacted for transcription repression (distal from DSB site). It is not clear which aspects of chromatin structure is regulated by diRNAs yet. There are several possibilities: (i) diRNAs could promote either open and closed chromatin structure around DSB sites. (ii) diRNAs could help make a boundary between closed and open chromatin structures around DSB sites. (iii) Compaction and/or decompaction of chromatin structures around DSBs by diRNAs may occur at different times during repair, or (iv) diRNAs may not directly regulate chromatin structure around DSBs but rather function through regulating the recruitment of DDR factors.
Histone Loss and Histone Gain During DNA Double-Strand Break Repair
It is highly likely that the changes to the chromatin structure and transcriptional states described above that occur in response to DSBs are accompanied by the removal and replacement of histone proteins from the DNA. Histones H3/H4 are located at the center of the nucleosome, and their removal from DNA or their placement onto the DNA necessitates the removal or replacement of the entire histone octamer. This appears to occur, at least to some degree during DNA repair, because newly-expressed histone H3.1 is recruited to sites of UV-induced and laser-irradiation-induced DNA damage in mammalian cells (Polo et al., 2006). The histone chaperone CAF-1 (chromatin assembly factor 1) is required for this incorporation of H3.1 onto the newly-repaired DNA, and CAF-1 physically localizes to sites of DNA repair (Polo et al., 2006). The histone chaperone complex of HIRA/UBN1/CABIN1 is also recruited to microirradiation sites, however, their function in DNA repair is not clear yet (Adamson et al., 2012). Anti-silencing function 1 homolog A/B (ASF1A/B), a histone chaperone for H3/H4 dimers, also colocalizes with replication fork-associated repair foci. By immunoprecipitation assay, ASF1A/B are precipitated with the MMS22L (methyl methanesulfonate-sensitivity protein 22-like) DNA repair protein, and ASF1A and ASF1B colocalize with RPA foci under camptothecin (a topoisomerase inhibitor) or hydroxyurea (ribonucleotide reductase inhibitor) treatment, indicating that ASF1A and ASF1B are also recruited to replication fork-associated DNA damage lesions (Duro et al., 2010). However, the function of ASF1A/B in DNA repair is not clear, because HR efficiency or RAD51 foci formation is not affected by knockdown of ASF1A and/or ASF1B (Duro et al., 2010). Why are histone chaperones recruited to DNA damage site? One possibility is that histone chaperones are necessary for the restoration of chromatin structure in the wake of DNA repair. In budding yeast, asf1 deletion mutants do not have a significant DNA repair deficiency (slightly delayed) but they do have reduced levels of histone H3 over the repaired DNA and defective recovery from checkpoint arrest after DNA repair (Chen et al., 2008). Similar results have been reported in a mammalian system. Depletion of ASF1A delays checkpoint recovery caused by UV irradiation (Battu et al., 2011).
Histones H2A/H2B occupy more peripheral positions on the nucleosome and they can be removed and replaced without the requirement for H3/H4 removal or replacement. Notably, PPM1G is recruited to DNA lesions in response to microirradiation (Beli et al., 2012), and it has been reported that PPM1G exerts H2A/H2B histone chaperone activity (Kimura et al., 2006). There is also evidence for the incorporation of variant versions of H2A in the vicinity of DNA breaks. For example, the DNA repair factor APLF functions as a histone chaperone, and is recruited to laser-microirradiation induced DSBs in a PARP-dependent manner (Mehrotra et al., 2011). One of two splice variants of macroH2A1, macroH2A1.1 (mH2A1.1) is recruited to DSBs, and its recruitment is dependent on its macrodomain [poly-ADP-ribose (PAR) binding domain] and the APLF histone chaperone (Timinszky et al., 2009; Mehrotra et al., 2011; Xu et al., 2012a). Chromatin structure around DSBs is rearranged by mH2A1.1 recruitment (Timinszky et al., 2009), even though it is not incorporated into nucleosome (Xu et al., 2012a). Depletion of mH2A1.1 leads to reduced 53BP1 foci and phospho-Chk2 levels but has no effect on γ-H2A.X (Xu et al., 2012a). Recently Xu et al. (2012b) reported that histone H2A.Z is rapidly deposited around DSBs after induction of DNA damage. This incorporation of H2A.Z around the DSB is required for H4 acetylation, RNF8-mediated ubiquitylation, and BRCA1-foci formation as well as Ku70/80 loading.
It is not clear yet how to connect histone chaperone activity and histone exchange with non-coding RNA transcription during DDR, especially around DNA damage sites. It is possible that the chromatin structure around the DNA damage site can affect non-coding RNA transcription and it should be further studied.
Novel Links Between DNA Damage, Non-Coding RNAs, and Chromatin in Cellular Senescence and Aging
Cellular senescence is mainly regulated by the p53/p21 and p16/pRB pathways. lin-4 was the first identified miRNA which can regulate lifespan in C. elegans (Boehm and Slack, 2005) and a large number of other miRNAs have now been linked to the aging or senescence processes, for example, via regulating the p53/p21 and p16/pRB pathways. Some miRNAs that are expressed specifically during aging are secreted outside of cells, termed circulatory miRNAs, and are being studied as potential biomarkers of aging or for the diagnosis of age-related diseases. The influence of miRNAs in senescence and aging has been covered in detail in multiple recent excellent reviews (Smith-Vikos and Slack, 2012; Kato and Slack, 2013; Xu and Tahara, 2013) and will not be discussed here. Instead, we will discuss recent studies that have uncovered aging-dependent changes in the global chromatin structure, and how this relates to persistent activation of the DNA damage checkpoint and the concomitant induction of non-coding RNAs from repetitive elements such as the human Alu elements.
Telomere length is highly connected with the replicative lifespan of metazoan cells. Telomeres shorten with each successive round of DNA replication until one telomere reaches a threshold of “shortness” that emits a chronic DNA damage signal that causes the cell to cease dividing and enter senescence. Mechanistically, the short telomeres are seen as chronic DNA damage by the cell, resulting in the activation of the DDR, which causes a G1 cell cycle arrest (d’Adda di Fagagna et al., 2003). Telomere-mediated activation of the DDR appears to be intimately linked to the chromatin structure. Specifically, it was shown that chronic DNA damage from the processed telomeres also affects histone expression leading to their depletion and to the depletion of the central histone chaperones (O’Sullivan et al., 2010). The telomeric chromatin was progressively destabilized by this histone depletion, leading to a boost in telomere-associated DDR with each successive cell cycle (O’Sullivan et al., 2010). In a related study, the senescence-inducing chronic DNA damage signal caused by genotoxic stress was accompanied by reduced levels of the histone H2A variants in human fibroblasts (Lopez et al., 2012). By analogy, the level of total histone proteins decreases dramatically during replicative aging of budding yeast, but in this instance the trigger is not shortened telomeres but instead it appears to be due to a defect in protein synthesis during aging (Feser et al., 2010).
Perhaps related to the global relaxation of chromatin during aging, silenced regions of the genome become expressed. In yeast, replicative aging has long been known to be accompanied by the loss of transcriptional silencing (Smeal et al., 1996). This loss of silencing during yeast aging is likely due to both a reduction in histone protein levels during aging (Feser et al., 2010) and the reduced level of the Sir2 silencing protein (Dang et al., 2009).
Not all regions of the genome show relaxation of chromatin structure during aging, because senescent cells show heterochromatin foci called senescence-associated heterochromatin foci (SAHF; Zhang et al., 2005). The formation of SAHF is dependent on the histone chaperones ASF1A and HIRA and SAHF have a repressive histone variant, termed macroH2A (Zhang et al., 2005,2007). It is not easy to distinguish when and where chromatin relaxation or condensation is occurred during cellular senescence. Besides, SAHF have not yet been observed in cells from old organisms although the levels of the essential factors to form SAHF (HIRA and macro H2A) are induced in cells from old organisms (Jeyapalan et al., 2007; Kreiling et al., 2011). Further studies are needed to determine how the chromatin structure changes over different regions of the genome during in vivo and in vitro aging (or senescence).
Increased transcription from the normally silenced Alu and SINE retrotransposons of human adult stem cells also occurs during aging (Wang et al., 2011). Whether this increased retrotransposon transcription is the consequence of the potential decay of the chromatin structure during aging is not yet known. Intriguingly, this Alu/SINE transcription appears to activate the DDR and subsequently causes senescence of adult human stem cells. This was all shown in an elegant study from the Lunyak lab, using ex vivo aging of human adipose derived mesenchymal stem cells where they noticed that the DNA damage foci that arose during aging and senescence localize to Alu retrotransposon/SINE elements and pericentromeric regions (Wang et al., 2011). Alu/SINE elements and pericentromeric chromatin are normally transcriptionally silenced. However during aging and senescence the Alu/SINEs were transcribed by RNA polymerase III (Wang et al., 2011). Furthermore Alu/SINE transcription may be required for the recruitment of 53BP1 to the DNA repair foci because a RNA pol III inhibitor blocked formation of 53BP1 foci (Wang et al., 2011). The age-dependent DNA damage foci at the pericentromeric DNA is accompanied by a localized failure to recruit cohesion and condensin 1 complexes during aging (Wang et al., 2011). Given that cohesion aids in DSB repair (Unal et al., 2004,2007), failure to recruit cohesin during aging could explain the persistent DNA damage foci. If this is the case, the retrotransposon transcripts rather than retrotransposon transcription appear to be important for cohesion complex recruitment, given that knockdown of the Alu/SINE RNAs in senescent human adult stem cells caused loss of the persistent DNA damage foci and even restored their proliferative properties (Wang et al., 2011). This leads to a model where low levels of the retrotransposon transcripts are important for maintaining a repressive chromatin structure that allows the recruitment of cohesion and condensin I complexes that are important for the maintenance of pericentromeric integrity (Figure 2). However, in response to a potential decay of the chromatin structure during aging, excess retrotransposon transcription prevents recruitment of cohesion and condensin, causing persistent DNA damage checkpoint activation and senescence.
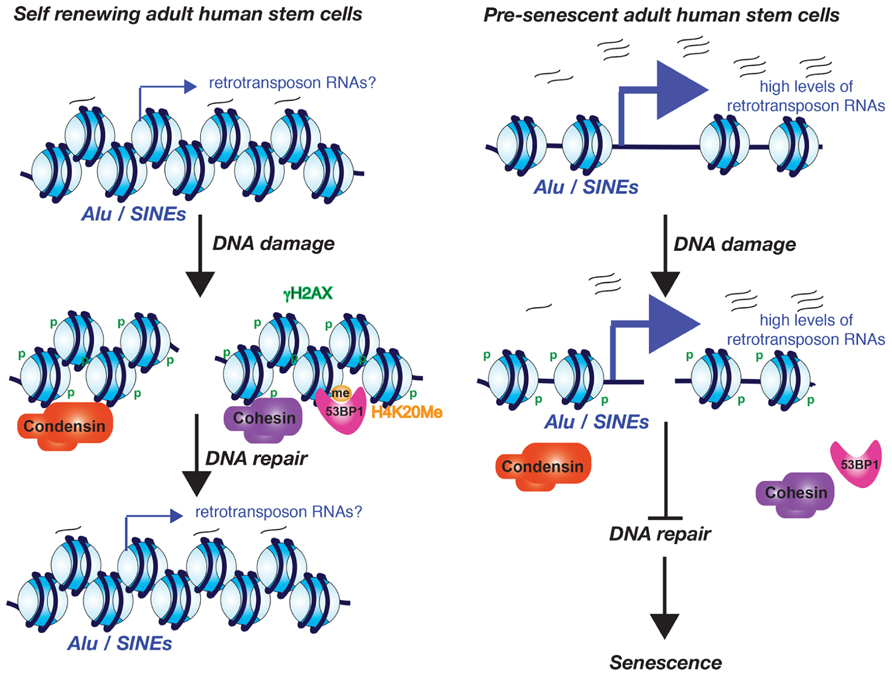
FIGURE 2. Model for the role of excessive Alu/SINE RNA transcription in driving cells into senescence. The model is based on the work from the Lunyak lab and is described in the text.
Remaining Questions
The recent papers discussed here have hinted at a complex interplay between the chromatin structure, transcription, DDR, and cellular senescence. However, many questions remain to be answered. These include: What is the function of non-coding RNAs in the DDR and cellular senescence? Are they involved in regulation of chromatin structures and/or recruitment of proteins to chromatin during the DDR and cellular senescence? What is the mechanism to induce non-coding RNAs during DDR and cellular senescence? Also, it will be interesting to determine whether the diRNAs that are transcribed from the vicinity of DSBs remain in the vicinity of the DSB. If so, this would be consistent with the diRNAs helping to physically establish a repressive chromatin structure? By contrast, if the diRNAs do not remain in the vicinity of the DSB, this would be suggestive that the act of transcription per se results in a more open chromatin structure, which would indirectly help recruit the DNA repair machinery such as 53BP1. Conversely, it is also a possibility that diRNA transcription around a DSB may be the consequence, not the cause, of local chromatin relaxation that occurs around the DSB. To determine if this is the case, it would be interesting to determine if the diRNAs are only produced over the domain that has lost histones or has H2AZ incorporation.
A better understanding of how diRNAs are induced in response to DNA damage may also be relevant for understanding how retrotransposon RNA transcription is induced during adult human stem cell aging. Is it a response to DNA breaks or a consequence of altered chromatin structure allowing access to the general transcription machinery? Do retrotransposon transcripts play a structural role in the maintenance of chromatin structure over these regions of the genome? Investigation of these and many other pertinent questions will doubtless provide mechanistic insight and more important discoveries.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adamson, B., Smogorzewska, A., Sigoillot, F. D., King, R. W., and Elledge, S. J. (2012). A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 14, 318–328. doi: 10.1038/ncb2426
Baker, D. J., Wijshake, T., Tchkonia, T., LeBrasseur, N. K., Childs, B. G., van de Sluis, B., et al. (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. doi: 10.1038/nature10600
Battu, A., Ray, A., and Wani, A. A. (2011). ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res. 39, 7931–7945. doi: 10.1093/nar/gkr523
Beli, P., Lukashchuk, N., Wagner, S. A., Weinert, B. T., Olsen, J. V., Baskcomb, L., et al. (2012). Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225. doi: 10.1016/j.molcel.2012.01.026
Boehm, M., and Slack, F. (2005). A developmental timing microRNA and its target regulate life span in C. elegans. Science 310, 1954–1957. doi: 10.1126/science.1115596
Botuyan, M. V., Lee, J., Ward, I. M., Kim, J.-E., Thompson, J. R., Chen, J., et al. (2006). Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373. doi: 10.1016/j.cell.2006.10.043
Bradshaw, V. A., and McEntee, K. (1989). DNA damage activates transcription and transposition of yeast Ty retrotransposons. Mol. Gen. Genet. 218, 465–474. doi: 10.1007/BF00332411
Braunstein, S., Badura, M. L., Xi, Q., Formenti, S. C., and Schneider, R. J. (2009). Regulation of protein synthesis by ionizing radiation. Mol. Cell. Biol. 29, 5645–5656. doi: 10.1128/MCB.00711-09
Campisi, J., and d’Adda di Fagagna, F. (2007). Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. doi: 10.1038/nrm2233
Celeste, A., Petersen, S., Romanienko, P. J., Fernandez-Capetillo, O., Chen, H. T., Sedelnikova, O. A., et al. (2002). Genomic instability in mice lacking histone H2AX. Science 296, 922–927. doi: 10.1126/science.1069398
Chen, C.-C., Carson, J. J., Feser, J., Tamburini, B., Zabaronick, S., Linger, J., et al. (2008). Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134, 231–243. doi: 10.1016/j.cell.2008.06.035
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. doi: 10.1016/j.molcel.2010.09.019
Coppede, F. (2012). Premature aging syndrome. Adv. Exp. Med. Biol. 724, 317–331. doi: 10.1007/978-1-4614-0653-2_24
Culligan, K. M., Robertson, C. E., Foreman, J., Doerner, P., and Britt, A. B. (2006). ATR and ATM play both distinct and additive roles in response to ionizing radiation. Plant J. 48, 947–961. doi: 10.1111/j.1365-313X.2006.02931.x
d’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., et al. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198. doi: 10.1038/nature02118
Dang, W., Steffen, K. K., Perry, R., Dorsey, J. A., Johnson, F. B., Shilatifard, A., et al. (2009). Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459, 802–807. doi: 10.1038/nature08085
Dang, Y., Yang, Q., Xue, Z., and Liu, Y. (2011). RNA interference in fungi: pathways, functions, and applications. Eukaryot. Cell 10, 1148–1155. doi: 10.1128/EC.05109-11
Deng, J., Harding, H. P., Raught, B., Gingras, A.-C., Berlanga, J. J., Scheuner, D., et al. (2002). Activation of GCN2 in UV-irradiated cells inhibits translation. Curr. Biol. 12, 1279–1286. doi: 10.1016/S0960-9822(02)01037-0
Duro, E., Lundin, C., Ask, K., Sanchez-Pulido, L., MacArtney, T. J., Toth, R., et al. (2010). Identification of the MMS22L–TONSL complex that promotes homologous recombination. Mol. Cell 40, 632–644. doi: 10.1016/j.molcel.2010.10.023
Feser, J., Truong, D., Das, C., Carson, J. J., Kieft, J., Harkness, T., et al. (2010). Elevated histone expression promotes life span extension. Mol. Cell 39, 724–735. doi: 10.1016/j.molcel.2010.08.015
Finn, K., Lowndes, N. F., and Grenon, M. (2012). Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci. 69, 1447–1473. doi: 10.1007/s00018-011-0875-3
Francia, S., Michelini, F., Saxena, A., Tang, D., de Hoon, M., Anelli, V., et al. (2012). Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 488, 231–235. doi: 10.1038/nature11179
Han, C., Wan, G., Langley, R. R., Zhang, X., and Lu, X. (2012). Crosstalk between the DNA damage response pathway and microRNAs. Cell. Mol. Life Sci. 69, 2895–2906. doi: 10.1007/s00018-012-0959-8
Hayflick, L. (1965). Limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37, 614–636. doi: 10.1016/0014-4827(65)90211-9
Herbig, U., Jobling, W. A., Chen, B. P., Chen, D. J., and Sedivy, J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501–513. doi: 10.1016/S1097-2765(04)00256-4
Howard, B. H. (1996). Replicative senescence: considerations relating to the stability of heterochromatin domains. Exp. Gerontol. 31, 281–293. doi: 10.1016/0531-5565(95)00022-4
Hu, H., and Gatti, R. A. (2011). MicroRNAs: new players in the DNA damage response. J. Mol. Cell Biol. 3, 151–158. doi: 10.1093/jmcb/mjq042
Huen, M. S. Y., Grant, R., Manke, I., Minn, K., Yu, X., Yaffe, M. B., et al. (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131, 901–914. doi: 10.1016/j.cell.2007.09.041
Huyen, Y., Zgheib, O., DiTullio, R. A., Gorgoulis, V. G., Zacharatos, P., Petty, T. J., et al. (2004). Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432, 406–411. doi: 10.1038/nature03114
Jeyapalan, J. C., Ferreira, M., Sedivy, J. M., and Herbig, U. (2007). Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 128, 36–44. doi: 10.1016/j.mad.2006.11.008
Kato, M., and Slack, F. J. (2013). Ageing and the small, non-coding RNA world. Ageing Res. Rev. 12, 429–435. doi: 10.1016/j.arr.2012.03.012
Kimura, H., Takizawa, N., Allemand, E., Hori, T., Iborra, F. J., Nozaki, N., et al. (2006). A novel histone exchange factor, protein phosphatase 2Cgamma, mediates the exchange and dephosphorylation of H2A-H2B. J. Cell Biol. 175, 389–400. doi: 10.1083/jcb.200608001
Kitano, H., and Imai, S. (1998). The two-process model of cellular aging. Exp. Gerontol. 33, 393–419. doi: 10.1016/S0531-5565(98)00008-4
Kreiling, J. A., Tamamori-Adachi, M., Sexton, A. N., Jeyapalan, J. C., Munoz-Najar, U., Peterson, A., et al. (2011). Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 10, 292–304. doi: 10.1111/j.1474-9726.2010.00666.x
Krietsch, J., Caron, M.-C., Gagné, J.-P., Ethier, C., Vignard, J., Vincent, M., et al. (2012). PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 40, 10287–10301. doi: 10.1093/nar/gks798
Kruhlak, M., Crouch, E. E., Orlov, M., Montano, C., Gorski, S. A., Nussenzweig, A., et al. (2007). The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447, 730–734. doi: 10.1038/nature05842
Lee, H.-C., Aalto, A. P., Yang, Q., Chang, S.-S., Huang, G., Fisher, D., et al. (2010). The DNA/RNA-dependent RNA polymerase QDE-1 enerates aberrant RNA and dsRNA for RNAi in a process requiring replication protein a and a DNA helicase. PLoS Biol. 8:e1000496. doi: 10.1371/journal.pbio.1000496
Lee, H. C., Chang, S. S., Choudhary, S., Aalto, A. P., Maiti, M., Bamford, D. H., et al. (2009). qiRNA is a new type of small interfering RNA induced by DNA damage. Nature 459, 274–277. doi: 10.1038/nature08041
Liu, W. M., Maraia, R. J., Rubin, C. M., and Schmid, C. W. (1994). Alu transcripts: cytoplasmic localisation and regulation by DNA methylation. Nucleic Acids Res. 22, 1087–1095. doi: 10.1093/nar/22.6.1087
Lopez, M. F., Tollervey, J., Krastins, B., Garces, A., Sarracino, D., Prakash, A., et al. (2012). Depletion of nuclear histone H2A variants is associated with chronic DNA damage signaling upon drug-evoked senescence of human somatic cells. Aging 4, 823–842.
Lou, Z., Minter-Dykhouse, K., Franco, S., Gostissa, M., Rivera, M. A., Celeste, A., et al. (2006). MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 21, 187–200. doi: 10.1016/j.molcel.2005.11.025
Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., et al. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900. doi: 10.1016/j.cell.2007.09.040
Mehrotra, P. V., Ahel, D., Ryan, D. P., Weston, R., Wiechens, N., Kraehenbuehl, R., et al. (2011). DNA repair factor APLF is a histone chaperone. Mol. Cell 41, 46–55. doi: 10.1016/j.molcel.2010.12.008
Morawetz, C. (1987). Effect of irradiation and mutagenic chemicals on the generation of ADH2-constitutive mutants in yeast: significance for the inducibility of Ty transposition. Mutat. Res. 177, 53–60. doi: 10.1016/0027-5107(87)90021-2
Morawetz, C., and Hagen, U. (1990). Effect of irradiation and mutagenic chemicals on the generation of ADH2- and ADH4-constitutive mutants in yeast: the inducibility of Ty transposition by UV and ethyl methanesulfonate. Mutat. Res. 229, 69–77. doi: 10.1016/0027-5107(90)90009-S
Morley, S. J., McKendrick, L., and Bushell, M. (1998). Cleavage of translation initiation factor 4G (eIF4G) during anti-Fas IgM-induced apoptosis does not require signalling through the p38 mitogen-activated protein (MAP) kinase. FEBS Lett. 438, 41–48. doi: 10.1016/S0014-5793(98)01269-1
Murr, R., Loizou, J. I., Yang, Y. G., Cuenin, C., Li, H., Wang, Z. Q., et al. (2006). Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8, 91–99. doi: 10.1038/ncb1343
O’Sullivan, R. J., Kubicek, S., Schreiber, S. L., and Karlseder, J. (2010). Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 17, 1218–1225. doi: 10.1038/nsmb.1897
Pankotai, T., Bonhomme, C., Chen, D., and Soutoglou, E. (2012). DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 19, 276–282. doi: 10.1038/nsmb.2224
Papamichos-Chronakis, M., and Peterson, C. L. (2013). Chromatin and the genome integrity network. Nat. Rev. Genet. 14, 62–75. doi: 10.1038/nrg3345
Polo, S. E., Roche, D., and Almouzni, G. (2006). New histone incorporation marks sites of UV repair in human cells. Cell 127, 481–493. doi: 10.1016/j.cell.2006.08.049
Powley, I. R., Kondrashov, A., Young, L. A., Dobbyn, H. C., Hill, K., Cannell, I. G., et al. (2009). Translational reprogramming following UVB irradiation is mediated by DNA-PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev. 23, 1207–1220. doi: 10.1101/gad.516509
Pryde, F., Khalili, S., Robertson, K., Selfridge, J., Ritchie, A. M., Melton, D. W., et al. (2005). 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J. Cell Sci. 118, 2043–2055. doi: 10.1242/jcs.02336
Reyes-Turcu, F. E., and Grewal, S. I. (2012). Different means, same end-heterochromatin formation by RNAi and RNAi-independent RNA processing factors in fission yeast. Curr. Opin. Genet. Dev. 22, 156–163. doi: 10.1016/j.gde.2011.12.004
Rudin, C. M., and Thompson, C. B. (2001). Transcriptional activation of short interspersed elements by DNA-damaging agents. Genes Chromosomes Cancer 30, 64–71. doi: 10.1002/1098-2264(2000)9999:9999
Salton, M., Lerenthal, Y., Wang S.-Y., Chen, D. J., and Shiloh, Y. (2010). Involvement of Matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle 9, 1568–1576. doi: 10.4161/cc.9.8.11298
Sedelnikova, O. A., Horikawa, I., Zimonjic, D. B., Popescu, N. C., Bonner, W. M., and Barrett, J. C. (2004). Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6, 168–170. doi: 10.1038/ncb1095
Shanbhag, N. M., Rafalska-Metcalf, I. U., Balane-Bolivar, C., Janicki, S. M., and Greenberg, R. A. (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. doi: 10.1016/j.cell.2010.04.038
Smeal, T., Claus, J., Kennedy, B., Cole, F., and Guarente, L. (1996). Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84, 633–642. doi: 10.1016/S0092-8674(00)81038-7
Smith-Vikos, T., and Slack, F. J. (2012). MicroRNAs and their roles in aging. J. Cell Sci. 125, 7–17. doi: 10.1242/jcs.099200
Spriggs, K. A., Bushell, M., and Willis, A. E. (2010). Translational regulation of gene expression during conditions of cell stress. Mol. Cell 40, 228–237. doi: 10.1016/j.molcel.2010.09.028
Stewart, G. S., Wang, B., Bignell, C. R., Taylor, A. M. R., and Elledge, S. J. (2003). MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421, 961–966. doi: 10.1038/nature01446
Stucki, M., Clapperton, J. A., Mohammad, D., Yaffe, M. B., Smerdon, S. J., and Jackson, S. P. (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226. Erratum in: Cell (2008), 133, 549.
Takai, H., Smogorzewska, A., and de Lange, T. (2003). DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556. doi: 10.1016/S0960-9822(03)00542-6
Tee, A. R., and Proud, C. G. (2000). DNA-damaging agents cause inactivation of translational regulators linked to mTOR signalling. Oncogene 19, 3021–3031. doi: 10.1038/sj.onc.1203622
Timinszky, G., Till, S., Hassa, P. O., Hothorn, M., Kustatscher, G., Nijmeijer, B., et al. (2009). A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16, 923–929. doi: 10.1038/nsmb.1664
Tsurumi, A., and Li, W. X. (2012). Global heterochromatin loss: a unifying theory of aging? Epigenetics 7, 680–688. doi: 10.4161/epi.20540
Unal, E., Arbel-Eden, A., Sattler, U., Shroff, R., Lichten, M., Haber, J. E., et al. (2004). DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell 16, 991–1002. doi: 10.1016/j.molcel.2004.11.027
Unal, E., Heidinger-Pauli, J. M., and Koshland, D. (2007). DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7). Science 317, 245–248. doi: 10.1126/science.1140637
Villeponteau, B. (1997). The heterochromatin loss model of aging. Exp. Gerontol. 32, 383–394. doi: 10.1016/S0531-5565(96)00155-6
Vorce, R. L., Lee, B., and Howard, B. H. (1994). Methylation- and mutation-dependent stimulation of Alu transcription in vitro. Biochem. Biophys. Res. Commun. 203, 845–851. doi: 10.1006/bbrc.1994.2260
Wakeman, T. P., Wang, Q., Feng, J., and Wang, X.-F. (2012). Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J. 31, 2169–2181. doi: 10.1038/emboj.2012.50
Wan, G., Mathur, R., Hu, X., Zhang, X., and Lu, X. (2011). miRNA response to DNA damage. Trends Biochem. Sci. 36, 478–484. doi: 10.1016/j.tibs.2011.06.002
Wang, J., Geesman, G. J., Hostikka, S. L., Atallah, M., Blackwell, B., Lee, E., et al. (2011). Inhibition of activated pericentromeric SINE/Alu repeat transcription in senescent human adult stem cells reinstates self-renewal. Cell Cycle 10, 3016–3030. doi: 10.4161/cc.10.17.17543
Wei, W., Ba, Z., Gao, M., Wu, Y., Ma, Y., Amiard, S., et al. (2012). A role for small RNAs in DNA double-strand break repair. Cell 149, 101–112. doi: 10.1016/j.cell.2012.03.002
Xu, C., Xu, Y., Gursoy-Yuzugullu, O., and Price, B. D. (2012a). The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. FEBS Lett. 586, 3920–3925. doi: 10.1016/j.febslet.2012.09.030
Xu, Y., Ayrapetov, M. K., Xu, C., Gursoy-Yuzugullu, O., Hu, Y., and Price, B. D. (2012b). Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 48, 723–733. doi: 10.1016/j.molcel.2012.09.026
Xu, D., and Tahara, H. (2013). The role of exosomes and microRNAs in senescence and aging. Adv. Drug Deliv. Rev. 65, 368–375. doi: 10.1016/j.addr.2012.07.010
Xu, Y., Sun, Y., Jiang, X., Ayrapetov, M. K., Moskwa, P., Yang, S., et al. (2010). The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J. Cell Biol. 191, 31–43. doi: 10.1083/jcb.201001160
Zhang, R., Chen, W., and Adams, P. D. (2007). Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell. Biol. 27, 2343–2358. doi: 10.1128/MCB.02019-06
Zhang, R., Poustovoitov, M. V., Ye, X., Santos, H. A., Chen, W., Daganzo, S. M., et al. (2005). Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 8, 19–30. doi: 10.1016/j.devcel.2004.10.019
Zhao, Q., Wang, Q.-E., Ray, A., Wani, G., Han, C., Milum, K., et al. (2009). Modulation of nucleotide excision repair by mammalian SWI/SNF chromatin-remodeling complex. J. Biol. Chem. 284, 30424–30432. doi: 10.1074/jbc.M109.044982
Keywords: non-coding RNA, chromatin, genomic instability, aging, senescence
Citation: Ohsawa R, Seol J-H and Tyler JK (2013) At the intersection of non-coding transcription, DNA repair, chromatin structure, and cellular senescence. Front. Genet. 4:136. doi: 10.3389/fgene.2013.00136
Received: 05 February 2013; Accepted: 26 June 2013;
Published online: 26 July 2013.
Edited by:
Philipp Oberdoerffer, National Cancer Institute, USAReviewed by:
Peter David Adams, University of Glasgow, UKRebecca C. Burgess, National Cancer Institute, USA
Copyright: © 2013 Ohsawa, Seol and Tyler. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Jessica K. Tyler, Department of Biochemistry and Molecular Biology, University of Texas MD Anderson Cancer Center, Unit 1000, 1515 Holcombe Boulevard, Houston, TX 77030, USA e-mail: jtyler@mdanderson.org