 Chiranjeevi Sandi
Chiranjeevi Sandi Madhavi Sandi
Madhavi Sandi Sara Anjomani Virmouni
Sara Anjomani Virmouni Sahar Al-Mahdawi
Sahar Al-Mahdawi Mark A. Pook
Mark A. Pook- Division of Biosciences, School of Health Sciences and Social Care, Brunel University London, Uxbridge, UK
Friedreich ataxia (FRDA) is a lethal autosomal recessive neurodegenerative disorder caused primarily by a homozygous GAA repeat expansion mutation within the first intron of the FXN gene, leading to inhibition of FXN transcription and thus reduced frataxin protein expression. Recent studies have shown that epigenetic marks, comprising chemical modifications of DNA and histones, are associated with FXN gene silencing. Such epigenetic marks can be reversed, making them suitable targets for epigenetic-based therapy. Furthermore, since FRDA is caused by insufficient, but functional, frataxin protein, epigenetic-based transcriptional re-activation of the FXN gene is an attractive therapeutic option. In this review we summarize our current understanding of the epigenetic basis of FXN gene silencing and we discuss current epigenetic-based FRDA therapeutic strategies.
Introduction
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative mitochondrial disorder caused primarily by a homozygous GAA repeat expansion mutation within intron 1 of the frataxin gene (FXN) located on chromosome 9q21.1 (Campuzano et al., 1996). Unaffected individuals have up to 43 GAA repeats, while affected individuals have 44–1700 GAA repeats, most commonly between 600 and 900 GAA repeats (Sharma et al., 2004; Pandolfo, 2008). The effect of the GAA repeat expansion is to decrease expression of the essential and ubiquitously expressed mitochondrial protein frataxin (Campuzano et al., 1997). However, asymptomatic carriers produce about 50% frataxin levels compared to unaffected individuals (Pianese et al., 2004). Therefore, drugs that induce frataxin expression, at least to the levels of healthy carriers, would be beneficial. The FXN gene spans 95 kb of genomic DNA and contains seven exons, 1–5a, 5b, and 6 (Campuzano et al., 1996). The first five exons are transcribed to produce a 210 amino acid major isoform of frataxin, but rarely exon 5b can be transcribed by alternative splicing to produce a 171 amino acid protein. In addition, a recent study has identified two novel tissue specific transcript variants, encoding two isoforms of frataxin that lack the mitochondrial targeting sequence and are therefore different from the canonical transcript (Xia et al., 2012).
Reduced levels of frataxin in FRDA patients are associated with defects of iron-sulfur cluster (ISC) biosynthesis (Bradley et al., 2000), mitochondrial iron accumulation in heart and dentate nucleus (Foury and Cazzalini, 1997; Waldvogel et al., 1999; Koeppen et al., 2007), and increased susceptibility to oxidative stress (Wong et al., 1999). The main pathological effects are loss of large sensory neurons in the dorsal root ganglia (DRG) and degenerative atrophy of the posterior columns of the spinal cord, contributing to symptoms of progressive ataxia, muscle weakness, and sensory deficit. There is also pathological involvement of non-neuronal tissues, with hypertrophic cardiomyopathy a common feature, and diabetes mellitus identified in approximately 10% of FRDA patients (Schulz et al., 2009). Affected individuals generally die in early adulthood from the associated heart disease and at present there is no effective therapy for FRDA. Therefore, there is a high unmet clinical need to develop a therapy for this devastating disorder.
Although the exact mechanism by which the GAA repeat expansion leads to decreased frataxin expression is not known, two hypotheses have been proposed (Figure 1). Firstly, evidence from in vitro and cell transfection studies suggests that the GAA repeat expansion may adopt abnormal non-B DNA structures (triplexes or “sticky DNA”) or DNA•RNA hybrid structures (R-loops), which impede the process of RNA polymerase II and thus reduce FXN gene transcription (Grabczyk et al., 2007; Wells, 2008). Secondly, there is evidence that GAA repeat expansions can produce heterochromatin-mediated gene silencing effects (Saveliev et al., 2003). Consistent with the latter hypothesis, several FRDA disease-related epigenetic changes have been identified in the immediate vicinity of the expanded GAA repeats of the FXN gene and these changes will be discussed further within this review.
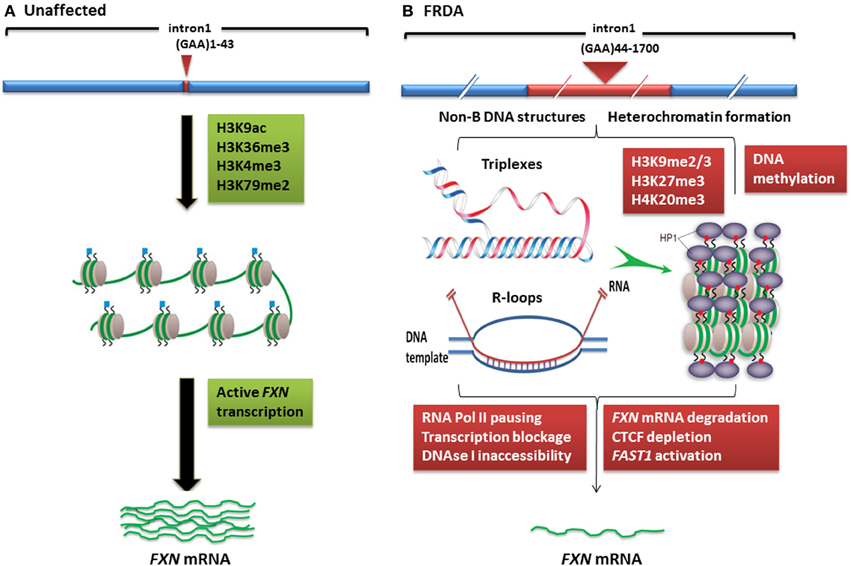
Figure 1. Models of FXN gene silencing in FRDA. (A) Unaffected individuals, who carry up to 43 GAA•TTC repeats, contain active histone marks of gene transcription initiation and elongation at the FXN promoter and intron 1 regions. (B) In FRDA patients, the presence of large GAA•TTC repeat expansion leads to FXN gene silencing by two potential mechanisms: (i) the GAA•TTC repeat may adopt abnormal non-B DNA structures (triplexes) or DNA•RNA hybrid structures (R loops), which impede the process of RNA polymerase and thus reduce FXN gene transcription, (ii) increased levels of DNA methylation and HP1 and significant enrichment of repressive histone marks at the FXN gene trigger heterochromatin formation that may lead to more pronounced FXN gene silencing. This image was adapted from Festenstein (2006); Wells (2008); Chan et al. (2013).
Epigenetic Changes Associated with FRDA
Epigenetic mechanisms, including DNA methylation and hydroxymethylation, post-translational histone modifications, chromatin remodeling, and non-coding RNA effects, produce effects on gene expression without involving changes in the primary DNA sequence. The potential role of epigenetic mechanisms in FRDA disease was first suggested by the finding that long GAA repeats could suppress the expression of a nearby heterochromatin-sensitive cell surface reporter gene in transgenic mice by position effect variegation (PEV) (Saveliev et al., 2003). Further studies of FRDA cells, tissues, and mouse models have subsequently led to the identification of epigenetic changes, including DNA methylation and hydroxymethylation, histone deacetylation, and histone methylation, which may be involved in FXN gene silencing in FRDA (Table 1).
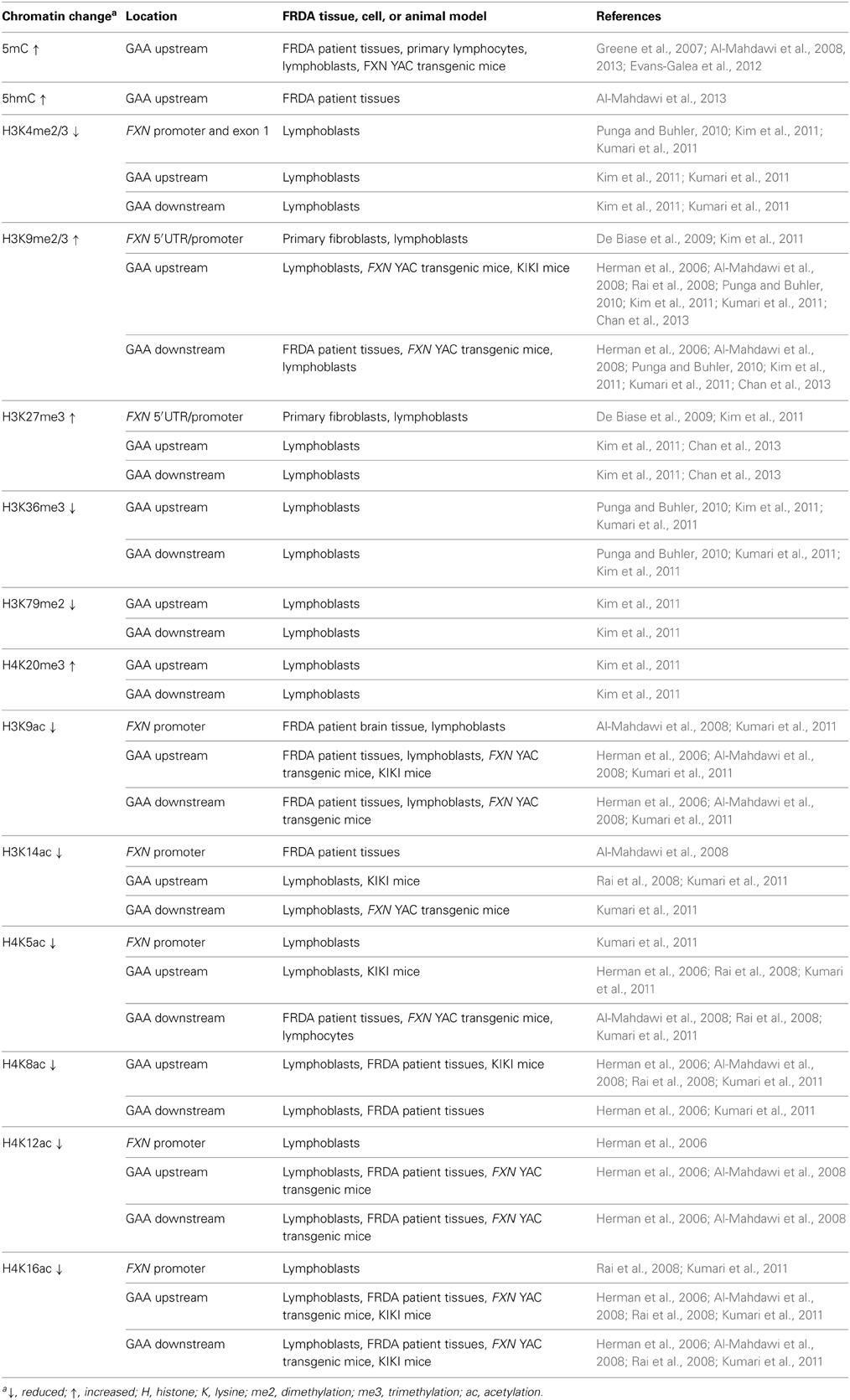
Table 1. Epigenetic changes in tissues, cells, and animal models of FRDA.
DNA Methylation and Hydroxymethylation
DNA methylation is carried out by DNA methyltransferase (DNMT) enzymes, which catalyze the conversion of cytosine to 5'-methylcytosine (5mC), predominantly within CpG dinucleotides (Robertson, 2001). In mammals, the DNMT family includes three functional proteins: DNMT1 preferentially methylates hemi-methylated DNA and is thus responsible for methylation during DNA replication (Pradhan et al., 1999), while DNMT3a and DNMT3b have an equal preference for hemi-methylated and non-methylated DNA and so have been classified as de novo methyltransferases (Okano et al., 1999). Studies investigating the DNA methylation profiles of transcriptionally silenced genes have revealed a strong correlation between promoter DNA methylation and transcriptional silencing. However, it has also been reported that intragenic DNA methylation can contribute to transcriptional gene silencing (Lorincz et al., 2004). More recent studies have revealed the existence of an alternative modification, 5'-hydroxymethylcytosine (5hmC), which is formed by oxidation of 5mC by ten-eleven translocation (TET) enzymes (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009). 5hmC may either be an intermediate in the removal of 5mC by an active demethylation process (Guo et al., 2011) or it may be an epigenetic modification in its own right, regulating chromatin or transcriptional factors (Szulwach et al., 2011). Therefore, it is possible that 5hmC may prove to be involved in epigenetic-based disease mechanisms, such as those proposed for FRDA.
Initial investigations of DNA methylation within the FXN gene by Usdin and colleagues revealed hypermethylation of specific CpG sites upstream of the GAA repeat sequence in FRDA patient-derived lymphoblastoid cells compared to cells derived from unaffected individuals (Greene et al., 2007). In particular, three out of 15 CpG residues that span a 700 bp region upstream of the GAA repeat were found to be unmethylated, or sparsely methylated, in unaffected cells, but highly methylated in FRDA cells. Studies by our laboratory also revealed significantly increased DNA methylation at the same upstream GAA repeat region in FRDA patient autopsy brain, heart, and cerebellum tissues, with similar findings in tissues of FRDA YAC transgenic mice (Al-Mahdawi et al., 2008). Subsequently, a study of FRDA patient blood samples showed that the degree of DNA methylation at the FXN upstream GAA repeat region correlates with the length of the GAA repeats and inversely correlates with the age of disease onset (Castaldo et al., 2008). Furthermore, analysis of blood and buccal cells from a large cohort of FRDA patients showed that the level of DNA methylation in FRDA patients is significantly elevated in the same upstream GAA repeat region and inversely correlated with FXN expression levels (Evans-Galea et al., 2012). More recently, our laboratory has shown that, for at least one CpG site within the upstream GAA repeat region, the increased level of DNA methylation predominantly comprises 5hmC rather than 5mC (Al-Mahdawi et al., 2013). Thus, FRDA can now be grouped together with other TNR expansion diseases in which an association with DNA methylation and hydroxymethylation has been observed (Table 1, Figure 2).
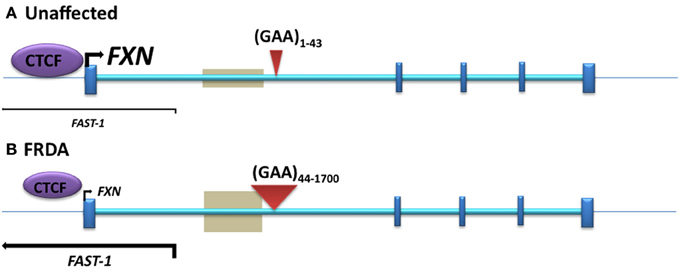
Figure 2. The position of DNA methylation, hydroxymethylation, and CTCF binding sites within the FXN gene. (A) Unaffected: normal-sized GAA repeat (B) FRDA: GAA repeat expansion. Gray boxes represent regions of disease-associated DNA methylation and hydroxymethylation. Arrow marks represent the directions and levels of transcription for FXN and FAST-1. Blue bars represent exons of the FXN gene. Red triangles indicate GAA repeats within intron 1 of the FXN gene.
Post-Translational Histone Modifications
The nucleosome consists of an octamer of four pairs of histone proteins, H2A, H2B, H3, and H4, surrounded by 147 bp of DNA (Kouzarides, 2007). Histone proteins contain a globular C-terminal domain and an unstructured N-terminal tail, which can contain many different modified residues. Histone acetylation at lysine residues is regulated by two distinct families of enzymes with opposing actions, histone acetyltransferases (HATs) and histone deacetylases (HDACs). Similarly, histone lysine methylation is controlled by histone methyltransferases (HMTases) and histone demethylases (HDMs), which have been linked to a number of cellular processes including transcriptional activation and repression (Kouzarides, 2007). HATs are diverse set of enzymes that are grouped into two different families based on their catalytic domains: Gcn5 N-acetyltransferases (GNATs, e.g., Gcn5 and PCAF) and MYST HATs (e.g., Morf and Ybf2) (Kimura et al., 2005). Each HAT is usually capable of modifying several different histone residues. Eighteen different HDACs have been identified in mammals and these have been divided into four classes. Class I consists of HDACs 1, 2, 3, and 8, which are similar to yeast RPD3 deacetylase. Class II consists of HDACs 4, 5, 7, and 9, which have homology to the yeast HDAC Had-1 gene. Class III HDACs, also known as sirtuins, which have homology to the Yeast Sir2 gene, include sirtuin 1–7. Lastly, class IV HDACs consists of only HDAC 11. HMTases are divided into lysine-specific and arginine-specific groups. They have great specificity and usually modify only one particular histone residue (Kouzarides, 2007). The HDMs are classified into two distinct enzyme families: the nuclear amine oxidase homologs (e.g., LSD1) and the JmjC-domain proteins (e.g., JHDM1) (Kouzarides, 2007).
In mammals, heterochromatin formation and gene silencing is associated with hypoacetylation of certain histone residues, particularly H3K9, together with increased methylation of histone residues, including H3K9me2, H3K9me3, H3K27me3, and H4K20me3 (Kourmouli et al., 2004; Martin and Zhang, 2005; Jenuwein, 2006). Such histone modifications have now been identified within the FXN gene in FRDA cells, tissues, and mouse models, predominantly at the region immediately upstream of the expanded GAA repeats, indicating that the FXN gene is subject to heterochromatin silencing (Sandi et al., 2013) (Table 1, Figure 1). Histone modifications at the FXN locus were first identified by Gottesfeld and colleagues, who reported lower levels of several acetylated H3 and H4 lysine residues, together with increased H3K9me2 and H3K9me3 in the upstream GAA regions of FRDA lymphoblastoid cells (Herman et al., 2006). Usdin and colleagues then reported increased H3K9me2 levels within FXN intron 1 in FRDA lymphoblastoid cells (Greene et al., 2007) and our laboratory reported changes of histone modifications at the FXN promoter, upstream, and downstream GAA repeat regions in FRDA patient and YAC transgenic mouse brain tissues (Al-Mahdawi et al., 2008). Subsequently, Bidichandani and colleagues reported that FRDA patient fibroblasts have significantly higher levels of H3K9me3 and H3K27me3 at the FXN 5' UTR region, coupled with elevated levels of heterochromatin protein 1 (HP1), compared to normal fibroblasts (De Biase et al., 2009). More recently, Chan and colleagues have identified increased H3K9me3 and H3K27me3 at the upstream and downstream GAA repeat regions in FRDA lymphoblasts (Chan et al., 2013).
In contrast, histone modifications such as H3K4me3, H3K36me3, and H3K79me3 are associated with a more open chromatin state and active gene expression. H3K4me3 is particularly associated with initiation of gene transcription, while H3K36me3 and H3K79me3 are associated with elongation of gene transcription. Recent studies have shown decreased levels of H3K36me3 and H3K79me3 at the upstream and downstream GAA repeat regions of the FXN gene in FRDA cells, indicating that there is a defect in transcription elongation (Punga and Buhler, 2010; Kim et al., 2011; Kumari et al., 2011). Decreased levels of H3K4me3 have also been identified at the upstream GAA repeat region, but not at the promoter region, which suggests a more pronounced defect of the post-initiation and elongation stages of FXN gene expression rather than an early transcription initiation defect (Punga and Buhler, 2010; Kim et al., 2011; Kumari et al., 2011) (Figure 1). In summary, there is good evidence that reduction of frataxin protein expression in FRDA is primarily caused by GAA repeat expansion-induced transcriptional silencing, which is associated with specific post-translational histone modifications.
Antisense Transcription and CTCF Binding
Antisense RNA transcripts, which play a role in regulation of gene expression, have previously been associated with TNR expansion diseases such as Huntington disease (HD) (Chung et al., 2011), FRAXA (Ladd et al., 2007; Khalil et al., 2008), DM1 (Cho et al., 2005), SCA7 (Sopher et al., 2011), and SCA8 (Moseley et al., 2006). Furthermore, the CCCTC-binding factor (CTCF) protein, which can prevent the spreading of DNA methylation (Filippova et al., 2005; Engel et al., 2006), has binding sites in the repeat expansion flanking regions of several TNR disorders, such as FRAXA (Ladd et al., 2007), DM1 (Filippova et al., 2001), and SCA7 (Libby et al., 2008), and loss of CTCF binding at the DM1 CTG expansion is associated with the spread of heterochromatin and DNA methylation (Cho et al., 2005). Bidichandani and colleagues have reported significantly increased levels of a frataxin antisense transcript 1 (FAST1) in FRDA fibroblast cells, associated with depletion of CTCF binding at the 5' UTR region of the FXN gene (Figure 2), suggesting involvement of these factors in the heterochromatin and FXN gene silencing processes of FRDA disease (De Biase et al., 2009). More recently, our laboratory has also reported increased expression of FAST1 in FRDA mouse fibroblasts (Sandi et al., 2014) and reduced CTCF binding at the 5' UTR region of the FXN gene in FRDA cerebellum tissue (Al-Mahdawi et al., 2013). Systematic genome-wide mapping by ChIP analysis has shown that CTCF binds to tens of thousands of genomic sites, often distal to transcription start sites (TSS) of genes, but also at promoter and enhancer sequences (Barski et al., 2007; Holwerda and De Laat, 2013). Thus, CTCF can direct the interaction of tissue-specific enhancers with different neighboring promoters resulting in the regulation of gene expression at a particular locus (Shen et al., 2012). The association of CTCF binding at the 5' UTR region of the FXN gene with high levels of FXN expression and low levels of FAST1 expression indicates that such CTCF control mechanisms might be in operation at the FXN locus. In a large proportion of cases, control of CTCF binding has been shown to be sensitive to DNA methylation (Wang et al., 2012). However, this is not likely to be the case for FRDA, because FRDA-specific changes in DNA methylation have not been detected at the CTCF-binding site within the 5' UTR region of the FXN gene (Al-Mahdawi et al., 2008; De Biase et al., 2009). Therefore, further consideration of antisense transcription, CTCF binding, and other associated factors are needed, since they are likely to be highly relevant to the development of an epigenetic-based therapy for FRDA.
Epigenetic-Based Therapies for FRDA
DNA Demethylation Therapies
Since FRDA is associated with increased levels of DNA methylation, one can propose the use of DNA demethylating agents to potentially activate a silenced FXN gene. DNA demethylating agents are generally divided into nucleoside analog DNMT inhibitors and non-nucleoside analog DNMT inhibitors. Nucleoside analog DNMT inhibitors, including 5-azacytidine (5-aza-CR or Vidaza), 5-aza-2'-deoxycytidine (5-aza-CdR or Decitabine), and Zebularine, are analogs of cytosine. 5-aza-CdR is an FDA-approved drug that is used to treat leukemia patients (Jain et al., 2009). Furthermore, treatment of lymphoblastoid cells from FRAXA patients with 5-aza-CdR, either alone (Chiurazzi et al., 1998) or in combination with HDAC inhibitors (Chiurazzi et al., 1999), efficiently reverses the FMR1 promoter hypermethylation and restores mRNA and protein levels to normal. Such studies have led to the consideration of DNA demethylating agents as a potential therapy for neurodegenerative disorders. Thus, far, there have been no reports of describing the use of DNA demethylating agents as a therapeutic approach for FRDA, mainly due to the lack of evidence for a causal link between DNA methylation and FXN gene silencing. However, DNA demethylating agents, such as 5-aza-CdR, Zebularine, or the oligonucleotide antisense inhibitor of DNMT1, MG98 (Plummer et al., 2009), may be considered as FRDA therapeutic options in the future.
HDAC Inhibitors
HDAC inhibitors can affect transcription by increasing acetylation of histones, transcription factors, and other proteins that regulate transcription (Butler and Bates, 2006). In view of the recent identification of histone acetylation changes at the FXN gene in FRDA, it has been proposed that the reversal of such histone modifications could represent a useful therapeutic approach for FRDA (Figure 3) (Festenstein, 2006; Herman et al., 2006). An initial study to screen for frataxin-increasing compounds by Sarsero and colleagues first demonstrated a small effect of the general HDAC inhibitor sodium butyrate on FXN gene activity using an EGFP reporter cell line (Sarsero et al., 2003). Subsequently, Gottesfeld and colleagues treated FRDA lymphoblastoid cells using a selection of commercially available HDAC inhibitors. They revealed that only the benzamide compound BML-210 produced a significant increase of FXN mRNA expression (Herman et al., 2006), although other HDAC inhibitors showed a more pronounced increase of histone acetylation without any increase in FXN expression, indicating a degree of compound specificity for FXN gene silencing (Table 2). An analog of BML-210, designated 4b, was then synthesized and was shown to directly modulate the histones associated with the FXN gene to increase FXN mRNA expression in FRDA primary lymphocytes (Herman et al., 2006). Compound 4b has subsequently been used to demonstrate amelioration of the HD phenotype in R6/2 mice without any discernible toxicity (Thomas et al., 2008). Further development of HDAC inhibitors by the Gottesfeld lab and the Repligen Corporation (Waltham, Massachusetts) identified three 2-aminobenzamide compounds, designated 106, 136, and 109, which can each significantly increase FXN mRNA and frataxin protein levels in FRDA cells with only a small effect on unaffected control cells (reviewed in Soragni et al., 2012). The exact mechanism of action of the 2-aminobenzamide HDAC inhibitors in FRDA is not known. However, compound 106 has been shown to act as selective slow-on, slow-off, tight-binding inhibitor of class I HDACs, with a preference for inhibition of HDAC3 (Chou et al., 2008; Xu et al., 2009). Compounds 106, 136, and 109 have undergone investigations to determine safety, efficacy and pharmacokinetic profile in short-term treatments of FRDA patient derived cells and KIKI knock-in mice (Chou et al., 2008; Rai et al., 2008, 2010; Soragni et al., 2008; Xu et al., 2009), and a long-term treatment in YG8R YAC transgenic mice (Sandi et al., 2011) (Table 2). Compound 109, which emerged as the most promising compound for FRDA treatment, has now been assessed in a phase I clinical trial as RG2833 (Soragni et al., 2012) and further related compounds are now under development.
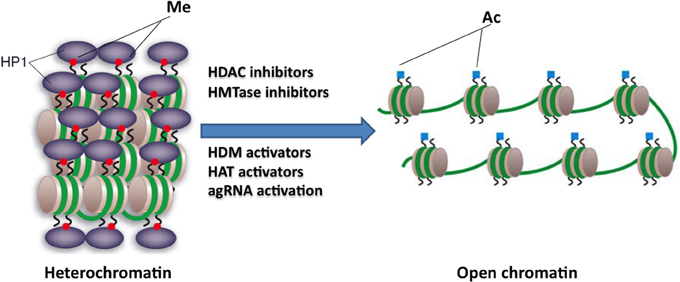
Figure 3. Potential epigenetic-based therapies for FRDA. Large GAA•TTC repeats in FRDA patients are associated with heterochromatin mediated FXN gene silencing. The use of specific HDAC inhibitors, HDM activators, HAT activators, HMTase inhibitors, or agRNA activation may reverse the heterochromatin to a more open chromatin structure, and may thus lead to active FXN gene transcription. This image was adapted from Festenstein (2006); Chan et al. (2013).
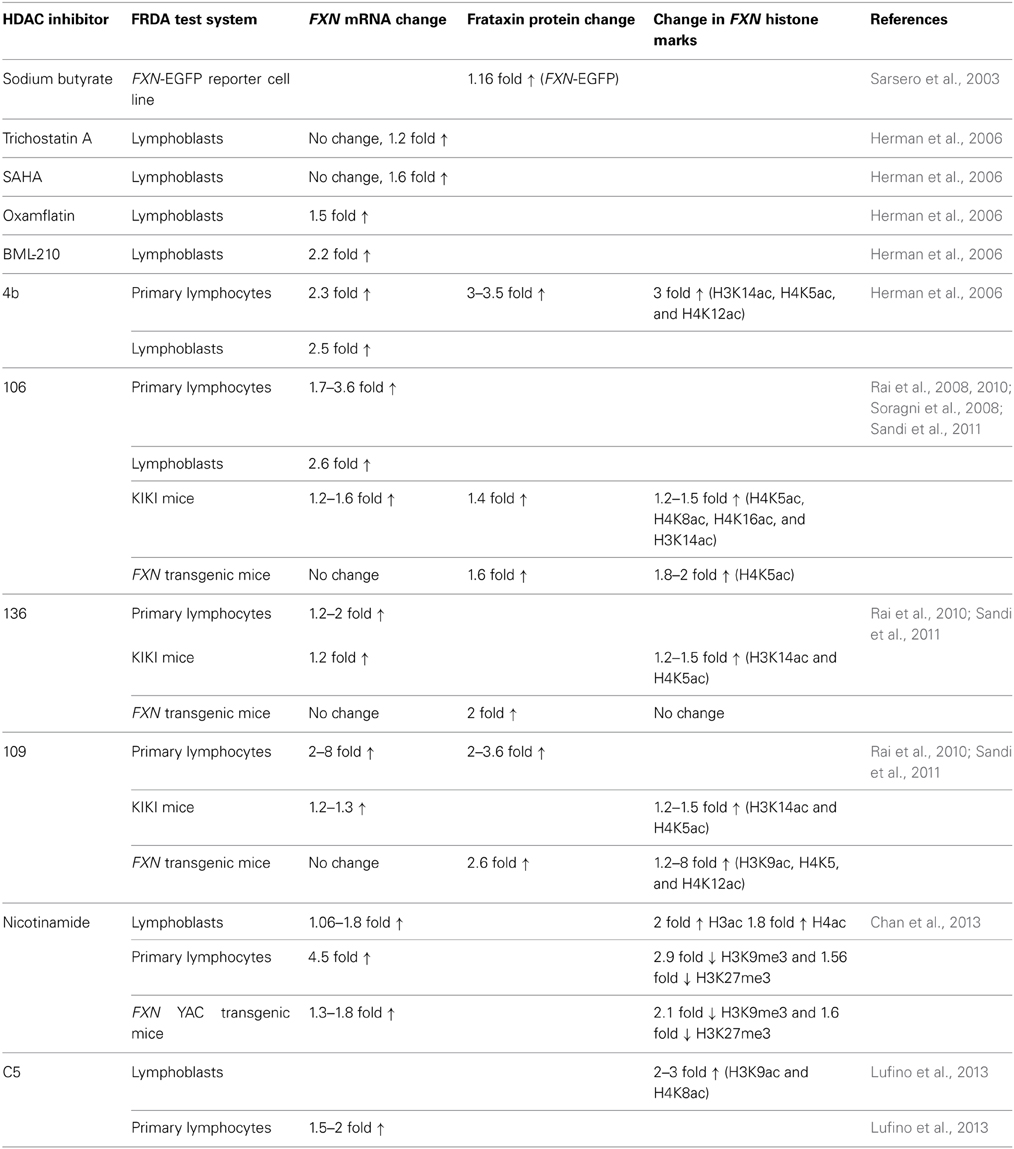
Table 2. HDAC inhibitor preclinical studies of FRDA cells and mice.
Recently, the class III HDAC inhibitor nicotinamide (Ghosh and Feany, 2004) has been shown to increase frataxin expression and decrease H3K9me3 and H3K27me3 at the FXN gene in FRDA cells and mouse models and this compound is now in early stage clinical trials (Chan et al., 2013) (Table 2). Furthermore, other HDAC inhibitors such as sirtinol (Ota et al., 2006), splitomicin (Biacsi et al., 2008), LBH589 (Garbes et al., 2009), and oxamflatin (Kim et al., 1999) have shown positive effects in other diseases including cancer and/or neurodegenerative disorders and these compounds may also be considered for future FRDA therapy. A number of research groups are also currently screening for novel frataxin-increasing compounds and one group has identified a novel HDAC inhibitor, designated C5, which upregulates FXN expression in FRDA patient primary lymphocytes and increases H3K9ac and H4K8ac in FRDA lymphoblasts (Lufino et al., 2013) (Table 2). Interestingly, it has also recently been reported that resveratrol, a sirtuin activator, can produce a marked increase in frataxin expression in a FXN-EGFP reporter cell line and in a FRDA mouse model (Li et al., 2013). Therefore, further studies are required to dissect the interplay of HDACs and compounds that modulate HDAC activity in the direct or indirect control of FXN gene expression. Epigenetic-based therapies for FRDA are also likely to have other off-target effects, which do not directly affect FXN gene regulation, but which may nevertheless have other positive or negative effects on FRDA disease. For example, recent studies have demonstrated that frataxin deficiency in a conditional knockout mouse model triggers SIRT3 inhibition, resulting in marked hyperacetylation of numerous cardiac mitochondrial proteins (Wagner et al., 2012) that may lead to an increased sensitivity to oxidative stress. This finding suggests that the compounds that specifically activate SIRT3 could be a beneficial FRDA therapy.
HMTase Inhibitors
HMTase inhibitors, which may induce a more open chromatin structure at the FXN gene, are now also being considered for FRDA therapy (Figure 3). For example GSK126 has been identified as a potent and highly specific inhibitor of EZH2, a major component of the Polycomb repressor complex 2 (PRC2) (McCabe et al., 2012). EZH2 trimethylates lysine 27 on the tail of histone H3 (H3K27) and has been linked to the repression of several genes, thus, inhibition of this repressive histone mark at active genes could have a beneficial therapeutic outcome. High throughput biochemical screening has revealed genome-wide loss of H3K27 methylation and activation of previously silenced genes with minimal off-target effects following treatment with GSK126 (McCabe et al., 2012). In addition, small cell lung cancer (SCLC) cells treated with GSK126 showed substantial inhibition of cellular growth and this inhibition was concomitant with the reduced levels of H3K27me3 (Sato et al., 2013). These findings have highlighted the fact that inhibition of EZH2 activity by small molecule inhibitors, like GSK126, has a therapeutic benefit. In FRDA, increased levels of H3K27me3 have been reported in lymphoblastoid cells and fibroblast cells (De Biase et al., 2009; Kim et al., 2011; Chan et al., 2013) (Table 1). Therefore, this compound, which can inhibit such repressive marks, could have a beneficial FRDA therapeutic effect by reactivating FXN gene transcription.
Other studies have shown that H3K9me2 can be erased from the promoters of reactivated tumor suppressor genes following treatment with DNMTs and/or HDAC inhibitors (McGarvey et al., 2006). Therefore, compounds which can reduce H3K9me2 levels are an attractive option where gene silencing involves increased H3K9me2 levels, as seen with FRDA (Sandi et al., 2013) (Table 1). This has led to the development of various compounds that can inhibit the G9a activity directly or indirectly. The initial screening of 125,000 compounds from a chemical library has identified BIX-01294 as a potent and specific inhibitor of G9a (Kubicek et al., 2007). BIX-01294 binds to the SET domain of GLP in the same groove at which the target lysine (H3K9) binds. This prevents the binding of the peptide substrate and, consequently, the deposition of methylation marks at H3K9 (Chang et al., 2009). Recent studies in FRDA lymphoblastoid cells using the HMTase inhibitor, BIX-01294, identified a significant reduction in H3K9me2/me3, but without increasing the FXN mRNA levels, suggesting a possible redundant role for H3K9me2/me3 in FXN gene silencing (Punga and Buhler, 2010). In contrast, Chan and colleagues have recently identified nicotinamide-induced reactivation of the FXN gene in association with decreased H3K9me3 and H3K27me3 (Chan et al., 2013). Recently, a new inhibitor for G9a and GLP, UNC0638, has been identified (Vedadi et al., 2011). Treatment of a variety of cell lines with UNC0638 resulted in lower global H3K9me2 levels and activated several previously silenced genes in vitro (Vedadi et al., 2011). In addition, treatment of U2OS and HeLa cells with UNC0638 has shown that it specifically inhibits H3K9me2, with minimal effect on H3K27me3 and H4Kme3 levels (Machleidt et al., 2011). ERG-associated protein with SET domain (ESET), a novel histone H3K9 methyltransferase, has been shown to mediate histone methylation (Rea et al., 2000). It is also proposed that ESET may have a role in epigenetic silencing of neuronal genes through its HMTase activity (Li et al., 2006). Importantly, the ESET levels were significantly elevated in HD patients and in R6/2 transgenic mice (Ryu et al., 2006). Subsequent treatment with mithramycin (or in combination with cystamine), resulted in substantial reduction of H3K9me3 and significantly improved the behavioral and neuropathological phenotype in R6/2 and 82Q mice of HD (Ryu et al., 2006; Stack et al., 2007). Mithramycin is a clinically approved guanosine-cytosine rich DNA antitumor antibiotic that interferes with the DNA binding of the Sp family transcription factors, but its effects in FRDA have not yet been studied. Ongoing preclinical studies of cell and mouse models using such HMTase inhibitors will determine if any of these compounds can be considered suitable to progress to FRDA clinical studies, as is the case now for HDAC inhibitors.
Antigene RNA-Based Therapies
Antigene RNAs (agRNAs) are small duplex RNAs of 19 bp in length that target gene promoters. Depending on the target sequence and cell type, agRNAs can either silence (Janowski et al., 2006) or activate the gene transcription (Janowski et al., 2007). Since agRNAs target in a sequence specific manner, it may be possible to modulate agRNAs to activate gene expression in disease-associated genes where gene activation is essential, as with FRDA. Importantly, agRNAs can target both sense or antisense strands, and coding or non-coding RNA transcripts (Watts et al., 2010). Therefore, the use of agRNA to activate the FXN gene by targeting the FXN promoter or the FAST1 transcript may prove useful, since both mechanisms may be involved in reversing FXN gene silencing and thus ameliorating FRDA disease. However, as with any macromolecule-based therapy, the major problem of how to achieve effective delivery to cells will first have to be overcome.
Potential Risks of Epigenetic Therapies
Epigenetic therapies to date have primarily focused on the use of DNA hypomethylating agents and HDAC inhibitors. The major risk associated with any epigenetic therapy is the potential lack of specificity. To the best of our knowledge it is not yet possible to silence or reactivate specific genes through use of DNMT inhibitors or HDAC inhibitors. Due to the general effects of these compounds, they tend to disrupt global DNA methylation and histone acetylation status. Genome-wide loss of DNA methylation has been shown to produce a high incidence of lymphomas in mouse models (Gaudet et al., 2003), which raises the concern of potential tumor formation in patients given DNA hypomethylating treatments. HDAC inhibitors also have off-target effects that may be difficult to detect, especially if they appear years after the initial therapy. This is important, because most FRDA patients are diagnosed before adulthood. Therefore, prolonged HDAC inhibitor treatment in childhood may induce tumor formation, for example, that is only apparent later in adulthood. However, studies to date show that HDAC inhibitors are actually reasonably well tolerated in the short term. They appear to have less toxicity when used for cancer treatment than classical cytotoxic chemotherapeutic agents, but they are still prone to induce primary clinical toxicities such as cardiac side effects, nausea, vomiting, fatigue, and reduced blood cell counts (Subramanian et al., 2010). The clinical use of HMTase inhibitors has not advanced as far as the use of HDAC inhibitors, but HMTase inhibitors are also expected to induce extensive gene expression changes, and hence potential off-target effects. Having said this, use of the HMTase inhibitor GSK126 for the treatment of lymphoma has been shown to exert minimal off-target effects despite genome-wide changes in H3K27 methylation, which is somewhat encouraging (McCabe et al., 2012).
Conclusions
Due to the identification of several associated epigenetic marks, FRDA can now be considered as an epigenetic disease and drug treatments are being developed to target these epigenetic changes in attempts to ameliorate the disease phenotype. However, epigenetic-based therapies are generally non-specific, with off-target effects, and the development of drugs to more specifically target the FXN locus may require further consideration. Epigenetic-based therapies for FRDA will also benefit from further understanding of how FXN gene expression is controlled. For example, it has been recently reported that two transcription factors, SRF and TFAP2, directly bind to the promoter region of the human FXN gene and alter frataxin mRNA and protein levels (Li et al., 2010). Small non-coding RNAs, such as microRNAs, have also been implicated in several neurodegenerative disorders including AD (Cogswell et al., 2008), HD (Lee et al., 2011), and FRDA (Mahishi et al., 2012). An elevated level of miR-886-3p in FRDA is associated with the downregulation of the FXN gene and the use of anti-miR-886-3p or the HDAC inhibitor 4b alone has been shown to partially reverse FXN gene repression by reducing miR-886-3p levels (Mahishi et al., 2012; Bandiera et al., 2013). Therefore, it would be interesting to investigate the combined effect of anti-microRNA and HDAC inhibitor compounds in the activation of FXN gene transcription. In general, it may also be useful to simultaneously administer two or more epigenetic-based drugs to examine synergistic treatment effects. In addition, to identify novel epigenetic-based FRDA therapeutic compounds, various drug-screening systems, cell and animal models have been developed and are currently being utilized by several labs (see review Martelli et al., 2012). Overall, considering the rapid progress that has been made to take HDAC inhibitor therapy from basic research to FRDA clinical trials, the next few years will hopefully see the emergence of at least one effective epigenetic-based therapy for FRDA.
Author Contributions
All authors contributed to draft the manuscript and all authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Chiranjeevi Sandi and Madhavi Sandi were supported by funding to Mark A. Pook from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement number 242193/EFACTS. Sara Anjomani Virmouni was supported by funding to Mark A. Pook from the Friedreich's Ataxia Research Alliance (FARA). Sahar Al-Mahdawi was supported by funding to Mark A. Pook from the Wellcome Trust (089757).
References
Al-Mahdawi, S., Pinto, R. M., Ismail, O., Varshney, D., Lymperi, S., Sandi, C., et al. (2008). The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 17, 735–746. doi: 10.1093/hmg/ddm346
Al-Mahdawi, S., Sandi, C., Mouro Pinto, R., and Pook, M. A. (2013). Friedreich ataxia patient tissues exhibit increased 5-hydroxymethylcytosine modification and decreased CTCF binding at the FXN locus. PLoS ONE 8:e74956. doi: 10.1371/journal.pone.0074956
Bandiera, S., Cartault, F., Jannot, A.-S., Hatem, E., Girard, M., Rifai, L., et al. (2013). Genetic variations creating microRNA target sites in the FXN 3'-UTR affect Frataxin expression in Friedreich ataxia. PLoS ONE 8:e54791. doi: 10.1371/journal.pone.0054791
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. doi: 10.1016/j.cell.2007.05.009
Biacsi, R., Kumari, D., and Usdin, K. (2008). SIRT1 inhibition alleviates gene silencing in Fragile X mental retardation syndrome. PLoS Genet. 4:e1000017. doi: 10.1371/journal.pgen.1000017
Bradley, J. L., Blake, J. C., Chamberlain, S., Thomas, P. K., Cooper, J. M., and Schapira, A. H. (2000). Clinical, biochemical and molecular genetic correlations in Friedreich's ataxia. Hum. Mol. Genet. 9, 275–282. doi: 10.1093/hmg/9.2.275
Butler, R., and Bates, G. P. (2006). Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat. Rev. Neurosci. 7, 784–796. doi: 10.1038/nrn1989
Campuzano, V., Montermini, L., Lutz, Y., Cova, L., Hindelang, C., Jiralerspong, S., et al. (1997). Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 6, 1771–1780. doi: 10.1093/hmg/6.11.1771
Campuzano, V., Montermini, L., Molto, M. D., Pianese, L., Cossee, M., Cavalcanti, F., et al. (1996). Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427. doi: 10.1126/science.271.5254.1423
Castaldo, I., Pinelli, M., Monticelli, A., Acquaviva, F., Giacchetti, M., Filla, A., et al. (2008). DNA methylation in intron 1 of the frataxin gene is related to GAA repeat length and age of onset in Friedreich ataxia patients. J. Med. Genet. 45, 808–812. doi: 10.1136/jmg.2008.058594
Chan, P. K., Torres, R., Yandim, C., Law, P. P., Khadayate, S., Mauri, M., et al. (2013). Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich's ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum. Mol. Genet. 22, 2662–2675. doi: 10.1093/hmg/ddt115
Chang, Y., Zhang, X., Horton, J. R., Upadhyay, A. K., Spannhoff, A., Liu, J., et al. (2009). Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 16, 312–317. doi: 10.1038/nsmb.1560
Chiurazzi, P., Pomponi, M. G., Pietrobono, R., Bakker, C. E., Neri, G., and Oostra, B. A. (1999). Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum. Mol. Genet. 8, 2317–2323. doi: 10.1093/hmg/8.12.2317
Chiurazzi, P., Pomponi, M. G., Willemsen, R., Oostra, B. A., and Neri, G. (1998). In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 7, 109–113. doi: 10.1093/hmg/7.1.109
Cho, D. H., Thienes, C. P., Mahoney, S. E., Analau, E., Filippova, G. N., and Tapscott, S. J. (2005). Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 20, 483–489. doi: 10.1016/j.molcel.2005.09.002
Chou, C. J., Herman, D., and Gottesfeld, J. M. (2008). Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 283, 35402–35409. doi: 10.1074/jbc.M807045200
Chung, D. W., Rudnicki, D. D., Yu, L., and Margolis, R. L. (2011). A natural antisense transcript at the Huntington's disease repeat locus regulates HTT expression. Hum. Mol. Genet. 20, 3467–3477. doi: 10.1093/hmg/ddr263
Cogswell, J. P., Ward, J., Taylor, I. A., Waters, M., Shi, Y., Cannon, B., et al. (2008). Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 14, 27–41. doi: 10.1016/j.jalz.2008.05.420
De Biase, I., Chutake, Y. K., Rindler, P. M., and Bidichandani, S. I. (2009). Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS ONE 4:e7914. doi: 10.1371/journal.pone.0007914
Engel, N., Thorvaldsen, J. L., and Bartolomei, M. S. (2006). CTCF binding sites promote transcription initiation and prevent DNA methylation on the maternal allele at the imprinted H19/Igf2 locus. Hum. Mol. Genet. 15, 2945–2954. doi: 10.1093/hmg/ddl237
Evans-Galea, M. V., Carrodus, N., Rowley, S. M., Corben, L. A., Tai, G., Saffery, R., et al. (2012). FXN methylation predicts expression and clinical outcome in Friedreich ataxia. Ann. Neurol. 71, 487–497. doi: 10.1002/ana.22671
Festenstein, R. (2006). Breaking the silence in Friedreich's ataxia. Nat. Chem. Biol. 2, 512–513. doi: 10.1038/nchembio1006-512
Filippova, G. N., Cheng, M. K., Moore, J. M., Truong, J. P., Hu, Y. J., Nguyen, D. K., et al. (2005). Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev. Cell 8, 31–42. doi: 10.1016/j.devcel.2004.10.018
Filippova, G. N., Thienes, C. P., Penn, B. H., Cho, D. H., Hu, Y. J., Moore, J. M., et al. (2001). CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat. Genet. 28, 335–343. doi: 10.1038/ng570
Foury, F., and Cazzalini, O. (1997). Deletion of the yeast homologue of the human gene associated with Friedreich's ataxia elicits iron accumulation in mitochondria. FEBS Lett. 411, 373–377. doi: 10.1016/S0014-5793(97)00734-5
Garbes, L., Riessland, M., Holker, I., Heller, R., Hauke, J., Trankle, C., et al. (2009). LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum. Mol. Genet. 18, 3645–3658. doi: 10.1093/hmg/ddp313
Gaudet, F., Hodgson, J. G., Eden, A., Jackson-Grusby, L., Dausman, J., Gray, J. W., et al. (2003). Induction of tumors in mice by genomic hypomethylation. Science 300, 489–492. doi: 10.1126/science.1083558
Ghosh, S., and Feany, M. B. (2004). Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum. Mol. Genet. 13, 2011–2018. doi: 10.1093/hmg/ddh214
Grabczyk, E., Mancuso, M., and Sammarco, M. C. (2007). A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 35, 5351–5359. doi: 10.1093/nar/gkm589
Greene, E., Mahishi, L., Entezam, A., Kumari, D., and Usdin, K. (2007). Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 35, 3383–3390. doi: 10.1093/nar/gkm271
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., and Song, H. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. doi: 10.1016/j.cell.2011.03.022
Herman, D., Jenssen, K., Burnett, R., Soragni, E., Perlman, S. L., and Gottesfeld, J. M. (2006). Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat. Chem. Biol. 2, 551–558. doi: 10.1038/nchembio815
Holwerda, S. J., and De Laat, W. (2013). CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20120369. doi: 10.1098/rstb.2012.0369
Jain, N., Rossi, A., and Garcia-Manero, G. (2009). Epigenetic therapy of leukemia: an update. Int. J. Biochem. Cell Biol. 41, 72–80. doi: 10.1016/j.biocel.2008
Janowski, B. A., Hu, J., and Corey, D. R. (2006). Silencing gene expression by targeting chromosomal DNA with antigene peptide nucleic acids and duplex RNAs. Nat. Protoc. 1, 436–443. doi: 10.1038/nprot.2006.64
Janowski, B. A., Younger, S. T., Hardy, D. B., Ram, R., Huffman, K. E., and Corey, D. R. (2007). Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat. Chem. Biol. 3, 166–173. doi: 10.1038/nchembio860
Jenuwein, T. (2006). The epigenetic magic of histone lysine methylation. FEBS J. 273, 3121–3135. doi: 10.1111/j.1742-4658
Khalil, A. M., Faghihi, M. A., Modarresi, F., Brothers, S. P., and Wahlestedt, C. (2008). A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS ONE 3:e1486. doi: 10.1371/journal.pone.0001486
Kim, E., Napierala, M., and Dent, S. Y. (2011). Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich's ataxia. Nucleic Acids Res. 39, 8366–8377. doi: 10.1093/nar/gkr542
Kim, Y. B., Lee, K. H., Sugita, K., Yoshida, M., and Horinouchi, S. (1999). Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene 18, 2461–2470. doi: 10.1038/sj.onc.1202564
Kimura, A., Matsubara, K., and Horikoshi, M. (2005). A decade of histone acetylation: marking eukaryotic chromosomes with specific codes. J. Biochem. 138, 647–662. doi: 10.1093/jb/mvi184
Koeppen, A. H., Michael, S. C., Knutson, M. D., Haile, D. J., Qian, J., Levi, S., et al. (2007). The dentate nucleus in Friedreich's ataxia: the role of iron-responsive proteins. Acta Neuropathol. 114, 163–173. doi: 10.1007/s00401-007-0220-y
Kourmouli, N., Jeppesen, P., Mahadevhaiah, S., Burgoyne, P., Wu, R., Gilbert, D. M., et al. (2004). Heterochromatin and tri-methylated lysine 20 of histone H4 in animals. J. Cell Sci. 117, 2491–2501. doi: 10.1242/jcs.01238
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. doi: 10.1126/science.1169786
Kubicek, S., O'Sullivan, R. J., August, E. M., Hickey, E. R., Zhang, Q., Teodoro, M. L., et al. (2007). Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25, 473–481. doi: 10.1016/j.molcel.2007.01.017
Kumari, D., Biacsi, R. E., and Usdin, K. (2011). Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J. Biol. Chem. 286, 4209–4215. doi: 10.1074/jbc.M110.194035
Ladd, P. D., Smith, L. E., Rabaia, N. A., Moore, J. M., Georges, S. A., Hansen, R. S., et al. (2007). An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 16, 3174–3187. doi: 10.1093/hmg/ddm293
Lee, S. T., Chu, K., Im, W. S., Yoon, H. J., Im, J. Y., Park, J. E., et al. (2011). Altered microRNA regulation in Huntington's disease models. Exp. Neurol. 227, 172–179. doi: 10.1016/j.expneurol.2010.10.012
Li, H., Rauch, T., Chen, Z. X., Szabo, P. E., Riggs, A. D., and Pfeifer, G. P. (2006). The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 281, 19489–19500. doi: 10.1074/jbc.M513249200
Li, K., Singh, A., Crooks, D. R., Dai, X., Cong, Z., Pan, L., et al. (2010). Expression of human Frataxin is regulated by transcription factors SRF and TFAP2. PLoS ONE 5:e12286. doi: 10.1371/journal.pone.0012286
Li, L., Voullaire, L., Sandi, C., Pook, M. A., Ioannou, P. A., Delatycki, M. B., et al. (2013). Pharmacological screening using an FXN-EGFP cellular genomic reporter assay for the therapy of Friedreich ataxia. PLoS ONE 8:e55940. doi: 10.1371/journal.pone.0055940
Libby, R. T., Hagerman, K. A., Pineda, V. V., Lau, R., Cho, D. H., Baccam, S. L., et al. (2008). CTCF cis-regulates trinucleotide repeat instability in an epigenetic manner: a novel basis for mutational hot spot determination. PLoS Genet. 4:e1000257. doi: 10.1371/journal.pgen.1000257
Lorincz, M. C., Dickerson, D. R., Schmitt, M., and Groudine, M. (2004). Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 11, 1068–1075. doi: 10.1038/nsmb840
Lufino, M. M., Silva, A. M., Nemeth, A. H., Alegre-Abarrategui, J., Russell, A. J., and Wade-Martins, R. (2013). A GAA repeat expansion reporter model of Friedreich's ataxia recapitulates the genomic context and allows rapid screening of therapeutic compounds. Hum. Mol. Genet. 22, 5173–5187. doi: 10.1093/hmg/ddt370
Machleidt, T., Robers, M. B., Hermanson, S. B., Dudek, J. M., and Bi, K. (2011). TR-FRET cellular assays for interrogating posttranslational modifications of histone H3. J. Biomol. Screen. 16, 1236–1246. doi: 10.1177/1087057111422943
Mahishi, L. H., Hart, R. P., Lynch, D. R., and Ratan, R. R. (2012). miR-886-3p levels are elevated in Friedreich ataxia. J. Neurosci. 32, 9369–9373. doi: 10.1523/JNEUROSCI.0059-12.2012
Martelli, A., Napierala, M., and Puccio, H. (2012). Understanding the genetic and molecular pathogenesis of Friedreich's ataxia through animal and cellular models. Dis. Model. Mech. 5, 165–176. doi: 10.1242/dmm.008706
Martin, C., and Zhang, Y. (2005). The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849. doi: 10.1038/nrm1761
McCabe, M. T., Ott, H. M., Ganji, G., Korenchuk, S., Thompson, C., Van Aller, G. S., et al. (2012). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. doi: 10.1038/nature11606
McGarvey, K. M., Fahrner, J. A., Greene, E., Martens, J., Jenuwein, T., and Baylin, S. B. (2006). Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 66, 3541–3549. doi: 10.1158/0008-5472.CAN-05-2481
Moseley, M. L., Zu, T., Ikeda, Y., Gao, W., Mosemiller, A. K., Daughters, R. S., et al. (2006). Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 38, 758–769. doi: 10.1038/ng1827
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. doi: 10.1016/S0092-8674(00)81656-6
Ota, H., Tokunaga, E., Chang, K., Hikasa, M., Iijima, K., Eto, M., et al. (2006). Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25, 176–185. doi: 10.1038/sj.onc.1209049
Pandolfo, M. (2008). Friedreich ataxia. Arch. Neurol. 65, 1296–1303. doi: 10.1001/archneur.65.10.1296
Pianese, L., Turano, M., Lo Casale, M. S., De Biase, I., Giacchetti, M., Monticelli, A., et al. (2004). Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers. J. Neurol. Neurosurg. Psychiatr. 75, 1061–1063. doi: 10.1136/jnnp.2003.028605
Plummer, R., Vidal, L., Griffin, M., Lesley, M., De Bono, J., Coulthard, S., et al. (2009). Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin. Cancer Res. 15, 3177–3183. doi: 10.1158/1078-0432.CCR-08-2859
Pradhan, S., Bacolla, A., Wells, R. D., and Roberts, R. J. (1999). Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 274, 33002–33010. doi: 10.1074/jbc.274.46.33002
Punga, T., and Buhler, M. (2010). Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol. Med. 2, 120–129. doi: 10.1002/emmm.201000064
Rai, M., Soragni, E., Chou, C. J., Barnes, G., Jones, S., Rusche, J. R., et al. (2010). Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich's ataxia patients and in a mouse model. PLoS ONE 5:e8825. doi: 10.1371/journal.pone.0008825
Rai, M., Soragni, E., Jenssen, K., Burnett, R., Herman, D., Coppola, G., et al. (2008). HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE 3:e1958. doi: 10.1371/journal.pone.0001958
Rea, S., Eisenhaber, F., O'Carroll, D., Strahl, B. D., Sun, Z. W., Schmid, M., et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. doi: 10.1038/35020506
Robertson, K. D. (2001). DNA methylation, methyltransferases, and cancer. Oncogene 20, 3139–3155. doi: 10.1038/sj.onc.1204341
Ryu, H., Lee, J., Hagerty, S. W., Soh, B. Y., McAlpin, S. E., Cormier, K. A., et al. (2006). ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 19176–19181. doi: 10.1073/pnas.0606373103
Sandi, C., Al-Mahdawi, S., and Pook, M. A. (2013). Epigenetics in Friedreich's ataxia: challenges and opportunities for therapy. Genet. Res. Int. 2013, 12. doi: 10.1155/2013/852080
Sandi, C., Pinto, R. M., Al-Mahdawi, S., Ezzatizadeh, V., Barnes, G., Jones, S., et al. (2011). Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol. Dis. 42, 496–505. doi: 10.1016/j.nbd.2011.02.016
Sandi, C., Sandi, M., Jassal, H., Ezzatizadeh, V., Anjomani-Virmouni, S., Al-Mahdawi, S., et al. (2014). Generation and characterisation of Friedreich ataxia YG8R mouse fibroblast and neural stem cell models. PLoS ONE 9:e89488. doi: 10.1371/journal.pone.0089488
Sarsero, J. P., Li, L., Wardan, H., Sitte, K., Williamson, R., and Ioannou, P. A. (2003). Upregulation of expression from the FRDA genomic locus for the therapy of Friedreich ataxia. J. Gene Med. 5, 72–81. doi: 10.1002/jgm.320
Sato, T., Kaneda, A., Tsuji, S., Isagawa, T., Yamamoto, S., Fujita, T., et al. (2013). PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci. Rep. 3, 1911. doi: 10.1038/srep01911
Saveliev, A., Everett, C., Sharpe, T., Webster, Z., and Festenstein, R. (2003). DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 422, 909–913. doi: 10.1038/nature01596
Schulz, J. B., Boesch, S., Burk, K., Durr, A., Giunti, P., Mariotti, C., et al. (2009). Diagnosis and treatment of Friedreich ataxia: a European perspective. Nat. Rev. Neurol. 5, 222–234. doi: 10.1038/nrneurol.2009.26
Sharma, R., De Biase, I., Gomez, M., Delatycki, M. B., Ashizawa, T., and Bidichandani, S. I. (2004). Friedreich ataxia in carriers of unstable borderline GAA triplet-repeat alleles. Ann. Neurol. 56, 898–901. doi: 10.1002/ana.20333
Shen, Y., Yue, F., McCleary, D. F., Ye, Z., Edsall, L., Kuan, S., et al. (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120. doi: 10.1038/nature11243
Sopher, B. L., Ladd, P. D., Pineda, V. V., Libby, R. T., Sunkin, S. M., Hurley, J. B., et al. (2011). CTCF regulates ataxin-7 expression through promotion of a convergently transcribed, antisense noncoding RNA. Neuron 70, 1071–1084. doi: 10.1016/j.neuron.2011.05.027
Soragni, E., Herman, D., Dent, S. Y., Gottesfeld, J. M., Wells, R. D., and Napierala, M. (2008). Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia. Nucleic Acids Res. 36, 6056–6065. doi: 10.1093/nar/gkn604
Soragni, E., Xu, C., Plasterer, H. L., Jacques, V., Rusche, J. R., and Gottesfeld, J. M. (2012). Rationale for the development of 2-aminobenzamide histone deacetylase inhibitors as therapeutics for Friedreich ataxia. J. Child Neurol. 27, 1164–1173. doi: 10.1177/0883073812448533
Stack, E. C., Del Signore, S. J., Luthi-Carter, R., Soh, B. Y., Goldstein, D. R., Matson, S., et al. (2007). Modulation of nucleosome dynamics in Huntington's disease. Hum. Mol. Genet. 16, 1164–1175. doi: 10.1093/hmg/ddm064
Subramanian, S., Bates, S. E., Wright, J. J., Espinoza-Delgado, I., and Piekarz, R. L. (2010). Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 3, 2751–2767. doi: 10.3390/ph3092751
Szulwach, K. E., Li, X., Li, Y., Song, C. X., Wu, H., Dai, Q., et al. (2011). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616. doi: 10.1038/nn.2959
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Thomas, E. A., Coppola, G., Desplats, P. A., Tang, B., Soragni, E., Burnett, R., et al. (2008). The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 105, 15564–15569. doi: 10.1073/pnas.0804249105
Vedadi, M., Barsyte-Lovejoy, D., Liu, F., Rival-Gervier, S., Allali-Hassani, A., Labrie, V., et al. (2011). A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7, 566–574. doi: 10.1038/nchembio.599
Wagner, G. R., Pride, P. M., Babbey, C. M., and Payne, R. M. (2012). Friedreich's ataxia reveals a mechanism for coordinate regulation of oxidative metabolism via feedback inhibition of the SIRT3 deacetylase. Hum. Mol. Genet. 21, 2688–2697. doi: 10.1093/hmg/dds095
Waldvogel, D., Van Gelderen, P., and Hallett, M. (1999). Increased iron in the dentate nucleus of patients with Friedrich's ataxia. Ann. Neurol. 46, 123–125.
Wang, H., Maurano, M. T., Qu, H., Varley, K. E., Gertz, J., Pauli, F., et al. (2012). Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 22, 1680–1688. doi: 10.1101/gr.136101.111
Watts, J. K., Yu, D., Charisse, K., Montaillier, C., Potier, P., Manoharan, M., et al. (2010). Effect of chemical modifications on modulation of gene expression by duplex antigene RNAs that are complementary to non-coding transcripts at gene promoters. Nucleic Acids Res. 38, 5242–5259. doi: 10.1093/nar/gkq258
Wells, R. D. (2008). DNA triplexes and Friedreich ataxia. FASEB J. 22, 1625–1634. doi: 10.1096/fj.07-097857
Wong, A., Yang, J., Cavadini, P., Gellera, C., Lonnerdal, B., Taroni, F., et al. (1999). The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 8, 425–430. doi: 10.1093/hmg/8.3.425
Xia, H., Cao, Y., Dai, X., Marelja, Z., Zhou, D., Mo, R., et al. (2012). Novel frataxin isoforms may contribute to the pathological mechanism of Friedreich ataxia. PLoS ONE 7:e47847. doi: 10.1371/journal.pone.0047847
Keywords: Friedreich ataxia, FRDA, frataxin, FXN, GAA repeat, DNA demethylation, HDAC inhibitor, HMTase inhibitor
Citation: Sandi C, Sandi M, Anjomani Virmouni S, Al-Mahdawi S and Pook MA (2014) Epigenetic-based therapies for Friedreich ataxia. Front. Genet. 5:165. doi: 10.3389/fgene.2014.00165
Received: 01 April 2014; Accepted: 19 May 2014;
Published online: 03 June 2014.
Edited by:
Steven G. Gray, St. James Hospital/Trinity College Dublin, IrelandReviewed by:
Antonius Plagge, University of Liverpool, UKRoberto Testi, University of Rome Tor Vergata, Italy
Copyright © 2014 Sandi, Sandi, Anjomani Virmouni, Al-Mahdawi and Pook. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark A. Pook, Division of Biosciences, School of Health Sciences and Social Care, Brunel University London, Kingston Lane, Uxbridge, UB8 3PH, UK e-mail: mark.pook@brunel.ac.uk
†Present address: Chiranjeevi Sandi, Uro-Oncology Research Group, Cancer Research UK-Cambridge Institute, University of Cambridge, Cambridge, UK