 Gaston K. Mazandu
Gaston K. Mazandu Nicola J. Mulder
Nicola J. Mulder- Computational Biology Group, Department of Clinical Laboratory Sciences, Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Cape Town, South Africa
With the advancement of new high throughput sequencing technologies, there has been an increase in the number of genome sequencing projects worldwide, which has yielded complete genome sequences of human, animals and plants. Subsequently, several labs have focused on genome annotation, consisting of assigning functions to gene products, mostly using Gene Ontology (GO) terms. As a consequence, there is an increased heterogeneity in annotations across genomes due to different approaches used by different pipelines to infer these annotations and also due to the nature of the GO structure itself. This makes a curator's task difficult, even if they adhere to the established guidelines for assessing these protein annotations. Here we develop a genome-scale approach for integrating GO annotations from different pipelines using semantic similarity measures. We used this approach to identify inconsistencies and similarities in functional annotations between orthologs of human and Drosophila melanogaster, to assess the quality of GO annotations derived from InterPro2GO mappings compared to manually annotated GO annotations for the Drosophila melanogaster proteome from a FlyBase dataset and human, and to filter GO annotation data for these proteomes. Results obtained indicate that an efficient integration of GO annotations eliminates redundancy up to 27.08 and 22.32% in the Drosophila melanogaster and human GO annotation datasets, respectively. Furthermore, we identified lack of and missing annotations for some orthologs, and annotation mismatches between InterPro2GO and manual pipelines in these two proteomes, thus requiring further curation. This simplifies and facilitates tasks of curators in assessing protein annotations, reduces redundancy and eliminates inconsistencies in large annotation datasets for ease of comparative functional genomics.
1. Introduction
The development of fast and relatively inexpensive sequencing technologies has yielded complete genome sequences of thousands of organisms. Several sequence databases store these sequences, including GenBank Benson et al. (2009), Ensembl Flicek et al. (2010); Fernández-Suárez and Schuster (2010); Spudich and Fernández-Suárez (2010), NCBI (Pruitt et al., 2005; Sayers et al., 2009) and the UniProt database, which is an integrated repository of protein sequence and function created by joining the information contained in the Swiss-Prot, TrEMBL, and PIR proteins databases (Jain et al., 2009; UniProt-Consortium, 2010). In these databases, an increased deficiency in functional annotation was observed for many sequenced proteins as approximately 20–50% of proteins within a genome were still labeled “unknown,” “uncharacterized” or “hypothetical” (Mazandu and Mulder, 2012). Thus, several annotation pipelines, including experimental and electronic, were developed to functionally characterize these proteins. Subsequently, the Gene Ontology (GO) (GO-Consortium, 2012) arose to organize and unify biology and information about genes and proteins shared by different organisms, and emerged as one of the dominant and most popular functional classification schemes for functional annotation of genes and their products.
Many annotation pipelines were developed to predict or assign functions to proteins using GO terms from the three different ontologies of GO, namely Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). These include electronic annotation methods, such as Ensembl Compara, InterPro, UniProtKB/Swiss-Prot Keywords (SPKW), UniProtKB-Subcellular Location (SPSL), UniPathway, Enzyme Commission (EC), and High-quality Automated and Manual Annotation of Microbial Proteomes (HAMAP), and manual annotation efforts, such as the Gene Ontology Annotation project, the Reference Genome Annotation Initiative (The Reference Genome Group of the Gene Ontology Consortium, 2009), and Cardiovascular (www.ebi.ac.uk/GOA/CVI) and Renal (www.ebi.ac.uk/GOA/kidney) Gene Ontology Annotation Initiatives. The Gene Ontology Annotation (GOA) project at the European Bioinformatics Institute (EBI) commits to integrating these protein annotations into a single set of high-quality electronic and manual associations (annotations) of GO terms to UniProt Knowledgebase (UniProtKB) entries. Most of the data is generated from the conversion maps, namely SPKW2GO, SPSL2GO, EC2GO, HAMAP2GO, UniPathway2GO and InterPro2GO, which themselves are manually curated to ensure high-accuracy annotations from the electronically inferred GO annotation set.
Producing high-quality and accurate GO annotations is challenging, as manual annotation is a slow and expensive process and the number of manual annotations available for a particular genome is usually far fewer than those produced by electronic annotation pipelines. To improve the annotation quality, the GOA project does manual GO curation following standards set by the GO Consortium (http://www.geneontology.org/GO.annotation.conventions.shtml). However, different annotation pipelines can differ widely in their specific procedures (annotation algorithms, confidence thresholds, etc.) and this increases the heterogeneity across a gene set, thus making curators' tasks difficult. Furthermore, considering the structure of GO, the set of associations produced by GOA is often redundant and subjected to potential mismatches and inconsistencies (Dolan et al., 2005). Fortunately, the hierarchical structure of the GO enables the assessment of the GO term closeness using semantic relationships between terms. These semantic relationships between terms have been used to set up semantic similarity tools that enable efficient exploitation of the enormous corpus of biological knowledge embedded in the GO directed acyclic graph (DAG) structure by comparing GO terms and proteins at the functional level (Mazandu and Mulder, 2013a).
The use of semantic relationships between GO terms enables the quantification of GO term specificity through measurements of information content (IC) values in the GO DAG (Mazandu and Mulder, 2013b). These GO term IC values are used to evaluate semantic similarity scores between GO terms and annotated proteins, reflecting the closeness between two concepts in the GO DAG. Several semantic similarity measures have been introduced (Mazandu and Mulder, 2012, 2013a,b) and used in different biological applications, such as gene clustering, gene expression data analysis, prediction and validation of molecular interactions, and disease gene prioritization (Mazandu and Mulder, 2013a). In the context of current high-throughput biological technologies, these measures may be used to set up novel bioinformatics approaches that enable the efficient use of the GO DAG structure for integrating annotations from multiple sources. Here we use these semantic similarity measures as tools to develop an efficient, large-scale approach that enables “optimal” integration of protein GO annotations from heterogeneous annotation pipelines or different sources, thus reducing redundancy and eliminating inconsistencies in the integrated annotation datasets for ease of comparative functional genomics.
2. Materials and Methods
In order to compare different annotation pipelines, to filter an annotation dataset or to integrate annotations from different pipelines, we define GO annotation quality assessment measures based on GO semantic similarity scores. These measures are applied to Drosophila melanogaster (fruitfly) and human GO annotations as a demonstration of the variety of applications. We assess GO annotations assigned by InterPro, which are electronically inferred with IEA (Inferred from Electronic Annotation) as the evidence code, in comparison with the manual and experimental annotation in FlyBase (Tweedie et al., 2008). In addition, we compare annotations in Flybase to orthologous proteins from human to assess the quality of GO annotation between different annotation groups and we integrate electronically inferred and experimental annotations for Drosophila melanogaster proteins.
2.1. GO Annotation Quality Assessment Measures
Currently electronic annotation pipelines dominate manual annotation in terms of number of annotations available for a particular genome and this is the most likely future trend. The GO annotations produced by these electronic annotation pipelines represent more than 98% of the GOA dataset, but the mapping filters used are, in most cases, manually curated, which increases confidence levels of these annotations, even though they still receive the IEA evidence code. In this context, the GO annotation quality for a proteome or a set of proteins should mainly be measured by the specificity or the level of detail of GO annotations used, the consistency of these annotations in terms of similarity between terms used and the non-redundancy of the annotation set. We used semantic similarity measures between terms in the GO DAG to derive these different GO annotation quality assessment measures, as described below.
2.1.1. Fuzzy non-redundant set of annotations for a gene
Given a gene g with TXg its set of GO terms in the ontology X, a term t∈ TXg is redundant in TXg when it contributes to the specification of its descendant term s∈ TXg based on the semantic similarity between them, in which case the score is greater than a given threshold δ ≥ 0. This threshold score provides the semantic similarity degree at which an ancestor term is considered to semantically reflect in the specification of its descendant or a child term consistently includes the ancestor term under consideration in its specification. The set 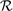 Xg of redundant annotations for the gene g is given by
Xg of redundant annotations for the gene g is given by
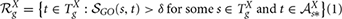
where 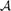 Xs* denotes the set of ancestors of the term s in the ontology X and
Xs* denotes the set of ancestors of the term s in the ontology X and 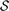 GO(s,t) is the semantic similarity score between GO terms s and t retrieved from the following formula (Mazandu and Mulder, 2013b):
GO(s,t) is the semantic similarity score between GO terms s and t retrieved from the following formula (Mazandu and Mulder, 2013b):
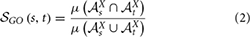
where Xx = Xx*∪{x}, μ(Xs ∩ Xt)≥ 0 and μ(Xs ∪ Xt) > 0 are measures of the commonality between and of the description of Xs and Xt, respectively.
The set 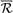 Xg of non-redundant terms annotating the gene g is the complementary set of Xg in TXg, i.e.,
Xg of non-redundant terms annotating the gene g is the complementary set of Xg in TXg, i.e.,

If δ = 0, the set Xg is referred to as the set of strict non-redundant terms annotating the gene g, which represents the set of terms annotating a gene at a high level. The functional redundancy score 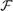 R(g) of the gene g is given by
R(g) of the gene g is given by
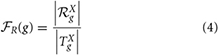
where |T| is the number of terms in the set T. R(g) represents the proportion of terms in the set of terms annotating the gene g that are ancestors implicitly included to other term specification in that set based on the semantic similarity between them. Thus, the term “fuzzy” is used to map the fact that the consideration of an ancestor term in the specification of its descendant is no longer a two-valued logic yes (1) or no (0) but now depends on the semantic similarity score between them.
2.1.2. Assessing functional specificity of a gene
A given gene can perform several functions or be involved in several processes. In this case, the gene is annotated by a set of GO terms. The specificity of each term assessing its informativeness depends on its position in the GO structure and the deeper the term is in the DAG structure the more specific or informative the term is. This indicates that the closer or the more similar to the leaf term (term without a child term) the more specific or informative the term is. The specificity score of an annotated gene depends on the specificity of terms used to annotate the gene and is the average of specificity scores of terms in the set of strict non-redundant terms annotating the gene. Thus, for a given gene g, its functional specificity score S(g) is assessed by measuring how similar its GO annotations are to leaf terms of the GO DAG connected to the set Xg of strict non-redundant terms annotating g. This functional specificity score is computed as follows:
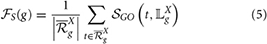
where 𝕃Xg is the set of all leaves of the GO DAG connected to all terms in Xg. The higher this score is the more informative the set of terms annotating the gene under consideration is.
2.1.3. Quantifying gene annotation consistency
In general, different annotations of a given protein are expected to be less diverse or more coherent (Defoin-Piatel et al., 2011) with minimum variability among them. In order to quantify the coherence among different annotations of a gene or protein, we compute the annotation consistency score 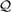 C(g) for a gene g as follows:
C(g) for a gene g as follows:
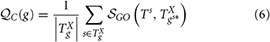
where Ts = {s} and TXgs* = TXg −{s} with TXg the set of GO terms in the ontology X annotating g and GO(Ts, TXgs*) is the semantic similarity between two sets of GO annotations, singleton Ts and TXgs*, and can be computed using any semantic similarity model (Mazandu and Mulder, 2013a). The gene annotation consistency score ranges between 0 and 1, and enables the detection of annotation inconsistencies. This score is set to 1 for a gene with one annotation and the lowest value of annotation consistency score is an indication of possible annotation error for the gene under consideration or alternatively it indicates that the gene is multi-functional at the molecular level or involved in several biological processes. Such gene annotations should be subjected to further investigation to check their accuracy.
2.2. Scoring Gene Annotation Matches and Mismatches Between Pipelines
Different annotation pipelines are likely to lead to different GO annotations for a given gene. This is due to several reasons (Dolan et al., 2005), including the GO structure itself, incomplete annotation and annotation errors. Difference in annotation for a gene or protein related to the GO structure or incomplete annotation is often a consequence of insufficient knowledge about either the protein being annotated or the term being used to annotate the protein under consideration. These differences and even those due to annotation error from manual annotation assignment can be resolved and corrected by the curator. However, if the annotation assignment has been inferred electronically, the annotation error may be hard to correct since the source of the error may not be local or the cause of the error is far from the point where it is being detected as a result of the propagation of annotation errors from protein databases (Devos and Valencia, 2000).
Currently, the level of gene or protein annotation matches or mismatches can be quantitatively scored using semantic similarity measures (Mazandu and Mulder, 2013a) by computing a semantic similarity score GO(Xp1, Xp2) between strict non-redundant sets of GO annotations Xp1 and Xp2 provided by different annotation pipelines for a protein p. Thus, the score of an annotation match ω(p) for a protein p is given by:
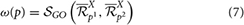
and the score of an annotation mismatch ω(p) is computed as follows:
for a given protein p, two annotation pipelines are completely in agreement if ω(p) = 1 or ω(p) = 0 and completely in disagreement if ω(p) = 1 or ω(p) = 0. The complete disagreement between two annotation pipelines about annotations assigned to a protein is an indication of an annotation error from one pipeline or a protein is multi-functional and the two pipelines under consideration are unable to identify multi-annotations or diversified annotations for multi-functional proteins, in which case further curation is required.
2.3. Comparing Different Annotation Pipelines
In this study, we use functional specificity, redundancy and consistency scores to quantitatively assess GO annotation quality of an annotation pipeline or dataset. The “optimal” pipeline or dataset is that producing good annotation quality scores, enabling comparison of different pipelines or datasets. In addition, when comparing different datasets, matches, mismatches and missing annotations are checked or identified. Here, we applied these different scores to human and fruitfly proteomes annotated by InterPro2GO and manual pipelines using the GO-universal metric (Mazandu and Mulder, 2012) as a semantic similarity model. However, this approach is applicable to any dataset and researchers can adapt it to any semantic similarity model. This is useful for curators and end-users of datasets to detect issues related to different datasets as quantitative quality assessment is crucial to the comparative functional genomics community.
2.4. Integrating GO Annotations and Filtering Gene Annotation Datasets
Integrating GO annotations from different pipelines or filtering gene annotation datasets consists essentially of reducing redundancy in the integrated dataset produced. There are two types of redundancy detected in a given genome or annotation data set. The first type, referred to as type I redundancy, is due to the “true path” rule in the hierarchical structure of the GO DAG according to which a child term contains all features of its ancestor terms. The second one, referred to as type II redundancy, is due to the GO evidence code or the source (reference) of the annotation. The type II redundancy occurs when the same annotation is generated by different pipelines (electronic or manual/experimental) for a given protein and this is mainly reflected in the size of the file storing the dataset produced. We illustrate these different types of redundancy on the protein E3 ubiquitin-protein ligase Topors (Q9NS56) with GO MF annotations retrieved from the GOA database and shown in Table 1. In this GO annotation set, the GO term DNA topoisomerase binding (GO:0044547) is a descendant of the term protein binding (GO:0005515), and thus contains all biological specifications of this ancestor and including this ancestor (GO:0005515) in the set produces a type I redundancy. The GO term ubiquitin-protein transferase activity (GO:0004842) or SUMO ligase activity (GO:0019789), for example, was assigned to this protein by different sources or by the same source using different methods and recorded in the annotation file. This yields a type II redundancy, which only reflects in the file storing these annotations but not in any analysis using these annotations.
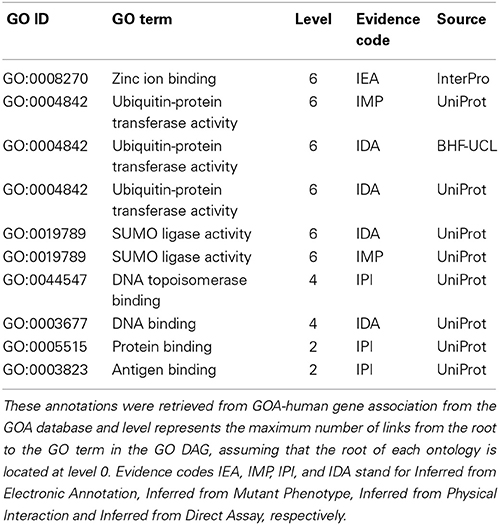
Table 1. GO annotations of the protein E3 ubiquitin-protein ligase Topors (Q9NS56).
Different sources may also annotate a protein with different GO terms but which are very similar in the context of the GO DAG, resulting in type I redundancy. One can annotate the protein to more specific terms in the same path or to sibling terms, i.e., sharing a direct parent. Lack of more complete biological knowledge about the protein under consideration results in the pipeline annotating it with a more general term. This redundancy issue can be solved through semantic similarity scores between terms in the GO-DAG, which are related when using a given ancestor term for its descendant specification (Mazandu and Mulder, 2012). Thus, the ancestor term is retained only when it is unable to directly contribute to its descendant term specification based on the semantic similarity between them, in which case the score is lower than the agreement level or threshold (Mazandu and Mulder, 2013a).
3. Results
In order to compare different annotation pipelines, to filter an annotation dataset or to integrate annotations from different pipelines, we define GO annotation quality assessment measures based on GO semantic similarity scores (see Materials and Methods Section). These quality assessment measures include the specificity or the level of detail of GO annotations used, the consistency of these annotations in terms of similarity between terms used, the non-redundancy of the annotation set and annotation match and mismatch scores. These measures are defined using an abstract semantic similarity measure, but here we are using the GO-universal metric (Mazandu and Mulder, 2012) when applied to the Drosophila melanogaster (fruitfly) and human proteomes with GO annotations manually assigned with the following GO evidence codes (Experimental category): Inferred from Experiment (EXP), Inferred from Direct Assay (IDA), Inferred from Physical Interaction (IPI), Inferred from Mutant Phenotype (IMP), Inferred from Genetic Interaction (IGI) and Inferred from Expression Pattern (IEP), and annotations originating from InterPro2GO, referred to as electronic inference.
For Drosophila melanogaster, available data from the UniProt database shows 60887 total entries (proteins coding genes), but only totals of 11046, 12048, and 10030 proteins are annotated with respect to the BP, MF, and CC ontologies, respectively, as extracted from the latest version of GOA UniProt (version 130), released on 15 April, 2014 (http://www.ebi.ac.uk/GOA/proteomes). From these totals, only 7299, 3495, and 4860 entries contain annotations manually assigned with respect the BF, MF, and CC ontologies, respectively. Among annotations inferred electronically, a total of 3195, 6055, and 2276 proteins have annotations inferred electronically using InterPro2GO mappings for BP, MF, and CC ontologies, respectively. Similarly, for 47592 total reviewed entries of human proteome from UniProt, 29844, 36177, and 31683 proteins are characterized with respect to the BP, MF, and CC ontologies, respectively, among which 6507, 8665, and 7416 entries contain annotations manually assigned, and 12422, 21989, and 8725 entries with annotations inferred electronically using InterPro2GO mappings.
3.1. Comparing Manual and Interpro Annotations
We analyzed the redundancy in the manual and InterPro2GO pipelines using different confidence levels (0.0, 0.3, and 0.7), the specificity, consistency of terms obtained from InterPro and manual pipelines, and annotation matches (mismatches) between these two pipelines using the GO-universal metric (Mazandu and Mulder, 2012) to compute similarity between terms in a given ontology. Results are shown in Table 2 for redundancy, Figures 1, 2 for annotation specificity and consistency, respectively, and Figure 3 for annotation matches in each organism under consideration.
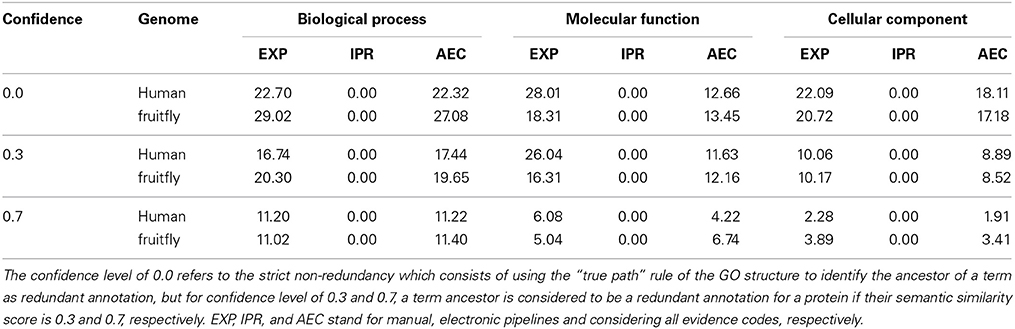
Table 2. Percentage redundancy of manual and electronic pipelines for different confidence levels.
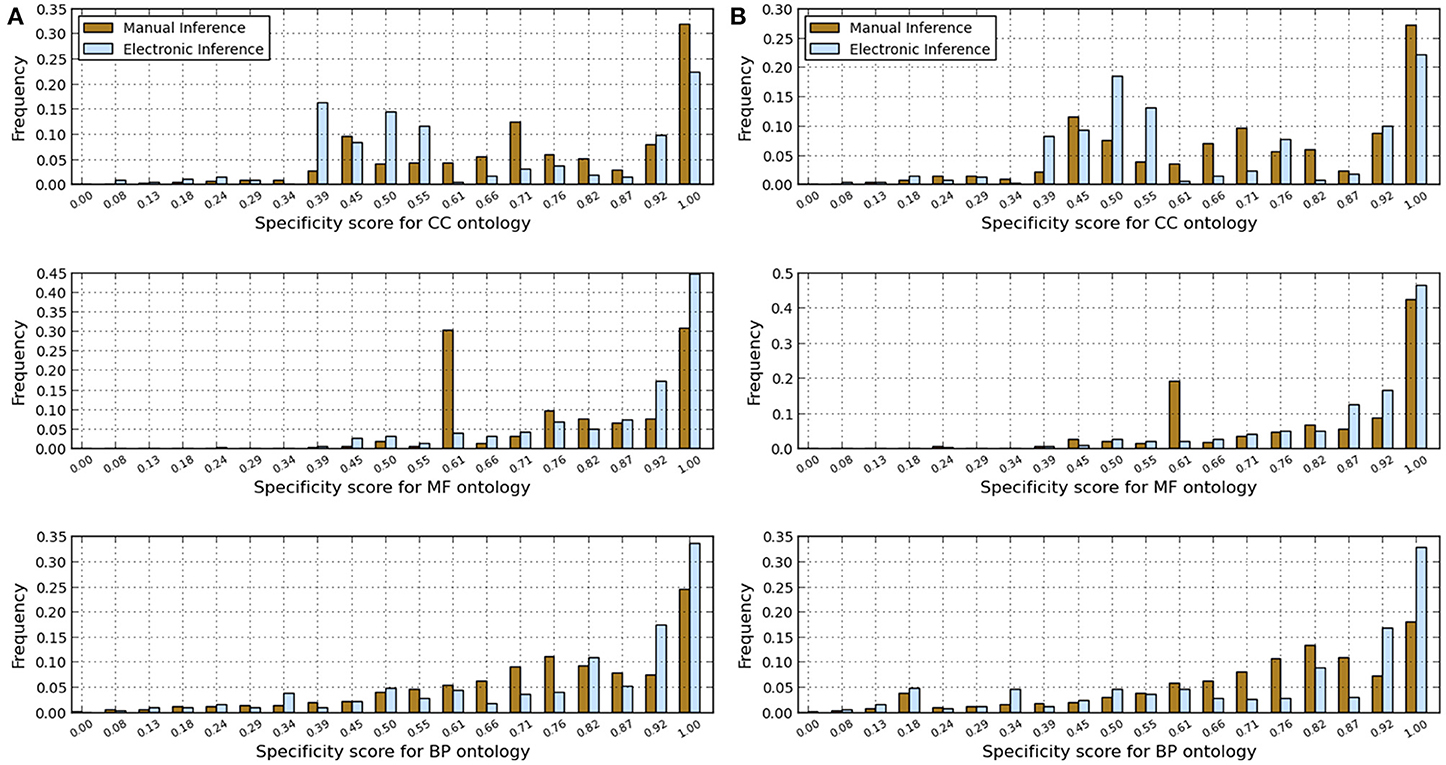
Figure 1. Comparison of annotations inferred manually and electronically in human and fruitfly genomes in terms of annotation specificity score computed using the GO-universal metric. (A) Human genome. (B) Fruitfly genome.
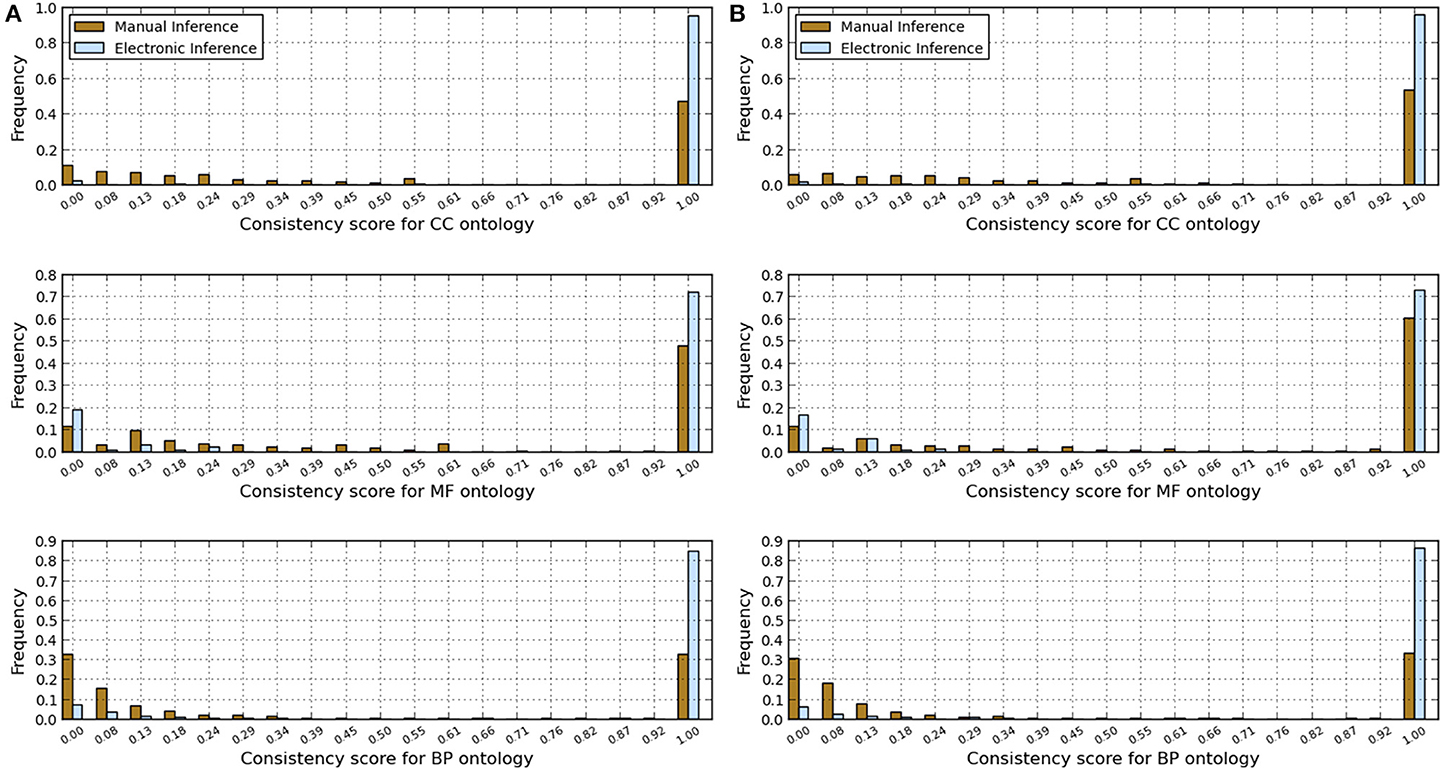
Figure 2. Comparison of annotations inferred manually and electronically in human and fruitfly genomes in terms of annotation consistency score computed using the GO-universal metric. (A) Human genome. (B) Fruitfly genome.
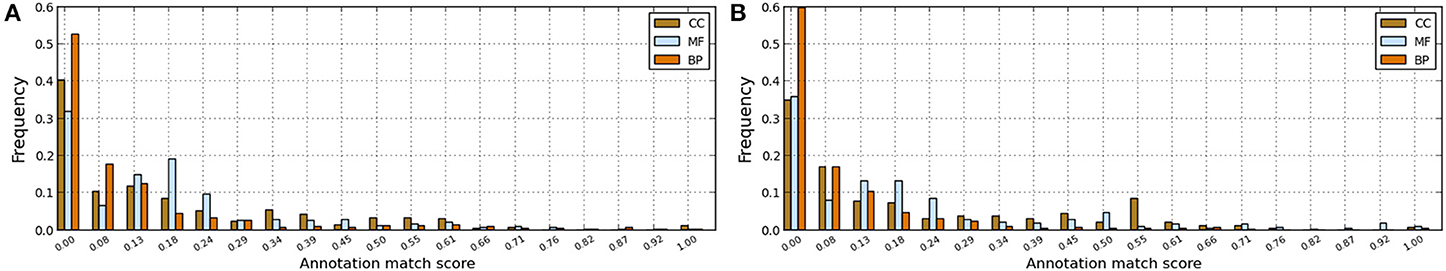
Figure 3. Annotation matches between manual and electronic pipelines for human and fruitfly genomes with scores computed using the GO-universal metric. (A) Human genome. (B) Fruitfly genome.
These results indicate that electronic inference (InterPro2GO) produces non-redundant and more consistent annotations than the manual pipeline for different ontologies (Table 2 and Figure 2), and this electronic pipeline also produces more specific annotations for BP and MF ontologies, but not for the CC ontology for which the manual pipeline provides more specific annotations compared to the electronic inference (Figure 1). However, it has been previously reported (Mazandu and Mulder, 2012) that electronic mapping annotations tend to be to higher level GO terms compared to manual pipelines. Furthermore, the lower annotation consistency displayed by the manual pipeline (Figure 2) may be due to the fact that this pipeline is better equipped to identify multi-annotations or diversified annotations for multi-functional proteins. This shows that the only effective way to assign annotations to uncharacterized proteins is the combination of manual and electronic inference (Mazandu and Mulder, 2011). These different results reveal that an optimal integration and filtration of annotations obtained from different pipelines may enable high-quality annotations for a genome annotation. For the two proteomes under consideration, the results in Table 2 show that using the GO-univesal metric for filtering these proteome annotations reduces redundancy in human annotation data up to 22.32, 12.66, and 18.11% and in fruitfly annotation data up to 27.08, 13.45, and 17.18% with respect to the BP, MF and CC ontologies, respectively.
In order to assess annotation coherence between electronic and manual pipelines, we compute functional similarity scores between annotations of a given protein annotated by these two pipelines. Thus, we used the formula (6) and the Best Match Average (BMA) measure (Mazandu and Mulder, 2012, 2013a,b) to score functional similarity between the two sets of annotations for a given protein and results are displayed in Figure 3. These results generally show low annotation matches for several proteins with annotations from the two pipelines (electronic and manual) in the two genomes under consideration for BP, MF, and CC ontologies. Possible reasons include protein mis-annotations, the inability of a given pipeline to identify an annotation for a given protein (missing annotations) and the use of more general GO terms for the manual pipeline and more specific terms for the electronic pipeline (see Figure 1). Once again, these results indicate that the integration of annotations from multiple pipelines can provide more accurate annotations and quality control for functional genomics data.
3.2. Assessing Fruitfly-Human Ortholog Functional Similarity
We compared annotations of ortholog proteins between fruitfly and human to check for annotation equivalence as ortholog proteins share common evolutionary processes and are thought to maintain similar functions (Mazandu and Mulder, 2012). Based on this principle, known as “ortholog” conjecture (Nehrt et al., 2011), electronic annotation pipelines, such as Ensembl Compara, arose in order to transfer annotations between ortholog proteins between different species. In this study, we check for matches and missing annotation for every ortholog protein pair. Ortholog protein pairs were retrieved from the Ensembl website (Flicek et al., 2010; Kinsella et al., 2011) at http://www.ensembl.org using biomart, and GO-association data were downloaded from the GOA site (Dimmer et al., 2012).
From the list of ortholog protein pairs, we have considered those pairs with high confidence according to Ensembl in lists of reviewed proteins (Swiss-Prot) for these two genomes from the UniProt database. We ended up with a list of 3346 ortholog protein pairs with 1988 and 2949 proteins in fruitfly and human proteomes (Ensembl uses a one to many ortholog relationship), respectively, and different results obtained are shown in Table 3 and Figure 4 for different ortholog annotation features and functional similarity scores. The main result in Table 3 is the missing annotations in both genomes for 371, 319, and 485 ortholog proteins in BP, MF and CC ontologies, respectively. This lack of annotations for ortholog proteins in one genome and characterized in another genome is partly to incomplete knowledge, and elicits the need for further curation of existing information about these specific organisms.
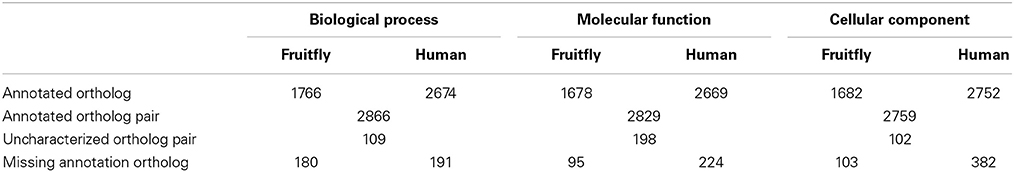
Table 3. General features of fruitfly-human ortholog proteins in terms total number of ortholog proteins with GO annotations in BP, MF, and CC ontologies for different genomes under consideration.
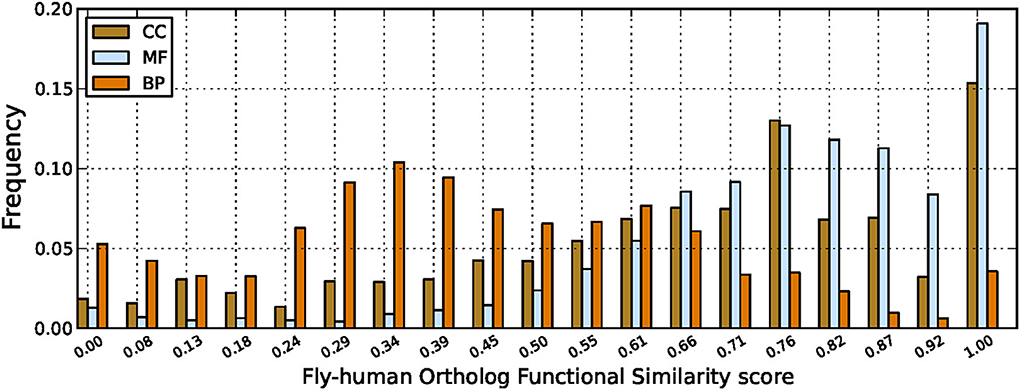
Figure 4. Fruitfly-human ortholog functional similarity scores. Comparing GO annotations for ortholog protein pairs between human and fruitfly genomes with scores computed using the GO-universal metric.
Results in Figure 4 show high functional similarity between protein orthologs as expected, especially for the MF and CC ontologies. Orthologs with very low functional similarity based on their GO annotations have also been detected, especially for the BP ontology, and this is not in agreement with the belief about function conservation between orthologs. There are several possible reasons for this, including protein mis-annotations, the use of more general GO terms for one and more specific terms for the other protein, or simply the lack of relevant biological knowledge about these proteins, thus requiring further curation of existing knowledge in these systems as pointed out previously. In particular, for the BP ontology describing broad biological goals for genes and proteins, this could be an indication of missing annotations in both orthologs as the differing terms may not be conflicting processes so it may be that the other terms are correct, but have just not yet been added, or they may be organism specific (Mazandu and Mulder, 2012).
3.3. Discussion
Worldwide DNA sequencing efforts have led to a rapid increase in sequence data in the public domain, but also the lack of functional annotations for many newly sequenced genes and their predicted proteins. Thus, several genome annotation pipelines were set up to assign or predict functions for the sequenced genes. Even though these annotation pipelines widely adopted the Gene Ontology (GO) as an efficient and standardized functional scheme, the specific procedures followed by each annotation pipeline varies. This leads to an increased heterogeneity created by different granularity of GO annotations across a genome or gene set, rendering curators' tasks tedious and challenging. In this study, we have introduced a large-scale approach that allows the integration of protein GO annotations from different pipelines using GO semantic similarity measures for efficient exploitation of biological knowledge embedded in the GO structure.
We have defined a set of annotation quality assessment measures using an abstract semantic similarity measure and producing a very general approach that can use any semantic similarity measure for filtering and integrating functional annotations from multiple pipelines. We have applied this approach to the manual and InterPro2GO annotation pipelines for the Drosophila melanogaster (fruitfly) and human proteomes for different ontologies. In the context of the Gene Ontology, several semantic similarity measures have been introduced and used in different biological applications. These measures are being used to set up novel bioinformatics approaches that enable the efficient use of the GO DAG structure in protein functional analysis, function prediction and assignment. Here, we used the GO universal metric as a semantic similarity measure and results showed that this integrative model may enable high-quality and accurate annotations for genome annotation, reducing redundancy and eliminating inconsistencies in the integrated annotation datasets for ease of comparative functional genomics.
We have assessed GO annotations manually assigned (manual pipeline) with the following GO evidence codes (Experimental category): Inferred from Experiment (EXP), Inferred from Direct Assay (IDA), Inferred from Physical Interaction (IPI), Inferred from Mutant Phenotype (IMP), Inferred from Genetic Interaction (IGI) and Inferred from Expression Pattern (IEP), and compared them to those originating from InterPro (InterPro2GO pipeline). Results revealed that, in general, the InterPro2GO (electronic) pipeline produces non-redundant and more consistent annotations than the manual pipeline due to the fact that the manual pipeline is possibly assigning diversified annotations to multi-functional proteins. This suggests that an efficient integration and filtration of annotations from different pipelines would enable high-quality non-redundant annotation in genome annotation. In the case of human and fruifly annotation datasets, an efficient integration of annotation pipelines can reduce redundancy up to 27 and 22% (see Table 2), respectively, and identify mismatches between some protein annotations from these pipelines (see Figure 3) and incoherence or missing annotations for some orthologs in the two proteomes under consideration (see Table 3 and Figure 4), requiring further curation of existing knowledge. Such an approach is useful for the comparative genomics community, simplifies tasks of curators and should advance comparative functional genomics research.
Author Contributions
Nicola J. Mulder conceived and supervised the project, and finalized the paper. Gaston K. Mazandu analyzed, designed, and implemented the method and wrote the paper. Gaston K. Mazandu and Nicola J. Mulder analyzed data and read and approved the final paper and Nicola J. Mulder approved the production of this paper.
Funding
This work has been supported by the National Research Foundation (NRF) in South Africa, University of Cape Town.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Any work dependent on open-source software owes debt to those who developed these tools. The authors thank everyone involved with free software, from the core developers to those who contributed to the documentation. Many thanks to the authors of the freely available libraries.
References
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Sayers, E. W. (2009). Genbank. Nucleic Acids Res. 37, D26–D31. doi: 10.1093/nar/gkn723
Defoin-Piatel, M., Hindle, M. M., Lysenko, A., Powers, S. J., Habash, D. Z., Rawlings, C. J., et al. (2011). AIGO: towards a unified framework for the Analysis and the Inter-comparison of GO functional annotations. BMC Bioinformatics 12:431. doi: 10.1186/1471-2105-12-431
Devos, D., and Valencia, A. (2000). Practical limits of function prediction. Proteins 41, 98–107. doi: 10.1002/1097-0134(20001001)41:1<98::AID-PROT120>3.0.CO;2-S
Dimmer, E. C., Huntley, R. P., Alam-Faruque, Y., Sawford, T., O'Donovan, C., and Martin, M. J. (2012). The UniProt-GO Annotation database in 2011. Nucleic Acids Res. 40, D565–D570. doi: 10.1093/nar/gkr1048
Dolan, M. E., Ni, L., Camon, E., and Blake, J. A. (2005). A procedure for assessing GO annotation consistency. Bioinformatics 21(Suppl. 1), i136–i143. doi: 10.1093/bioinformatics/bti1019
Fernández-Suárez, X. M., and Schuster, M. K. (2010). Using the Ensembl genome server to browse genomic sequence data. Curr. Protoc. Bioinformatics. Chapter 1:Unit 1.15. doi: 10.1002/0471250953.bi0115s16
Flicek, P., Aken, B. L., Ballester, B., Beal, K., Bragin, E., Brent, S., et al. (2010). Ensembl's 10th year. Nucleic Acids Res. 38, D557–D562. doi: 10.1093/nar/gkp972
GO-Consortium. (2012). The gene ontology: enhancements for 2011. Nucleic Acids Res. 40, D559–D564. doi: 10.1093/nar/gkr1028
Jain, E., Bairoch, A., Duvaud, S., Phan, I., Redaschi, N., Suzek, B. E., et al. (2009). Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics 10:136. doi: 10.1186/1471-2105-10-136
Kinsella, R. J., Kähäri, A., Haider, S., Zamora, J., Proctor, G., Spudich, G., et al. (2011). Ensembl biomarts: a hub for data retrieval across taxonomic space. Database 2011:bar030. doi: 10.1093/database/bar030
Mazandu, G. K., and Mulder, N. J. (2011). Using the underlying biological organization of the Mycobacterium tuberculosis functional network for protein function prediction. Infect. Genet. Evol. 12, 922–932. doi: 10.1016/j.meegid.2011.10.027
Mazandu, G. K., and Mulder, N. J. (2012). A topology-based metric for measuring term similarity in the Gene Ontology. Adv. Bioinformatics 2012:975783. doi: 10.1155/2012/975783
Mazandu, G. K., and Mulder, N. J. (2013a). DaGO-Fun: tool for Gene Ontology-based functional analysis using term information content measures. BMC Bioinformatics 14:284. doi: 10.1186/1471-2105-14-284
Mazandu, G. K., and Mulder, N. J. (2013b). Information content-based gene ontology semantic similarity approaches: toward a unified framework theory. BioMed Res. Int. 2013:292063. doi: 10.1155/2013/292063
Nehrt, N. L., Clark, W. T., Radivojac, P., and Hahn, M. W. (2011). Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS Comput. Biol. 7:e1002073. doi: 10.1371/journal.pcbi.1002073
Pruitt, K. D., Tatusova, T., and Maglott, D. R. (2005). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33(Suppl. 1), D501–D504. doi: 10.1093/nar/gki025
Sayers, E. W., Barrett, T., Benson, D. A., Bryant, S. H., Canese, K., Chetvernin, V., et al. (2009). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 37, D5–D15. doi: 10.1093/nar/gkp382
Spudich, G. M., and Fernández-Suárez, X. M. (2010). Touring Ensembl: a practical guide to genome browsing. BMC Genomics 11:295. doi: 10.1186/1471-2164-11-295
The Reference Genome Group of the Gene Ontology Consortium. (2009). The gene ontology's reference genome project: a unified framework for functional annotation across species. PLoS Comput. Biol. 5:e1000431. doi: 10.1371/journal.pcbi.1000431
Tweedie, S., Ashburner, M., Falls, K., Leyland, P., McQuilton, P., Marygold, S., et al. (2008). FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 37, D555–D559. doi: 10.1093/nar/gkn788
Keywords: functional annotation, Gene Ontology annotation, annotation pipeline, manual annotation, electronic annotation
Citation: Mazandu GK and Mulder NJ (2014) The use of semantic similarity measures for optimally integrating heterogeneous Gene Ontology data from large scale annotation pipelines. Front. Genet. 5:264. doi: 10.3389/fgene.2014.00264
Received: 30 May 2014; Accepted: 18 July 2014;
Published online: 06 August 2014.
Edited by:
Prashanth Suravajhala, Bioclues.org, DenmarkReviewed by:
David John Studholme, University of Exeter, UKMikhail P. Ponomarenko, Institute of Cytology and Genetics of Siberian Branch of Russian Academy of Sciences, Russia
Copyright © 2014 Mazandu and Mulder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola J. Mulder, Computational Biology Group, Department of Clinical Laboratory Sciences, Institute of Infectious Disease and Molecular Medicine, University of Cape Town Faculty of Health Sciences, Anzio Rd., Observatory, Cape Town 7925, South Africa e-mail: nicola.mulder@uct.ac.za