 Liliya Euro1
Liliya Euro1 Svetlana Konovalova1Jorge Asin-Cayuela2
Svetlana Konovalova1Jorge Asin-Cayuela2 Már Tulinius3Helen Griffin4Rita Horvath4
Már Tulinius3Helen Griffin4Rita Horvath4 Robert W. Taylor5Patrick F. Chinnery4Ulrike Schara6
Robert W. Taylor5Patrick F. Chinnery4Ulrike Schara6 David R. Thorburn7
David R. Thorburn7 Anu Suomalainen1,8
Anu Suomalainen1,8 Joseph Chihade9
Joseph Chihade9 Henna Tyynismaa1,10*
Henna Tyynismaa1,10*- 1Research Programs Unit, Molecular Neurology, Biomedicum Helsinki, University of Helsinki, Helsinki, Finland
- 2Department of Clinical Chemistry, University of Gothenburg, Sahlgrenska University Hospital, Gothenburg, Sweden
- 3Department of Pediatrics, Queen Silvia Children’s Hospital, University of Gothenburg, Gothenburg, Sweden
- 4Institute of Genetic Medicine, Wellcome Trust Centre for Mitochondrial Research, Newcastle University, Newcastle upon Tyne, UK
- 5Institute of Neuroscience, Wellcome Trust Centre for Mitochondrial Research, Newcastle University, Newcastle upon Tyne, UK
- 6Department of Neuropediatrics, Developmental Neurology and Social Pediatrics, University of Essen, Essen, Germany
- 7Murdoch Childrens Research Institute, Royal Children’s Hospital and Department of Paediatrics, University of Melbourne, Melbourne, VIC, Australia
- 8Department of Neurology, Helsinki University Central Hospital, Helsinki, Finland
- 9Department of Chemistry, Carleton College, Northfield, MN, USA
- 10Department of Medical Genetics, Haartman Institute, University of Helsinki, Helsinki, Finland
The accuracy of mitochondrial protein synthesis is dependent on the coordinated action of nuclear-encoded mitochondrial aminoacyl-tRNA synthetases (mtARSs) and the mitochondrial DNA-encoded tRNAs. The recent advances in whole-exome sequencing have revealed the importance of the mtARS proteins for mitochondrial pathophysiology since nearly every nuclear gene for mtARS (out of 19) is now recognized as a disease gene for mitochondrial disease. Typically, defects in each mtARS have been identified in one tissue-specific disease, most commonly affecting the brain, or in one syndrome. However, mutations in the AARS2 gene for mitochondrial alanyl-tRNA synthetase (mtAlaRS) have been reported both in patients with infantile-onset cardiomyopathy and in patients with childhood to adulthood-onset leukoencephalopathy. We present here an investigation of the effects of the described mutations on the structure of the synthetase, in an effort to understand the tissue-specific outcomes of the different mutations. The mtAlaRS differs from the other mtARSs because in addition to the aminoacylation domain, it has a conserved editing domain for deacylating tRNAs that have been mischarged with incorrect amino acids. We show that the cardiomyopathy phenotype results from a single allele, causing an amino acid change R592W in the editing domain of AARS2, whereas the leukodystrophy mutations are located in other domains of the synthetase. Nevertheless, our structural analysis predicts that all mutations reduce the aminoacylation activity of the synthetase, because all mtAlaRS domains contribute to tRNA binding for aminoacylation. According to our model, the cardiomyopathy mutations severely compromise aminoacylation whereas partial activity is retained by the mutation combinations found in the leukodystrophy patients. These predictions provide a hypothesis for the molecular basis of the distinct tissue-specific phenotypic outcomes.
Introduction
Mitochondrial protein synthesis produces key subunits of the oxidative phosphorylation (OXPHOS) complexes that generate the majority of the ATP for our cells. The protein synthesis in mitochondria requires careful synchrony between two genomes and cellular compartments, because the protein components of its machinery are encoded by the nuclear genome and imported into mitochondria, whereas the RNA components (2 rRNAs and 22 tRNAs) are encoded by mitochondrial DNA (Hallberg and Larsson, 2014). Among the necessary proteins are the aminoacyl-tRNA synthetases (ARSs) that recognize specific tRNAs and charge them with cognate amino acids, thus contributing to the initiation and accuracy of the protein synthesis. Nineteen ARSs function in mammalian mitochondria (mtARSs), one for each amino acid, except glutamine, which is charged by an alternative pathway (Nagao et al., 2009).
Defects of mitochondrial translation are a major cause of diseases that lead to mitochondrial OXPHOS dysfunction (Rotig, 2011). Since 2007, the mtARS genes have been recognized as a new group of disease genes affecting mitochondrial protein synthesis (Konovalova and Tyynismaa, 2013; Diodato et al., 2014a; Schwenzer et al., 2014). Proteins need to be synthesized in mitochondria in all cell types, with the exception of red blood cells, and thus the tissue-specificity of the phenotypes caused by mutations in the different mtARS genes has been unexpected. Now that pathogenic mutations have been reported in 15 genes encoding mtARSs (and in the two genes GARS and KARS encoding dual localized synthetases for cytoplasmic and mitochondrial translation), we can, however, conclude that the majority of the mtARS defects affect the central nervous system. Nevertheless, the range of CNS phenotypes is also remarkably wide, affecting specific neurons in many cases, and including patients with infantile-, early- and late-onset of disease.
Central nervous system involvement as the main or sole clinical presentation is found in defects of the mtARSs for aspartate (DARS2; Scheper et al., 2007), glutamate (EARS2; Steenweg et al., 2012), methionine (MARS2; Bayat et al., 2012), phenylalanine (FARS2; Elo et al., 2012), arginine (RARS2; Edvardson et al., 2007), valine (VARS2; Diodato et al., 2014b), threonine (TARS2; Diodato et al., 2014b), proline (PARS2; Sofou et al., 2015), asparagine (NARS2; Sofou et al., 2015), and cysteine (CARS2; Hallmann et al., 2014). Exceptions are the mutations in the genes for histidyl-tRNA (HARS2) and leucyl-tRNA (LARS2) synthetases that cause sensorineural hearing loss and ovarian dysgenesis (Pierce et al., 2011, 2013), for tyrosyl-tRNA synthetase (YARS2) in a combination of myopathy and anemia (Riley et al., 2010), and for seryl-tRNA synthetase (SARS2; Belostotsky et al., 2011) in HUPRA, for hyperuricemia, pulmonary hypertension, renal failure in infancy, and alkalosis, syndrome. A common feature for all the defects mentioned above is that so far each of them has been described in one tissue-specific phenotype or in one syndrome.
We identified mutations in the gene for mitochondrial alanyl-tRNA synthetase (AARS2) in a phenotype that differs from the other reported mtARS defects, being clinically a fatal early onset cardiomyopathy (Gotz et al., 2011). Our patients died within the first 10 months of life with severe cardiomyopathy, having a significant reduction in OXPHOS complexes in the heart and partially also in the skeletal muscle and brain. Two other studies have later reported patients with fatal infantile-onset AARS2 mutations (Calvo et al., 2012; Taylor et al., 2014). Although cardiomyopathy is not a usual feature of the other mtARS defects, it is typical in early infancy in many other mitochondrial protein synthesis defects, such as in those caused by mutations in genes encoding for mitoribosome subunits MRPL44 and MRPL3 (Galmiche et al., 2011; Carroll et al., 2013), in tRNA-modifiers MTO1 (Ghezzi et al., 2012) and GTPBP3 (Kopajtich et al., 2014), and in RNA processing enzyme ELAC2 (Haack et al., 2013). Remarkably, a recent study reported AARS2 mutations in patients who developed leukoencephalopathy and ovarian failure, with onset from childhood to adulthood, and no signs of a cardiomyopathy (Dallabona et al., 2014). AARS2 has thus been an example of mutations in the same mtARS gene leading to two very different diseases with dissimilar tissue involvement.
The mitochondrial alanyl-tRNA (mtAlaRS) synthetase encoded by AARS2 differs itself from the other mtARSs, because it has in addition to the aminoacylation domain a conserved editing domain for deacylating mischarged tRNAs (Beebe et al., 2003; Guo et al., 2009b; Gotz et al., 2011). The mischarging results from the inability of the enzyme’s aminoacylation domain to discriminate alanine from two other small amino acids, serine and glycine. In addition, the synthetase has a C-terminal domain, which is required for cooperative binding of both the aminoacylation and editing domains to the tRNA (Guo et al., 2009a). We have previously hypothesized that in our cardiomyopathy patients, the AARS2 mutation resulting in R592W change, which is located in the editing domain of the synthetase, might interfere with tRNA binding, thus preventing editing, and resulting in mistranslation (Gotz et al., 2011). Based on homology modeling, we now present structural predictions of all AARS2 missense mutations in the two different phenotypes, and show that the tissue-specific defects affect distinct domains of the synthetase but compromise the aminoacylation with different outcomes. Furthermore, we show that the AARS2 cardiomyopathy mutation (R592W) is a common founder mutation and carried by all the identified patients with the severe infantile-onset phenotype.
Materials and Methods
Haplotype Analysis
Nine single-nucleotide polymorphisms (SNPs) flanking AARS2 gene were selected for haplotype analysis, based on the haplotype blocks presented previously (Taylor et al., 2014). Genomic DNA was amplified by PCR using flanking oligonucleotides (sequences available on request) and Sanger sequenced.
Structure Prediction
For human mtAlaRS homology modeling we used the recently solved structure of full-length alanyl-tRNA synthetase (AlaRS) from Archaeoglobus fulgidus as a template. This bacterial AlaRS is a homodimer with one subunit crystallized with bound tRNA substrate and an alanyl-adenylate analog in so-called “closed” conformation, while the second subunit is in “open,” substrate free conformation (Naganuma et al., 2014; PDB id 3WQY). “Closed” conformation of human mtAlaRS was modeled using A chain in solved A. fulgidus AlaRS structure (3WQY_A), and the “open” substrate free form was modeled using B chain of the same structure (3WQY_B). Sequence alignment of human mtAlaRS (985 aa) and template A. fulgidus AlaRS (906 aa) was done using similarity matrix BLOSUM62. Multiple sequence alignments of human mtAlaRS and template sequence with a number of prokaryote and eukaryote homologs were done using PROMALS3D server. Obtained alignment was then submitted for homology modeling to the SWISS-MODEL server. Modeling was performed using alignment mode using either 3WQY_A or 3WQY_B structure as template. Resulting “open” and “closed” structures were analyzed using Discovery Studio v4.1 (Accelrys) software. Overall architecture and arrangement of secondary structure elements in the obtained models was similar to those in the template structures except for two loops, encompassing residues 211–231 and 540–561, which were omitted in the analysis. Docking of the bacterial tRNAAla and alanyl-adenylate into the model was done after superimposition of template chain A with the modeled human mtAlaRS using Discovery Studio v4.1 (Accelrys) software.
Results
The AARS2 c.1774C > T (R592W) is a Founder Mutation and Carried by Every Patient with the Cardiomyopathy Phenotype
Seven patients with infantile-onset cardiomyopathy and AARS2 mutations have been reported (Gotz et al., 2011; Taylor et al., 2014) and one who died in utero was described to have myopathy, hypotonia and multiple fractures (Calvo et al., 2012). In addition, we report here three additional patients of Swedish or German origin.
The Swedish patient was a boy born as the second child of healthy, non-consanguineous parents. An older sibling was healthy. The pregnancy and delivery were normal. Birth weight was 3990 g. The Apgar Scores were 2, 5, and 7 at 1, 5, and 10 min. At birth the boy was floppy, had decreased movements and presented breathing difficulties needing ventilatory treatment from the age of 1 h. He was diagnosed with a hypertrophic cardiomyopathy involving both the left and right ventricles, and with lactic acidosis. He was referred for further investigation at the age of 13 days. A skeletal muscle biopsy performed at the age of 14 days showed a mitochondrial myopathy with ragged red fibers, which were cytochrome-c oxidase negative, and with increased lipid droplets. Investigation of isolated skeletal muscle mitochondria showed decreased activities of complexes I-IV. The cardiomyopathy was progressive and the boy died at the age of 7 weeks of cardio-respiratory failure. An autopsy was performed and showed a severely hypertrophic heart that weighed 60 g.
The German patients were twin girls of healthy, non-consanguineous parents. The pregnancy and delivery were normal. Birth weights of the first and second twin were 1890 g and 2150 g, respectively. The Apgar Scores were 9, 10, 10, and 6, 8, 9, respectively. Onset of disease in the first twin was at birth with breathing difficulties needing ventilatory treatment from the age of 8 h. The second twin was stable and did not need ventilatory support. The first twin was diagnosed with a hypertrophic cardiomyopathy involving mostly the left ventricle and with lactic acidosis at the age of 8 weeks. A skeletal muscle biopsy performed at the age of 11 weeks showed a mitochondrial myopathy with cytochrome-c oxidase negative fibers and perimysial fibrosis as well as reduced activity of coenzyme Q. Investigation of respiratory chain complexes disclosed reduced activities for complexes I–IV. The cardiomyopathy was progressive and the girl died at the age of 16 weeks of cardio-respiratory failure. The symptoms in the second twin started a few weeks later and showed the same clinical course, she died at the age of 20 weeks.
Out of these eleven patients six were homozygous for the c.1774C > T (R592W) mutation and five had compound heterozygous AARS2 mutations with each of them carrying the same c.1774C > T (R592W) mutation in one allele (Table 1).
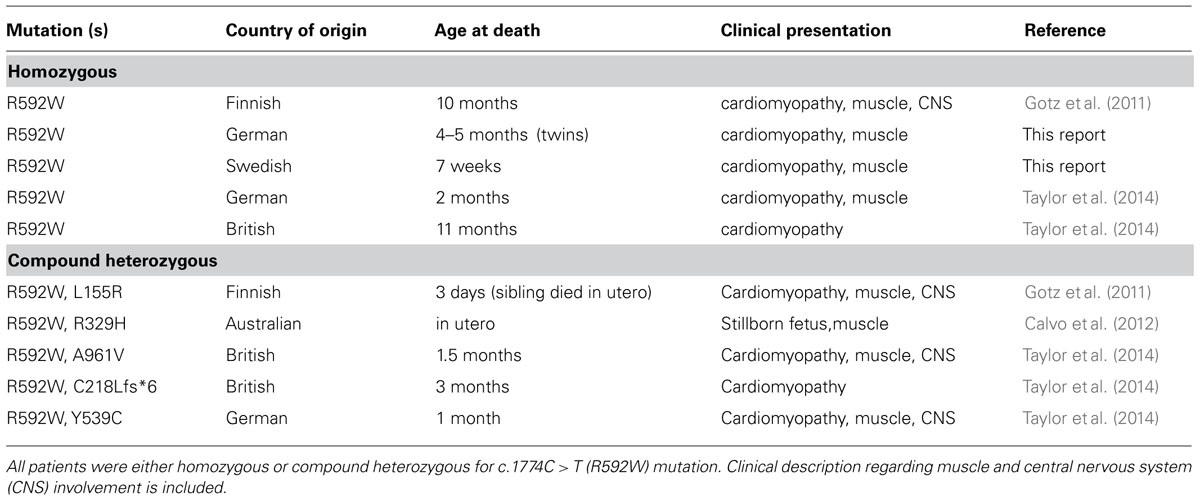
TABLE 1. List of all patients reported with mutations in AARS2 that presented with infantile-onset cardiomyopathy.
Patients with British or German origin were recently suggested to share a common disease haplotype for R592W (Taylor et al., 2014). We present here the haplotypes surrounding this mutation for patients from Finland, Sweden, and Australia in comparison to the British and German patients, which suggest that the shared mutation described in each AARS2 cardiomyopathy patient originates from a common founder (Figure 1).
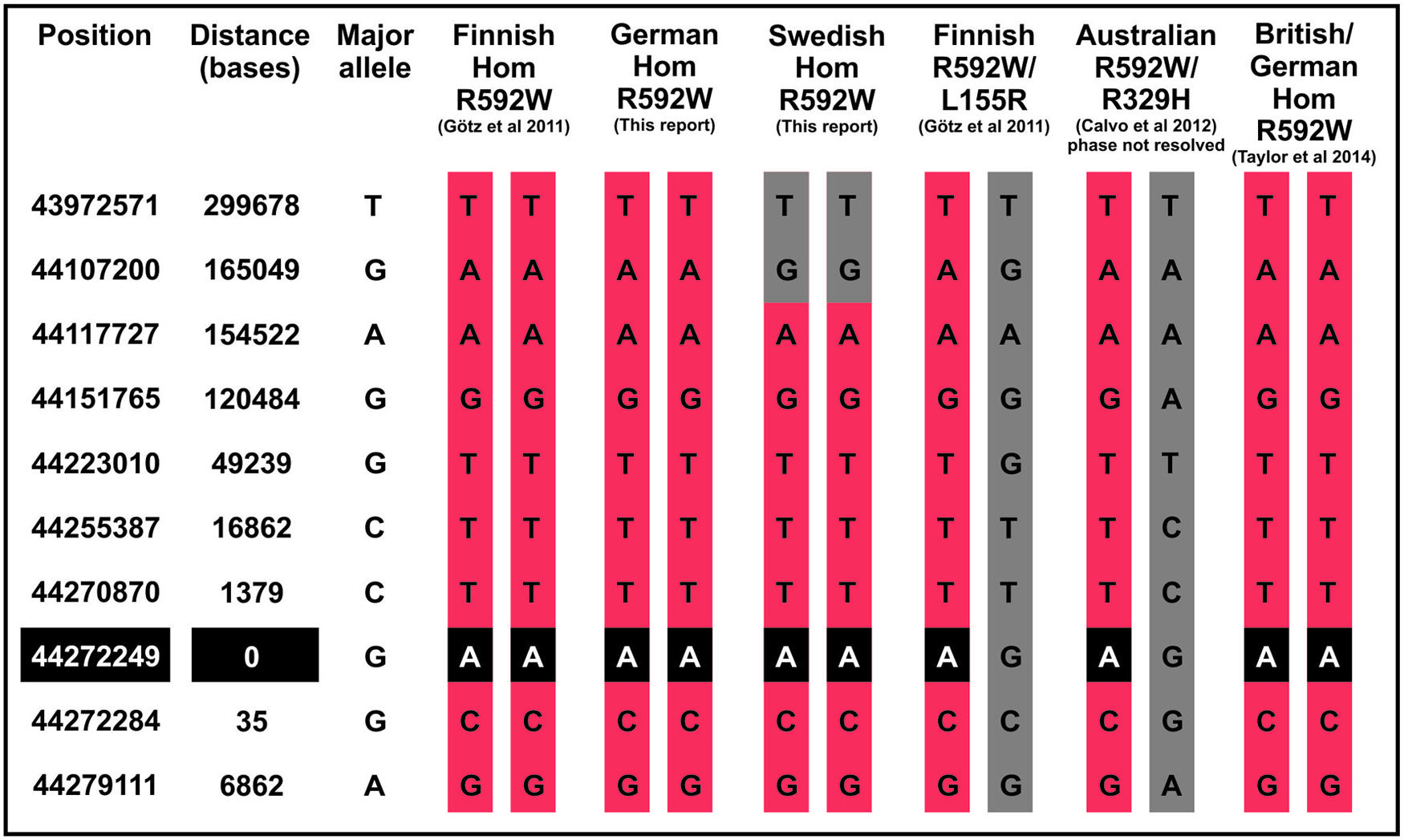
FIGURE 1. Patient haplotypes around the AARS2 mutation. The haplotype of the Swedish patient shows recombination 165 kilobases upstream of the mutation site.
Carriers of the c.1774C > T (R592W) mutation can be found in exome sequencing databases of different populations (Table 2), with the highest minor allele frequency of 0.0006 in Finland, where the first AARS2 cardiomyopathy patients were identified.
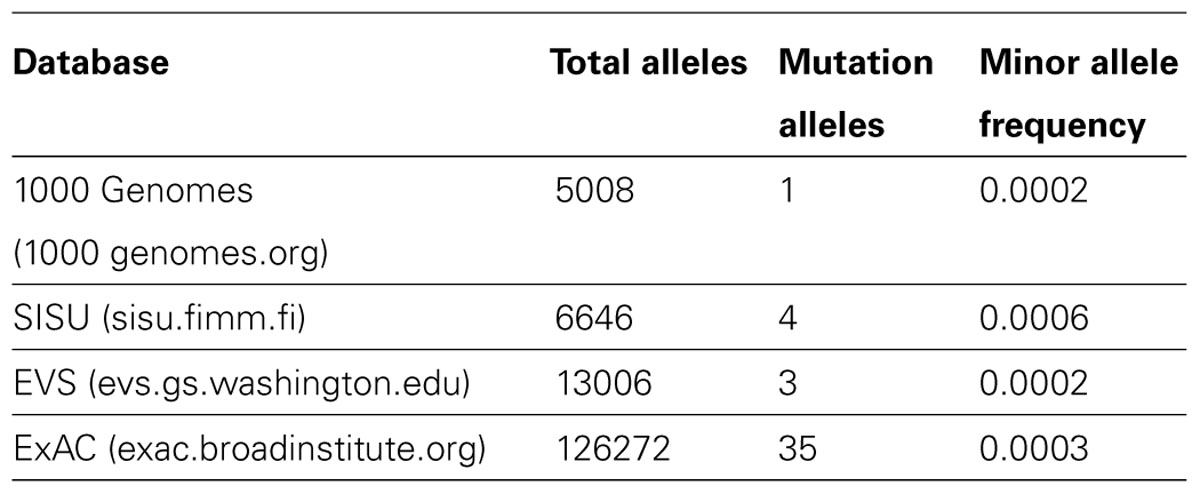
TABLE 2. Allele frequency of the mutation leading to amino acid change R592W in different exome/genome databases. SISU and 1000 Genomes contribute to the larger ExAC database.
The Distribution of the Mutated Amino Acids in Cardiomyopathy and Leukodystrophy Suggests Differential Molecular Pathogenesis
The R592W amino acid change locates to the editing domain of the mtAlaRS and we initially hypothesized that it causes mitochondrial mistranslation, which can be expected to harm OXPHOS assembly and function by amino acid misincorporation in the subunits that are synthesized by mitoribosomes (Gotz et al., 2011). The new AARS2 mutations reported in patients with leukodystrophy seemed, at first glance, to be scattered along the entire AARS2 gene, with some located in the aminoacylation domain and some in the editing domain (Dallabona et al., 2014). All of the leukodystrophy patients had compound heterozygous mutations. To clarify the range and localization of the mutations in both phenotypes, we analyzed them in the actual combinations in which they were found in the patients (Figure 2). The cardiomyopathy patients were either homozygous for the editing domain mutation R592W or had it in combination with a mutation located in the aminoacylation, editing or the C-terminal domain or with a truncating mutation. Mutations that cause premature termination or frameshift can be expected to be total loss-of-function alleles with no stable synthetase present. The mutation spectrum was very different for the leukodystrophy patients because these presented combinations of two missense mutations in the aminoacylation domain or an aminoacylation domain missense mutation with a truncating mutation, with one exception, which was a combination of missense mutations in the aminoacylation domain and the C-terminal domain. Thus none of the missense mutations in leukodystrophy patients were in the editing domain, which was in striking contrast to all of the cardiomyopathy patients (Figure 2).
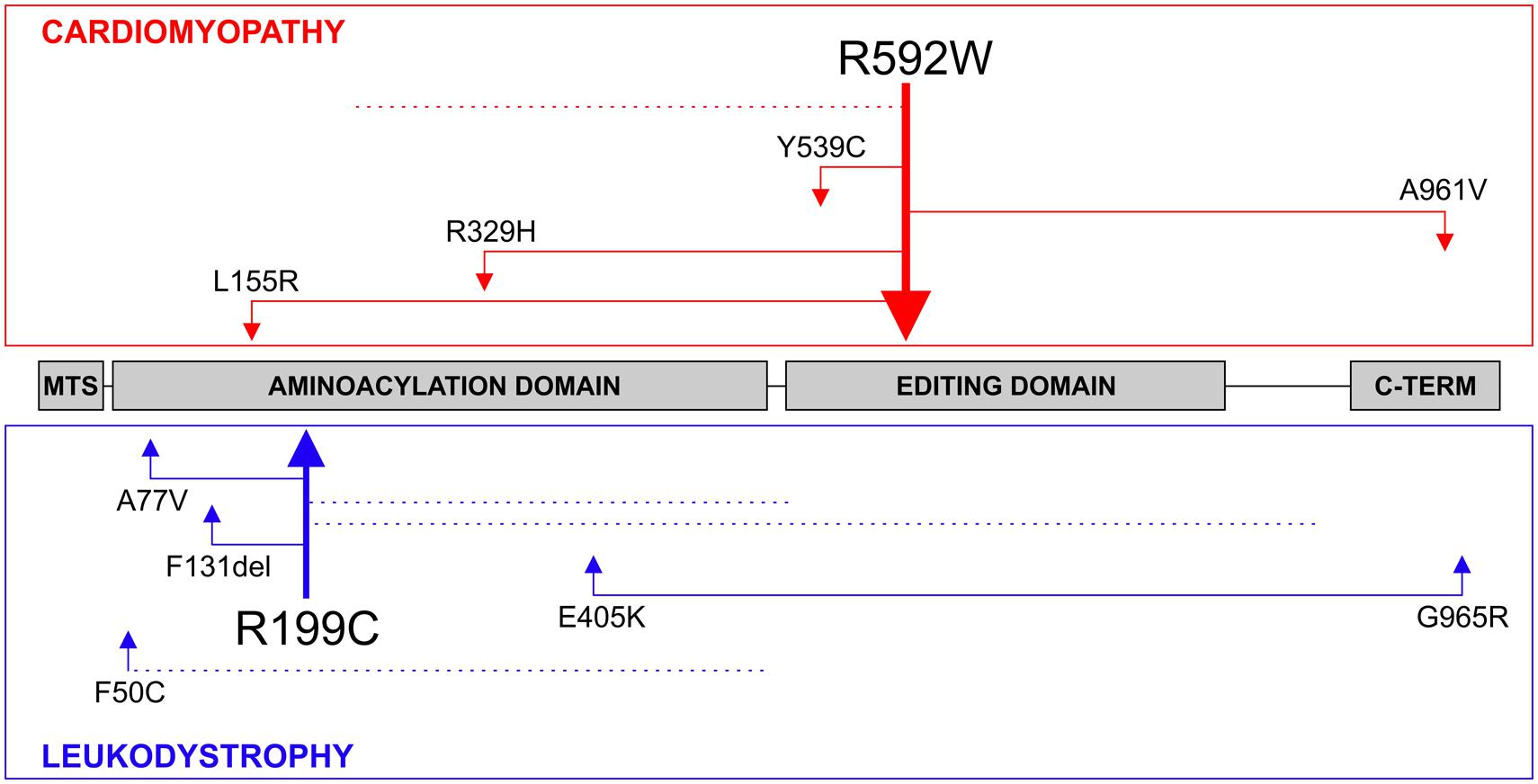
FIGURE 2. AARS2 mutations. The compound heterozygous mutations found in patients with cardiomyopathy or leukodystrophy are illustrated. Truncating mutations that are predicted to destabilize the entire synthetase are marked with dotted lines. The thicker arrows demonstrate recurring mutations. The AARS2 mutation leading to the R592W amino acid change has been the only one identified in a homozygous state.
Overview of the Structural Model of Human mtAlaRS
The recent crystal structure of a full-length length AlaRS (Naganuma et al., 2014) made possible our current complete structural model of the human mtAlaRS, which includes the arrangement of all three domains: aminoacylation, editing and C-terminal. The human mtAlaRS and template A. fulgidus AlaRS sequences shared overall 17.5% identity and 30.9% similarity (Figure 3). Domain-specific sequence identity and similarity was 17.5 and 31.2% for the aminoacylation domain, 25.0 and 39.8% for the editing domain and 14.7 and 26.5% for the C-terminal domain, respectively. Each domain can be further divided into two subdomains: the aminoacylation domain contains subdomains for aminoacylation and tRNA recognition, the editing domain consists of β-barrel and editing core, and the C-terminal or so-called C-Ala domain consists of helical and globular subdomains (Figure 4).
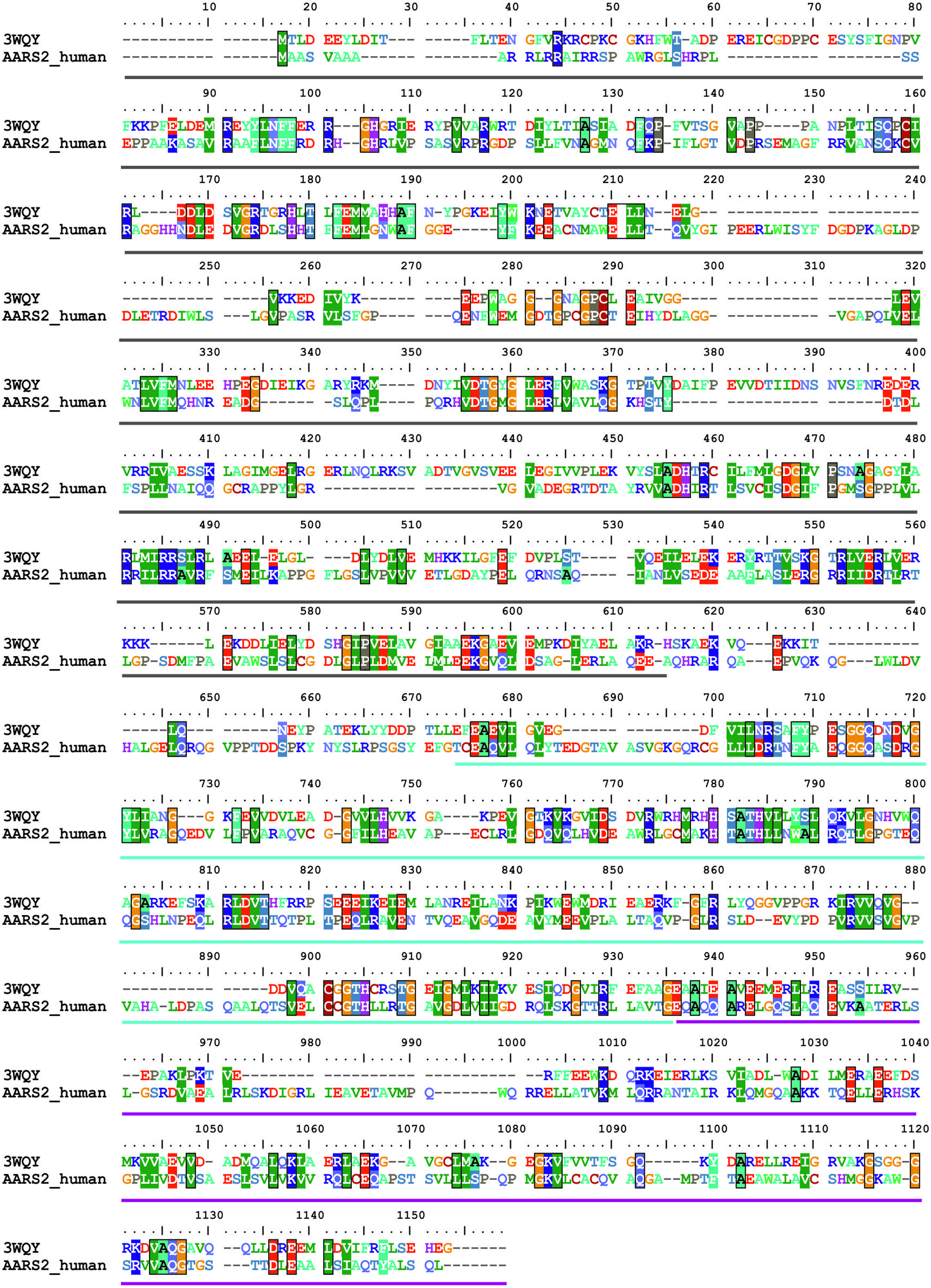
FIGURE 3. Sequence alignment between human mtAlaRS and AlaRS from A. fulgidus (3WQY). The alignment was extracted from multiple sequence alignment of AlaRS homologs from eukaryotes and prokaryotes using BioEdit software. The aminoacylation domain is marked with dark gray bar, the editing domain with cyan, and the C-terminal domain with magenta bars. Identical/similar amino acid residues in the same position are marked with colored boxes.
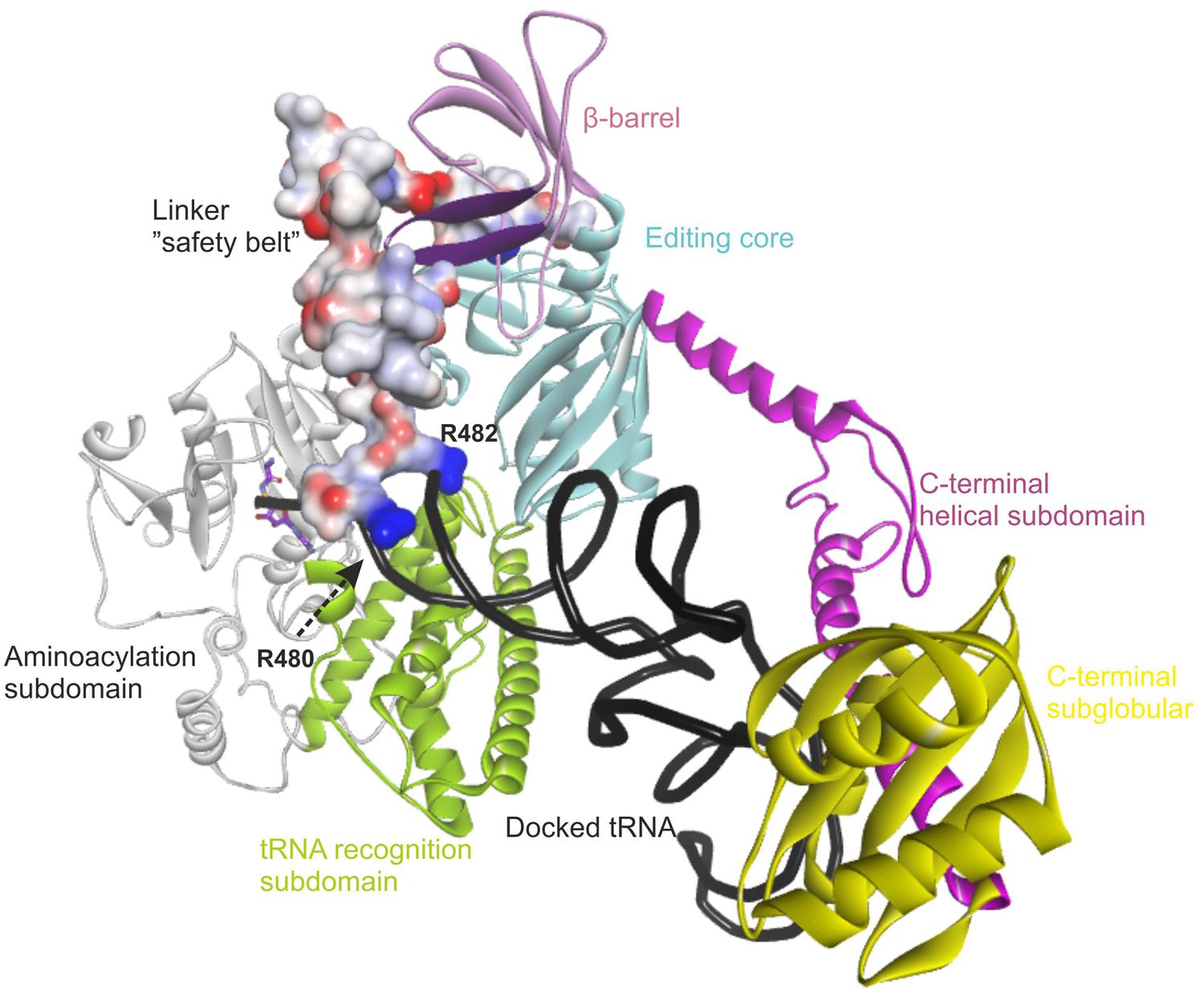
FIGURE 4. Overview of the modeled structure of human AlaRS. Aminoacylation subdomain (24–312 aa) is in silver, tRNA recognition subdomain (313–477 aa) of aminoacylation domain is in green, linker (“safety belt” – 478–529 aa) between tRNA recognition and editing domain is presented as solvent interpolated charge surface, β-barrel (530–621 aa) and editing core (622–783 aa) subdomains of editing domain are in pink and cyan, respectively. C-Ala domain or C-terminal domain is predicted to have helical (784–874 aa in magenta) and globular (875–985 aa in yellow) subdomains. R480 and R482 from the linker are predicted to stabilize bound tRNA within the aminoacylation domain. Backbone of docked tRNA is shown in dark gray. Docked alanyl-adenylate in the aminoacylation site is shown as stick model with carbon atoms labeled with magenta. Part of β-barrel subdomain (592–604 aa) of the editing domain proposed to interact with the linker is marked with purple (see text).
Since the template structure of AlaRS of A. fulgidus was resolved as a dimer, with one subunit in a substrate bound “closed” state and the other in a substrate free “open” state (Naganuma et al., 2014), it was possible to use bacterial tRNAAla and alanyl-adenylate bound to the template structure (chain A in 3WQY) for docking into our model and explore intermolecular interactions within the tertiary complex (Figure 4). The homology modeling suggests that human mtAlaRS shares with A. fulgidus AlaRS the unique structural features inherent for all AlaRSs. Firstly, it has a large tRNA-binding cavity whereas other ARSs adopt cognate tRNAs on their surface. Secondly, all three domains are involved in the binding of tRNA undergoing aminoacylation. Previous studies have shown that tRNA binding and the rate of aminoacylation are enhanced by the C-terminal domain as it has the highest tRNA binding affinity (Guo et al., 2009a). The structure of A. fulgidus AlaRS revealed that the C-terminal globular subdomain holds the elbow of the L-shaped tRNA while the acceptor stem is fixed within the aminoacylation domain by concerted binding to the so-called “safety belt” (the linker between aminoacylation and editing domains), and to the tRNA recognition subdomain and the surface of editing core (Naganuma et al., 2014). We predict the same type of tRNA binding for aminoacylation also in the modeled human mtAlaRS (Figure 4). Close inspection of the A. fulgidus AlaRS structure as well as the modeled tertiary complex of mtAlaRS revealed that the “safety belt” folds along the surface of the β-barrel in the editing domain and establishes different interactions with its surface exposed residues in “open” and “closed” states (Figure 5). This suggests that the β-barrel may serve as a “buckle” for fastening the “safety belt” when the tRNA is bound in the aminoacylation site. In human mtAlaRS the interacting surfaces of the linker and β-barrel are comprised of different residues than in the template, but comparison of “open” and “closed” conformations shows rearrangements in their interactions upon tRNA binding (Figure 6). Interestingly, the linker has an amino acid sequence stretch, L494-Q505, which is highly conserved between mitochondrial homologs (Figure 6). Modeled structure shows that L502 from this stretch is involved in hydrophobic interactions predominantly with residues from β-hairpin (R592–A604) of the β-barrel. Since this motif is also very conserved between mitochondrial homologs and oriented toward the linker it is likely that these two fragments extend their interaction by establishing transient contacts upon sliding of the linker on the surface of the β-barrel during transition from “open” to “closed” conformations of the enzyme. Therefore, we propose that the editing domain of mtAlaRS, in addition to its proofreading activity of deacylating mischarged tRNAs, may also be involved in the aminoacylation activity of the enzyme by being involved in the binding of tRNAAla for aminoacylation and stabilizing position of its acceptor stem within the catalytic site.
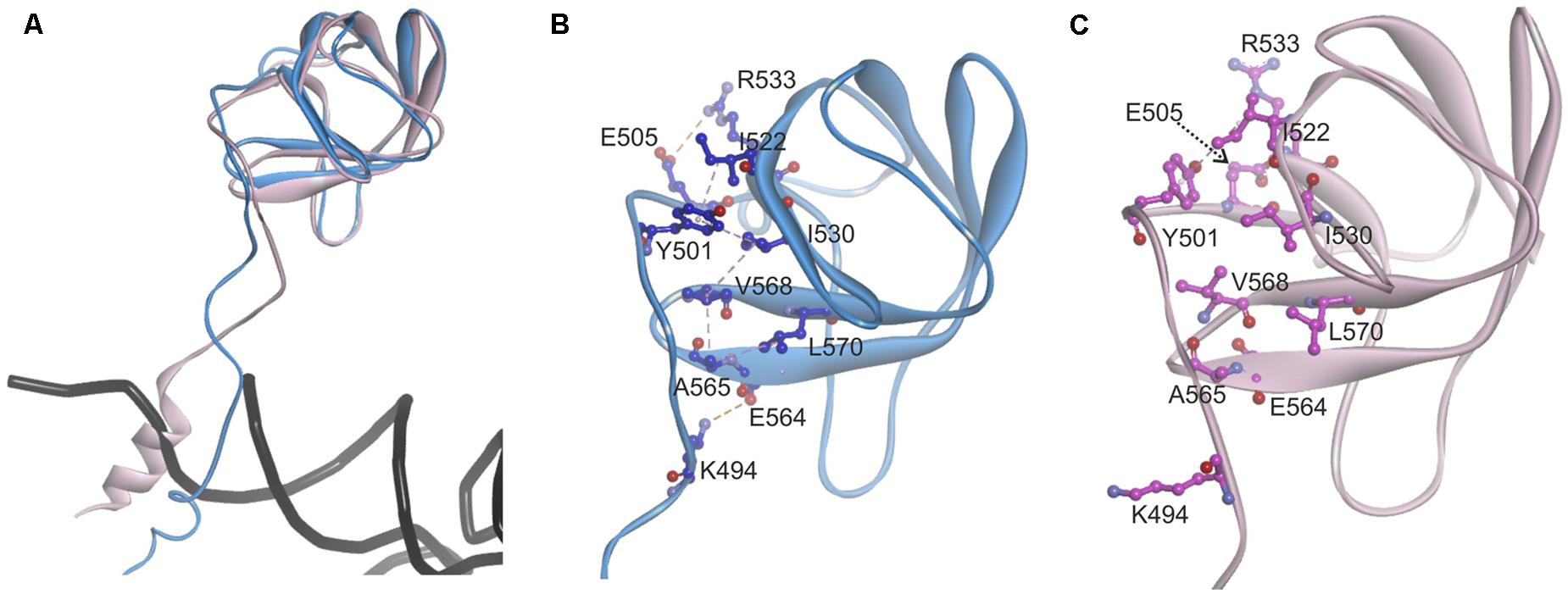
FIGURE 5. Analysis of the contact surface between linker (“safety belt”) and β-barrel of the editing domain in A. fulgidus AlaRS structure (3WQY), which was used as template for modeling. (A) Superimposed chains A “closed” form (blue) and B “open” form (pink). Fragments encompassing residues K474–D589 are shown. For superimposition T582–V613 fragment was used as tether. (B) Residues from the linker (“safety belt”) interacting with the surface exposed residues on the β-barrel in “closed” conformation. Carbon atoms of interacting amino acid residues are in blue. (C) The position of the same residues in “open” conformation (carbon atoms in magenta). Hydrophobic interactions are marked with magenta dashed lines, electrostatic interactions with orange dashed lines.
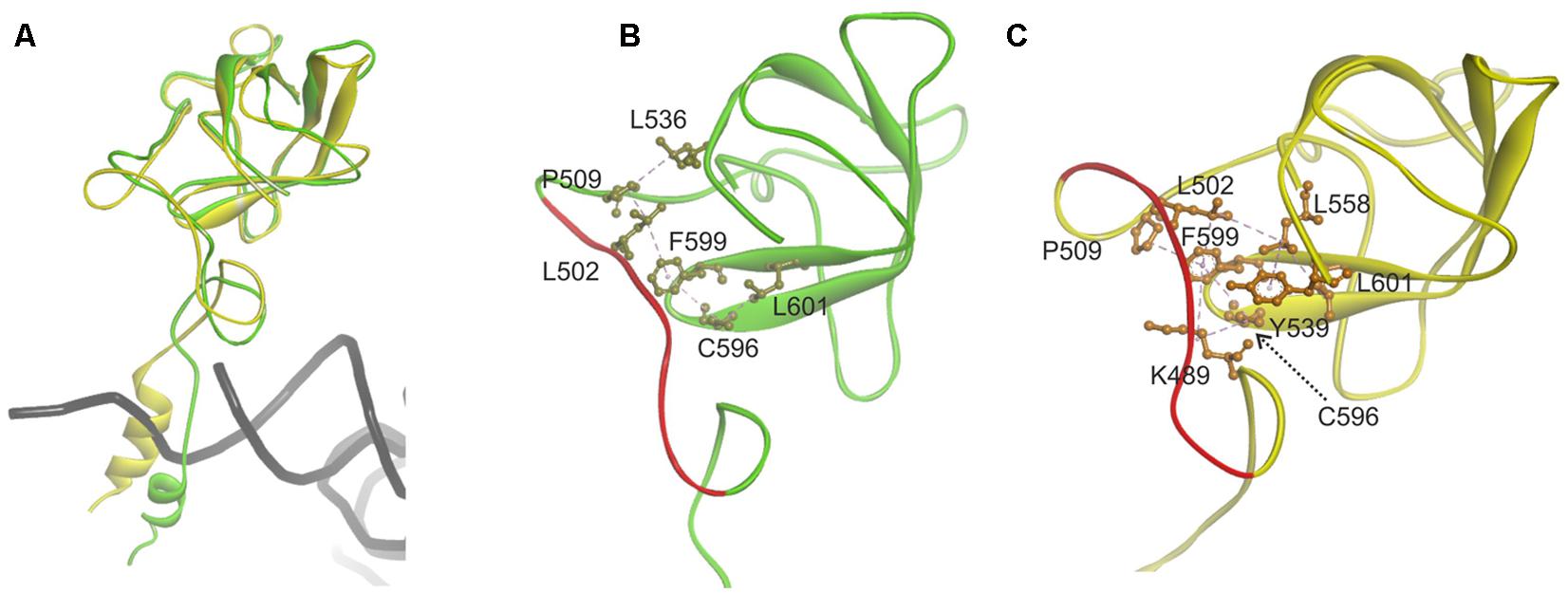
FIGURE 6. Analysis of the contact surface between linker (“safety belt”) and β-barrel of the editing domain in modeled human mitochondrial AlaRS. (A) Superimposed chains A “closed” form (green) and B “open” form (yellow). Fragments encompassing residues G468-D621 are shown. For superimposition Q615–L646 fragment was used as tether. (B) Interacting residues from the linker (”safety belt”) and surface exposed residues on the β-barrel in ”closed” conformation (shown in khaki). (C) Interacting residue in the “open” conformation (in orange). Hydrophobic interactions are marked with magenta dashed lines. Fragment L494–Q505 conserved between mitochondrial AlaRS homologs is marked with red.
Structural Analysis of AARS2 Mutations
We utilized our full-length mtAlaRS homology model to localize all amino acid changes found in mtARS in patients with cardiomyopathy (L155R, R329H, Y539C, R592W, and A961V) and leukodystrophy (F50C, A77V, R199C, E405K, and G965R; Figure 7) to predict their effects on synthetase structure and function (Table 3). The missense mutations could be divided into three groups based on the effect on the aminoacylation activity: loss-of-function, severe, and moderate.
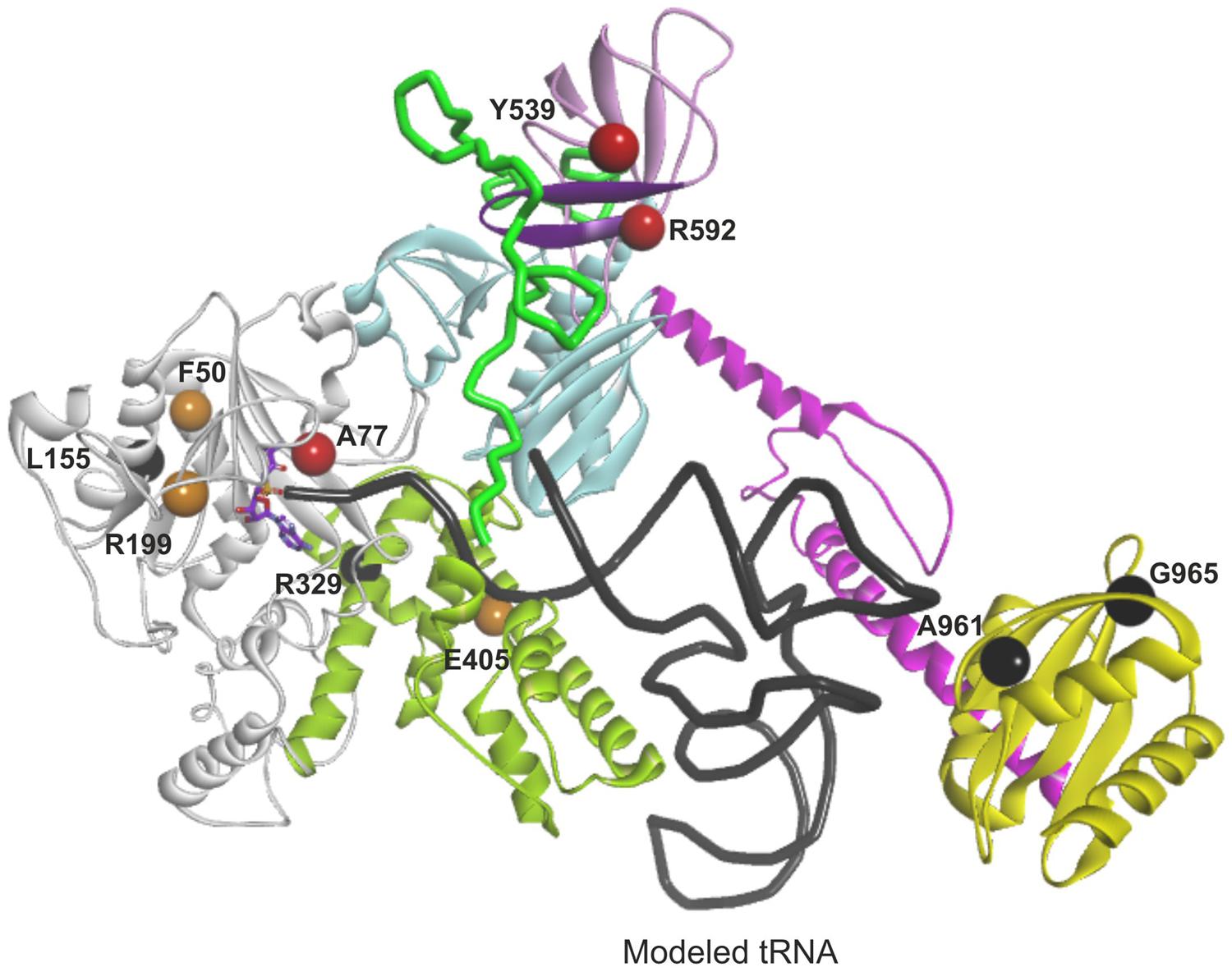
FIGURE 7. Mapping and function predictions of AARS2 mutations associated with cardiomyopathy and leukodystrophy. Domains are color-coded as in Figure 4. Structural loss-of-function mutations are marked in black, mutations affecting substrate binding and resulting in severe reduction in aminoacylation activity are in red, and mutations resulting in moderate decrease of aminoacylation activity are in orange.
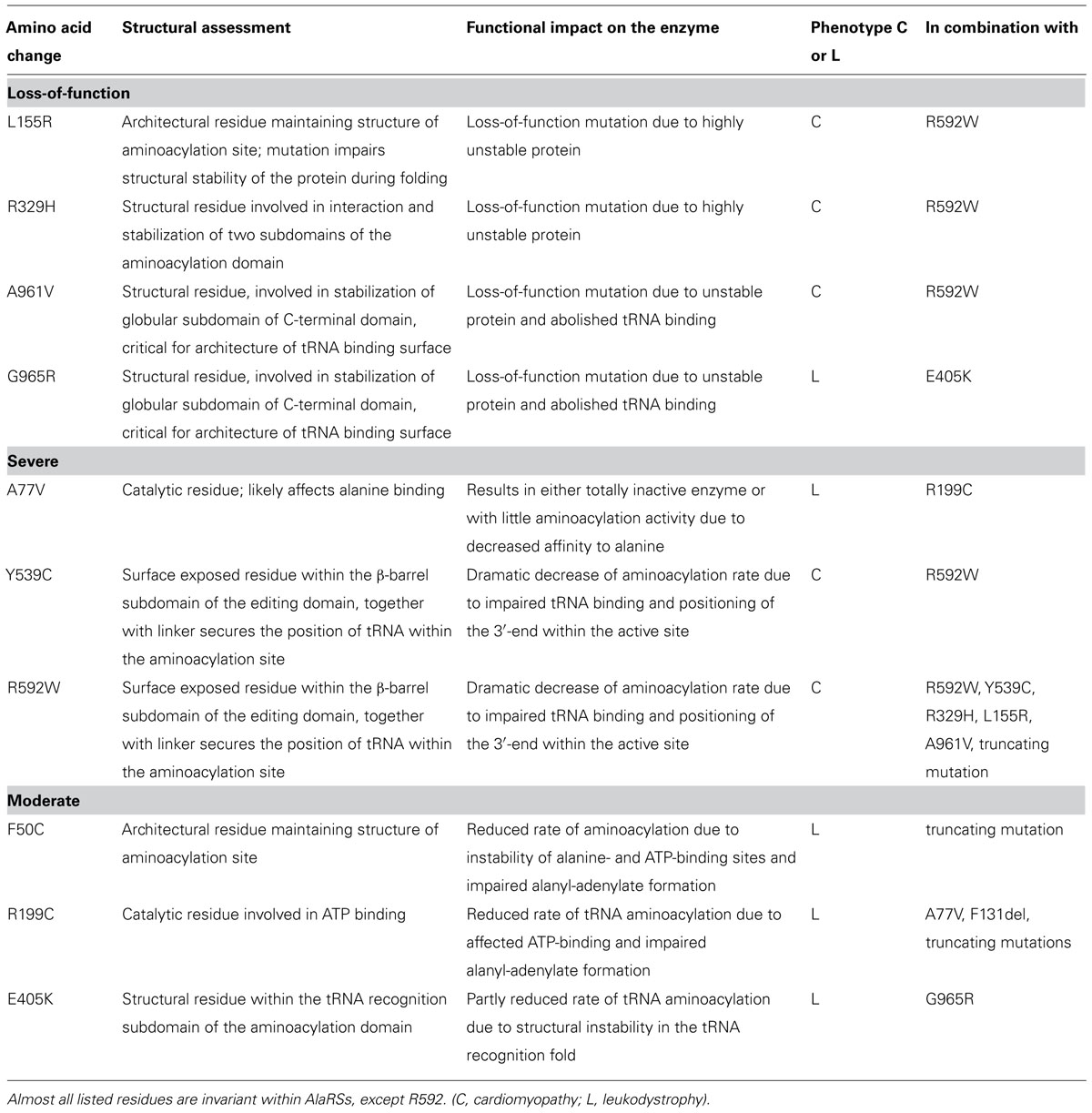
TABLE 3. Structural analysis of cardiomyopathy and leukodystrophy mutations in AARS2 and assessment of their impact on the synthetase function based on modeled protein structure.
Loss of function
The amino acid changes L155R, R329H, A961V, and G965R (residues L173, R345, A880, and V884 in A. fulgidus, respectively) are predicted to impair protein folding and stability resulting in loss of aminoacylation activity (black color-coded in Figure 7). Most of these residues are invariant between cytoplasmic, mitochondrial, and bacterial homologs of AlaRS and critical for maintaining the structure. Accordingly, the change L155R was recently modeled in yeast and had a deleterious effect on cell growth suggesting complete absence of aminoacylation activity (Dallabona et al., 2014). Residues A961 and G965 are important for the fold and stability of C-globular subdomain, and therefore for the architecture of the tRNA binding site. The C-terminal globular domain is separated from the main body of the enzyme, so its disruption may not result in overall misfolding. However, even if the aminoacylation and editing domains remain stable, disruption of the C-terminal domain’s crucial role in tRNA binding and orientation is likely to result in severely reduced rates of aminoacylation.
Severe
The A77V, Y539C, and R592W (residues A99, E525, and D561 in A. fulgidus, respectively) changes are not predicted to affect protein stability, as they are all surface exposed (red color-coded in Figure 7). However, each of these mutations is predicted to result in dramatic reduction of aminoacylation rate due to effects on substrate binding. In case of A77V (invariant residue in all AlaRSs) the binding and activation of alanine is likely to be affected. Y539C and R592W are predicted to impair tRNA binding and stabilization within the aminoacylation site by affecting interactions between the linker and β-barrel that we propose to be important for binding and securing acceptor stem of tRNAAla within the aminoacylation domain.
Moderate
The F50C, R199C, and E405K (residues F73, E202, and K416 in A. fulgidus, respectively) changes (orange color-coded in Figure 7) affect structural residues and are predicted to cause local structural disturbances within the aminoacylation domain leading to partial reduction of aminoacylation activity. The F50C mutant modeled in yeast showed partial reduction of tRNAAla charging and protein instability, which became noticeable only at a higher temperature (Dallabona et al., 2014). These results are in line with our structural predictions.
In conclusion, our analysis of the homology model predicts that the missense mutations in the first two categories, affecting overall structure and substrate binding, severely compromise the aminoacylation by the synthetase, which is also true for the truncating mutations, whereas the missense mutations in the third category result in reduced function but with some retained aminoacylation activity. Combining the data of the compound heterozygous mutations in each patient clearly shows that all patients with leukodystrophy have combinations where one allele is in one of the two severe categories or is a truncating mutation and the other allele is in the third, moderate category, whereas all mutations of the cardiomyopathy patients are severe (Figure 8).
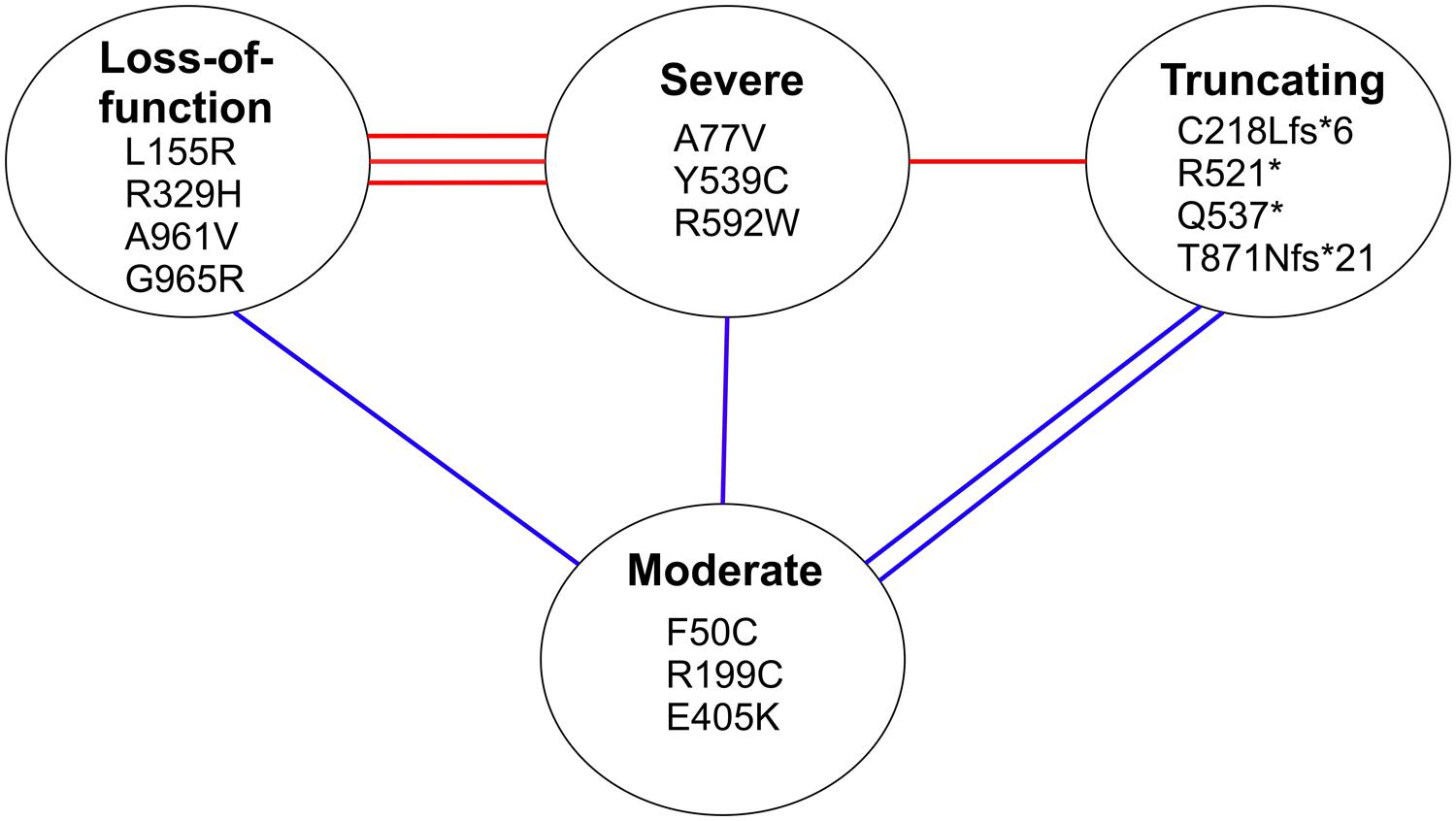
FIGURE 8. Classification of mutations based on structural predictions. The lines between mutation categories connect the compound heterozygous mutations identified in individual cardiomyopathy patients (with red lines), and leukodystrophy patients (with blue lines). The three categories at the top are predicted to have a severe effect on the aminoacylation function of the synthetase, whereas the mutations in the lower category are predicted to retain partial catalytic activity.
Discussion
The tissue-specificity of mitochondrial diseases is puzzling. The pathogenic mechanisms behind the tissue-specific manifestations are likely to involve cell type-specific mitochondrial functions and metabolite requirements, in addition to defects of ATP production by oxidative phosphorylation (Nunnari and Suomalainen, 2012). Differential basal mRNA expression levels of mtARSs have been suggested to contribute to the tissue-specificity, as brain, muscle, and heart were found to have low levels of mtARS mRNAs compared to other tissues (Florentz et al., 2013). The example of AARS2, however, shows that mutations in the same mtARS can also give rise to highly different tissue-specific diseases.
Our previous hypothesis for the cardiomyopathy-related R592W was that it affected tRNA binding in the editing domain and thus interfered with the proofreading of mischarged tRNAs. The homology modeling of human mtAlaRS, based on the new resolved structure of AlaRS of A. fulgidus, suggested instead that all domains of the synthetase, including the editing domain, have an important role in tRNA binding for aminoacylation. We speculate whether this unique way of cooperative tRNA binding for aminoacylation in AlaRSs might explain why the mtAlaRS has a preserved editing domain whereas most other mitochondrial ARSs do not. For example, editing activity was lost during the evolution of mtPheRS (Roy et al., 2005; Elo et al., 2012), and the editing active site of mtLeuRS is not operational (Lue and Kelley, 2005). Although the change at residue R592 does not appear to affect the editing active site, we cannot rule out an effect on editing activity, but we propose that the crucial consequence is on tRNA binding for aminoacylation.
We suggest that the two distinct phenotypes caused by AARS2 mutations are simply caused by differential effects on aminoacylation, with cardiomyopathy resulting from highly severe reduction in aminoacylation activity, and leukodystrophy resulting from only partial reduction in activity. This conclusion could be reached only by careful analysis of the combined mutations found in the patients. The clinical feature of the cardiomyopathy patients was the heart defect, although OXPHOS defects could be seen also in muscle and brain. This suggests that the patients had a combined tissue-involvement that can be expected when mitochondrial protein synthesis is significantly compromised, and that other tissues would have also suffered, had the patients lived longer.
We have previously proposed that severe defects of mitochondrial translation manifest in the heart immediately or soon after birth (Ahola et al., 2014), most often leading to early death. However, those few patients who survived the early severe cardiac disease (Carroll et al., 2013; Haack et al., 2013; Ahola et al., 2014) stabilized for their heart function after a few years of age, but later tended to develop a brain disease. These findings suggest a special requirement for mitochondrial protein synthesis in the heart in the early life, which can be partially compensated for after a critical window of sensitivity, only to be followed by manifestations in the sensitive organ of later childhood-adulthood years, the brain. This hypothesis is also supported by our current findings: the severe AARS2 mutations manifested in the heart soon after birth – or even in late pregnancy – whereas the partial reduction of the synthetase function manifested from 2 to 40 years of age in the brain (Dallabona et al., 2014).
In the future, in vivo studies on the partial retained activity of the leukodystrophy mutations – sufficient for heart function but limiting for brain function during later years – are warranted, to get further insight into the particular events taking place in the heart and in the brain, constituting ‘the narrow permissive window for disease manifestation’ (Steenweg et al., 2012; Ahola et al., 2014). The roles of amino acid availability and varying mtARS expression levels in different developmental stages of tissues, as well as the indispensable level of mitochondrial protein synthesis in each stage and cell type, are of interest.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Riitta Lehtinen for technical assistance and the Academy of Finland (to Liliya Euro, Anu Suomalainen, and Henna Tyynismaa), Jane and Aatos Erkko Foundation (to Anu Suomalainen), Sigrid Jusélius Foundation and University of Helsinki (to Anu Suomalainen and Henna Tyynismaa) for funding support.
References
Ahola, S., Isohanni, P., Euro, L., Brilhante, V., Palotie, A., Pihko, H.,et al. (2014). Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy. Neurology 83, 743–751. doi: 10.1212/WNL.0000000000000716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bayat, V., Thiffault, I., Jaiswal, M., Tetreault, M., Donti, T., Sasarman, F.,et al. (2012). Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 10:e1001288. doi: 10.1371/journal.pbio.1001288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beebe, K., Ribas De Pouplana, L., and Schimmel, P. (2003). Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J. 22, 668–675. doi: 10.1093/emboj/cdg065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belostotsky, R., Ben-Shalom, E., Rinat, C., Becker-Cohen, R., Feinstein, S., Zeligson, S.,et al. (2011). Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 88, 193–200. doi: 10.1016/j.ajhg.2010.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calvo, S. E., Compton, A. G., Hershman, S. G., Lim, S. C., Lieber, D. S., Tucker, E. J.,et al. (2012). Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 4:118ra10. doi: 10.1126/scitranslmed.3003310
Carroll, C. J., Isohanni, P., Poyhonen, R., Euro, L., Richter, U., Brilhante, V.,et al. (2013). Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J. Med. Genet. 50, 151–159. doi: 10.1136/jmedgenet-2012-101375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dallabona, C., Diodato, D., Kevelam, S. H., Haack, T. B., Wong, L. J., Salomons, G. S.,et al. (2014). Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 82, 2063–2071. doi: 10.1212/WNL.0000000000000497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Diodato, D., Ghezzi, D., and Tiranti, V. (2014a). The mitochondrial aminoacyl tRNA synthetases: genes and syndromes. Int. J. Cell. Biol. 2014:787956. doi: 10.1155/2014/787956
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Diodato, D., Melchionda, L., Haack, T. B., Dallabona, C., Baruffini, E., Donnini, C.,et al. (2014b). VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum. Mutat. 35, 983–989. doi: 10.1002/humu.22590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edvardson, S., Shaag, A., Kolesnikova, O., Gomori, J. M., Tarassov, I., Einbinder, T.,et al. (2007). Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 81, 857–862. doi: S0002-9297(07)63063-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elo, J. M., Yadavalli, S. S., Euro, L., Isohanni, P., Gotz, A., Carroll, C. J.,et al. (2012). Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet. 21, 4521–4529. doi: 10.1093/hmg/dds294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Florentz, C., Pütz, J., Jühling, F., Schwenzer, H., Stadler, P. F., Lorber, B.,et al. (2013). “Translation in mammalian mitochondria: order and disorder linked to tRNAs and aminoacyl-tRNA synthetases,” in Translation in Mitochondria and Other Organelles, ed. A.-M. Duchêne (Berlin Heidelberg: Springer), 55–83.
Galmiche, L., Serre, V., Beinat, M., Assouline, Z., Lebre, A. S., Chretien, D.,et al. (2011). Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 32, 1225–1231. doi: 10.1002/humu.21562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghezzi, D., Baruffini, E., Haack, T. B., Invernizzi, F., Melchionda, L., Dallabona, C.,et al. (2012). Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am. J. Hum. Genet. 90, 1079–1087. doi: 10.1016/j.ajhg.2012.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gotz, A., Tyynismaa, H., Euro, L., Ellonen, P., Hyotylainen, T., Ojala, T.,et al. (2011). Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 88, 635–642. doi: 10.1016/j.ajhg.2011.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, M., Chong, Y. E., Beebe, K., Shapiro, R., Yang, X. L., and Schimmel, P. (2009a). The C-Ala domain brings together editing and aminoacylation functions on one tRNA. Science 325, 744–747. doi: 10.1126/science.1174343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guo, M., Chong, Y. E., Shapiro, R., Beebe, K., Yang, X. L., and Schimmel, P. (2009b). Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature 462, 808–812. doi: 10.1038/nature08612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haack, T. B., Kopajtich, R., Freisinger, P., Wieland, T., Rorbach, J., Nicholls, T. J.,et al. (2013). ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 93, 211–223. doi: 10.1016/j.ajhg.2013.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hallberg, B. M., and Larsson, N. G. (2014). Making proteins in the powerhouse. Cell Metab. 20, 226–240. doi: 10.1016/j.cmet.2014.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hallmann, K., Zsurka, G., Moskau-Hartmann, S., Kirschner, J., Korinthenberg, R., Ruppert, A. K.,et al. (2014). A homozygous splice-site mutation in CARS2 is associated with progressive myoclonic epilepsy. Neurology 83, 2183–2187 doi: 10.1212/WNL.0000000000001055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konovalova, S., and Tyynismaa, H. (2013). Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol. Genet. Metab. 108, 206–211. doi: 10.1016/j.ymgme.2013.01.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kopajtich, R., Nicholls, T. J., Rorbach, J., Metodiev, M. D., Freisinger, P., Mandel, H.,et al. (2014). Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am. J. Hum. Genet. 95, 708–720. doi: 10.1016/j.ajhg.2014.10.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lue, S. W., and Kelley, S. O. (2005). An aminoacyl-tRNA synthetase with a defunct editing site. Biochemistry 44, 3010–3016. doi: 10.1021/bi047901v
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Naganuma, M., Sekine, S., Chong, Y. E., Guo, M., Yang, X. L., Gamper, H.,et al. (2014). The selective tRNA aminoacylation mechanism based on a single G*U pair. Nature 510, 507–511. doi: 10.1038/nature13440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagao, A., Suzuki, T., Katoh, T., Sakaguchi, Y., and Suzuki, T. (2009). Biogenesis of glutaminyl-mt tRNAGln in human mitochondria. Proc. Natl. Acad. Sci. U.S.A. 106, 16209–16214. doi: 10.1073/pnas.0907602106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pierce, S. B., Chisholm, K. M., Lynch, E. D., Lee, M. K., Walsh, T., Opitz, J. M.,et al. (2011). Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 6543–6548. doi: 10.1073/pnas.1103471108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pierce, S. B., Gersak, K., Michaelson-Cohen, R., Walsh, T., Lee, M. K., Malach, D.,et al. (2013). Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 92, 614–620. doi: 10.1016/j.ajhg.2013.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Riley, L. G., Cooper, S., Hickey, P., Rudinger-Thirion, J., McKenzie, M., Compton, A.,et al. (2010). Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am. J. Hum. Genet. 87, 52–59. doi: 10.1016/j.ajhg.2010.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rotig, A. (2011). Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta 1807, 1198–1205. doi: 10.1016/j.bbabio.2011.06.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roy, H., Ling, J., Alfonzo, J., and Ibba, M. (2005). Loss of editing activity during the evolution of mitochondrial phenylalanyl-tRNA synthetase. J. Biol. Chem. 280, 38186–38192. doi: M508281200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scheper, G. C., van der Klok, T., van Andel, R. J., van Berkel, C. G., Sissler, M., Smet, J.,et al. (2007). Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 39, 534–539. doi: 10.1038/ng2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schwenzer, H., Zoll, J., Florentz, C., and Sissler, M. (2014). Pathogenic implications of human mitochondrial aminoacyl-tRNA synthetases. Top. Curr. Chem. 344, 247–292. doi: 10.1007/128_2013_457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sofou, K., Kollberg, G., Holmström, M., Dávila, M., Darin, N., Gustafsson, C. M.,et al. (2015). Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol. Genet. Genomic Med. 3, 59–68. doi: 10.1002/mgg3.115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steenweg, M. E., Ghezzi, D., Haack, T., Abbink, T. E., Martinelli, D., van Berkel, C. G.,et al. (2012). Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain 135, 1387–1394. doi: 10.1093/brain/aws070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Taylor, R. W., Pyle, A., Griffin, H., Blakely, E. L., Duff, J., He, L.,et al. (2014). Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312, 68–77. doi: 10.1001/jama.2014.7184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mitochondrial disease, aminoacyl-tRNA synthetases, alanyl-tRNA synthetase, tissue-specificity, structural modeling
Citation: Euro L, Konovalova S, Asin-Cayuela J, Tulinius M, Griffin H, Horvath R, Taylor RW, Chinnery PF, Schara U, Thorburn DR, Suomalainen A, Chihade J and Tyynismaa H (2015) Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Front. Genet. 6:21. doi: 10.3389/fgene.2015.00021
Received: 28 November 2014; Accepted: 15 January 2015;
Published online: 06 February 2015.
Edited by:
Daniele Ghezzi, Istituto Neurologico Carlo Besta, ItalyReviewed by:
Lluís Ribas De Pouplana, IRB Barcelona, SpainMichal Minczuk, Medical Research Council, UK
Daria Diodato, Istituto Neurologico Carlo Besta, Italy
Copyright © 2015 Euro, Konovalova, Asin-Cayuela, Tulinius, Griffin, Horvath, Taylor, Chinnery, Schara, Thorburn, Suomalainen, Chihade and Tyynismaa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henna Tyynismaa, Research Programs Unit, Molecular Neurology, Biomedicum Helsinki, University of Helsinki, r.C520A, Haartmaninkatu 8, 00290 Helsinki, Finland e-mail: henna.tyynismaa@helsinki.fi