 Masaaki Murakami1,2,3 Toshio Hirano1,2,3,4*
Masaaki Murakami1,2,3 Toshio Hirano1,2,3,4*- 1 Laboratory of Developmental Immunology, JST-CREST, Graduate School of Frontier Biosciences, Osaka University, Osaka, Japan
- 2 Laboratory of Developmental Immunology, JST-CREST, Graduate School of Medicine, Osaka University, Osaka, Japan
- 3 WPI Immunology Frontier Research Center, Osaka University, Osaka, Japan
- 4 Laboratory of Cytokine Signaling, RIKEN Research Center for Allergy and Immunology, Yokohama, Japan
It is commonly thought that autoimmune diseases are caused by the breakdown of self-tolerance, which suggests the recognition of specific antigens by autoreactive CD4+ T cells contribute to the specificity of autoimmune diseases (Marrack et al., 2001; Mathis and Benoist, 2004). In several cases, however, even for diseases associated with class II major histocompatibility complex (MHC) alleles, the causative tissue-specific antigens recognized by memory/activated CD4+ T cells have not been established (Mocci et al., 2000; Skapenko et al., 2005). Rheumatoid arthritis (RA) and arthritis in F759 knock-in mice (F759 mice) are such examples (Atsumi et al., 2002; Brennan et al., 2002; Falgarone et al., 2009). These include associations with class II MHC and CD4 molecules; increased numbers of memory/activated CD4+ T cells; and improved outcomes in response to suppressions and/or deficiencies in class II MHC molecules, CD4+ T cells, and the T cell survival cytokine IL-7. Regarding the development of arthritis in F759 mice, it is not only the immune system, but also non-immune tissue that are involved, indicating that the importance of their interactions (Sawa et al., 2006, 2009; Ogura et al., 2008; Hirano, 2010; Murakami et al., 2011). Furthermore, we have shown that local events such as microbleeding together with an accumulation of activated CD4+ T cells in a manner independent of tissue antigen-recognitions induces arthritis in the joints of F759 mice (Murakami et al., 2011). For example, local microbleeding-mediated CCL20 expression induce such an accumulation, causing arthritis development via chronic activation of an IL-17A-dependent IL-6 signaling amplification loop in type 1 collagen+ cells that is triggered by CD4+ T cell-derived cytokine(s) such as IL-17A, which leads to the synergistic activation of STAT3 and NFκB in non-hematopoietic cells in the joint (Murakami et al., 2011). We named this loop the IL-6-mediated inflammation amplifier, or IL-6 amplifier for short (Ogura et al., 2008; Hirano, 2010; Murakami et al., 2011). Thus, certain class II MHC-associated, tissue-specific autoimmune diseases, including some RA subtypes, may be induced by local events that cause an antigen-independent accumulation of effector CD4+ T cells followed by the induction of the IL-6 amplifier in the affected tissue. In other words, in certain cases, the target tissue itself may determine the specificity of the autoimmune disease via activation of the IL-6 amplifier. To explain this hypothesis, we have proposed a four-step model for MHC class II-associated autoimmune diseases (Murakami et al., 2011): (1) T cell activation regardless of antigen specificity; (2) local events inducing a tissue-specific accumulation of activated T cells; (3) transient activation of the IL-6 amplifier; and (4) enhanced sensitivity to cytokines in the target tissue. The interaction of these events results in chronic activation of the IL-6 amplifier and subsequent manifestation of autoimmune diseases. Thus, the IL-6 amplifier, which is chronically activated by these four events, is a critical regulator of chronic inflammations in tissue-specific MHC class II-associated autoimmune diseases.
Tissue-Specific MHC Class II-Associated Autoimmune Diseases and Antigen-Recognitions by CD4+ T Cells
It has been proposed that autoimmune diseases are caused by a breakdown of self-tolerance due to multiple genetic and/or environmental factors (Marrack et al., 2001; Mathis and Benoist, 2004), suggesting the dysregulation of immune responses is fundamental to autoimmune diseases. This agrees with the theory that certain autoimmune diseases like rheumatoid arthritis (RA) develop in specific tissues as a result of cognate antigen-recognition by CD4+ T cells, particularly when these diseases are associated with class II major histocompatibility complex (MHC) alleles (Steinman, 2001; Zhang et al., 2008; Imboden, 2009). Consistent with this, joint-specific antigenic peptides such as derivatives of aggrecan, fibrillin, and collagen have been identified in humans (Polgár et al., 2003; Chapuy-Regaud et al., 2005; Takizawa et al., 2006; Van Steendam et al., 2010), while immunodominant MHC class II peptides in an animal model have been found to match those seen in human RA (Andersson et al., 2010). However, it is unclear whether these peptides are a result or cause of joint damage. Despite the evidence for antigen-specific T cell activation in some RA patients, tissue-specific self or non-self antigens recognized by activated CD4+ T cells in many class II MHC-associated diseases and even a majority of RA cases have not been well-established (Mocci et al., 2000; Skapenko et al., 2005). This raises the possibility that a breakdown in CD4+ T cell tolerance for a tissue-specific antigen is not always necessary for tissue-specific autoimmune diseases. Instead, it may be the consequence of local events that are initiated by inflammation triggered by certain genetic and/or environmental factors (Hirano, 1998, 2010; Matsumoto et al., 1999; Marrack et al., 2001; Sawa et al., 2006) such that the specificity of an autoimmune disease could be determined by the non-immune target tissue itself (Brennan et al., 2002; Hirano, 2002). In these cases, CD4+T cells may act as the source for a variety of inflammatory cytokines (Brennan et al., 2002). In fact, various subsets of effector CD4+ T cells – e.g., T helper 1 (Th1) cells, Th2 cells, and Th17 cells, which produce IFNγ, IL-4, and IL-17A, respectively (Mosmann and Coffman, 1989; Glimcher and Murphy, 2000; Cua et al., 2003; Harrington et al., 2005; Park et al., 2005; Veldhoen et al., 2006; Zhu et al., 2006; Bettelli et al., 2007; Nishihara et al., 2007) – may initiate and drive the progression of disease. This may help explain why RA is more common in older populations, as there exists an age-dependent increase in memory/activated CD4+ T cells resulting from a homeostatic proliferation that is mediated by a reduction in T cell input from the thymus (Surh and Sprent, 2000). Moreover, it has been reported that an age-dependent reduction in naive CD4+ T cells in peripheral second lymphoid organs increases the likelihood of (i) weak interactions between TCRs and peptides presented by self-class II MHC molecules including auto-antigenic peptides involved in positive selections in the thymus and (ii) cytokine consumption per CD4+ T cell including the T cell survival factor IL-7 (Surh and Sprent, 2000). This could help explain the occurrence of other diseases too, as the homeostatic proliferation of CD4+ T cells has been shown to be involved in the development of diabetes, arthritis, and Omenn syndrome (King et al., 2004; Jang et al., 2006; Sawa et al., 2006; Khiong et al., 2007). This process is also associated with a specific cytokine profile that includes the IL-17A and IFNγ produced by CD4+ T cells (Gudmundsdottir and Turka, 2001; Khiong et al., 2007; Nishihara et al., 2007).
IL-6 in Autoimmune Diseases
IL-6 is a pleiotropic cytokine that regulates multiple biological processes including the development of the nervous and hematopoietic systems, acute-phase responses, inflammation, and immune responses (Hirano, 1998). To date, 10 IL-6 family cytokines have been identified: IL-6, oncostatin M, LIF, CNTF, CT-1, NNT-1, neuropoietin, IL-11, IL-27, and IL-31 (Kamimura et al., 2003; Murakami et al., 2004; Suthaus et al., 2010). All of these share gp130 as the signal transducer in their receptor complexes. Upon IL-6 stimulation, gp130 transduces two major signaling pathways: the JAK–signal transducer and activator of transcription 3 (STAT3) pathway, which is mediated by the YxxQ motif of gp130, and the SHP2–Gab-Ras-Erk–MAPK pathway, which is regulated by Y759, a cytoplasmic SOCS3 binding residue in gp130 (Fukada et al., 1996; Ohtani et al., 2000; Kamimura et al., 2003). Additionally, a number of studies have suggested IL-6 has an important role in autoimmune diseases (Hirano, 1998, 2010; O’Shea et al., 2002; Sakaguchi and Sakaguchi, 2005; Awasthi and Kuchroo, 2009). The F759 knock-in mouse line (F759), for example, which expresses a mutant variant of gp130 where Y759 is substituted for phenylalanine (F), shows enhanced IL-6-mediated STAT3 activation due to a lack of SOCS3-mediated suppression (Ohtani et al., 2000). As these mice age, they spontaneously develop a RA-like tissue-specific disease, indicating that constitutive activation of IL-6 signaling is involved in the development of certain autoimmune diseases (Atsumi et al., 2002). Moreover, patients with RA show high synovial concentrations of IL-6 (Hirano et al., 1988), while anti-IL-6 receptor therapy is effective for some RA patients (Nakagawa et al., 2010). These observations support the use of the F759 mouse as a murine model for RA. Furthermore, we have previously shown that MHC II-restricted CD4+ T cells, but not CD8+ T cells and B cells, are involved in the development of arthritis in F759 mice (Sawa et al., 2006), while a subset of CD8+ T cells that express Foxp3 and are induced by IL-6 signaling suppress it (Nakagawa et al., 2010).
An IL-17A-Dependent IL-6 Signaling Amplification Loop in Type 1 Collagen+ Cells, the IL-6 Amplifier, Plays a Role in the Development of MHC Class II-Associated Autoimmune Diseases
While CD4+T cells are required for F759 arthritis, bone marrow transplantation experiments have demonstrated the F759 mutation in non-hematopoietic cells is sufficient for F759 arthritis without any accompanying mutation in the CD4+ T cells (Sawa et al., 2006). This shows that an interaction between non-immune tissues/cells and the immune system plays a critical role this and most likely several other autoimmune and chronic inflammatory diseases (Hirano, 1998, 2010). Our detailed experiments utilizing bone marrow chimera mice and knock-out mice further demonstrate that excess IL-6 signaling in non-hematopoietic cells, particularly type 1 collagen+ cells, due to the F759 mutation induces an enhanced production of the T cell survival factor IL-7, which increases memory/activated CD4+ T cells via an increase in homeostatic proliferation – a process that is critical for arthritis development in F759 mice (Sawa et al., 2006). Furthermore, IL-17A-expressing CD4+ T cells show a memory/activated phenotype in vivo in F759 mice, while IL-17A regulates their arthritis (Ogura et al., 2008; Murakami et al., 2011). Thus, it is plausible that the age-dependent increase in homeostatic proliferation via IL-6-mediated IL-7 expression plays a role in the accumulation of antigen-experienced, memory/activated CD4+ T cells expressing IL-17A. This is especially true for those F759 mice that show excess IL-6 signaling. We previously showed that IL-17A-triggerred positive feedback of IL-6 signaling, which results in synergistic hyper-expressions of chemokines and IL-6 itself in type 1 collagen+ cells, is enhanced in a manner dependent on NF-κB and STAT3, which our themselves stimulated by IL-17A in the presence of an IL-6 signal (Ogura et al., 2008; Figure 1). We named this IL-17A-dependent IL-6 signaling amplification loop in type 1 collagen+ cells the IL-6-mediated inflammation amplifier, or IL-6 amplifier for short (Ogura et al., 2008; Hirano, 2010). Furthermore, activation of the IL-6 amplifier is critical not only for the development of arthritis in F759 mice but also for MOG antigen-specific, T cell-mediated experimental autoimmune encephalomyelitis (EAE; Ogura et al., 2008). These results further support the idea that interactions between the immune system and non-immune tissue play roles in the development of autoimmune diseases and that the IL-6 amplifier makes a major contribution to this interaction.
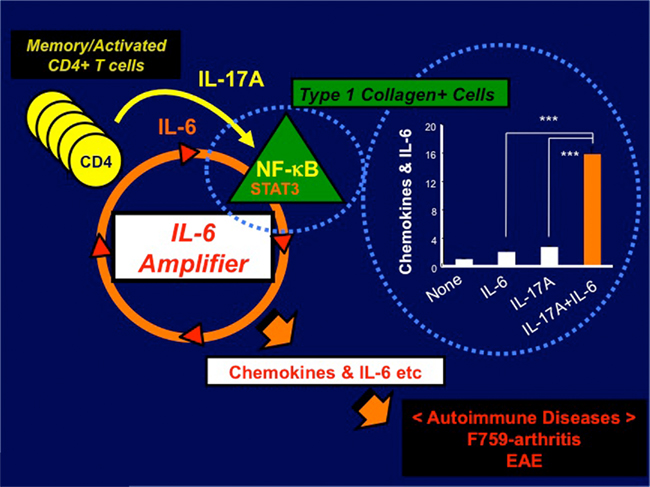
Figure 1. IL-6 amplifier activation plays a role in the development of autoimmune diseases such as arthritis in F759 mice and EAE. IL-17A-triggerred positive feedback of IL-6 signaling, which results in synergistic hyper-expressions of chemokines and IL-6 itself in type 1 collagen+ cells, is enhanced in a manner dependent on NF-κB and STAT3, which our themselves stimulated by IL-17A in the presence of an IL-6 signal (bar graph). We named this IL-17A-dependent IL-6 signaling amplification loop in type 1 collagen+ cells the IL-6-mediated inflammation amplifier, or IL-6 amplifier for short. Importantly, activation of the IL-6 amplifier is critical not only for the development of arthritis in F759 mice but also for MOG antigen-specific, T cell-mediated experimental autoimmune encephalomyelitis (EAE; Ogura et al., 2008).
A Four-Step Model Explains the Chronic Activation of the IL-6 Amplifier that is Followed by the Development of MHC Class II-Associated Autoimmune Diseases
Because CD4+ T cells bearing a single TCR that recognizes antigens not related to joint tissue induces arthritis in Rag2 deficient mice that have the F759 mutation, we concluded that cognate antigen-recognition by effector CD4+ T cells is not necessary for tissue specificity in F759 mice (Murakami et al., 2011). From this, we hypothesized that disease specificity may be determined by the tissue itself such that local events in the joint may determine and initiate the disease via the IL-6 amplifier. For example, intravenous transfer of in vitro polarized Th17 cells into young F759 mice does not develop arthritis within 3 months. This sharply contrasts with the case where MOG antigen-specific Th17 transfer induces EAE. However, if Th17 cell transfer is done before inducing experimental microbleeding in one leg of F759 mice, then this leg and only this leg will develop arthritis. Even Th17 cells derived from TCR transgenic mice induced arthritis in the microbleeding-induced leg of F759 mice. These findings are consistent with the idea that local events determine the disease specificity even if activation of tissue antigen-specific T cells does not occur. We further observed that T cells accumulate in the joint where arthritis occurs. This microbleeding-induced accumulation of Th17 cells is dependent on the production of CCL20, a target of the IL-6 amplifier, in the joint. Disease induction requires T cell produced IL-17A, IL-6, and enhanced STAT3 signaling in type I collagen-expressing cells (Murakami et al., 2011). Based on these results, we propose that certain class II MHC-associated autoimmune diseases such as RA arise through a series of at least four steps (Figure 2): (1) T cell activation regardless of antigen specificity; (2) local events inducing a tissue-specific accumulation of activated T cells; (3) transient activation of the IL-6 amplifier, which is triggered by CD4+ T cell-derived cytokines such as IL-17A; and (4) enhanced sensitivity to T cell-derived cytokines and/or IL-6 in type 1 collagen+ cells in the target tissue. After these four steps, chronic activation of the IL-6 amplifier followed by the development of an autoimmune disease occurs. It is likely that each step interacts with the others, and the degree of the contribution of each to the pathogenesis varies with the disease. Our four-step model provides a plausible explanation for why tissue-specific antigens recognized by activated CD4+ T cells have not been identified in several autoimmune diseases, especially those associated with class II MHC molecules. It is likely that in diseases where tissue antigen-specific T cells play roles, tissue antigen-specific recognition by T cells would bypass the requirement of local events, even though these local events can still affect the accumulation of tissue antigen-specific T cells in the target tissue. Our four-step model, therefore, should be applicable to a wide range of autoimmune and other chronic inflammatory diseases (see last section).
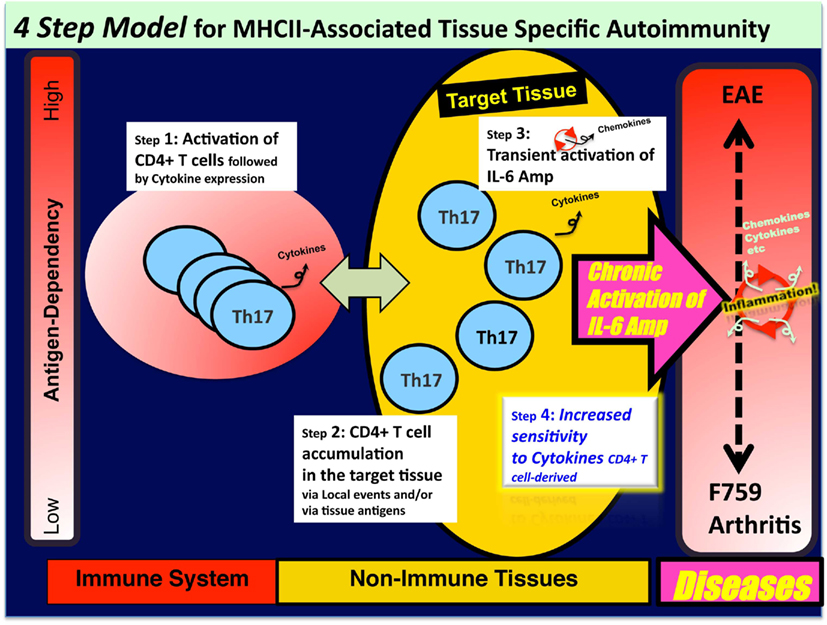
Figure 2. A four-step model for MHC class II-associated autoimmune diseases. Certain class II MHC-associated autoimmune diseases arise through a series of four steps: (1) T cell activation regardless of antigen specificity; (2) local events inducing a tissue-specific accumulation of activated T cells; (3) transient activation of the IL-6 amplifier, which is triggered by CD4+ T cell-derived cytokines such as IL-17A; and (4) enhanced sensitivity to T cell-derived cytokines and/or IL-6 in type 1 collagen+ cells in the target tissue. Following these four steps, chronic activation of the IL-6 amplifier followed by the development of an autoimmune disease occurs. It is likely that each step interacts with the others, and the degree of the contribution of each to the pathogenesis varies with the disease.
Chronic Activation of the IL-6 Amplifier Can Override Homeostasis in Target Tissues
The number of cytokine-secreting effector/memory CD4+ T cells increases with age due to an accumulation of pathogen-specific memory T cells and homeostatic proliferation of CD4+ T cells. These CD4+ T cells are increasingly localized in parenchymal organs like the alimentary tract, lung, and liver, rather than the lymphoid organs, meaning at steady state, memory/activated CD4+ T cells may migrate to and/or stay in non-lymphoid tissues that are at high risk for autoimmune diseases. Consistent with this, these diseases are more prevalent in older patients who have a larger population of memory/activated CD4+ T cells, some of which secrete cytokines because of homeostatic proliferation and/or chronic inflammations in other tissues (Hasler and Zouali, 2005; Larbi et al., 2008). Therefore, the first three steps will occur to some extent even in healthy subjects, although the degree will differ among individuals (see Figure 2). Autoimmune diseases like RA, however, do not develop in all individuals. Therefore, the relatively low rates of these disorders may reflect the fact that multiple genetic and environmental factors make a significant contribution, particularly at the fourth step, for disease development.
Potential Factors that Accelerate Tissue Sensitivity to Cytokines Involved in Autoimmune Diseases
Because the activation of the IL-6 amplifier is mediated by the synergistic activation of NF-κB and STAT3 molecules, we hypothesize factors that stimulate the signaling pathways regulating these two molecules in non-immune tissues/cells play a role in the development of MHC class II-associated autoimmune diseases and possibly other chronic inflammatory diseases (Hasler and Zouali, 2005; Larbi et al., 2008). These factors and the role of their cognate recognitions in CD4+ T cells are discussed below.
Virus/Bacteria Products and Exogenous TLR Stimulators
One example is products made by viruses or bacteria (Münz et al., 2009). For instance, HTLV1 infection is a significant risk factor for arthritis (Ishihara et al., 2004), while the transgenic expression of p40 Tax, a product of HTLV1 that activates NF-κB, causes a RA-like disease in mice (Iwakura et al., 1991). Indeed, forced expression of p40 Tax in F759 mice has been seen to enhance disease development (Ishihara et al., 2004). Moreover, many viral proteins, including the hepatitis C virus Core protein and EBNA2 from the Epstein–Barr virus, are strong STAT3 activators (Yoshida et al., 2002; Muromoto et al., 2009). Products from pathogens are also known to stimulate Toll-like receptors that lead to NF-κB activation. Since viruses and bacteria also have their own preferential target cells and/or tissues, their infections could determine the tissue specificity of a disease by enhancing cytokine sensitivity in the given tissue. Consistent with this, autoimmune diseases are sometimes induced after infections that also increase the number of activated, pathogen-specific, cytokine-secreting CD4+ T cells (Kivity et al., 2009).
Microbleeding and Mechanical Stress in the Tissues
Microbleeding in the joints may result in the accumulation of many different cell types including red blood cells, neutrophils, macrophages, and dendritic cells as well as memory/activated CD4+ T cells including Th17 cells. Here we focused on IL-17A expression from Th17 cells accumulating in the joints. However, one can argue that microbleeding can accelerate the inflammatory reaction by other means. For example, the environment of the joint synovium or the presence of dead cells may lead to the release of intracellular stimulants such as heme and/or danger-associated molecular patterns (DAMPs; Zhang et al., 2010). These stimulants secondarily enhance cell death by heme’s toxic effect. They might also induce IL-6 and/or CCL20 expression based on the fact that DAMPs can activate the NF-κB pathway (Bianchi, 2007; Sims et al., 2010).
Tissue sensitivity to mechanical stress that arises with age is another potential trigger or enhancer for autoimmune diseases. We have shown that an experimental compression enhances arthritis development in F759 mice in the presence of Th17 cell transfer (unpublished data), suggesting that such stress can induce local events like microbleeding and/or IL-6 expression via activation of NF-κB.
Genetic Factors/Mutations Affecting Signaling Molecules in NF-κB and STAT3 Pathways
Moreover, MHC class II-associated autoimmune diseases might be associated with various genetic aberrations including a somatic mutation in gp130 molecules that induces STAT3 hyperactivation, and mutations in NF-κB and its regulators that lead to dysregualted NF-κB signaling (Lenz et al., 2008; Compagno et al., 2009; Rebouissou et al., 2009). This may not apply in humans, however, as to date we have not identified mutations in the cytoplasmic region of gp130 in patients. Nevertheless, there still remains evidence that STAT3 abnormalities are involved, as demonstrated in the B cells of patients with hyper-immunoglobulin E syndrome (Minegishi et al., 2007). Because F759 mice lack SOCS3-mediated negative feedback only in the gp130 signaling pathway, it is reasonable to speculate that specifically deleting SOCS3 in non-hematopoietic cells could also increase the risk of autoimmune and/or other chronic inflammatory diseases. Consistent with this notion, SOCS3 deficiency in liver cells increased the degree of liver fibrosis (Ogata et al., 2006). Although this phenotype is should not be classified specifically as an autoimmune syndrome, the fibrosis likely mirrors the effects of NF-κB/STAT3 mutations. Finally, dysregulated NF-κB/STAT3 activation in non-immune cells such as type I collagen+ fibroblasts may trigger a feedback loop that increases IL-6 expression to induce inflammation like that seen in F759 mice.
Role of Other Cytokines Derived from CD4+ T Cells
Cytokines other than IL-17A may also contribute to class II MHC-associated diseases by enhancing IL-6 signaling in affected tissues and cells like type 1 collagen+ cells (the fourth step), as non-polarized activated CD4+ T cells, Th1, and IL-17A−/− Th17 cells too induce a mild form of arthritis in F759 mice (Murakami et al., 2011). TNFα, for example, may contribute to localized class II MHC-associated autoimmune diseases via NF-κB activation followed by IL-6 amplifier activation. In support of this idea, numerous studies have demonstrated the efficacy of targeting TNFα when treating RA and other chronic autoimmune diseases, the majority of which involve class II MHC molecules (Feldmann and Maini, 2001). Furthermore, activated CD4+ T cells are known to express TNFα (Cherwinski et al., 1987; Constant and Bottomly, 1997; Brennan et al., 2002; Williams et al., 2008), while we have found a lack of TNFα attenuates arthritis in F759 mice (unpublished data). Moreover, it is interesting that LPS administration around the joints induces arthritis in mice that have an excess number of Th1 cells (Nickdel et al., 2009). This may suggest that LPS-mediated IL-6 production induces a local accumulation of Th1 cells followed by activation of the IL-6 amplifier.
Role of Cognate Recognitions by CD4+ T Cells
Activation of the IL-6 amplifier is also involved in the development of EAE (Ogura et al., 2008). Because the model for EAE is dependent on tissue specific, MOG-derived peptides, these results suggest that antigen specificity by effector CD4+ T cells and IL-6 amplifier activation in the affected tissue do not always function independently of each other in tissue-specific autoimmune diseases. If this is the case, cognate antigen-recognition by effector CD4+ T cells could occur upstream of the enhanced IL-6 signaling in the affected tissue such that the antigen specificity of the effector CD4+ T cells functions initially to target CD4+ T cells around said tissues. In other words, the antigen specificity might bypass initial local events (like microbleeding) to induce tissue-specific accumulation of activated T cells, although some local events might increase the efficacy of the tissue accumulation even in diseases like EAE (step 2 in Figure 2). The resulting pool of activated CD4+ T cells around the affected tissues could enhance local IL-6 signaling, which would then act as a source for cytokines via the IL-6 amplifier. Thus, regardless of the stimulus for the local accumulation of effector CD4+ T cells, the resulting inflammatory disease is associated with class II MHC molecules if cytokines from the activated CD4+ T cells are involved in the disease development.
The IL-6 Amplifier Beyond MHC Class II-Associated Diseases and Disorders
Indeed, MHC class II genes are associated with a number of human diseases and disorders that extend beyond typical autoimmune diseases including metabolic syndrome, psychotic illnesses, and other inflammatory diseases. Diseases and disorders in the Genetic Association Database at the National Institute of Aging (http://geneticassociationdb.nih.gov/cgi-bin/index.cgi) found to be associated with the MHC class II genes described above are wide and varied (Table 1). These associations and our four-step model suggest that at least some subfamilies of these diseases and disorders might be affected by the activation status of the IL-6 amplifier, which is triggered by cytokines from CD4+ T cells. However, we also hypothesize that the importance of the IL-6 amplifier activation extends beyond MHC class II-associated diseases and disorders to include those that arise from inflammation induction. One reason is the amplifier’s synergistic activation of NF-κB and STAT3 in type 1 collagen+ cells. In other words, it is possible that not all NF-κB and/or STAT3 stimulators are supplied by activated CD4+ T cells. Therefore, chronic activation of NF-κB and STAT3 induced by genetic and/or environmental factors may give rise to similar effects exerted by chronic activation of the IL-6 amplifier. It is likely that activation of NF-κB and STAT3 by genetic and environmental factors other than T cell products triggers certain chronic inflammatory diseases that are not readily apparent to be associated with MHC class II genes. Examples include adult Still’s disease and Castleman’s disease. Indeed, these diseases do associate with the IL-18 gene (Sugiura et al., 2002), an NF-κB stimulator, and with the IL-6 gene, suggesting perhaps that IL-18- and IL-6-mediated IL-6 amplifier activation plays a role. In these cases, local events can determine the tissue specificity for the development of diseases like F759 arthritis even in the absence of tissue antigen-recognition by activated T cells (Hirano, 2010; Murakami et al., 2011).
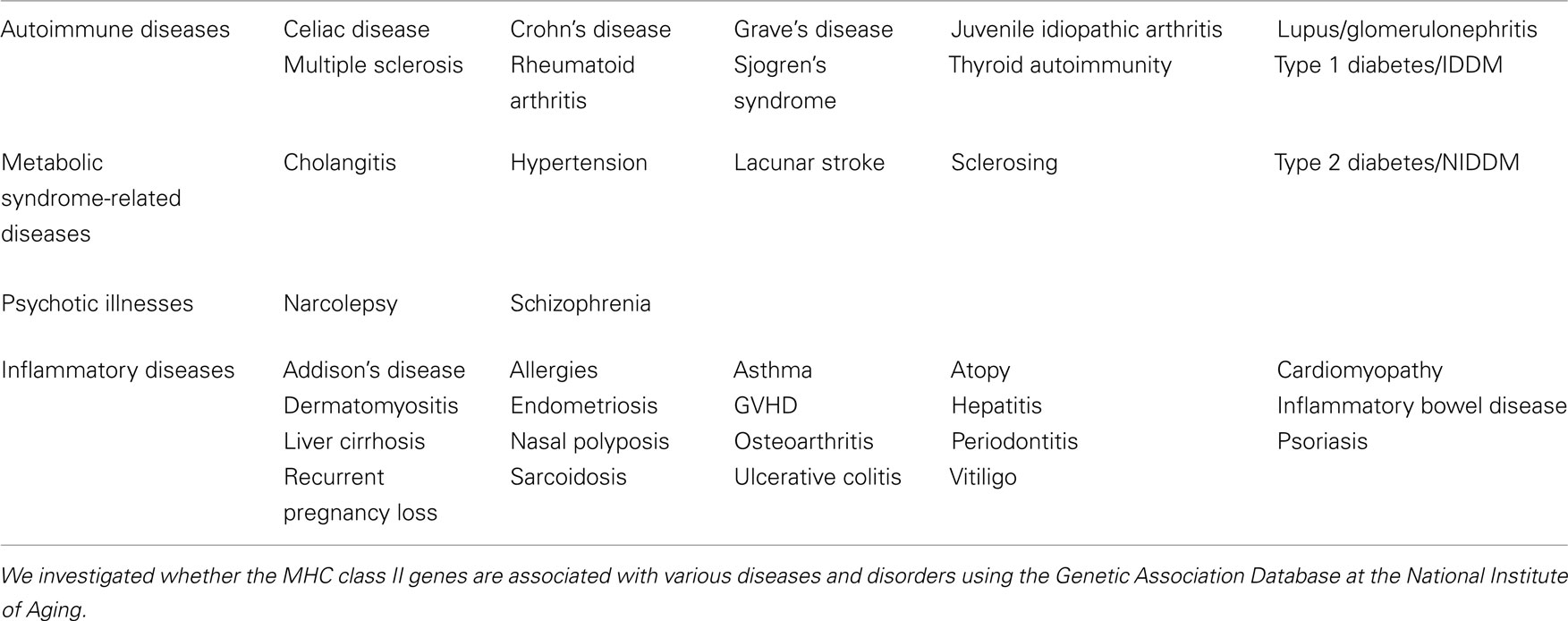
Table 1. The MHC class II genes are associated with various diseases and disorders.
To summarize, we have investigated how MHC class II-associated tissue-specific autoimmune arthritis develops and propose a four-step model to explain the process. This model provides a possible explanation for why tissue-specific antigens recognized by activated CD4+ T cells have not been identified in many tissue-specific autoimmune diseases associated with class II MHC molecules including RA. This may be explained by our four-step model, which highlights the idea that the tissue itself can determine the tissue specificity of the autoimmune disease. Additionally, genetic and environmental factors affecting the target tissue are involved. These results have led us to propose that direct activation of the IL-6 amplifier by STAT3 and NF-κB can result in chronic inflammatory diseases that are not apparently associated with MHC class II. Thus, we expect our four-step model will provide new and important insights on the immunological mechanisms that underlie autoimmune disease as well as other chronic inflammatory diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs D. Kamimura, H. Ogura, and especially P. Karagiannis (Osaka University, Osaka, Japan) for carefully reading the manuscript. This work was supported by KAKENHI (Masaaki Murakami and Toshio Hirano), the JST-CREST program (Masaaki Murakami and Toshio Hirano), and the Osaka Foundation for the Promotion of Clinical Immunology (Masaaki Murakami).
References
Andersson, I. E., Batsalova, T., Dzhambazov, B., Edvinsson, L., Holmdahl, R., Kihlberg, J., and Linusson, A. (2010). Oxazole-modified glycopeptides that target arthritis-associated class II MHC A(q) and DR4 proteins. Org. Biomol. Chem. 8, 2931–2940.
Atsumi, T., Ishihara, K., Kamimura, D., Ikushima, H., Ohtani, T., Hirota, S., Kobayashi, H., Park, S., Saeki, Y., Kitamura, Y., and Hirano, T. (2002). A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J. Exp. Med. 196, 979–990.
Awasthi, A., and Kuchroo, V. K. (2009). Th17 cells: from precursors to players in inflammation and infection. Int. Immunol. 21, 489–498.
Bettelli, E., Oukka, M., and Kuchroo, V. K. (2007). T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 345–350.
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5.
Brennan, F. M., Hayes, A. L., Ciesielski, C. J., Green, P., Foxwell, B. M., and Feldmann, M. (2002). Evidence that rheumatoid arthritis synovial T cells are similar to cytokine-activated T cells: involvement of phosphatidylinositol 3-kinase and nuclear factor kappaB pathways in tumor necrosis factor alpha production in rheumatoid arthritis. Arthritis Rheum. 46, 31–41.
Chapuy-Regaud, S., Sebbag, M., Baeten, D., Clavel, C., Foulquier, C., De Keyser, F., and Serre, G. (2005). Fibrin deimination in synovial tissue is not specific for rheumatoid arthritis but commonly occurs during synovitides. J. Immunol. 174, 5057–5064.
Cherwinski, H. M., Schumacher, J. H., Brown, K. D., and Mosmann, T. R. (1987). Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J. Exp. Med. 166, 1229–1244.
Compagno, M., Lim, W. K., Grunn, A., Nandula, S. V., Brahmachary, M., Shen, Q., Bertoni, F., Ponzoni, M., Scandurra, M., Califano, A., Bhagat, G., Chadburn, A., Dalla-Favera, R., and Pasqualucci, L. (2009). Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 459, 717–721.
Constant, S. L., and Bottomly, K. (1997). Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu. Rev. Immunol. 166, 297–322.
Cua, D. J., Sherlock, J., Chen, Y., Murphy, C. A., Joyce, B., Seymour, B., Lucian, L., To, W., Kwan, S., Churakova, T., Zurawski, S., Wiekowski, M., Lira, S. A., Gorman, D., Kastelein, R. A., and Sedgwick, J. D. (2003). Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748.
Falgarone, G., Semerano, L., Rullé, S., and Boissier, M. C. (2009). Targeting lymphocyte activation to treat rheumatoid arthritis. Joint Bone Spine. 76, 327–332.
Feldmann, M., and Maini, R. N. (2001). Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu. Rev. Immunol. 19, 163–196.
Fukada, T., Hibi, M., Yamanaka, Y., Takahashi-Tezuka, M., Fujitani, Y., Yamaguchi, T., Nakajima, K., and Hirano, T. (1996). Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity 5, 449–460.
Glimcher, L. H., and Murphy, K. M. (2000). Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 14, 1693–1711.
Gudmundsdottir, H., and Turka, L. A. (2001). A closer look at homeostatic proliferation of CD4+ T cells: costimulatory requirements and role in memory formation. J. Immunol. 167, 3699–3707.
Harrington, L. E., Hatton, R. D., Mangan, P. R., Turner, H., Murphy, T. L., Murphy, K. M., and Weaver, C. T. (2005). Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132.
Hasler, P., and Zouali, M. (2005). Immune receptor signaling, aging, and autoimmunity. Cell. Immunol. 233, 102–108.
Hirano, T. (2002). Revival of the autoantibody model in rheumatoid arthritis. Nat. Immunol. 3, 342–344.
Hirano, T. (2010). Interleukin 6 in autoimmune and inflammatory diseases: a personal memoir. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86, 717–730.
Hirano, T., Matsuda, T., Turne, M., Miyasaka, N., Buchan, G., Tang, B., Sato, K., Shimizu, M., Maini, R., Feldmann, M., and Kishimoto, T. (1988). Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur. J. Immunol. 18, 1797–1801.
Imboden, J. B. (2009). The immunopathogenesis of rheumatoid arthritis. Annu. Rev. Pathol. 4, 417–434.
Ishihara, K., Sawa, S., Ikushima, H., Hirota, S., Atsumi, T., Kamimura, D., Park, S., Murakami, M., Kitamura, Y., Iwakura, Y., and Hirano, T. (2004). The point mutation of tyrosine 759 of the IL-6 family cytokine receptor gp130 synergizes with HTLV-1 pX in promoting rheumatoid arthritis-like arthritis. Int. Immunol. 16, 455–465.
Iwakura, Y., Tosu, M., Yoshida, E., Takiguchi, M., Sato, K., Kitajima, I., Nishioka, K., Yamamoto, K., Takeda, T., Hatanaka, M., Yamamoto, H., and Sekiguchi, T. (1991). Induction of inflammatory arthropathy resembling rheumatoid arthritis in mice transgenic for HTLV-I. Science 253, 1026–1028.
Jang, E., Kim, H. R., Cho, S. H., Paik, D. J., Kim, J. M., Lee, S. G., and Youn, J. (2006). Prevention of spontaneous arthritis by inhibiting homeostatic expansion of autoreactive CD4+ T cells in K/BxN mouse model. Arthritis Rheum. 54, 492–498.
Kamimura, D., Ishihara, K., and Hirano, T. (2003). IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev. Physiol. Biochem. Pharmacol. 149, 1–38.
Khiong, K., Murakami, M., Kitabayashi, C., Ueda, N., Sawa, S., Sakamoto, A., Kotzin, B. L., Rozzo, S. J., Ishihara, K., Verella-Garcia, M., Kappler, J., Marrack, P., and Hirano, T. (2007). Homeostatically proliferating CD4 T cells are involved in the pathogenesis of an Omenn syndrome murine model. J. Clin. Invest. 117, 1270–1281.
King, C., Ilic, A., Koelsch, K., and Sarvetnick, N. (2004). Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell 117, 265–277.
Kivity, S., Agmon-Levin, N., Blank, M., and Shoenfeld, Y. (2009). Infections and autoimmunity–friends or foes? Trends Immunol. 30, 409–414.
Larbi, A., Fülöp, T., and Pawelec, G. (2008). Immune receptor signaling, aging and autoimmunity. Adv. Exp. Med. Biol. 640, 312–324.
Lenz, G., Davis, R. E., Ngo, V. N., Lam, L., George, T. C., Wright, G. W., Dave, S. S., Zhao, H., Xu, W., Rosenwald, A., Ott, G., Muller-Hermelink, H. K., Gascoyne, R. D., Connors, J. M., Rimsza, L. M., Campo, E., Jaffe, E. S., Delabie, J., Smeland, E. B., Fisher, R. I., Chan, W. C., and Staudt, L. M. (2008). Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679.
Marrack, P., Kappler, J., and Kotzin, B. (2001). Autoimmune disease: why and where it occurs. Nat. Med. 7, 899–905.
Matsumoto, I., Staub, A., Benoist, C., and Mathis, D. (1999). Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 286, 1732–1735.
Minegishi, Y., Saito, M., Tsuchiya, S., Tsuge, I., Takada, H., Hara, T., Kawamura, N., Ariga, T., Pasic, S., Stojkovic, O., Metin, A., and Karasuyama, H. (2007). Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448, 1058–1062.
Mocci, S., Lafferty, K., and Howard, M. (2000). The role of autoantigens in autoimmune disease. Curr. Opin. Immunol. 12, 725–730.
Mosmann, T. R., and Coffman, R. L. (1989). TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7, 145–173.
Münz, C., Lünemann, J. D., Getts, M. T., and Miller, S. D. (2009). Antiviral immune responses: triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 9, 246–258.
Murakami, M., Kamimura, D., and Hirano, T. (2004). New IL-6 (gp130) family cytokine members, CLC/NNT1/BSF3 and IL-27. Growth Factors 22, 75–77.
Murakami, M., Okuyama, Y., Ogura, H., Asao, S., Arima, Y., Tsuruoka, M., Harada, M., Kanamoto, M., Iwakura, Y., Takatsu, K., Kamimura, D., and Hirano, T. (2011). Local microbleeding facilitates IL-6– and IL-17–dependent arthritis in the absence of tissue antigen recognition by activated T cells. J. Exp. Med. 208, 103–114.
Muromoto, R., Ikeda, O., Okabe, K., Togi, S., Kamitani, S., Fujimuro, M., Harada, S., Oritani, K., and Matsuda, T. (2009). Epstein-Barr virus-derived EBNA2 regulates STAT3 activation. Biochem. Biophys. Res. Commun. 378, 439–443.
Nakagawa, T., Tsuruoka, M., Ogura, H., Okuyama, Y., Arima, Y., Hirano, T., and Murakami, M. (2010). IL-6 positively regulates Foxp3+CD8+ T cells in vivo. Int. Immunol. 22, 129–139.
Nickdel, M. B., Conigliaro, P., Valesini, G., Hutchison, S., Benson, R., Bundick, R. V., Leishman, A. J., McInnes, I. B., Brewer, J. M., and Garside, P. (2009). Dissecting the contribution of innate and antigen-specific pathways to the breach of self-tolerance observed in a murine model of arthritis. Ann. Rheum. Dis. 68, 1059–1066.
Nishihara, M., Ogura, H., Ueda, N., Tsuruoka, M., Kitabayashi, C., Tsuji, F., Aono, H., Ishihara, K., Huseby, E., Betz, U. A., Murakami, M., and Hirano, T. (2007). IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 19, 695–702.
Ogata, H., Chinen, T., Yoshida, T., Kinjyo, I., Takaesu, G., Shiraishi, H., Iida, M., Kobayashi, T., and Yoshimura, A. (2006). Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene 25, 2520–2530.
Ogura, H., Murakami, M., Okuyama, Y., Tsuruoka, M., Kitabayashi, C., Kanamoto, M., Nishihara, M., Iwakura, Y., and Hirano, T. (2008). Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 29, 628–636.
Ohtani, T., Ishihara, K., Atsumi, T., Nishida, K., Kaneko, Y., Miyata, T., Itoh, S., Narimatsu, M., Maeda, H., Fukada, T., Itoh, M., Okano, H., Hibi, M., and Hirano, T. (2000). Dissection of signaling cascades through gp130 in vivo: reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity 12, 95–105.
O’Shea, J. J., Ma, A., and Lipsky, P. (2002). Cytokines and autoimmunity. Nat. Rev. Immunol. 2, 37–45.
Park, H., Li, Z., Yang, X. O., Chang, S. H., Nurieva, R., Wang, Y. H., Wang, Y., Hood, L., Zhu, Z., Tian, Q., and Dong, C. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6, 1133–1141.
Polgár, A., Falus, A., Koó, E., Ujfalussy, I., Seszták, M., Szuts, I., Konrád, K., Hodinka, L., Bene, E., Mészáros, G., Ortutay, Z., Farkas, E., Paksy, A., and Buzás, E. I. (2003). Elevated levels of synovial fluid antibodies reactive with the small proteoglycans biglycan and decorin in patients with rheumatoid arthritis or other joint diseases. Rheumatology (Oxford) 42, 522–527.
Rebouissou, S., Amessou, M., Couchy, G., Poussin, K., Imbeaud, S., Pilati, C., Izard, T., Balabaud, C., Bioulac-Sage, P., and Zucman-Rossi, J. (2009). Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 457, 200–204.
Sakaguchi, S., and Sakaguchi, N. (2005). Animal model of arthritis caused by systemic alteration of the immune system. Curr. Opin. Immunol. 17, 589–594.
Sawa, S., Kamimura, D., Jin, G. H., Morikawa, H., Kamon, H., Nishihara, M., Ishihara, K., Murakami, M., and Hirano, T. (2006). Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J. Exp. Med. 12, 1459–1470.
Sawa, Y., Arima, Y., Ogura, H., Kitabayashi, C., Jiang, J. J., Fukushima, T., Kamimura, D., Hirano, T., and Murakami, M. (2009). Hepatic interleukin-7 expression regulates T cell responses. Immunity 30, 447–457.
Sims, G. P., Rowe, D. C., Rietdijk, S. T., Herbst, R., and Coyle, A. J. (2010). HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 24, 267–388.
Skapenko, A., Leipe, J., Lipsky, P. E., and Schulze-Koops, H. (2005). The role of the T cell in autoimmune inflammation. Arthritis Res. Ther. 7, S4–S14.
Sugiura, T., Kawaguchi, Y., Harigai, M., Terajima-Ichida, H., Kitamura, Y., Furuya, T., Ichikawa, N., Kotake, S., Tanaka, M., Hara, M., and Kamatani, N. (2002). Association between adult-onset Still’s disease and interleukin-18 gene polymorphisms. Genes Immun. 3, 394–399.
Surh, C. D., and Sprent, J. (2000). Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J. Exp. Med. 192, F9–F14.
Suthaus, J., Tillmann, A., Lorenzen, I., Bulanova, E., Rose-John, S., and Scheller, J. (2010). Forced homo- and heterodimerization of all gp130-type receptor complexes leads to constitutive ligand-independent signaling and cytokine-independent growth. Mol. Biol. Cell 21, 2797–2807.
Takizawa, Y., Suzuki, A., Sawada, T., Ohsaka, M., Inoue, T., Yamada, R., and Yamamoto, K. (2006). Citrullinated fibrinogen detected as a soluble citrullinated autoantigen in rheumatoid arthritis synovial fluids. Ann. Rheum. Dis. 65, 1013–1020.
Van Steendam, K., Tilleman, K., De Ceuleneer, M., De Keyser, F., Elewaut, D., and Deforce, D. (2010). Citrullinated vimentin as an important antigen in immune complexes from synovial fluid of rheumatoid arthritis patients with antibodies against citrullinated proteins. Arthritis Res. Ther. 12, R132.
Veldhoen, M., Hocking, R., Atkins, C., Locksley, R., and Stockinger, B. (2006). TGF in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189.
Williams, M. A., Ravkov, E. V., and Bevan, M. J. (2008). Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity 28, 533–545.
Yoshida, T., Hanada, T., Tokuhisa, T., Kosai, K., Sata, M., Kohara, M., and Yoshimura, A. (2002). Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J. Exp. Med. 196, 641–653.
Zhang, L., Nakayama, M., and Eisenbarth, G. S. (2008). Insulin as an autoantigen in NOD/human diabetes. Curr. Opin. Immunol. 20, 111–118.
Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal, T., Junger, W., Brohi, K., Itagaki, K., and Hauser, C. J. (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107.
Keywords: MHC class II association, autoimmune diseases, inflammation, IL-6-mediated inflammation amplifier, cytokines, chemokines, Th17 cells
Citation: Murakami M and Hirano T (2011) A four-step model for the IL-6 amplifier, a regulator of chronic inflammations in tissue-specific MHC class II-associated autoimmune diseases. Front. Immun. 2:22. doi: 10.3389/fimmu.2011.00022
Received: 27 April 2011;
Paper pending published: 24 May 2011;
Accepted: 06 June 2011;
Published online: 16 June 2011.
Edited by:
Anna Rubartelli, National Cancer Research Institute, ItalyReviewed by:
Massimo Gadina, National Institute of Arthritis Musculoskeletal and Skin diseases-National Institutes of Health, USAMarco Gattorno, G. Gaslini Institute for Children, Italy
Copyright: © 2011 Murakami and Hirano. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Toshio Hirano, Laboratory of Developmental Immunology, JST-CREST, Graduate School of Frontier Biosciences, Osaka University, Osaka, Japan. e-mail: hirano@molonc.med.osaka-u.ac.jp