
- 1 Immunology Research Unit, Carmel Medical Center, The Ruth and Bruce Rappaport Faculty of Medicine, Technion, Haifa, Israel
- 2 Ischemia-shock Research Laboratory, Carmel Medical Center, The Ruth and Bruce Rappaport Faculty of Medicine, Technion, Haifa, Israel
Monocytes and Macrophages (Mo/Mɸ) exhibit great plasticity, as they can shift between different modes of activation and, driven by their immediate microenvironment, perform divergent functions. These include, among others, patrolling their surroundings and maintaining homeostasis (resident Mo/Mɸ), combating invading pathogens and tumor cells (classically activated or M1 Mo/Mɸ), orchestrating wound healing (alternatively activated or M2 Mo/Mɸ), and restoring homeostasis after an inflammatory response (resolution Mɸ). Hypoxia is an important factor in the Mɸ microenvironment, is prevalent in many physiological and pathological conditions, and is interdependent with the inflammatory response. Although Mo/Mɸ have been studied in hypoxia, the mechanisms by which hypoxia influences the different modes of their activation, and how it regulates the shift between them, remain unclear. Here we review the current knowledge about the molecular mechanisms that mediate this hypoxic regulation of Mɸ activation. Much is known about the hypoxic transcriptional regulatory network, which includes the master regulators hypoxia-induced factor-1 and NF-κB, as well as other transcription factors (e.g., AP-1, Erg-1), but we also highlight the role of post-transcriptional and post-translational mechanisms. These mechanisms mediate hypoxic induction of Mɸ pro-angiogenic mediators, suppress M1 Mɸ by post-transcriptionally inhibiting pro-inflammatory mediators, and help shift the classically activated Mɸ into an activation state which approximate the alternatively activated or resolution Mɸ.
Introduction
Many physiological and pathological processes (e.g., inflammation, wound healing, acute myocardial infarction, retinopathies, atherosclerosis, solid tumors, and more) are characterized by both low oxygen tensions (hypoxia) and presence of monocytes/macrophages (Mo/Mɸ). Mo/Mɸ are essential regulators of inflammation and central participants in hypoxia-driven processes, and the mediators they express and secrete recruit other cells and orchestrate their activity. The role of Mo/Mɸ and the effects hypoxia exerts on them were studied mostly in solid tumors, but the same molecular mechanisms apply in other clinical scenarios. We review here the different molecular mechanisms exerted by hypoxia, which regulate Mo/Mɸ functions. We elaborate on the hypoxia-induced transcriptional network in Mɸ, which is driven mostly by the hypoxia-induced factors (HIFs) and NF-κB, the two master regulators of the hypoxic response. We also draw attention to additional post-transcriptional, translational, and post-translational mechanisms that enable hypoxia to activate or suppress gene and protein expression. Finally, we review the relatively few studies on the mechanisms and effects of hypoxia on differently activated Mo/Mɸ, and discuss how these mechanisms help shift the pro-inflammatory activated Mɸ toward an anti-inflammatory, pro-angiogenic phenotype.
We do not expand on how hypoxia-induced mechanisms affect the different Mɸ functions, as these aspects were extensively reviewed before (Lewis et al., 1999; Murdoch et al., 2004, 2008; Bosco et al., 2008; Martinez et al., 2008; Coffelt et al., 2009; Walmsley et al., 2009; Qian and Pollard, 2010). Because the investigation of mechanisms is most often performed in vitro, we limit our discussion to these studies, and only briefly mention the increasing number of evidences pointing to the crucial role hypoxia plays in vivo in the pathophysiology of many diseases. Moreover, although hypoxia is physiologically and pathologically followed by re-oxygenation, we focus only on the isolated effects of hypoxic stress on Mo/Mɸ. Likewise, the effects of reduced pH, low glucose levels, or increased lactate, conditions that also accompany hypoxia and ischemia, are not discussed.
Different Modes of Macrophage Activation
Monocytes (Mo) migrate into tissues and differentiate into Macrophages (Mɸ) to perform many functions needed in the tissue. These include resident Mɸ that patrol their surroundings and maintain homeostasis, Mɸ that combat invading pathogens and tumor cells and protect the tissue, Mɸ that orchestrate the process of wound healing, and Mɸ that resolve inflammation. To perform these multiple tasks, Mɸ phagocytose pathogens, secrete cytokines, chemokines, and growth factors, present antigenic determinants to T cells and activate them, scavenge dead cells and necrotic debris and deposit matrix proteins. Since one cell cannot perform all these divergent tasks simultaneously, it was suggested that Mɸ can be differently activated, depending on the signals received from the microenvironment. Thus, Mɸ exhibit an enormous plasticity (Stout and Suttles, 2004), and dynamically shift from one form of activation to another according to the conditions in the changing microenvironment (Stout et al., 2009). This concept, as well as the evidences supporting it and the markers that characterize each Mɸ phenotype, has been extensively reviewed (Mosser, 2003; Martinez et al., 2008; Mosser and Edwards, 2008; Murdoch et al., 2008; Gordon and Martinez, 2010; Qian and Pollard, 2010), and will be only briefly mentioned here.
In the continuum of Mɸ phenotypes, two main sub-populations have been described. In one extreme, the classically activated Mɸ, or M1 Mɸ, is responsible for the multi-stage process of recognizing pathogens, phagocytosing, and degrading them to present their antigenic determinant to helper T cells in the context of the MHC class II molecules, whose expression is elevated (Mosser, 2003; Martinez et al., 2008). This process is accompanied by secretion of IL-12, which is necessary to support a Th1 response. M1 Mɸ are activated by ligands of different receptor families such as the toll-like receptor (TLR) ligands (e.g., lipopolysaccharides – LPS), and pro-inflammatory cytokines (e.g., interferon-Γ – IFNΓ, tumor necrosis factor-α – TNFα, and interleukin-1β – IL-1β). They secrete high amounts of pro-inflammatory mediators that kill the invading pathogens or tumor cells, such as the cytotoxic TNFα and nitric oxide (NO), the latter (and the enzyme producing it, inducible nitric oxide synthase – iNOS) serving as the hallmark of this Mɸ subset. They also secrete chemokines, which attract more neutrophils and Mo/Mɸ to the inflamed site, thus amplifying the pro-inflammatory response. Secretion of proteolytic enzymes, such as matrix metalloproteinases (MMPs) helps degrade components of the extracellular matrix (ECM) and allows migration of leukocytes to the inflamed tissue.
In the other extreme we find Mɸ that are activated by and secrete anti-inflammatory mediators (e.g., IL-10, IL-13, tumor growth factor beta – TGFβ, and prostaglandin E2 – PGE2), which generate a microenvironment that suppresses the activity of M1 macrophage. They remove cellular debris and perform phagocytosis which is mediated by receptors different than those expressed by M1 Mɸ, such as the mannose receptor CD206; contribute to tissue remodeling by depositing ECM proteins; and express high levels of arginase-1, which produces ornithine, a precursor for the synthesis of the ECM protein collagen. Arginase-1 also competes with iNOS for their common substrate L-arginine and thus prevents NO production. Thus, these alternatively activated or M2 Mɸ are involved mainly in homeostasis and wound healing. M2 Mɸ also secrete proteolytic enzymes that help them move about, and as they are activated by IL-4 and IL-13, they are particularly suited to present helminthes antigens to helper T cells and initiate a Th2 response (Martinez et al., 2009), so that the term “anti-inflammatory” refers, in fact, to a different type of inflammation.
A third subset of Mɸ is responsible for immune regulation, and may include several subtypes. Some of these cells are activated by ligands of TLRs in combination with immune complexes, and some are activated by anti-inflammatory signals, such as adenosine or phagocytosed apoptotic cells (Mosser, 2003; Mosser and Edwards, 2008). Immature Mɸ, which compose some of the myeloid-derived suppressor cells (MDSCs) population, also belong to regulatory Mɸ. MDSCs are triggered by a combination of IFNΓ and IL-13, and secrete IFNΓ, IL-13, IL-10, and TGFβ, which help them suppress Th1 cell-mediated immune response, induce regulatory T cells and inhibit M1 Mɸ (Bronte, 2009; Gabrilovich and Nagaraj, 2009; Ostrand-Rosenberg and Sinha, 2009).
A different subset of Mɸ isolated at the resolution-phase of inflammation (resolution or rMɸ) is often activated by mediators released from apoptotic cells, such as sphingosine-1-phosphate (S1P) or TGFβ. These rMɸ secrete predominantly the anti-inflammatory IL-10, very low levels of pro-inflammatory cytokines and chemokines and express arginase-1 and CD206, as well as the pro-inflammatory iNOS and COX-2 (Bystrom et al., 2008). Moreover, in a model of self-resolving peritonitis, rMɸ were further divided into CD11bhigh and CD11blow cells (Schif-Zuck et al., 2011). CD11bhigh rMɸ engulf apoptotic neutrophils and stay at the inflammatory site, where they express markers of both M1 and M2 Mɸ (e.g., iNOS, arginase-1, COX-2, and MMP-9). However, once they engulf enough apoptotic cells (threshold determined at 7 cells), they convert into CD11blow cells, induce the expression of the enzyme 12/15-lipoxygenase which produces pro-resolving lipid mediators, and emigrate to lymphoid organs to convey resolution signals to lymphocytes (Schif-Zuck et al., 2011).
Collectively, all these regulatory Mɸ, which secrete anti-inflammatory mediators designed to resolve inflammation, are positioned between M1 and M2 Mɸ, and share markers with these two sub-populations.
Malignant tumors are characterized by repeated cycles of hypoxia and reoxygenation, and several Mɸ subsets have been found located in different regions of the tumor (Lewis and Pollard, 2006). These include the tumor-associated macrophages (TAMs) and the Tie-2 expressing macrophages (TEMs), which support tumor growth and metastasis, and are both an important source of pro-angiogenic factors. Whereas some TEMs reside close to blood vessels (peri-endothelial; Venneri et al., 2007), TAMs infiltrate deeper into the tumor and are located in perinecrotic areas where oxygen tensions are very low. Regulatory MDSCs are present within tumors, as well as in lymphoid organs, such as the bone marrow and spleen, and expand proportionally to the tumor burden. All these subtypes share activation markers with both M2 and regulatory Mɸ, thus placing them between these two subsets (De Palma et al., 2007; Mosser and Edwards, 2008; Murdoch et al., 2008). Of note, the principle of Mɸ plasticity extends even to TAMs subset, as they exhibit divergent phenotypes (Lewis and Pollard, 2006), depending on the type of the tumor, the stage of its development, the interactions with both the ECM and other neighboring cells, and the level of oxygen tensions they are exposed to.
Moving from Mɸ to Mo, we find the monocytes to have similar but not identical classifications. Circulatory Mo are divided into three subsets according to their CD14 and CD16 expression (in humans) or Ly6C expression (in mice). The Classical or M1 Mo express high levels of CD14 and no CD16 (denoted either CD14++CD16− or CD14+CD16− in human, and Ly6Chigh in mice), intermediate Mo, in humans only, express intermediate levels of CD14 and CD16 (CD14++CD16+ or CD14+CD16+), and resident, M2 or non-classical monocytes express very low levels of CD14 and high levels of CD16 (CD14dimCD16+ or CD14−CD16+ in human, and Ly6Clow in mice) (Ziegler-Heitbrock, 2007; van de Veerdonk and Netea, 2010). However, the function of these subsets as cells secreting either pro- or anti-inflammatory cytokines is still controversial. It is assumed that M1 and M2 Mo are the counterparts of M1 and M2 Mɸ, but this is not always accurate. For example, sorted Mo subsets that were stimulated ex vivo with LPS increased IL-10 secretion from CD14+CD16+ cells, whereas LPS-stimulated CD14dimCD16+ cells showed increased TNFα with little IL-10 secretion (Skrzeczynska-Moncznik et al., 2008). In a different study, CD14−CD16+ cells showed patrolling characteristics with weak ability for phagocytosis and low production of ROS and cytokines when challenged by bacterial ligands of TLRs, but secreted high amounts of the pro-inflammatory TNFα and IL-1β cytokines upon stimulation with viral ligands or nucleic acids (Cros et al., 2010). These results suggest that non-classical or M2 Mo can behave as M1 Mo under certain circumstances. Thus, Mo classification does not fully match the M1-M2 Mɸ taxonomy.
During inflammation differently activated Mo/Mɸ enter the tissue, first in order to eliminate pathogens or tumor cells (M1 Mo/Mɸ), and later to heal the damaged tissue and to restore homeostasis (M2 Mo/Mɸ and rMɸ; Nahrendorf et al., 2010). This requires precise signals that regulate this sequential migration. For example, the chemokines CCL2 and CX3CL1 and their receptors, expressed on CD14+CD16− and both CD16+ monocytes, respectively, were shown to mediate these responses (Geissmann et al., 2003; Auffray et al., 2009). Of note, this specific sequence of events may not always apply, and clinical scenarios which favor a Th2 response may differentially recruit non-classical Mo. Furthermore, whether classically activated Mo mature to M1 Mɸ in the tissue and are later skewed to become M2 Mɸ, or whether they die on site and are replaced by newly recruited M2 Mo, remains unclear, and the effects of the hypoxic microenvironment in this context must be further explored.
According to the danger model (Matzinger, 2002), the direction and magnitude of the inflammatory response depend not only on the nature of the stimulus (e.g., types of bacteria, site of entry, size of the bacterial inoculation), but also on the type and extent of expression of co-stimulatory molecules (e.g., CD80 vs. CD86) and specific signals received from the microenvironment, such as danger-associated molecular patterns (DAMPs). Necrotic and apoptotic cells that release their content into the microenvironment, are picked up by Mo/Mɸ, processed and presented to T cells in the context of MHC class II and co-stimulatory molecules. Thus, microenvironmental stimuli can affect Mo/Mɸ functions, directly or via their effect on other cells.
These examples reveal the high plasticity of Mo/Mɸ, and emphasize that their heterogeneity ranges beyond the M1–M2 polarization models, and extends to new phenotypes we may yet discover. Thus, Mo/Mɸ classification and the distinction between their different activation modes and differentiation status is an active area of study.
Inflammation and Hypoxia
Low oxygen tensions (hypoxia) are relevant in many physiological and pathological conditions. For example, hypoxia may arise due to occlusion of a blood vessel (e.g., during myocardial infarction or pulmonary embolism), significant blood loss or dilated blood vessels which instigate reduced blood flow and lack of oxygen (e.g., in burns, trauma, and sepsis), or simply when oxygen demands are not met (e.g., physical exercise, high altitudes, tumors). Hypoxia has been demonstrated in vivo in a variety of acute and chronic inflammatory sites, including the synovium in RA patients, the arterial intima in atherosclerotic lesions, myocardial infarcts, wounds, and sites of bacterial infection (reviewed in Murdoch et al., 2005). Since even healthy tissues exhibit a wide range of oxygen tensions, no one value can represent hypoxia, and hypoxia is, therefore, functionally defined as the inability of oxygen delivery to meet oxygen demands of the tissue (Papandreou et al., 2005). According to this definition, hypoxia occurs during inflammation, as more cells that require oxygen infiltrate the site and increase oxygen consumption, but only few capillaries can supply it. Thus, different oxygen tensions can be measured in different areas of tumors, wounds, or in tissues affected by chronic inflammation, generating a gradient of hypoxia, where levels as low as 5–10 mmHg were measured (Crowther et al., 2001). Of note, other microenvironmental factors, such as increased lactate concentrations and reduced pH, are linked to hypoxia, as they result from the shift to anaerobic metabolism. Therefore, investigating the role of prolonged hypoxia actually looks into the combined effect of all of these stress factors. Most in vitro studies measure the percent oxygen in the atmosphere flowing into the hypoxic chamber, but usually even in anoxic atmosphere (0% O2) some oxygen remains dissolved in the medium (20–30 mmHg).
Depending on its duration and severity, hypoxia can drive inflammation and aggravate cellular and tissue injury. For example, humans exposed to the scarce oxygen available in high altitudes or mice exposed to ambient hypoxia experience reduced arterial partial oxygen tensions, and develop pulmonary edema and increased release of pro-inflammatory cytokines (Hartmann et al., 2000; Grocott et al., 2009; Rosenberger et al., 2009). Hypoxia shifts cells toward anaerobic metabolism, leading to exclusive use of glycolysis as the means for ATP production, instead of Krebs cycle. As a result, lactate accumulates in the cells, causing cellular acidosis, production of ROS is increased, and lipids are peroxidated leading to membranal damage. This, combined with the lack of ATP, impairs the function of ion channels, increases Ca+2 influx, and membrane permeability, leading to spillage of cellular content (including proteolytic enzymes, hydrolases, and increased lactate), thereby increasing acidosis and tissue damage (Minko et al., 2005; Behn et al., 2007). Necrotic cells and increased tissue acidosis recruit more leukocytes into the area, and trigger inflammation. Hence, hypoxia and inflammation are interdependent, as chronic inflammation is accompanied by hypoxia and prolonged hypoxia leads to inflammation (Eltzschig, 2011).
Role of HIFs
Like other cell types, the macrophage response depends on the severity and the duration of the hypoxic insult. In recent years, the search for the way cells sense differences in oxygen tensions was focused primarily on the prolyl hydroxylases (PHD)–HIF pathway outlined below.
Hypoxia-induced factors are heterodimeric transcription factors consisting of a constitutively expressed β subunit (aryl hydrocarbon nuclear translocator – ARNT), and an α subunit, which is constitutively transcribed but immediately degraded in normoxia. Three α subunits (HIF-1α, HIF-2α, and HIF-3α) have been identified, which together with HIF-1β compose three isoforms that are differently expressed in various tissues. HIF-3α (also called inhibitory PAS domain protein – IPAS) is a transcriptional inhibitor and functions as a negative regulator of HIF-1α and HIF-2α, although its biology is still unclear (Heikkila et al., 2011), whereas HIF-1α and HIF-2α were more extensively studied. HIF-1α and HIF-2α are homologous, especially in their DNA binding domains, allowing their binding to the same hypoxia response element (HRE).
The binding of HIF-1 and HIF-2 to many HRE-containing promoters can induce the expression of a myriad of genes, some with opposing effects. By means of over-expressing the HIF-α subunits, or by ablating them in knockouts or by siRNA knock-down, the role of each subunit in determining Mɸ phenotypes and response to hypoxic stress was established. Thus, HIF proteins were shown to regulate all aspects of Mɸ functions in response to hypoxia, including the shift to anaerobic glycolysis, mitochondrial impairment, angiogenesis, invasion, and immune suppression (Coffelt et al., 2009; Walmsley et al., 2009; Werno et al., 2010a). Moreover, HIFs are also induced in normoxia, and play a central role in immune recognition, phagocytosis, bacterial killing, and pro-inflammatory cytokine production (Cramer et al., 2003; Peyssonnaux et al., 2007).
HIFs Expression in Different Mɸ Subsets
The two HIF isoforms, HIF-1α and HIF-2α are differently expressed in Mo and Mɸ, as hypoxia could not induce the expression of HIF-1α or HIF-2α in human Mo, but did induce them in human monocyte-derived macrophages (hMDM; Elbarghati et al., 2008). Differentiated human and mouse MDM show different kinetics of HIFs expression in response to hypoxia, with higher stability for HIF-2α expression (Elbarghati et al., 2008; Takeda et al., 2010). This suggests that HIF-α subunits could, at least partially, affect Mɸ activation modes. Furthermore, arginine metabolism was enhanced toward NO production in a HIF-1α-dependent manner upon LPS or IFNΓ stimulation, but NO production was reduced by IL-4 due to increase in HIF-2α-induced arginase-1 activity (Takeda et al., 2010). Conflicting evidences show higher accumulation of HIF-1α in hypoxic hMDM (Burke et al., 2002), or higher expression of HIF-2α under similar conditions (Griffiths et al., 2000). One study showed almost undetectable HIF-2α mRNA after prolonged exposure to hypoxia, but induced HIF-1α mRNA that was even further elevated by LPS in thioglycollate (TG)-elicited peritoneal Mɸ (Acosta-Iborra et al., 2009), whereas another study showed constitutive HIF-2α mRNA levels in these cells (Takeda et al., 2010). TAMs present in human breast carcinomas show high expression of HIF-2α, which correlates with increased micro-vessel density and high tumor grade (Patel and Simon, 2008). TAMs in different tumors in vivo show high expression of both HIF-1α and HIF-2α (Lewis and Pollard, 2006). The importance of exposure time is demonstrated in a study that compared acute (24 h) and chronic (5 days) exposures to hypoxia, and reported that hMDM subjected to chronic hypoxia increased HIF-1α and HIF-2α proteins relative to hMDM that were subjected to normoxia or to acute hypoxia (Staples et al., 2010). However, there is still no clear perspective regarding the role of the different HIFs in hypoxic Mɸ, and different periods of time of hypoxic exposure or different oxygen levels may have different effects on HIFs’ expression.
Regulation of HIFs in Normoxia and Hypoxia
Protein stability
Hypoxia-induced factor proteins are regulated primarily by protein stability. HIF-α subunits are constitutively transcribed and translated, but immediately directed for degradation in normoxia. This is achieved by the hydroxylation of proline residues (402 and 564 on HIF-1α and 405 and 532 on HIF-2α) by the three PHDs, which depend on oxygen, 2-oxoglutarate, Fe+2, and ascorbate as substrates and cofactors for their activity. Hydroxylation recruits the von Hippel Lindau (VHL), which forms a complex together with elongin B, elongin C, cullin-2, and ring-box 1 that has an E3 ubiquitin ligase activity. This complex ubiquinates HIF-α subunits and targets them for proteosomal degradation (Nizet and Johnson, 2009; Walmsley et al., 2009). Hypoxia inactivates PHDs due to the limited oxygen substrate, and therefore stabilizes the HIF-α subunits, allowing their heterodimerization with the HIF-1β subunit. Hypoxia also impairs electron transport in the mitochondria, leading to increased production of ROS, which oxidizes Fe+2 to Fe+3 and further inhibit PHDs. Additional proteins contribute to this regulation, and details on their activity are reviewed elsewhere (Yee Koh et al., 2008).
Although HIF-α subunits are typically induced in hypoxia, they can also be expressed in normoxia upon inflammatory stimulation. For example, high levels of succinate (the product of 2-oxoglutarate) or increased ROS production that oxidizes Fe+2 may cause PHDs inactivation and HIF-1α stabilization (Denko, 2008). Likewise, high concentrations of NO in normoxia can stabilize HIF-1α by inhibiting the activity PHDs, probably by targeting their Fe+2 catalytic site (Weigert and Brune, 2008). Ligation of TLRs, such as the binding of LPS to TLR4, stimulate HIF-1α expression in normoxia through the activity of NF-κB, and hypoxia in the presence of LPS shows a synergistic effect (Frede et al., 2006). Additional pathways can contribute to HIF-1α expression, including the Phosphatidylinositol 3-kinase (PI3K), Mitogen-activated protein kinases (MAPK) and the Ca2+/calmodulin-dependent protein kinases (CaMKII) pathways, and their inhibitors reduce HIF-1α accumulation as well as the expression of its targets (Westra et al., 2010). Thus, in addition to hypoxia, other stimuli, including bacterial infection, hormones, growth factors, cytokines, or RNS can stabilize and activate HIF-1α in normoxia.
Translation and transcription of HIFs
The main known level of HIFs regulation is post-translational, as described above. However other possible regulatory checkpoints, such as transcription and translation, are under-investigated. Although HIF-α subunits are constitutively transcribed in many cell types, their mRNA levels are increased in hypoxia. HIF-1α promoter consists of Sp1 sites, several HRE sites (suggesting a positive feedback loop), as well as NF-κB, Egr-1, and Stat3 binding sites that mediate increased transcription in hypoxia (Niu et al., 2008; Galban and Gorospe, 2009). Evidences show differences between Mo and Mɸ, as hypoxia inhibits HIF-1α transcription in human Mo (Bosco et al., 2006), but increased HIF-1α mRNA accumulation in hMDM by mechanisms other than increasing mRNA stability (Staples et al., 2010). Likewise, HIF-1α mRNA was increased during differentiation of THP-1 cells or peripheral blood Mo into Mɸ (Oda et al., 2006). This suggests a negative feedback designed to control the systemic hypoxic response.
Translational regulation of HIFs is an important checkpoint in both normoxia and hypoxia, which was investigated mainly in cancer cells. Increased HIF-1α protein levels in hypoxia can be observed without a parallel increase in the mRNA levels. This is mediated by the binding of the two RNA-binding proteins (RBP) polypyrimidine tract-binding protein (PTB) and human antigen R (HuR) to the 3′-UTR and 5′-UTR, respectively, as their knock-down through RNA interference inhibited HIF-1α accumulation (Galban et al., 2008). Iron response proteins (IRP) bind to their elements located in the 5′-UTR region of the HIF-2α mRNA and inhibit translation of the mRNA in normoxia, while hypoxia releases this binding and enhances HIF-2α translation (Galban and Gorospe, 2009). Furthermore, specific microRNAs (the miR-17-92 cluster and miR-199a) target HIF-1α mRNA in normoxia and inhibit its translation, whereas hypoxia reduces their levels and release HIF-1α mRNA translation (Galban and Gorospe, 2009). The mechanisms for translational controls have not yet been described in Mo/Mɸ, and this warrants more investigation.
Regulation of HIFs’ activity
Hypoxia-induced factors activity as transcription factors that bind to HREs and to the co-activators CBP/p300 is regulated by oxygen-dependent protein–protein interactions. Factor-inhibiting HIF-1 (FIH-1), another oxygen-dependent hydroxylase, can hydroxylate asparagine residues on HIF-α subunits (803 on HIF-1α and 847 on HIF-2α), resulting in proteins that cannot bind to the co-activators CBP/p300 (Walmsley et al., 2008; Nizet and Johnson, 2009). Additionally, VHL also acts as an adapter that facilitates the binding of FIH to HIF-α subunits in normoxia, while hypoxia disrupts this interaction (Li et al., 2011), allowing for the accumulation of HIFs and their binding to HRE-containing promoters. FIH-1 also plays a role in normoxia, as the cytoplasmic tail of MT1-MMP/MMP-14 provides a platform to which both FIH-1 and its inhibitor mint3/APBA3 bind. This limits the complex localization to the Golgi apparatus and allows mint3 to inhibit FIH-1 ability to hydroxylate HIF-1α, resulting in enhanced accumulation of HIF-1α in normoxia (Sakamoto and Seiki, 2010). It is yet unclear whether a similar mechanism operates on PHDs localization to render them inactive and inhibit HIFs degradation in normoxic Mɸ.
Post-translational modifications regulate stabilization, degradation, and activity of HIF-α subunits by affecting their protein–protein interactions, especially other transcription factors or components of the transcriptional machinery. These were investigated mostly in cancer cell lines and fibroblasts. For example, HIF-α is directly phosphorylated by the ERK1/2 MAPK, leading to its transport to the nucleus and increased activity (Mylonis et al., 2006). Two components of the PI3K/Akt pathway were shown to oppositely regulate HIF-1α. The mTOR kinase phosphorylates HIF-1α in its oxygen-dependent degradation domain, leading to its increased stabilization and enhanced transactivation (Hudson et al., 2002), whereas GSK-3-mediated phosphorylation induces HIF-1α destabilization and proteosomal degradation (Flugel et al., 2007). Recently, PHD3 was found to hydroxylate pyruvate kinase M2, leading to its enhanced interaction with both HIF-1α and p300 in hypoxia and promoting their transactivation of HRE-containing target genes (Luo et al., 2011).
Effects of HIF-1α S-nitrosylation are controversial, as it could increase or decrease its ability to interact with its co-activator p300 (Sumbayev et al., 2003; Cho et al., 2007; Li et al., 2007). However, this mechanism could be relevant only in normoxic induction of HIF-1α, as iNOS is inactivated in hypoxia (see Intracellular Trafficking and Protein–Protein Interactions). Hypoxia was shown to induce SUMOylation of HIF-1α, either promoting its binding to VHL and degradation (Cheng et al., 2007) or promoting its stabilization and transcriptional activity (Carbia-Nagashima et al., 2007). These apparent contradictions may reflect use of different cell types or different hypoxic regimes, and the effects of such post-translational modifications in hypoxic Mɸ should be studied.
Role of NF-κB
NF-κB are a family of proteins consisting of NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel that can heterodimerize. The role of the NF-κB family members as master regulators of many pro-inflammatory associated gene is widely accepted (Vallabhapurapu and Karin, 2009).
NF-κB in different Mɸ subsets
Canonical NF-κB activation is indispensible for M1 activation, although cooperation with other transcription factors such as Stat1 or the MAPK/AP-1 pathway is essential (Biswas and Lewis, 2010; Lawrence and Fong, 2010). NF-κB also regulates anti-inflammatory and pro-angiogenic gene expression (e.g., IL-10, VEGF, COX-2, TGFβ, and MMPs) and inhibits M1 Mɸ (Lawrence and Fong, 2010). In part, this is associated with over-expression of p50 and formation of p50:p50 homodimers, which inhibit transcription of pro-inflammatory cytokines while enhancing transcription of anti-inflammatory genes (Saccani et al., 2006; Biswas and Lewis, 2010; Mancino and Lawrence, 2010). Furthermore, deletion of IKKβ in myeloid cells shifts Mɸ toward M1 activation, increasing the expression of IL-12, MHC class II and iNOS, and revealing the inhibitory role of IKKβ on Stat1 (Fong et al., 2008).
Tumor-associated macrophages derived from human and murine tumors and cultured in normoxia demonstrate defective NF-κB activation, high IL-10 expression and reduced IL-12 and TNFα production (Sica et al., 2000). However, as distinct regions can be identified within solid tumors, TAMs could be activated both as M1 and M2 Mɸ and fluctuate between them, depending on the type of the tumor, its stage, Mɸ localization within the tumor, and the signal received from the microenvironment, including hypoxia (Mancino and Lawrence, 2010). Therefore, the role of NF-κB in specific Mɸ subsets is very complex.
Regulation of NF-κB in normoxia and hypoxia – links to the HIF pathway
Activation of NF-κB occurs in normoxia when TLRs or receptors for pro-inflammatory cytokines recruit the MyD88 adapter protein, which activates a complex signaling cascade that is based on protein–protein interactions and culminates in the phosphorylation, ubiquitination, and degradation of IκB proteins and translocation of NF-κB to the nucleus.
Hypoxia has long been shown to simulate NF-κB signaling. In vivo, hypoxia activates NF-κB expression in a tissue-specific manner, as transgenic mice harboring the luciferase gene under the control of NF-κB expressed it in the hearts and lungs (Fitzpatrick et al., 2011). In vitro there are many examples for hypoxia-induced expression of pro-inflammatory genes. Hypoxic RAW 264.7 cells induced MIP-2 expression, and inhibition of the ERK1/2 or PI3K pathways decreased p65 activity (Zampetaki et al., 2004). Increased production of ROS due to hypoxia was demonstrated in Mo/Mɸ to be responsible for NF-κB activation and for the synergetic effects of hypoxia and LPS (Chandel et al., 2000; Kim et al., 2010). Recently, PDHs were demonstrated as regulators of NF-κB signaling, as their inhibition suppressed LPS-induced expression of TNFα in Mɸ (Takeda et al., 2009). IKKβ was discovered as an additional substrate that PDHs can hydroxylate, leading to its activation and to IkB phosphorylation (Cummins et al., 2006). Additionally, ankyrin repeats on both IκBα and the p105 subunit of NF-κB can be hydroxylated by FIH-1 (Cockman et al., 2006; Cummins et al., 2006; Frede et al., 2006), explaining the effect of ROS on NF-κB activation. In fact, PDHs and FIH-1 involvement in NF-κB regulation suggests that both HIF and NF-κB proteins are redox-sensitive proteins regulated by the same oxygen sensors, in addition to their role as master regulators of inflammation and hypoxia.
Many hypoxia-induced genes are targeted by both NF-κB and HIF proteins. Indeed, a link between the HIF and NF-κB pathways is also provided by the presence of an NF-κB binding site in the HIF-1α promoter (but not HIF-2α), and the ability of p50:p65 heterodimers to bind to it and drive its expression in hypoxia (Belaiba et al., 2007), while siRNA for p65 inhibits this expression (Fitzpatrick et al., 2011). For example, COX-2 was induced by hypoxia in a manner dependent on both HIF-1 and NF-κB (Fitzpatrick et al., 2011), suggesting that the pro-inflammatory response to hypoxia is regulated by NF-κB both by direct binding to promoters of relevant genes and by inducing HIF-1α expression (Fitzpatrick et al., 2011).
Mice lacking IKKβ show impaired accumulation of HIF-1α both in hypoxia and upon bacterial infection (Rius et al., 2008), demonstrating that NF-κB transcriptionally regulates HIF-1α in both hypoxia and normoxia. In addition, a physical interaction between IKKΓ/NEMO and HIF-2α, but not HIF-1α, was described to enhance HIF-2α transcriptional activity in reporter assays (Patel and Simon, 2008), but the specific gene targets remain unknown. This link may be bidirectional, as prolonged hypoxia of hMDM increased the mRNA expression and phosphorylation of the several components in the NF-κB pathway (e.g., IKKβ, IKKΓ, IkBα, RelA/p65) in a manner that was dependent on both HIF-1α and HIF-2α, as demonstrated by knocking down their expression using siRNA (Fang et al., 2009).
Interactions with Other Transcription Factors
Hypoxia-induced factor and NF-κB can cooperate not only with each other, but also with additional transcription factors in normoxia and hypoxia, to achieve better target specificity or maximal activity while forming a large protein complex at the promoters of target genes. Activation of many transcription factors is managed by the crosstalk between signaling pathways. For example, ligation of TLRs or pro-inflammatory cytokines can recruit the adapter protein MyD88 that activates both the IKK–NF-κB and the MAPK pathways, leading to activation of NF-κB and AP-1. The TRIF adapter protein may activate IRF3 and initiate delayed expression of IFNβ, which activates Stat1 in an autocrine manner. Cooperation between NF-κB, AP-1, Stat1, HIF-1, and HIF-2 is required for M1 Mɸ activation (Biswas and Lewis, 2010; Lawrence and Fong, 2010). In contrast, anti-inflammatory cytokines (e.g., IL-4, IL-10, IL-13) activate Stat3 and Stat6, which are needed to induce anti-inflammatory gene expression (e.g., arginase-1, suppressor of cytokine signaling 3 – SOCS-3), and inhibit Stat1 and NF-κB.
The integration of all the different microenvironmental signals determines the balance between M1 and M2 activation modes, suggesting a complex interplay between the transcription factors. Thus, different combinations of transcription factors that cooperate on specific target promoters may determine the activation phenotype of the Mɸ (Biswas and Lewis, 2010; Lawrence and Fong, 2010). Signal transduction is transient, and different pathways can be activated with different kinetics, and may influence the complexity of the transcriptional network. This aspect warrants further study in differently activated Mɸ. Table 1 details some of the interactions between HIFs/NF-κB and several of the central transcription factors that are induced in hypoxia.
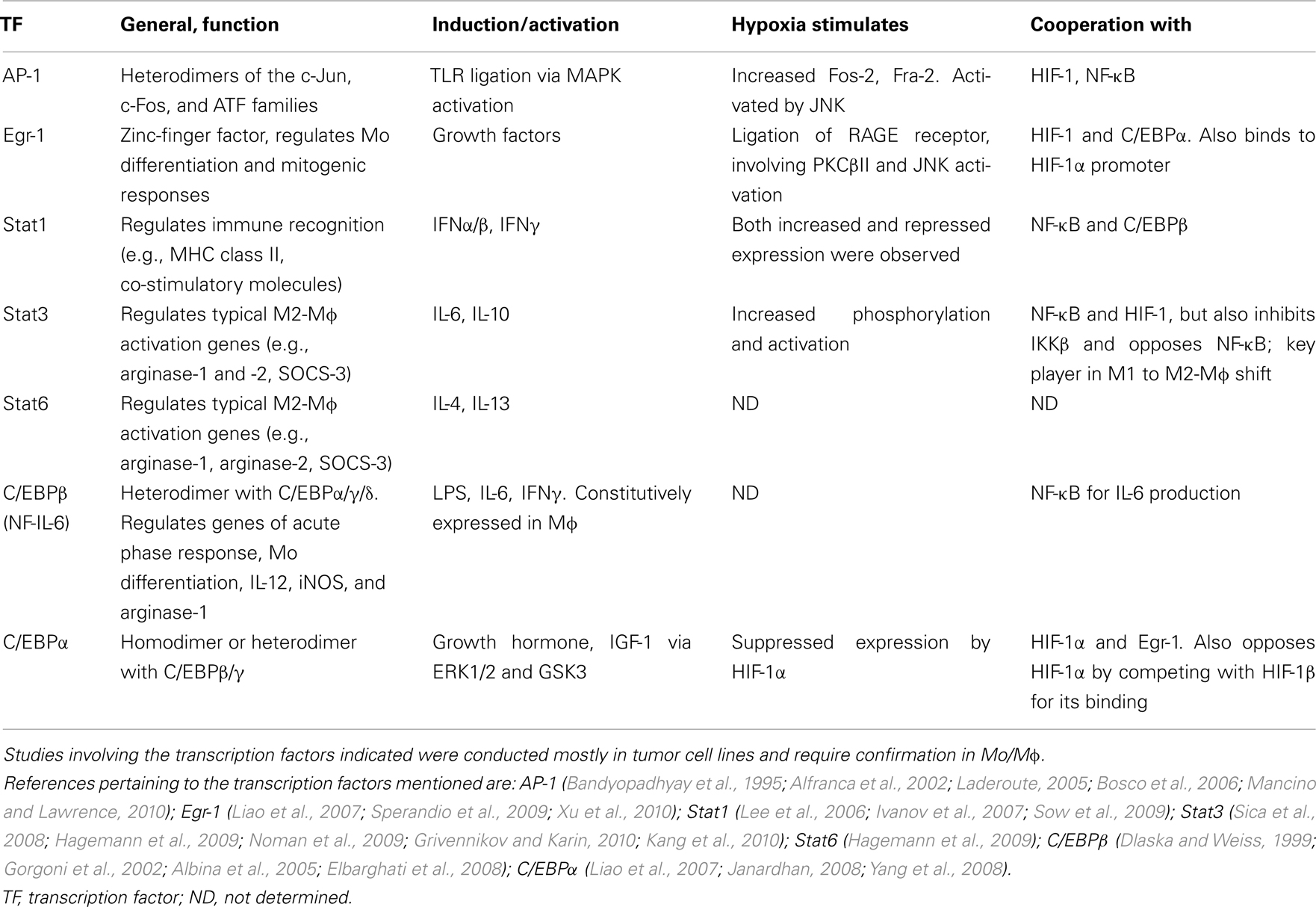
Table 1. Transcription factors other than HIFs and NF-κB, which are involved in hypoxic regulation of gene expression.
Transcriptional Inhibition
Generally, HIF and NF-κB are considered activators of gene transcription, although the NF-κB p50:p50 homodimers can be negative regulators, as mentioned above. Hypoxia can also selectively inhibit gene transcription by other means. For example, in human Mo hypoxia inhibited the constitutive expression of MCP-1/CCL2 mRNA and its induction by IFNΓ and LPS by a dual mechanism that included inhibition of transcription and enhancement of mRNA destabilization (Bosco et al., 2004). As MCP-1/CCL2 is a central chemoattractant for Mo, its inhibition may represent a negative regulatory mechanism that controls recruitment of M1-activated Mo/Mɸ into inflamed tissues. In support of this finding, hypoxia can also inhibit gene expression of CCR5, cathepsin C, 2.5-oligoadenylate synthetase, and the Ras family member Rab7 (Bosco et al., 2006). TIMP-2, the endogenous inhibitor of MMPs, is constitutively expressed in Mo, and hypoxia down-regulates it through the involvement of Sp1 phosphorylation (Lahat et al., 2011).
Surprisingly, in several cases HIF-1 plays a role as a suppressor. In fibroblasts and epithelial cells HIF-1α was implicated in the down-regulation of anti-angiogenic targets such as thrombospondin-1 (Laderoute et al., 2000) and the reversion-inducing cysteine-rich protein with Kazal motifs (RECK) together with histone deacetylase 1 (HDAC1; Lee et al., 2010). In Mɸ, the scavenger receptor MRS1 was down-regulated in hypoxia. Over-expression of HIF-1α suppressed MSR1 mRNA expression, and or its depletion by siRNA restored MSR1 expression, demonstrating the ability of HIF-1 to convey transcriptional inhibition (Shirato et al., 2009). Furthermore, hypoxia was able to suppress gene expression indirectly through HIF-1α, as it induced the expression of the transcription repressor BACH1, which regulates the expression of heme oxygenase-1 in human primary monocytes (Bosco et al., 2006). However, the mechanisms that allow HIF-1α to play a dual role as both an activator and suppressor are not yet known, and this needs to be further explored.
Post-Transcriptional Mechanisms of Hypoxic Regulation
Alternative Splicing and mRNA Stability
Like many other genes, the murine HIF-1α mRNA is alternatively spliced, generating the HIF-1αI.1 and HIF-1αI.2 isoforms, which have alternative first exons and promoters. Stimulation of TG-elicited peritoneal Mɸ and RAW 264.7 cells with TLR4 and A2AR agonists synergistically increased the expression of both isoforms with different kinetics. However, only HIF-1-αI.1 was involved in cytokine production and down-regulated the LPS-induced production of pro-inflammatory cytokines (e.g., TNFα, IL-1β, IL-6, IL-12p40, MIP-1α, MCP-1, and MIP-2), but not IL-1 or VEGF, as was demonstrated in HIF-1αI.1−/− cells. Hypoxia had no effect on the expression of both isoforms (Ramanathan et al., 2009).
The only case that we are aware of hypoxia-induced alternative splicing is of CD80, where hypoxia reduced its surface expression on RAW 264.7 cells while increasing the concentrations of the soluble protein in the supernatants. As amplification of the total mRNA coding for CD80 remained unchanged, but the mRNA coding for the transmembranal region was reduced, it was concluded that hypoxia triggers alternative splicing to generate soluble CD80 (Lahat et al., 2003).
Stability of mRNAs is mediated through the AU-rich elements (AREs) located on the 3′-UTRs of many labile mRNAs and their interaction with different RBP. Presence of AREs is often correlated with rapid mRNA degradation, as occurs with many pro-inflammatory genes such as cytokines (e.g., TNFα, GM-CSF, IL-8), growth factors (e.g., VEGF), transcription factors (e.g., HIF-1α), and other genes (e.g., iNOS, COX-2, uPA, MMPs; Khabar, 2010). Efficient AREs consist of overlapping repetitions of the pentamer AUUUA, giving rise to one or several UUAUUUAUU nonamers, and can assume either a linear or a stem-and-loop conformation. The length of the 3′-UTR and of the ARE itself affects mRNA stability, and longer AREs are associated with shorter half-lives of the mRNAs.
Several RBPs bind to 3′-UTR AREs and target the mRNAs for degradation, including tristetraprolin (TTP) and K-homology splicing-regulatory protein (KSRP), while the HuR promote their stabilization. HuR is mostly a nuclear protein, which upon stimulation translocates to the cytoplasm where it binds to selected mRNAs and stabilizes them. Alternatively, phosphorylation of TTP and KSRP reduce their affinity to AREs and thus stabilize the mRNA. In its unphosphorylated form, TTP recruits factors that mediate decapping, deadenylation, and mRNA destruction. Thus, there is a complex interplay between RBPs that bind to the AREs and determine the mRNA half-life (Khabar, 2010).
The 3′-UTR of the HIF-1α mRNA contains six scattered pentamer AREs and one nonamer ARE, suggesting that TTP could bind to the HIF-1α 3′-UTR and regulate its stability. In several cancer cell lines, hypoxia increased TTP expression, and the protein directly bound to the HIF-1α mRNA AREs. Consistently, over-expression of TTP resulted in decreased HIF-1α mRNA, suggesting that hypoxic cells use TTP to modulate HIF-1α expression (Shin et al., 2010). Although this was not tested in Mo/Mɸ, another study demonstrated that prolonged hypoxia and LPS increased the expression of the dephosphorylated form of TTP in RAW 264.7 cells, and reduced the stability of TNFα, IL-6, MIP-2, and GM-CSF. Knock-down of TTP by siRNA abolished this destabilization (Werno et al., 2010b). On the other hand, the expression of CXCR4 mRNA was stabilized in hypoxia, although the mechanism was not studied (Schioppa et al., 2003).
Translational Regulation: Riboswitches and microRNAs
Hypoxia leads to immediate shut-down of general protein translation. However, selected proteins, including the HIF proteins and their targets, are nonetheless translated. How these proteins escape global inhibition of translation remains unclear.
Translational regulation is achieved by several mechanisms, and structural elements in the non-coding regions of messenger RNA were shown to modulate gene expression. An example of such an element is the riboswitch, a regulatory element that binds small molecules or ribonucleicprotein (RNP) complexes that induce its conformational change. Riboswitches are usually identified in bacteria, but recently a similar element was identified in the VEGF-A mRNA. This transcript is equally transcribed in IFNΓ-induced U937 cells in normoxia and hypoxia, but the protein translation is inhibited after 24 h in normoxia, whereas hypoxia abolishes this effect (Ray et al., 2009). The VEGF-A transcript contains a 126-nt ARE called hypoxia stability region (HSR) that mediates the mRNA stability in hypoxia. It includes two cis-elements, one is a 21-nt CA-rich element (CARE) that binds with high affinity to the heterogeneous nuclear ribonucleoprotein L (hnRNP L), and the other is a 29-nt IFNΓ-activated inhibitor of translation (GAIT) element that binds the GAIT complex and silences inflammatory gene expression. The binding to these two elements is mutually exclusive. Thus, in normoxia IFNΓ stimulates the binding of GAIT complex to its element, thereby silencing VEGF-A gene expression, whereas in hypoxia the HSR undergoes a conformational change, which favors binding of hnRNP L and overrides the suppressive effects of IFNΓ and the GAIT complex (Ray et al., 2009).
Additional translational regulation is exerted by microRNAs (miRNAs), small non-coding RNA molecules that bind with imperfect complementarity to specific mRNA target sequences, usually located in the 3′-UTR of target mRNA, and rapidly and reversibly inhibit their translation. As miRNA are usually independently transcribed, and some contain HRE in their promoter, hypoxic stress can stimulate their transcription, as was shown in different cell types (Pocock, 2011). For example, miR-210 whose promoter contains an HRE is induced in hypoxic tumor and endothelial cells, and can bind to target genes that are crucial in the mitochondria electron transport, thus diverting the cells away from oxidative phosphorylation and toward glycolysis. Alternatively, miR-210 can repress normoxic target genes that are not required for the hypoxic response (Pocock, 2011).
In Mo/Mɸ several miRNAs were identified as regulators of the inflammatory response, in particular miR-155 (O’Connell et al., 2007), miR-146a, and miR-132 (Taganov et al., 2006). LPS markedly induced miR-146a expression in Mo/Mɸ in a prolonged kinetics, and negatively regulated IRAK-1 and TRAF-6 expression leading to down-regulation of the NF-κB pathway, as well as to LPS tolerance and cross tolerance (activation by other TLR ligands) in LPS-primed Mo/Mɸ (Nahid et al., 2009, 2011).
H2O2-induced oxidative stress in RAW 264.7 Mɸ activated the NF-κB pathway and down-regulated the expression of a unique set of miRNA*s (miRNA expressed at low levels relative to the miRNA in the opposite arm of a hairpin; i.e., miR-27a*, miR-27b*, miR-29b*, miR-24-2*, and miR-21*), implicating them, and miR-27b* in particular, in NF-κB regulation (Thulasingam et al., 2011). Regulation by both miRNA and RBPs is primarily executed on the 3′-UTR region in overlapping sequences, indicating possible crosstalk. Indeed, the translation of VEGF-A mRNA is inhibited by the binding of several miRNAs to the CARE, particularly miR-297 and miR-299 in normoxic monocytic cells (even without additional stimulation with IFNΓ). However, hypoxia induces translocation of hnRNP L to the cytoplasm, where it binds the VEGF-A mRNA and prevents miRNA silencing (Jafarifar et al., 2011).
Regulation of protein translation by miRNAs is even further complicated by the finding that miRNA can oscillate between translational repression and activation (which is distinct from alleviating translational repression), depending on their binding location and/or on the conditions in the microenvironment. For example, in proliferating cells TNFα translation is inhibited by the binding of miRNAs to its ARE located in the 3′-UTR. However, upon serum-starvation and cell cycle arrest, HEK293 cells up-regulated TNFα-ARE expression in a manner that depended on miR-369-3. This was mediated by the increased expression of miR-369-3, which recruited the fragile X mental retardation-related protein 1 (FXR1), found exclusively in the activating complex, and Ago2, found in both repressing and activating complexes, into a complex that executed translation activation (Vasudevan et al., 2007). However, as TNFα is mainly produced in Mo/Mɸ, and its 3′-UTR ARE is representative of other pro-inflammatory cytokines with short half-life, it is very important that these results be confirmed in Mo/Mɸ cells, and the precise microenvironmental conditions that activate this switch be defined. These results suggest a new multi-input signal integration mechanism, which could be advantageous particularly in Mo/Mɸ that must adapt to the changing microenvironment.
Intracellular Trafficking and Protein–Protein Interactions
The ability of Mo/Mɸ to migrate through the ECM is mediated by the balanced secretion of different MMPs, particularly MMP-9, and their endogenous inhibitors TIMPs. Hypoxia inhibits migration of human monocyte-derived dendritic cells by disrupting this balance, as TIMP-1 was increased whereas MMP-9 and MT1-MMP were decreased (Qu et al., 2005). These inhibitory effects of hypoxia may have been mediated by the adenosine receptor A2B in a cAMP and PKC dependent manner (Zhao et al., 2008). Hypoxia also inhibited the TNFα-induced secretion of MMP-9 from U937 cells and primary Mo, thus markedly inhibiting their migration (Rahat et al., 2006). This was not regulated at the transcriptional level, as MMP-9 mRNA was unchanged, or at the translational levels, as the amounts of the intracellular enzyme were increased in hypoxia. Instead, confocal microscopy revealed that in hypoxia MMP-9 was attenuated in secretory vesicles due to the effects of hypoxia on the cytoskeleton. Indeed, secretion of proMMP-9 was reduced by the addition of cytochalasin B or nocodazole, which inhibits the polymerization of actin and tubulin fibers, or by the addition of the Rho kinase inhibitor Y27632, suggesting the involvement of the cytoskeleton and the Rho GTPases in the process of enzyme secretion (Rahat et al., 2006).
Reduced MMP-9 in hypoxia could explain the immobilization of Mo/Mɸ in hypoxic regions, in addition to other mechanisms that were reviewed elsewhere (Murdoch et al., 2004; Bosco et al., 2008). Another example of intracellular trafficking is mentioned later (see TNFa Trafficking).
The hallmark of M1-activated Mɸ is iNOS expression and high NO production, which is crucial for the killing abilities of the Mɸ, whereas low levels are considered pro-angiogenic (Weigert and Brune, 2008). The main regulatory checkpoint for iNOS is transcriptional, and NF-κB, IRF-1, Stat1, HIF-1α, and C/EBPβ are implicated in its regulation (Pautz et al., 2010). In hypoxia, induction of iNOS transcription and protein expression is demonstrated in differently activated Mɸ (e.g., peritoneal TG-elicited Mɸ, wound Mɸ, spleen Mɸ). However, the enzyme is inactivated and may produce only very low amounts of NO (Melillo et al., 1996; Daniliuc et al., 2003). This inactivation was attributed to the lack of the oxygen substrate during hypoxic stress, however, hypoxic Mɸ lysates failed to produce NO in an a-cellular system in normoxia, where all cofactors and substrates were abundantly present, suggesting a more intricate regulation. Further investigation revealed that in normoxia iNOS is associated with α-actinin-4, a protein responsible for the cross-linking of actin fibers. This interaction recruits iNOS to the sub-membranal area and ensures its activity. However, hypoxia disrupts these interactions, interferes with iNOS localization and causes its inactivation (Daniliuc et al., 2003).
These two examples highlight a special role for the cytoskeleton in regulating enzyme activity and Mɸ phenotype. As cytoskeleton fibers are responsible for the cellular protein trafficking, their disruption may attenuate vesicles within the Mɸ and prevent secretion of their content, or inhibit delivery of proteins to their destination. For example, hypoxia increased tubulin stabilization and changed vesicle trafficking in breast carcinoma cells (Yoon et al., 2005). This is further supported by recent findings linking actin cytoskeleton to HIF-1α stability (Shin et al., 2010).
The Effects of Previous Mɸ Activation on the Hypoxic Stimulation – the TNFα Example
TNFα Trafficking
In addition to the MMP-9 example, where hypoxia attenuated secretory vesicles, hypoxia also affects TNFα transport. In TG-elicited peritoneal Mɸ and in RAW 264.7 cells, hypoxia did not change the LPS-induced TNFα mRNA levels relative to normoxia, whereas the membranal and intracellular amounts of the protein were reduced, suggesting degradation of TNFα. The proteososmal inhibitor MG132 did not change the hypoxic down-regulation of TNFα, whereas the lysosome inhibitor bafilomycin A1 dose-dependently inhibited degradation of intracellular TNFα, suggesting that TNFα is degraded at the lysosome. Using several Rab proteins as markers for different vesicles, TNFα trafficking was mapped to the endosomal pathway, and its presence was demonstrated in recycling endosomes, early endosomes, lysosome, and secretory lysosomes. The increased hypoxic co-localization of TNFα and LAMP-1, the lysosome marker, and the ability of bafilomycin A1 to increase intracellular TNFα to normoxic values, suggested that TNFα is directed to the lysosome and that hypoxia enhances its degradation (Lahat et al., 2008).
Effects of Previous Mɸ Activation on the Hypoxic Stimulation
The overall effects of hypoxia on the expression of TNFα are controversial. It is widely accepted that hypoxia and LPS increase Mɸ TNFα secretion (Table 2). However, in some cases hypoxia can inhibit TNFα secretion. In our own hands hypoxia exerted opposing effects on LPS-induced secretion of TNFα, as it enhanced it from human monocytes (Lahat et al., 2003), but down-regulated its secretion from peritoneal TG-elicited Mɸ and RAW 264.7 cells (Lahat et al., 2008). We have demonstrated that hypoxia enhances the lysosomal degradation of TNFα, thus causing its reduction in pro-inflammatory induced RAW 264.7 cells (Lahat et al., 2008). Werno et al. (2010b) have also shown that hypoxia reduced TNFα secretion from pro-inflammatory RAW 264.7 cells, although they suggested a mechanism of reduced mRNA stability, which also affected other pro-inflammatory mediators such as MIP-2 and IL-6, and is not mutually exclusive with lysosomal degradation.
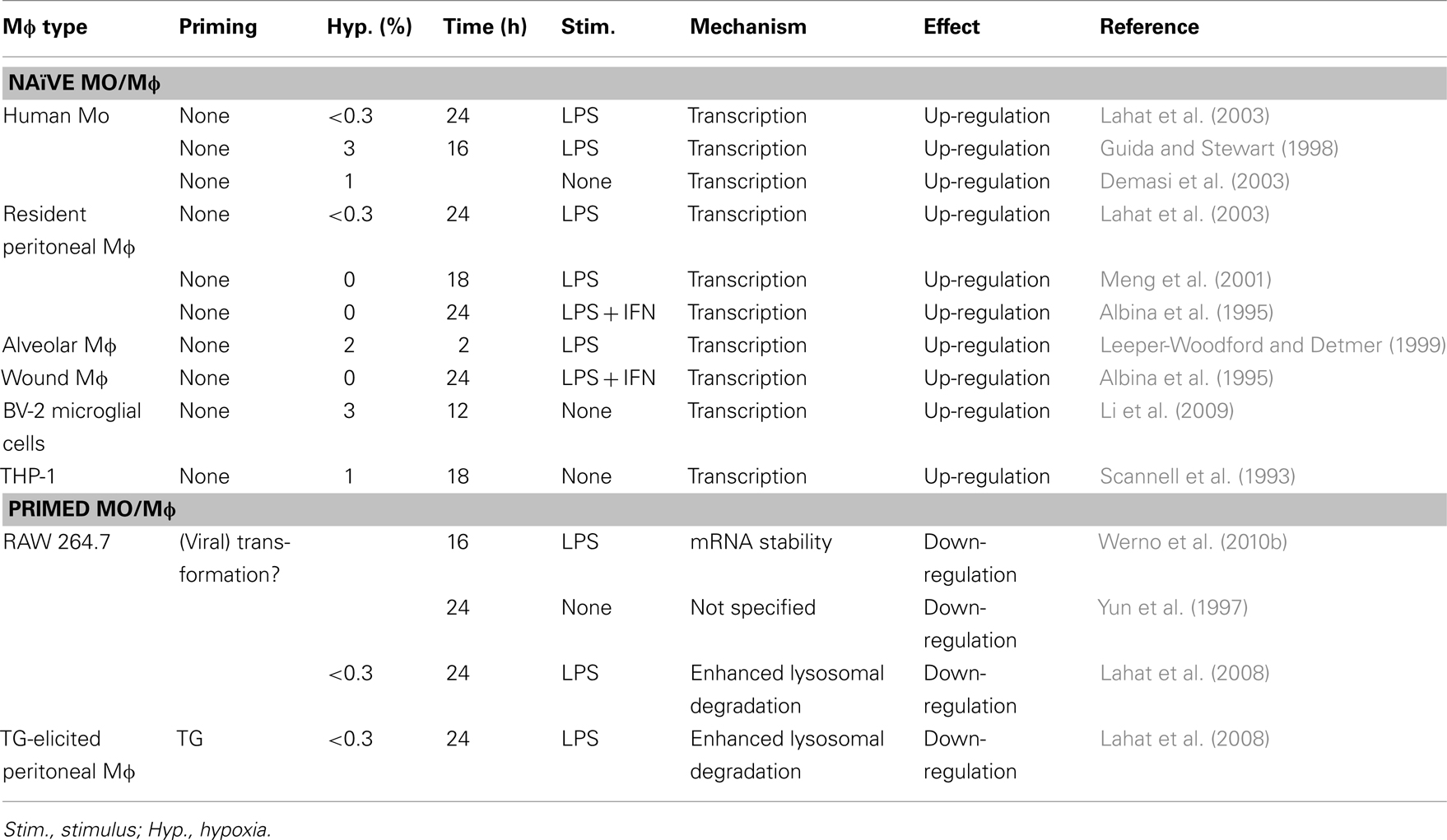
Table 2. Controversial effects of hypoxia on TNFα secretion from Mɸ.
This apparent contradiction between the enhancing and inhibiting effects of hypoxia on TNFα secretion may be resolved by taking into account the priming state of the Mɸ. Priming signals may by themselves be insufficient to stimulate Mɸ, but they sensitize it for further triggering. Traditionally, IFNΓ is used to prime Mɸ that consequently encounter TLR ligands (such as LPS) and become fully M1 activated, although the sequence of these events is insignificant (Schroder et al., 2006). We suggest that “naïve” Mɸ that were not previously primed respond to a combination of hypoxia and LPS by elevating TNFα secretion, whereas Mɸ that are already primed or M1-activated react by down-regulating TNFα secretion, relative to activation with LPS alone. In this sense, RAW 264.7 cells behaved like primed cells, and we speculate that as the cell line was obtained from a tumor induced by the Abelson murine leukemia virus, the viral transformation primes the macrophage.
To further make the point, we compared resident peritoneal Mɸ with TG-elicited Mɸ, by exposing them for 48 h to hypoxia, with or without addition of LPS. Figure 1 shows the opposing effects of hypoxia on these Mɸ, although they originate from the same organ. Given the wide use of TG-elicited Mɸ, it is important to note that these cells are derived from a site of ongoing inflammation, and are therefore at least partially activated and should be considered primed in comparison to the “naïve” resident peritoneal Mɸ (as stated in Takeda et al., 2010).
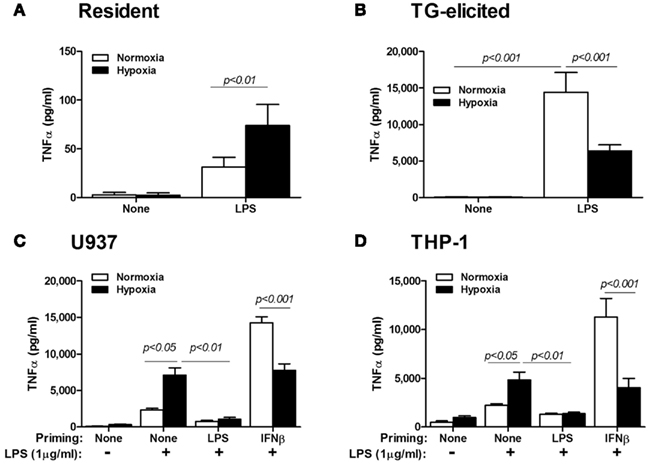
Figure 1. Priming of Mɸ affects the hypoxic response. (A) Resident peritoneal Mɸ (n = 14) or (B) TG-elicited peritoneal Mɸ (n = 16) were subjected to hypoxia (<0.3% O2) for 24 h, with or without stimulation with LPS (1 μg/ml). Hypoxia inhibited the secretion of high amounts of TNFα that were produced in normoxia in primed TG-elicited Mɸ, while an opposite response to hypoxia was observed for the naïve resident Mɸ. The human monocytic cell lines (C) U937 (n = 12) and (D) THP-1 (n = 18) were primed with either LPS (100 ng/ml) or IFNβ (10 U/ml) for 48 h in normoxia, before LPS (1 μg/ml) was added and cells were incubated for additional 24 h in normoxia or hypoxia. Hypoxia increased TNFα secretion in naïve Mo, but inhibited it in IFNβ-primed cells. TNFα was determined in the supernatants using ELISA.
Likewise, when we primed U937 or THP-1 cells by incubating them or 48 h with LPS (simulating gram negative bacterial activation) or with IFNβ (simulating viral activation), and then exposed them for additional 24 h to LPS (at a higher concentration) and hypoxia, we again observed the inhibitory effects of hypoxia on IFNβ-primed cells, but not on “naïve” cells (Figure 1). Note that the lack of response when cells were primed with LPS suggests the involvement of LPS tolerance, which is mediated by the NF-κB p50:p50 homodimers (Frede et al., 2009). It may also imply that IFNβ-priming is mediated by a different mechanism. Thus, the inhibited secretion of TNFα, a typical pro-inflammatory marker, from primed M1-activated Mɸ is inhibited by hypoxia, suggesting that hypoxia may act to restrain the pro-inflammatory Mɸ.
Concluding Remarks and Perspectives
Generally, the hypoxic response depends on cell type, duration of exposure, severity of the hypoxic insult, and as demonstrated for TNFα, previous priming of the Mɸ. Thus, hypoxia can activate Mɸ in a pro-inflammatory way, or skew the pro-inflammatory Mɸ toward a phenotype approximating the M2 or resolution phenotypes. Since hypoxic Mɸ display a mixture of markers of both M1 and M2 activation, classifying them is not straightforward, and they may represent a unique mode of activation which has several common markers with both M1 and M2 activation. In this respect, hypoxia may serve to restrain the pro-inflammatory Mɸ and prevent exacerbation of tissue damage, or alternatively ensure that Mɸ retained in the hypoxic tissue for long periods of time can gradually become M2 activated and engage in processes such as wound healing and/or resolution. In physiological scenarios (e.g., acute inflammation) this has a favorable outcome, whereas in chronic inflammation (e.g., in autoimmune diseases or solid tumors) the inhibitory effects of hypoxia may have devastating results.
Hypoxia profoundly changes Mo/Mɸ activation mode by regulating gene expression. Most of the known regulatory mechanisms are transcriptional, and rely on the cooperation between the master regulator NF-κB and HIFs to mediate this induction of gene expression. However, in some cases HIF can also act as a transcriptional suppressor. Determination of Mɸ activation mode further rely on the cooperation with additional transcriptional factors that are expressed or modified according to the signals received by the microenvironment. The effects of hypoxia on differently activated Mɸ and the molecular mechanisms it exerts (summarized in Figure 2) were not investigated enough. For example, it seems reasonable to assume that differently activated Mɸ located in divergent microenvironments would express different HIF isoforms in different proportions and with different kinetics. However, this assumption merits further study.
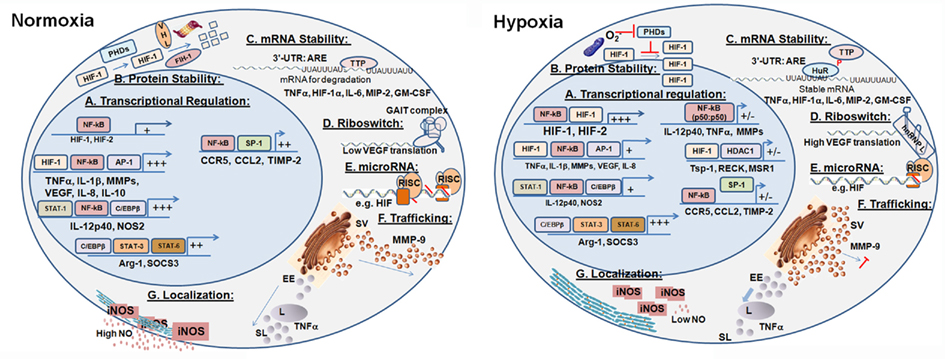
Figure 2. Molecular mechanisms mediating the regulatory effects of hypoxia. Hypoxia affects the production, secretion, and activity of key Mɸ proteins in several regulatory checkpoints, generally keeping the pro-inflammatory Mɸ in check. (A) Transcriptional regulation in the M1-activated Mɸ in normoxia is based on the collaboration between NF-kB, low level of HIF-1, and additional transcription factors. In hypoxia, more HIFs are produced, and genes involved in angiogenesis, metabolism, and survival are triggered. (B) HIFs protein stability in hypoxia is increased, as the prolyl hydroxylases that target them for proteosomal degradation in normoxia are inhibited. (C) Stability of mRNA is regulated by RNA-binding proteins to the AU-rich elements (ARE) located in the 3′-UTR. (D) Post-transcriptional regulation – Riboswitch: The VEGF example illustrates how hypoxia changes secondary structures of 3′-UTR elements, and the binding of protein complexes to them, thus increasing their stability and translation. (E) Post-transcriptional regulation – microRNAs: Hypoxia modulates the expression of microRNAs that bind selected transcripts, thereby inhibiting or alleviating inhibition of their translation. (F) Post-translational regulation – Trafficking: Hypoxia retains secretory vesicles by inhibiting the actin cytoskeleton, thus attenuating the secretion of proteins such as MMP-9. Hypoxia also enhances the lysosomal degradation of other proteins, such as TNFα, which are secreted via the endosomal pathway. (G) Post-translational regulation – Localization: Hypoxia disrupts the interactions between iNOS and α-actinin-4, which anchors it to the cortical cytoskeleton, resulting in its inactivation. SV, secretory vesicles; EE, early endosomes; L, lysosome; SL, secretory lysosomes. +/- indicated reduced or inhibited transcription.
Additional levels of post-transcriptional and post-translational regulation are gradually revealed. These include regulation of mRNA stability, regulation of translation via RBP and microRNA, protein trafficking and protein–protein interactions which may be regulated via reorganization of the cytoskeleton. We now begin to understand how some of these mechanisms allow Mɸ to integrate the different microenvironmental signals and to determine the Mɸ activation mode. Therefore, these mechanisms should be further studied in the context of Mɸ activation, to provide better insights and new approaches to manipulate Mɸ in the hypoxic microenvironment while taking advantage of their selective ability to home to and accumulate in hypoxic regions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a grant from the Rappaport Family Institute for Research in the Medical Sciences and by Research Grant 5343 from the Chief Scientist Office of the Israeli Ministry of Health. We would like to thank Amir Rahat for critical reading.
References
Acosta-Iborra, B., Elorza, A., Olazabal, I. M., Martin-Cofreces, N. B., Martin-Puig, S., Miro, M., Calzada, M. J., Aragones, J., Sanchez-Madrid, F., and Landazuri, M. O. (2009). Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-gamma production through the HIF-1 alpha transcription factor. J. Immunol. 182, 3155–3164.
Albina, J. E., Henry, W. L. Jr., Mastrofrancesco, B., Martin, B. A., and Reichner, J. S. (1995). Macrophage activation by culture in an anoxic environment. J. Immunol. 155, 4391–4396.
Albina, J. E., Mahoney, E. J., Daley, J. M., Wesche, D. E., Morris, S. M. Jr., and Reichner, J. S. (2005). Macrophage arginase regulation by CCAAT/enhancer-binding protein beta. Shock 23, 168–172.
Alfranca, A., Gutierrez, M. D., Vara, A., Aragones, J., Vidal, F., and Landazuri, M. O. (2002). c-Jun and hypoxia-inducible factor 1 functionally cooperate in hypoxia-induced gene transcription. Mol. Cell. Biol. 22, 12–22.
Auffray, C., Sieweke, M. H., and Geissmann, F. (2009). Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 27, 669–692.
Bandyopadhyay, R. S., Phelan, M., and Faller, D. V. (1995). Hypoxia induces AP-1-regulated genes and AP-1 transcription factor binding in human endothelial and other cell types. Biochim. Biophys. Acta 1264, 72–78.
Behn, C., Araneda, O. F., Llanos, A. J., Celedon, G., and Gonzalez, G. (2007). Hypoxia-related lipid peroxidation: evidences, implications and approaches. Respir. Physiol. Neurobiol. 158, 143–150.
Belaiba, R. S., Bonello, S., Zahringer, C., Schmidt, S., Hess, J., Kietzmann, T., and Gorlach, A. (2007). Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 18, 4691–4697.
Biswas, S. K., and Lewis, C. E. (2010). NF-kappaB as a central regulator of macrophage function in tumors. J. Leukoc. Biol. 88, 877–884.
Bosco, M. C., Puppo, M., Blengio, F., Fraone, T., Cappello, P., Giovarelli, M., and Varesio, L. (2008). Monocytes and dendritic cells in a hypoxic environment: spotlights on chemotaxis and migration. Immunobiology 213, 733–749.
Bosco, M. C., Puppo, M., Pastorino, S., Mi, Z., Melillo, G., Massazza, S., Rapisarda, A., and Varesio, L. (2004). Hypoxia selectively inhibits monocyte chemoattractant protein-1 production by macrophages. J. Immunol. 172, 1681–1690.
Bosco, M. C., Puppo, M., Santangelo, C., Anfosso, L., Pfeffer, U., Fardin, P., Battaglia, F., and Varesio, L. (2006). Hypoxia modifies the transcriptome of primary human monocytes: modulation of novel immune-related genes and identification of CC-chemokine ligand 20 as a new hypoxia-inducible gene. J. Immunol. 177, 1941–1955.
Bronte, V. (2009). Myeloid-derived suppressor cells in inflammation: uncovering cell subsets with enhanced immunosuppressive functions. Eur. J. Immunol. 39, 2670–2672.
Burke, B., Tang, N., Corke, K. P., Tazzyman, D., Ameri, K., Wells, M., and Lewis, C. E. (2002). Expression of HIF-1alpha by human macrophages: implications for the use of macrophages in hypoxia-regulated cancer gene therapy. J. Pathol. 196, 204–212.
Bystrom, J., Evans, I., Newson, J., Stables, M., Toor, I., Van Rooijen, N., Crawford, M., Colville-Nash, P., Farrow, S., and Gilroy, D. W. (2008). Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood 112, 4117–4127.
Carbia-Nagashima, A., Gerez, J., Perez-Castro, C., Paez-Pereda, M., Silberstein, S., Stalla, G. K., Holsboer, F., and Arzt, E. (2007). RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell 131, 309–323.
Chandel, N. S., Trzyna, W. C., Mcclintock, D. S., and Schumacker, P. T. (2000). Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J. Immunol. 165, 1013–1021.
Cheng, J., Kang, X., Zhang, S., and Yeh, E. T. (2007). SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 131, 584–595.
Cho, H., Ahn, D. R., Park, H., and Yang, E. G. (2007). Modulation of p300 binding by posttranslational modifications of the C-terminal activation domain of hypoxia-inducible factor-1alpha. FEBS Lett. 581, 1542–1548.
Cockman, M. E., Lancaster, D. E., Stolze, I. P., Hewitson, K. S., Mcdonough, M. A., Coleman, M. L., Coles, C. H., Yu, X., Hay, R. T., Ley, S. C., Pugh, C. W., Oldham, N. J., Masson, N., Schofield, C. J., and Ratcliffe, P. J. (2006). Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. U.S.A. 103, 14767–14772.
Coffelt, S. B., Hughes, R., and Lewis, C. E. (2009). Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim. Biophys. Acta 1796, 11–18.
Cramer, T., Yamanishi, Y., Clausen, B. E., Forster, I., Pawlinski, R., Mackman, N., Haase, V. H., Jaenisch, R., Corr, M., Nizet, V., Firestein, G. S., Gerber, H. P., Ferrara, N., and Johnson, R. S. (2003). HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657.
Cros, J., Cagnard, N., Woollard, K., Patey, N., Zhang, S. Y., Senechal, B., Puel, A., Biswas, S. K., Moshous, D., Picard, C., Jais, J. P., D’cruz, D., Casanova, J. L., Trouillet, C., and Geissmann, F. (2010). Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 33, 375–386.
Crowther, M., Brown, N. J., Bishop, E. T., and Lewis, C. E. (2001). Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J. Leukoc. Biol. 70, 478–490.
Cummins, E. P., Berra, E., Comerford, K. M., Ginouves, A., Fitzgerald, K. T., Seeballuck, F., Godson, C., Nielsen, J. E., Moynagh, P., Pouyssegur, J., and Taylor, C. T. (2006). Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. U.S.A. 103, 18154–18159.
Daniliuc, S., Bitterman, H., Rahat, M. A., Kinarty, A., Rosenzweig, D., and Lahat, N. (2003). Hypoxia inactivates inducible nitric oxide synthase in mouse macrophages by disrupting its interaction with alpha-actinin 4. J. Immunol. 171, 3225–3232.
De Palma, M., Murdoch, C., Venneri, M. A., Naldini, L., and Lewis, C. E. (2007). Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 28, 519–524.
Demasi, M., Cleland, L. G., Cook-Johnson, R. J., Caughey, G. E., and James, M. J. (2003). Effects of hypoxia on monocyte inflammatory mediator production: dissociation between changes in cyclooxygenase-2 expression and eicosanoid synthesis. J. Biol. Chem. 278, 38607–38616.
Denko, N. C. (2008). Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705–713.
Dlaska, M., and Weiss, G. (1999). Central role of transcription factor NF-IL6 for cytokine and iron-mediated regulation of murine inducible nitric oxide synthase expression. J. Immunol. 162, 6171–6177.
Elbarghati, L., Murdoch, C., and Lewis, C. E. (2008). Effects of hypoxia on transcription factor expression in human monocytes and macrophages. Immunobiology 213, 899–908.
Fang, H. Y., Hughes, R., Murdoch, C., Coffelt, S. B., Biswas, S. K., Harris, A. L., Johnson, R. S., Imityaz, H. Z., Simon, M. C., Fredlund, E., Greten, F. R., Rius, J., and Lewis, C. E. (2009). Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 114, 844–859.
Fitzpatrick, S. F., Tambuwala, M. M., Bruning, U., Schaible, B., Scholz, C. C., Byrne, A., O’connor, A., Gallagher, W. M., Lenihan, C. R., Garvey, J. F., Howell, K., Fallon, P. G., Cummins, E. P., and Taylor, C. T. (2011). An intact canonical NF-kappaB pathway is required for inflammatory gene expression in response to hypoxia. J. Immunol. 186, 1091–1096.
Flugel, D., Gorlach, A., Michiels, C., and Kietzmann, T. (2007). Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol. Cell. Biol. 27, 3253–3265.
Fong, C. H., Bebien, M., Didierlaurent, A., Nebauer, R., Hussell, T., Broide, D., Karin, M., and Lawrence, T. (2008). An antiinflammatory role for IKKbeta through the inhibition of “classical” macrophage activation. J. Exp. Med. 205, 1269–1276.
Frede, S., Stockmann, C., Freitag, P., and Fandrey, J. (2006). Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem. J. 396, 517–527.
Frede, S., Stockmann, C., Winning, S., Freitag, P., and Fandrey, J. (2009). Hypoxia-inducible factor (HIF) 1alpha accumulation and HIF target gene expression are impaired after induction of endotoxin tolerance. J. Immunol. 182, 6470–6476.
Gabrilovich, D. I., and Nagaraj, S. (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174.
Galban, S., and Gorospe, M. (2009). Factors interacting with HIF-1alpha mRNA: novel therapeutic targets. Curr. Pharm. Des. 15, 3853–3860.
Galban, S., Kuwano, Y., Pullmann, R. Jr., Martindale, J. L., Kim, H. H., Lal, A., Abdelmohsen, K., Yang, X., Dang, Y., Liu, J. O., Lewis, S. M., Holcik, M., and Gorospe, M. (2008). RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1alpha. Mol. Cell. Biol. 28, 93–107.
Geissmann, F., Jung, S., and Littman, D. R. (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82.
Gordon, S., and Martinez, F. O. (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604.
Gorgoni, B., Maritano, D., Marthyn, P., Righi, M., and Poli, V. (2002). C/EBP beta gene inactivation causes both impaired and enhanced gene expression and inverse regulation of IL-12 p40 and p35 mRNAs in macrophages. J. Immunol. 168, 4055–4062.
Griffiths, L., Binley, K., Iqball, S., Kan, O., Maxwell, P., Ratcliffe, P., Lewis, C., Harris, A., Kingsman, S., and Naylor, S. (2000). The macrophage – a novel system to deliver gene therapy to pathological hypoxia. Gene Ther. 7, 255–262.
Grivennikov, S. I., and Karin, M. (2010). Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 21, 11–19.
Grocott, M. P., Martin, D. S., Levett, D. Z., Mcmorrow, R., Windsor, J., and Montgomery, H. E. (2009). Arterial blood gases and oxygen content in climbers on Mount Everest. N. Engl. J. Med. 360, 140–149.
Guida, E., and Stewart, A. (1998). Influence of hypoxia and glucose deprivation on tumour necrosis factor-alpha and granulocyte-macrophage colony-stimulating factor expression in human cultured monocytes. Cell. Physiol. Biochem. 8, 75–88.
Hagemann, T., Biswas, S. K., Lawrence, T., Sica, A., and Lewis, C. E. (2009). Regulation of macrophage function in tumors: the multifaceted role of NF-kappaB. Blood 113, 3139–3146.
Hartmann, G., Tschop, M., Fischer, R., Bidlingmaier, C., Riepl, R., Tschop, K., Hautmann, H., Endres, S., and Toepfer, M. (2000). High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine 12, 246–252.
Heikkila, M., Pasanen, A., Kivirikko, K. I., and Myllyharju, J. (2011). Roles of the human hypoxia-inducible factor (HIF)-3alpha variants in the hypoxia response. Cell. Mol. Life Sci. [Epub ahead of print].
Hudson, C. C., Liu, M., Chiang, G. G., Otterness, D. M., Loomis, D. C., Kaper, F., Giaccia, A. J., and Abraham, R. T. (2002). Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 22, 7004–7014.
Ivanov, S. V., Salnikow, K., Ivanova, A. V., Bai, L., and Lerman, M. I. (2007). Hypoxic repression of STAT1 and its downstream genes by a pVHL/HIF-1 target DEC1/STRA13. Oncogene 26, 802–812.
Jafarifar, F., Yao, P., Eswarappa, S. M., and Fox, P. L. (2011). Repression of VEGFA by CA-rich element-binding microRNAs is modulated by hnRNP L. EMBO J. 30, 1324–1334.
Kang, S. H., Yu, M. O., Park, K. J., Chi, S. G., Park, D. H., and Chung, Y. G. (2010). Activated STAT3 regulates hypoxia-induced angiogenesis and cell migration in human glioblastoma. Neurosurgery 67, 1386–1395; discussion 1395.
Khabar, K. S. (2010). Post-transcriptional control during chronic inflammation and cancer: a focus on AU-rich elements. Cell. Mol. Life Sci. 67, 2937–2955.
Kim, D., Kim, Y. J., Koh, H. S., Jang, T. Y., Park, H. E., and Kim, J. Y. (2010). Reactive oxygen species enhance TLR10 expression in the human monocytic cell line THP-1. Int. J. Mol. Sci. 11, 3769–3782.
Laderoute, K. R. (2005). The interaction between HIF-1 and AP-1 transcription factors in response to low oxygen. Semin. Cell Dev. Biol. 16, 502–513.
Laderoute, K. R., Alarcon, R. M., Brody, M. D., Calaoagan, J. M., Chen, E. Y., Knapp, A. M., Yun, Z., Denko, N. C., and Giaccia, A. J. (2000). Opposing effects of hypoxia on expression of the angiogenic inhibitor thrombospondin 1 and the angiogenic inducer vascular endothelial growth factor. Clin. Cancer Res. 6, 2941–2950.
Lahat, N., Bitterman, H., Engelmayer-Goren, M., Rosenzweig, D., Weiss-Cerem, L., and Rahat, M. A. (2011). Reduced TIMP-2 in hypoxia enhances angiogenesis. Am. J. Physiol. Cell Physiol. 300, C557–C566.
Lahat, N., Rahat, M. A., Ballan, M., Weiss-Cerem, L., Engelmayer, M., and Bitterman, H. (2003). Hypoxia reduces CD80 expression on monocytes but enhances their LPS-stimulated TNF-alpha secretion. J. Leukoc. Biol. 74, 197–205.
Lahat, N., Rahat, M. A., Kinarty, A., Weiss-Cerem, L., Pinchevski, S., and Bitterman, H. (2008). Hypoxia enhances lysosomal TNF-alpha degradation in mouse peritoneal macrophages. Am. J. Physiol., Cell Physiol. 295, C2–C12.
Lawrence, T., and Fong, C. (2010). The resolution of inflammation: anti-inflammatory roles for NF-kappaB. Int. J. Biochem. Cell Biol. 42, 519–523.
Lee, K. J., Lee, K. Y., and Lee, Y. M. (2010). Downregulation of a tumor suppressor RECK by hypoxia through recruitment of HDAC1 and HIF-1alpha to reverse HRE site in the promoter. Biochim. Biophys. Acta 1803, 608–616.
Lee, M. Y., Joung, Y. H., Lim, E. J., Park, J. H., Ye, S. K., Park, T., Zhang, Z., Park, D. K., Lee, K. J., and Yang, Y. M. (2006). Phosphorylation and activation of STAT proteins by hypoxia in breast cancer cells. Breast 15, 187–195.
Leeper-Woodford, S. K., and Detmer, K. (1999). Acute hypoxia increases alveolar macrophage tumor necrosis factor activity and alters NF-kappaB expression. Am. J. Physiol. 276, L909–L916.
Lewis, C. E., and Pollard, J. W. (2006). Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66, 605–612.
Lewis, J. S., Lee, J. A., Underwood, J. C., Harris, A. L., and Lewis, C. E. (1999). Macrophage responses to hypoxia: relevance to disease mechanisms. J. Leukoc. Biol. 66, 889–900.
Li, F., Sonveaux, P., Rabbani, Z. N., Liu, S., Yan, B., Huang, Q., Vujaskovic, Z., Dewhirst, M. W., and Li, C. Y. (2007). Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 26, 63–74.
Li, P., Lu, J., Kaur, C., Sivakumar, V., Tan, K. L., and Ling, E. A. (2009). Expression of cyclooxygenase-1/-2, microsomal prostaglandin-E synthase-1 and E-prostanoid receptor 2 and regulation of inflammatory mediators by PGE(2) in the amoeboid microglia in hypoxic postnatal rats and murine BV-2 cells. Neuroscience 164, 948–962.
Li, S. H., Chun, Y. S., Lim, J. H., Huang, L. E., and Park, J. W. (2011). von Hippel-Lindau protein adjusts oxygen sensing of the FIH asparaginyl hydroxylase. Int. J. Biochem. Cell Biol. 43, 795–804.
Liao, H., Hyman, M. C., Lawrence, D. A., and Pinsky, D. J. (2007). Molecular regulation of the PAI-1 gene by hypoxia: contributions of Egr-1, HIF-1alpha, and C/EBPalpha. FASEB J. 21, 935–949.
Luo, W., Hu, H., Chang, R., Zhong, J., Knabel, M., O’Meally, R., Cole, R. N., Pandey, A., and Semenza, G. L. (2011). Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744.
Mancino, A., and Lawrence, T. (2010). Nuclear factor-kappaB and tumor-associated macrophages. Clin. Cancer Res. 16, 784–789.
Martinez, F. O., Helming, L., and Gordon, S. (2009). Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 27, 451–483.
Martinez, F. O., Sica, A., Mantovani, A., and Locati, M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461.
Melillo, G., Taylor, L. S., Brooks, A., Cox, G. W., and Varesio, L. (1996). Regulation of inducible nitric oxide synthase expression in IFN-gamma-treated murine macrophages cultured under hypoxic conditions. J. Immunol. 157, 2638–2644.
Meng, X., Ao, L., Shames, B. D., and Harken, A. H. (2001). Inhibition of cyclic-3′,5′-nucleotide phosphodiesterase abrogates the synergism of hypoxia with lipopolysaccharide in the induction of macrophage TNF-alpha production. J. Surg. Res. 101, 210–215.
Minko, T., Wang, Y., and Pozharov, V. (2005). Remediation of cellular hypoxic damage by pharmacological agents. Curr. Pharm. Des. 11, 3185–3199.
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969.
Murdoch, C., Giannoudis, A., and Lewis, C. E. (2004). Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 104, 2224–2234.
Murdoch, C., Muthana, M., Coffelt, S. B., and Lewis, C. E. (2008). The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 8, 618–631.
Murdoch, C., Muthana, M., and Lewis, C. E. (2005). Hypoxia regulates macrophage functions in inflammation. J. Immunol. 175, 6257–6263.
Mylonis, I., Chachami, G., Samiotaki, M., Panayotou, G., Paraskeva, E., Kalousi, A., Georgatsou, E., Bonanou, S., and Simos, G. (2006). Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J. Biol. Chem. 281, 33095–33106.
Nahid, M. A., Pauley, K. M., Satoh, M., and Chan, E. K. (2009). miR-146a is critical for endotoxin-induced tolerance: implication in innate immunity. J. Biol. Chem. 284, 34590–34599.
Nahid, M. A., Satoh, M., and Chan, E. K. (2011). Mechanistic role of microRNA-146a in endotoxin-induced differential cross-regulation of TLR signaling. J. Immunol. 186, 1723–1734.
Nahrendorf, M., Pittet, M. J., and Swirski, F. K. (2010). Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121, 2437–2445.
Niu, G., Briggs, J., Deng, J., Ma, Y., Lee, H., Kortylewski, M., Kujawski, M., Kay, H., Cress, W. D., Jove, R., and Yu, H. (2008). Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol. Cancer Res. 6, 1099–1105.
Nizet, V., and Johnson, R. S. (2009). Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 9, 609–617.
Noman, M. Z., Buart, S., Van Pelt, J., Richon, C., Hasmim, M., Leleu, N., Suchorska, W. M., Jalil, A., Lecluse, Y., El Hage, F., Giuliani, M., Pichon, C., Azzarone, B., Mazure, N., Romero, P., Mami-Chouaib, F., and Chouaib, S. (2009). The cooperative induction of hypoxia-inducible factor-1 alpha and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J. Immunol. 182, 3510–3521.
O’Connell, R. M., Taganov, K. D., Boldin, M. P., Cheng, G., and Baltimore, D. (2007). MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U.S.A. 104, 1604–1609.
Oda, T., Hirota, K., Nishi, K., Takabuchi, S., Oda, S., Yamada, H., Arai, T., Fukuda, K., Kita, T., Adachi, T., Semenza, G. L., and Nohara, R. (2006). Activation of hypoxia-inducible factor 1 during macrophage differentiation. Am. J. Physiol. Cell Physiol. 291, C104–C113.
Ostrand-Rosenberg, S., and Sinha, P. (2009). Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 182, 4499–4506.
Papandreou, I., Powell, A., Lim, A. L., and Denko, N. (2005). Cellular reaction to hypoxia: sensing and responding to an adverse environment. Mutat. Res. 569, 87–100.
Patel, S. A., and Simon, M. C. (2008). Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ. 15, 628–634.
Pautz, A., Art, J., Hahn, S., Nowag, S., Voss, C., and Kleinert, H. (2010). Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide 23, 75–93.
Peyssonnaux, C., Cejudo-Martin, P., Doedens, A., Zinkernagel, A. S., Johnson, R. S., and Nizet, V. (2007). Cutting edge: essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J. Immunol. 178, 7516–7519.
Pocock, R. (2011). Invited review: decoding the microRNA response to hypoxia. Pflugers Arch. 461, 307–315.
Qian, B. Z., and Pollard, J. W. (2010). Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51.
Qu, X., Yang, M. X., Kong, B. H., Qi, L., Lam, Q. L., Yan, S., Li, P., Zhang, M., and Lu, L. (2005). Hypoxia inhibits the migratory capacity of human monocyte-derived dendritic cells. Immunol. Cell Biol. 83, 668–673.
Rahat, M. A., Marom, B., Bitterman, H., Weiss-Cerem, L., Kinarty, A., and Lahat, N. (2006). Hypoxia reduces the output of matrix metalloproteinase-9 (MMP-9) in monocytes by inhibiting its secretion and elevating membranal association. J. Leukoc. Biol. 79, 706–718.
Ramanathan, M., Luo, W., Csoka, B., Hasko, G., Lukashev, D., Sitkovsky, M. V., and Leibovich, S. J. (2009). Differential regulation of HIF-1alpha isoforms in murine macrophages by TLR4 and adenosine A(2A) receptor agonists. J. Leukoc. Biol. 86, 681–689.
Ray, P. S., Jia, J., Yao, P., Majumder, M., Hatzoglou, M., and Fox, P. L. (2009). A stress-responsive RNA switch regulates VEGFA expression. Nature 457, 915–919.
Rius, J., Guma, M., Schachtrup, C., Akassoglou, K., Zinkernagel, A. S., Nizet, V., Johnson, R. S., Haddad, G. G., and Karin, M. (2008). NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453, 807–811.
Rosenberger, P., Schwab, J. M., Mirakaj, V., Masekowsky, E., Mager, A., Morote-Garcia, J. C., Unertl, K., and Eltzschig, H. K. (2009). Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 10, 195–202.
Saccani, A., Schioppa, T., Porta, C., Biswas, S. K., Nebuloni, M., Vago, L., Bottazzi, B., Colombo, M. P., Mantovani, A., and Sica, A. (2006). p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 66, 11432–11440.
Sakamoto, T., and Seiki, M. (2010). A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J. Biol. Chem. 285, 29951–29964.
Scannell, G., Waxman, K., Kaml, G. J., Ioli, G., Gatanaga, T., Yamamoto, R., and Granger, G. A. (1993). Hypoxia induces a human macrophage cell line to release tumor necrosis factor-alpha and its soluble receptors in vitro. J. Surg. Res. 54, 281–285.
Schif-Zuck, S., Gross, N., Assi, S., Rostoker, R., Serhan, C. N., and Ariel, A. (2011). Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur. J. Immunol. 41, 366–379.
Schioppa, T., Uranchimeg, B., Saccani, A., Biswas, S. K., Doni, A., Rapisarda, A., Bernasconi, S., Saccani, S., Nebuloni, M., Vago, L., Mantovani, A., Melillo, G., and Sica, A. (2003). Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 198, 1391–1402.
Schroder, K., Sweet, M. J., and Hume, D. A. (2006). Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology 211, 511–524.
Shin, I. J., Park, B. K., Ahn, Y. T., Kim, Y., and An, W. G. (2010). Actin disruption inhibits hypoxia inducible factor-1alpha expression via inactivity of Mdm2-mediated p70S6K. Mol. Med. Report 3, 815–819.
Shirato, K., Kizaki, T., Sakurai, T., Ogasawara, J. E., Ishibashi, Y., Iijima, T., Okada, C., Noguchi, I., Imaizumi, K., Taniguchi, N., and Ohno, H. (2009). Hypoxia-inducible factor-1alpha suppresses the expression of macrophage scavenger receptor 1. Pflugers Arch. 459, 93–103.
Sica, A., Allavena, P., and Mantovani, A. (2008). Cancer related inflammation: the macrophage connection. Cancer Lett. 267, 204–215.
Sica, A., Saccani, A., Bottazzi, B., Polentarutti, N., Vecchi, A., Van Damme, J., and Mantovani, A. (2000). Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J. Immunol. 164, 762–767.
Skrzeczynska-Moncznik, J., Bzowska, M., Loseke, S., Grage-Griebenow, E., Zembala, M., and Pryjma, J. (2008). Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand. J. Immunol. 67, 152–159.
Sow, F. B., Alvarez, G. R., Gross, R. P., Satoskar, A. R., Schlesinger, L. S., Zwilling, B. S., and Lafuse, W. P. (2009). Role of STAT1, NF-kappaB, and C/EBPbeta in the macrophage transcriptional regulation of hepcidin by mycobacterial infection and IFN-gamma. J. Leukoc. Biol. 86, 1247–1258.
Sperandio, S., Fortin, J., Sasik, R., Robitaille, L., Corbeil, J., and De Belle, I. (2009). The transcription factor Egr1 regulates the HIF-1alpha gene during hypoxia. Mol. Carcinog. 48, 38–44.
Staples, K. J., Sotoodehnejadnematalahi, F., Pearson, H., Frankenberger, M., Francescut, L., Ziegler-Heitbrock, L., and Burke, B. (2010). Monocyte-derived macrophages matured under prolonged hypoxia transcriptionally up-regulate HIF-1alpha mRNA. Immunobiology 216, 832–839.
Stout, R. D., and Suttles, J. (2004). Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J. Leukoc. Biol. 76, 509–513.
Stout, R. D., Watkins, S. K., and Suttles, J. (2009). Functional plasticity of macrophages: in situ reprogramming of tumor-associated macrophages. J. Leukoc. Biol. 86, 1105–1109.
Sumbayev, V. V., Budde, A., Zhou, J., and Brune, B. (2003). HIF-1 alpha protein as a target for S-nitrosation. FEBS Lett. 535, 106–112.
Taganov, K. D., Boldin, M. P., Chang, K. J., and Baltimore, D. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 12481–12486.
Takeda, K., Ichiki, T., Narabayashi, E., Inanaga, K., Miyazaki, R., Hashimoto, T., Matsuura, H., Ikeda, J., Miyata, T., and Sunagawa, K. (2009). Inhibition of prolyl hydroxylase domain-containing protein suppressed lipopolysaccharide-induced TNF-alpha expression. Arterioscler. Thromb. Vasc. Biol. 29, 2132–2137.
Takeda, N., O’dea, E. L., Doedens, A., Kim, J. W., Weidemann, A., Stockmann, C., Asagiri, M., Simon, M. C., Hoffmann, A., and Johnson, R. S. (2010). Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 24, 491–501.
Thulasingam, S., Massilamany, C., Gangaplara, A., Dai, H., Yarbaeva, S., Subramaniam, S., Riethoven, J. J., Eudy, J., Lou, M., and Reddy, J. (2011). miR-27b*, an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in RAW 264.7 cells. Mol. Cell. Biochem. 352, 181–188.
Vallabhapurapu, S., and Karin, M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733.
van de Veerdonk, F. L., and Netea, M. G. (2010). Diversity: a hallmark of monocyte society. Immunity 33, 289–291.
Vasudevan, S., Tong, Y., and Steitz, J. A. (2007). Switching from repression to activation: microRNAs can up-regulate translation. Science 318, 1931–1934.
Venneri, M. A., De Palma, M., Ponzoni, M., Pucci, F., Scielzo, C., Zonari, E., Mazzieri, R., Doglioni, C., and Naldini, L. (2007). Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 109, 5276–5285.
Walmsley, S. R., Chilvers, E. R., and Whyte, M. K. (2009). Hypoxia. Hypoxia, hypoxia inducible factor and myeloid cell function. Arthritis Res. Ther. 11, 219.
Walmsley, S. R., Mcgovern, N. N., Whyte, M. K., and Chilvers, E. R. (2008). The HIF/VHL pathway: from oxygen sensing to innate immunity. Am. J. Respir. Cell Mol. Biol. 38, 251–255.
Weigert, A., and Brune, B. (2008). Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide 19, 95–102.
Werno, C., Menrad, H., Weigert, A., Dehne, N., Goerdt, S., Schledzewski, K., Kzhyshkowska, J., and Brune, B. (2010a). Knockout of HIF-1alpha in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis 31, 1863–1872.
Werno, C., Schmid, T., Schnitzer, S. E., Peters, K., Milke, L., and Brune, B. (2010b). A combination of hypoxia and lipopolysaccharide activates tristetraprolin to destabilize proinflammatory mRNAs such as tumor necrosis factor-alpha. Am. J. Pathol. 177, 1104–1112.
Westra, J., Brouwer, E., Van Roosmalen, I. A., Doornbos-Van Der Meer, B., Van Leeuwen, M. A., Posthumus, M. D., and Kallenberg, C. G. (2010). Expression and regulation of HIF-1alpha in macrophages under inflammatory conditions; significant reduction of VEGF by CaMKII inhibitor. BMC Musculoskelet. Disord. 11, 61.
Xu, Y., Toure, F., Qu, W., Lin, L., Song, F., Shen, X., Rosario, R., Garcia, J., Schmidt, A. M., and Yan, S. F. (2010). Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J. Biol. Chem. 285, 23233–23240.
Yang, L., Jiang, Y., Wu, S. F., Zhou, M. Y., Wu, Y. L., and Chen, G. Q. (2008). CCAAT/enhancer-binding protein alpha antagonizes transcriptional activity of hypoxia-inducible factor 1 alpha with direct protein-protein interaction. Carcinogenesis 29, 291–298.
Yee Koh, M., Spivak-Kroizman, T. R., and Powis, G. (2008). HIF-1 regulation: not so easy come, easy go. Trends Biochem. Sci. 33, 526–534.
Yoon, S. O., Shin, S., and Mercurio, A. M. (2005). Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 65, 2761–2769.
Yun, J. K., Mccormick, T. S., Villabona, C., Judware, R. R., Espinosa, M. B., and Lapetina, E. G. (1997). Inflammatory mediators are perpetuated in macrophages resistant to apoptosis induced by hypoxia. Proc. Natl. Acad. Sci. U.S.A. 94, 13903–13908.
Zampetaki, A., Mitsialis, S. A., Pfeilschifter, J., and Kourembanas, S. (2004). Hypoxia induces macrophage inflammatory protein-2 (MIP-2) gene expression in murine macrophages via NF-kappaB: the prominent role of p42/p44 and PI3 kinase pathways. FASEB J. 18, 1090–1092.
Zhao, P., Li, X. G., Yang, M., Shao, Q., Wang, D., Liu, S., Song, H., Song, B., Zhang, Y., and Qu, X. (2008). Hypoxia suppresses the production of MMP-9 by human monocyte-derived dendritic cells and requires activation of adenosine receptor A2b via cAMP/PKA signaling pathway. Mol. Immunol. 45, 2187–2195.
Keywords: low oxygen tension, inflammation, post-transcriptional regulation, post-translational regulation, M1 macrophages, M2 macrophages
Citation: Rahat MA, Bitterman H and Lahat N (2011) Molecular Mechanisms Regulating Macrophage Response to Hypoxia. Front. Immun. 2:45. doi: 10.3389/fimmu.2011.00045
Received: 30 June 2011;
Paper pending published: 22 July 2011;
Accepted: 29 August 2011;
Published online: 16 September 2011.
Edited by:
Amiram Ariel, University of Haifa, IsraelReviewed by:
Francesco Saverio Di Giovine, University of Sheffield, UKSeth Lucian Masters, Trinity College Dublin, Ireland
Copyright: © 2011 Rahat, Bitterman and Lahat. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Michal A. Rahat, Immunology Research Unit, Carmel Medical Center, 7 Michal Street, Haifa 34362, Israel. e-mail: rahat_miki@clalit.org.il