
 Michael Molls1
Michael Molls1

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 January 2012
Sec. Inflammation
volume 2 - 2011 | https://doi.org/10.3389/fimmu.2011.00098
This article is part of the Research Topic The immunology of cellular stress proteins View all 11 articles
Chronic inflammatory mediators exert pleiotropic effects in the development of cancer. On the one hand, inflammation favors carcinogenesis, malignant transformation, tumor growth, invasion, and metastatic spread; on the other hand inflammation can stimulate immune effector mechanisms that might limit tumor growth. The link between cancer and inflammation depends on intrinsic and extrinsic pathways. Both pathways result in the activation of transcription factors such as NF-κB, STAT-3, and HIF-1 and in accumulation of tumorigenic factors in tumor and microenvironment. STAT-3 and NF-κB interact at multiple levels and thereby boost tumor-associated inflammation which can suppress anti-tumor immune responses. These factors also promote tumor growth, progression, and metastatic spread. IL-1, IL-6, TNF, and PGHS-2 are key mediators of an inflammatory milieu by modulating the expression of tumor-promoting factors. In this review we concentrate on the crucial role of pro-inflammatory mediators in inflammation-driven carcinogenesis and outline molecular mechanisms of IL-1 signaling in tumors. In addition, we elucidate the dual roles of stress proteins as danger signals in the development of anti-cancer immunity and anti-apoptotic functions.
Based on the presence of leukocytes in cancerous lesions, Rudolf Virchow, the founder of cellular pathology, speculated about an association between chronic inflammation and development of cancer already in 1863 (Virchow, 1863). In line with this observation, epidemiological studies indicate that apart from hereditary predisposition, inflammation serves as a potential risk factor for the development of cancer. Nowadays it is generally accepted that up to 25% of human malignancies are related to chronic inflammation and to viral and bacterial infections (Hussain and Harris, 2007). Table 1 provides an overview on inflammatory and pathogenic conditions that are considered to be associated with malignant transformation.
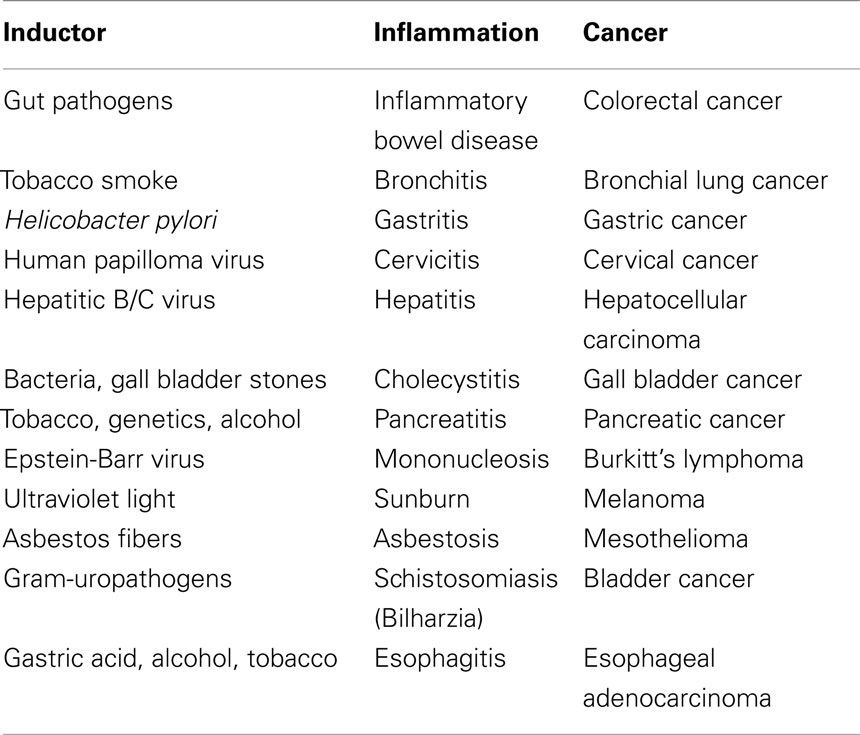
Table 1. Inflammation and their related cancers.
Cancer-related chronic inflammation facilitates unlimited replicative potential, independence of growth factors, resistance to growth inhibition, escape of programmed cell death, enhanced angiogenesis, tumor extravasation, and metastasis (Hanahan and Weinberg, 2000). Cancer-related inflammation represents the seventh hallmark in the development of cancer (Colotta et al., 2009). Persistent microbial infections induced by parasites, bacteria, and viruses and physical and/or chemical stimuli can cause inflammation (Coussens and Werb, 2002). Bacterial infections following surgical removal of primary tumors can promote metastatic growth in mice (Pidgeon et al., 1999) and humans (Taketomi et al., 1997). This process is mediated most likely by endotoxins altering the critical balance between cell growth and angiogenesis (Pidgeon et al., 1999). Moreover, chronic inflammation induced by non-infectious agents can also contribute to carcinogenesis and act as a driving force in tumor development. Apart from toxins, oncoproteins and growth factors can affect the host via an activation of pattern recognition receptors (PRR) that interact with pathogen-associated molecular patterns (PAMP). These receptors comprise to members of the Toll-like receptor (TLR) family, nucleotide-binding oligomerization domain-like (NOD-like) receptors (NLR), C-type lectin receptors (CLR), triggering receptors on myeloid cells (TREM), and retinoic acid inducible gene-I-like receptors (RLR; Kawai and Akira, 2011). Binding of PAMP to these receptors leads to an initiation of the host’s immune response by activation of inflammatory cells. The engagement of PRR triggers the induction of intracellular signaling pathways that induce the activation of numerous transcription factors such as NF-κB, STAT, and FOXO. These factors regulate the expression of several genes involved in the innate and adaptive immunity (Akira et al., 2006; Karin et al., 2006). Inadequate pathogen eradication, recurring tissue injury, prolonged inflammatory signaling, and failure of anti-inflammatory mechanisms can cause chronic inflammation which as a result supports tumorigenesis.
Numerous studies provide evidence that chronic inflammation increases the risk of cancer, promotes tumor progression, and supports metastatic spread (Mantovani et al., 2008; Aggarwal and Gehlot, 2009). In the initial phase of tumor development, inflammatory mediators such as cytokines, reactive oxygen species (ROS), and reactive nitrogen species (RNS) derived from tumor-infiltrating immune cells induce epigenetic alterations in pre-malignant lesions and silence tumor suppressor genes (Grivennikov and Karin, 2010). During tumor promotion, immune cells secrete cytokines and chemokines that act as survival and proliferation factors for malignant cells. The angiogenic switch is critical for an adequate supply of tumor cells with oxygen, nutrition, growth, and survival factors (Zumsteg and Christofori, 2009). During tumor progression and metastasis, both tumor and immune cells produce cytokines and chemokines leading to an increase in cell survival, motility, and invasiveness (DeNardo et al., 2008). Epithelial–mesenchymal transition (EMT), a crucial process in tumor invasiveness and metastasis, is also promoted (Yang and Weinberg, 2008). EMT refers to the loss of carcinoma epithelial phenotype and the acquisition of mesenchymal features (Zeisberg and Neilson, 2009). The group of Mehta recently found that aberrant tissue transglutaminase (TG2) expression induces EMT in epithelial cells (Kumar et al., 2010). This finding, in conjunction with the observation that inflammatory signals (e.g., TGF-β, TNF, and NF-κB) which induce EMT, also induce TG2 expression (Kawata et al., 2011), suggests a possible link between TG2, inflammation, and cancer progression presumably yielding novel therapeutic targets for improved patient outcomes. Other typical markers of EMT are cadherin-11 and fibroblast-specific protein (FSP)-1 which are associated with an increased motility (Zeisberg and Neilson, 2009). Twist is necessary to repress the transcription of E-cadherin (Thiery et al., 2009; Zeisberg and Neilson, 2009).
According to Mantovani et al. (2008), the connection between tumorigenesis and inflammation is mediated via intrinsic and extrinsic pathways. The intrinsic pathway is activated by genetic alterations causing inflammation and neoplasia. These alterations comprise mutation-driven proto-oncogene activation, chromosomal rearrangement/amplification, and inactivation of tumor suppressor genes. Transformed cells secrete inflammatory mediators and thus generate an inflammatory microenvironment. The extrinsic pathway is driven by inflammation or infections that increase the risk for the development of cancer in organs at risk such as the prostate, pancreas, colon, lung, and skin. Both pathways interfere in tumor cells and induce the activation of several transcription factors such as NF-κB, STAT-3, and HIF-1 that result in the formation of pro-inflammatory factors including chemokines, cytokines, and PGHS-2. These molecules recruit and activate various leukocyte populations such as macrophages, mast cells, eosinophils, and neutrophils into the tumor microenvironment like stromal and endothelial cells as well as infiltrating cells. This concerted action of tumor and micromilieu results in a more pronounced generation of inflammatory mediators that drives the progression of a positive amplification loop which further triggers tumor growth and invasiveness.
Proto-oncogene activation represents a critical component in the intrinsic pathway of cancer-related inflammation. In this context, mutations in RAS genes play an important role in tumorigenesis. Overall, up to 30% of all human tumors harbor mutations in canonical RAS genes (KRAS, HRAS, NRAS). Remarkably, these oncogenic mutations predominantly affect the KRAS locus, with oncogenic KRAS mutations being detected in 25–30% of all screened tumor samples (Forbes et al., 2011). The high frequency of KRAS mutations and their appearance in early tumor stages argue for a causative role of the K-Ras protein in human tumorigenesis (Fernandez-Medarde and Santos, 2011). More than 30 years ago the founding members of the RAS gene superfamily (HRAS, NRAS, KRAS) were discovered in human tumors as the first proto-oncogenes. Members of the RAS family are crucial for the connection of up-stream signals to down-stream effector pathways that are functionally related to cell cycle progression, growth, migration, cytoskeletal changes, apoptosis, and senescence. In tumor cells, activation of mutated RAS is followed by the induction of several intracellular signaling pathways. Signaling cascades induced by mutated RAS comprise the RAF/MEK/ERK kinase cascade, the PI3K/AKT pathway, and RalGDS proteins (Downward, 2009), the latter belonging to the family of nucleotide-exchange factors activating small GTPases such as RalB. Via the exocyst complex, an octameric protein complex implicated in tethering of vesicles to membranes (Yamashita et al., 2010), RalB stimulates the TANK-binding kinase-1 (TBK-1) resulting in NF-κB activation by IκBα phosphorylation. In cancer cells, a constitutive activation of this pathway, via chronic RalB activation, restricts the initiation of apoptosis after oncogenic stress (Chien et al., 2006). Beside NF-κB activation, TBK-1 activates the transcription factors IRF-3 and IRF-7 (Hacker and Karin, 2006) leading to the production of growth and inflammatory mediators. Previously it has been shown that K-Ras is a direct inducer of pro-inflammatory IL-6 and pro-angiogenic IL-8 required for the initiation of tumor-associated inflammation and neovascularization and promoting tumor growth. In these studies knock-down of IL6, genetic ablation of the IL6 gene, or treatment with a neutralizing IL-6 antibody retarded K-Ras-driven tumorigenesis (Ancrile et al., 2007). Over-expression of oncogenic K-Ras in tumorigenic HeLa cells induced IL-8 secretion, while IL-8 inhibition reduced growth of these cells and the number of CD31+ cells in a xenograft tumor model (Sparmann and Bar-Sagi, 2004). Moreover, TBK-1 and NF-κB signaling have been identified as being essential in K-Ras mutant tumors (Barbie et al., 2009). Regarding these observations it was assumed that targeting the NF-κB signaling pathway might be effective in treating RAS-mutated tumors (Downward, 2009). Meylan et al. (2009) demonstrated that inhibition of the NF-κB pathway in lung tumors resulted in significantly reduced tumor growth.
Tumor-associated inflammation requires the presence and activation of inflammatory cells such as macrophages and granulocytes in the tumor microenvironment, formation of inflammatory mediators by tumor and stromal cells, tumor remodeling, and angiogenesis (Kundu and Surh, 2008; Colotta et al., 2009). Accumulation of microbial pathogens and tissue necrosis activate transcription factors that are necessary for the expression of, e.g., pro-angiogenic factors (IL-8, VEGF), growth factors (IL-6, GM-CSF), anti-apoptotic factors (Bcl-XL, c-FLIP), invasion-promoting factors (MMP-2, MMP-7, MMP-9, uPA), inflammatory enzymes (PGHS-2, LOX), prostaglandins, iNOS, chemokines (CCL2, CCL20, IL-8), and pro-inflammatory cytokines (IL-1, IL-6, IL-23, TNF, TGF-β, EGF) that support the malignant phenotype. All molecules mentioned above are regulated by the transcription factor NF-κB, a key orchestrator in innate immunity and inflammation that has emerged as a crucial tumor promoter (Karin, 2006). The presence of constitutively active NF-κB was found to be associated with poor clinical outcome (for an overview see Aggarwal and Gehlot, 2009). NF-κB activation in inflammatory cells in response to infectious pathogens, pro-inflammatory mediators as well as necrotic cell products results in the generation of secretable factors that support growth, survival, and vascularization of pre-malignant and malignant cells (Karin, 2006). Activation of NF-κB up-regulates cell cycle mediators (cyclin D1, c-Myc), anti-apoptotic (c-FLIP, survivin, Bcl-XL) and adhesion molecules (ICAM-1, ELAM-1, VCAM-17), proteolytic enzymes (e.g., MMP, uPA), and pro-inflammatory factors (PGHS-2, cytokines) that promote an invasive phenotype (Aggarwal and Gehlot, 2009). iNOS is another important inflammatory mediator that causes the production of NO by macrophages that links chronic inflammation and tumorigenesis. Elevated levels of NO have been found in numerous pre-cancerous and malignant lesions such as Barrett’s mucosa (Wilson et al., 1998), prostate cancer (Aaltoma et al., 2001), breast cancer (De Paepe et al., 2002), and gastrointestinal carcinomas (Wink et al., 1998; Jaiswal et al., 2001). The iNOS product NO contributes to inflammation-associated tumorigenesis by inducing DNA damage, suppression of DNA repair, modification of oncoproteins, inhibition of apoptosis, promotion of tumor growth, angiogenesis, and metastasis as well as suppression of anti-tumor immunity (De Paepe et al., 2002). The NO-mediated inhibition of DNA repair enables cells harboring epigenetic alterations to escape from apoptosis. This results in clonal expansion of pre-malignant cells and subsequently to carcinogenesis (Sawa and Ohshima, 2006). Furthermore, NO promotes tumor growth by a transactivation of HIF-1α (Sandau et al., 2000), induces the expression of pro-angiogenic VEGF (Ravi et al., 2000), and down-regulates the tumor suppressor protein p53 (Ambs et al., 1998).
RANKL, a member of the TNF superfamily of cytokines, was originally found in T and dendritic cells (DC). RANKL supports differentiation and survival of effector cells (Anderson et al., 1997). Moreover, it is essential for the differentiation of bone-resorbing osteoclasts derived from monocyte–macrophage precursors, and enables survival and function of mature osteoclasts (Li et al., 2000; Teitelbaum, 2000). Recent studies documented an expression of RANKL in a variety of other cell types, including tumor cells. Breast cancer cells are able to produce RANKL (Park et al., 2003; Cross et al., 2006) and stimulate osteoclast differentiation when co-cultured with bone marrow stromal cells (Park et al., 2003). HIF-1α-induced expression of RANKL initiates an increased migration of breast cancer cells via the PI3K/AKT pathway (Tang et al., 2011). The expression of RANKL in prostate cancer cells was found to be associated with an increased appearance of bone metastases (Brown et al., 2001; Chen et al., 2006). In head and neck squamous cell carcinoma RANKL expression promotes EMT and tumor progression by inducing VEGF-independent angiogenesis (Yamada et al., 2011). Moreover, the activity of RANKL was found to be involved in the pathophysiology of osteosarcoma (Mori et al., 2007a,b), giant cell tumors of the bone (Ng et al., 2010), Paget’s sarcomas (Sun et al., 2006), and vascular diseases (Hofbauer and Schoppet, 2004). The expression of RANKL increases in response to pro-inflammatory mediators, such as IL-1 (Fernandez et al., 2010; Jurado et al., 2010). In fibroblast-like synoviocytes, murine osteoblastic, and fibroblastic cells, IL-23 was found to induce an up-regulation of RANKL via STAT-3 and NF-κB signaling pathways (Li et al., 2010; Mori et al., 2011). An exposure of these cells to pro-inflammatory cytokines such as IL-1, TNF, and IL-6 resulted into a direct or indirect activation of STAT-3 in a feed-forward loop. Further evidence for a crucial role of STAT-3 in the regulation of RANKL is shown by Schulze et al. (2010) who found that osteolytic prostate cancer cells induce the expression of RANKL in a STAT-3/5-dependent manner. Together these data highlight the significance of a STAT-3/5-mediated cytokine production in tumor cell migration and the formation of distant metastases.
Elevated levels of IL-1 have been identified in several human tumor entities such as melanoma, head and neck, colon, lung, and breast cancer. Overall, patients harboring IL-1-positive tumors have markedly worse prognoses (Lewis et al., 2006). Due to its pleiotropic nature, IL-1 promotes tumor growth and metastasis in an autocrine/paracrine manner. IL-1 is produced by tumor, stromal and endothelial cells, and the host’s infiltrating immune cells (Lewis et al., 2006). Depending on its subcellular location, different IL-1 isoforms mediate different functions. Membrane-bound IL-1α which is expressed on malignant cells induces anti-tumor immune responses, whereas, intracellular residing precursors of IL-1α control homeostatic functions including gene expression, differentiation, and cell growth. In contrast, low concentrations of secreted IL-1β down-regulate inflammatory responses and immune mechanisms, whereas high concentrations promote inflammation-associated tissue damage and tumor invasiveness (Apte et al., 2006). IL-1 can stimulate other cell types to produce pro-angiogenic and pro-metastatic mediators and thus plays an important role in inflammation-associated carcinogenesis (Lin and Karin, 2007; Voronov et al., 2007). In pancreatic cancer IL-1 confers chemoresistance via an up-regulation of PGHS-2 (Angst et al., 2008) and promotes angiogenesis during tumor progression (Shchors et al., 2006).
IL-1α and IL-1β exert identical agonist actions by binding to the IL-1 receptor type I (IL-1RI). After ligation, IL-1/IL-1RI associates with the IL-1 receptor accessory protein (IL-1RAcP) leading to activation of intracellular signal transduction cascades. This complex recruits a number of intracellular adapter molecules including MyD88 (Watters et al., 2007; Gay et al., 2011) to activate signal transduction pathways such as AP-1, p38MAPK, JNK, and NF-κB (Figure 1). In particular NF-κB provides a mechanistic link between inflammation and tumorigenesis. NF-κB is a major factor which controls apoptosis-based tumor immune surveillance mechanisms of pre-neoplastic and malignant cells. NF-κB also regulates tumor angiogenesis and invasiveness (Karin, 2006), and may contribute to chemoresistance of tumor cells (Fahy et al., 2004). A detailed description of the IL-1 signaling pathway is visualized schematically in Figure 1.
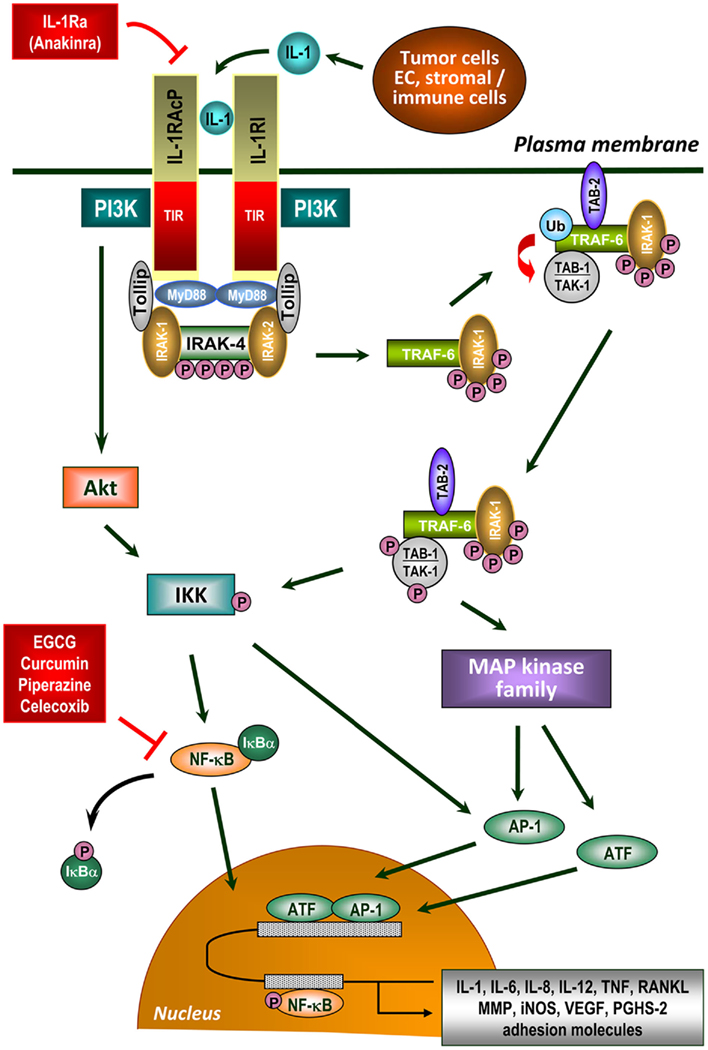
Figure 1. IL-1 signaling in the tumor microenvironment. IL-1 is a critical molecule in inflammation-associated carcinogenesis produced directly by tumor cells or cells of the tumor microenvironment. IL-1 signal transduction is initiated by binding of either form of IL-1 to IL-1 receptor type I (IL-1RI), which undergoes a conformational change allowing the IL-1 receptor accessory protein (IL-1RAcP) to recognize the ligated IL-1RI. IL-1RAcP does not recognize IL-1 but represents an essential component in the IL-1 signaling pathway (Wesche et al., 1997b; Radons et al., 2002). The naturally occurring IL-1 receptor antagonist (IL-1Ra) also binds to IL-1RI without leading to its activation. Ligand-mediated heterodimerization of the receptor complex leads to recruitment of dimeric myeloid differentiation protein 88 (MyD88) via its TIR domain (Muzio et al., 1997; Wesche et al., 1997a; Radons et al., 2003) followed by complex formation between IRAK-4, MyD88, and IL-1RAcP and subsequent phosphorylation of IRAK-4 (Cahill and Rogers, 2008). After recruitment of IRAK-1/Tollip to the complex, IRAK-1 is initially phosphorylated by IRAK-4 (Born et al., 1998; Dunne and O’Neill, 2003). Subsequently, IRAK-1 (and possibly IRAK-2) becomes hyperphosphorylated and dissociates into the cytoplasm where it binds TNF receptor-associated factor 6 (TRAF-6; Cao et al., 1996). IRAK-1 interacts with membrane-bound TAK-binding protein 2 (TAB-2) as well as TAK-1/TAB-1 complex (Dower and Qwarnstrom, 2003) followed by translocation of TAB-2 from the plasma membrane to the signalosome and subsequent partial activation of TAK-1 by TAB-2. IRAK-1, presumably as dimer or oligomer, enables dimerization of TRAF-6 resulting in its ubiquitination and activation. In close proximity to TAB-2, TAK-1 is partially activated followed by complete activation through polyubiquitinated TRAF-6 (Kishimoto et al., 2000; Martin and Wesche, 2002) enabling activation of numerous signaling cascades. Polyubiquitination of TRAF-6 obviously occurs through IRAK-2 (Keating et al., 2007). On the one hand, TAK-1 activates certain members of the MAP kinase family leading to activation of AP-1 and ATF (Ninomiya-Tsuji et al., 1999; O’Neill, 2000; Hefler et al., 2005; Blanco et al., 2008) the latter augmenting NF-κB-mediated transcription via transactivation (Jefferies and O’Neill, 2000; Cahill and Rogers, 2008). On the other hand, TAK-1 phosphorylates and activates IKK resulting in phosphorylation and inactivation of IκBα (Wang et al., 2001). Afterward, IκBα dissociates from the complex with NF-κB and undergoes proteasomal degradation. After phosphorylation, NF-κB translocates to the nucleus and activates NF-κB-dependent gene transcription (Chen and Greene, 2004). Inhibitors of NF-κB activation are indicated that suppress the inflammatory network in cancer development. IL-1 signaling also involves recruitment of PI3-kinase (PI3K) to the IL-1 receptor complex via the p85 regulatory subunit of PI3K (Reddy et al., 1997) and subsequent activation of AKT/PKB leading to IKK-dependent activation of NF-κB and AP-1 (Cahill and Rogers, 2008). Receptor ligation can also activate numerous G proteins resulting in activation of AP-1 and ATF mediated by several MAP kinases and an IκBα-independent transactivation of NF-κB (Singh et al., 1999; Jefferies and O’Neill, 2000). IL-1 signaling finally regulates gene expression of a great variety of tumorigenic factors including pro-angiogenic factors (IL-8, VEGF), growth factors (IL-6, GM-CSF), anti-apoptotic factors (Bcl-XL, c-FLIP), invasion-promoting factors (MMP-2, MMP-7, MMP-9, uPA), inflammatory enzymes (PGHS-2, LOX), prostaglandins, iNOS, chemokines (CCL2, CCL20, IL-8), and pro-inflammatory cytokines (IL-1, IL-6, IL-23, TNF, TGF-β, EGF, RANKL).
A third ligand, the naturally occurring IL-1 receptor antagonist (IL-1Ra), also binds to IL-1RI and acts as a true receptor antagonist. Because of its collagenase and prostaglandin-inhibiting properties, IL-1Ra (anakinra™) is approved for the treatment of chronic inflammatory diseases including rheumatoid arthritis (Dinarello, 1996) and systemic onset juvenile idiopathic arthritis (Hedrich et al., 2011). It has also been identified as being powerful in reverting IL-1 effects in numerous pathological settings (Dinarello, 1996). Actually, anakinra was successfully used in treating the rare lymphoproliferative disorder Castleman’s disease (El-Osta et al., 2010) as well as in myeloma (Lust et al., 2009) rendering the use of anakinra and other IL-1-blocking agents such as canakinumab™ (anti-IL-1β antibody) or rilonacept™ (construct of the two extracellular chains of IL-1RI/IL-1RAcP complex fused to the Fc segment of IgG) promising therapeutic approaches in human metastatic diseases. The last two agents have been approved for the treatment of the cryopyrin-associated periodic syndrome (CAPS; Hoffman et al., 2008; Lachmann et al., 2009a), a grouping of familial cold auto-inflammatory syndrome, Muckle–Wells syndrome, and neonatal onset multi-inflammatory disease. As summarized by Dinarello (2010), there are two meaningful reasons for the use of IL-1-blocking agents in the treatment of metastatic diseases. On the one hand, none of the above mentioned agents have been found as being associated with any organ toxicities, gastrointestinal, or hematological abnormalities. On the other hand, unlike TNF-blocking agents IL-1-inhibiting treatments lack opportunistic infections although routine bacterial and upper airway infections are observed. Due to the safety of IL-1 blockage and the availability of the three therapeutics in limiting IL-1 actions, clinical trials are encouraged. An NIH trial of anakinra in the treatment of cutaneous melanoma is ongoing because IL-1 plays a pivotal role in angiogenesis by inducing/up-regulating pro-angiogenic IL-8 and VEGF contributing to the pathogenesis of, e.g., multiple melanoma (Dinarello, 2010).
As a pleiotropic cytokine, IL-1 harbors numerous intensifying effects on the physiological functions of diverse innate and immunocompetent cells (Mizel, 1982), IL-12-mediated induction of Th1 development (Weaver et al., 1988), and induction of Th17 cells (Sutton et al., 2006). IL-1 effects on immune tolerance are also reported (Nakata et al., 1995). For instance, IL-1β stimulates function of memory T cells and impairs that of Treg cells (O’Sullivan et al., 2006). This brief overview highlights the complexity of the mechanisms by which IL-1 regulates all types of immune responses including tumor cell eradication.
IL-1β is first synthesized as biologically inactive precursor (pro-IL-1β) that is further processed by caspase-1, also known as IL-1-converting enzyme (ICE), to the mature form, while pro-IL-1α is cleaved by calpain. Although IL-1β contributes to growth and metastatic spread in experimental and human cancers, the molecular mechanisms regulating the conversion of pro-IL-1β to the secreted and active cytokine remains to be elucidated.
An elaborate multi-protein complex, the so-called “inflammasome,” is responsible for the recruitment and activation of caspase-1 (Martinon et al., 2002). Each inflammasome consists of different members of the nucleotide oligomerization domain-like receptor (NLR) family of proteins. Two of the best characterized human inflammasomes are NALP (NACHT, LRR, and pyrin domain-containing protein) 1 inflammasome and NALP2/3 inflammasome (Franchi et al., 2009). It was shown previously that in several auto-inflammatory diseases constitutive activation of NALP3 inflammasome leads to sustained local and systemic inflammation mediated by IL-1β (Goldbach-Mansky et al., 2006; Lachmann et al., 2009b). Recently, constitutively activated inflammasome was found in human melanoma cells (Okamoto et al., 2010). In this study human melanoma cells from the late stage of the disease spontaneously secrete biologically active IL-1β in the absence of exogenous stimuli because of constitutive activation of the inflammasome and IL-1 receptor (IL-1R) signaling. From these findings it can be concluded that IL-1-mediated autoinflammation contributes to the development and progression of human melanoma suggesting that inhibiting the inflammasome pathway or reducing IL-1 activity can be a therapeutic option for melanoma patients. The inflammasome also plays a substantial role in environmental cancer. It has been shown previously that silica and asbestos both activate the NALP3 inflammasome resulting in an increased IL-1β production and causing lung inflammation (Dostert et al., 2008). Chronic exposure to asbestos has been identified as being a high-risk factor for the development of mesothelioma implying a crucial contribution of inflammasome-mediated inflammation to the pathogenesis of mesothelioma. Controversely, in an animal model of colitis-associated cancer (CAC) the NALP3 inflammasome was found to be protective against CAC (Allen et al., 2010). The NALP3 inflammasome in DC obviously plays a crucial role by linking innate and adaptive immune responses against dying tumors (Ghiringhelli et al., 2009). Based on these observations one can hypothesize that constitutively active NALP3 inflammasome as can be found in certain tumors produces large amounts of IL-1 contributing to cancer-related inflammation and thus promoting tumor growth and invasiveness, whereas activation of the inflammasome in tumor-infiltrating immune cells might be beneficial in inducing anti-tumor immunity. According to Menu and Vince (2011), the NALP3 inflammasome can be considered as a triple-function agent (“the good, the bad, and the ugly”) in human malignancies.
The pleiotropic cytokine TNF plays a dual role in tumorigenesis. At high concentrations TNF is destructive to tumor vasculature and induces necrosis. On the other hand, its critical role in chronic inflammation and its tumor-promoting capacity are well documented (Lin and Yeh, 2005; Mocellin et al., 2005). An increased expression of TNF was found in human bladder, breast, colorectal, and prostate cancer as well as in leukemia and lymphoma (Balkwill and Mantovani, 2001). TNF is also produced by cells of the tumor microenvironment. Binding of TNF to the TNF receptor 1 (TNFR1) activates signaling cascades of NF-κB and c-Jun N-terminal kinase (JNK) which lead to an up-regulation of several pro-inflammatory, pro-angiogenic and invasiveness-promoting factors, and to the induction of anti-apoptotic molecules such as the caspase-8 inhibitor c-FLIP. Activation of NF-κB in response to TNFR1 terminates the activity of JNK (Kamata et al., 2005). The ubiquitin ligase Itch is a substrate of JNK that enables the degradation of c-FLIP (Chang et al., 2006). Inhibition of JNK via NF-κB-mediated blockage leads to an inactivation of Itch. This prevents degradation of c-FLIP and ensures tumor cell survival. Apart from its role in tumor initiation, TNF promotes angiogenesis and impairs immune surveillance by affecting T cell responses and the activity of macrophages (Elgert et al., 1998). The tumor-promoting role of TNF was confirmed in animal models. In the absence of TNF mice do not develop hepatocellular carcinoma in response to cholestatic hepatitis (Pikarsky et al., 2004). TNF and TNFR-deficient mice were also found to be resistant to chemically induced carcinogenesis of the skin (Arnott et al., 2004). These findings indicate that pro-inflammatory activity of TNF functions as a pivotal mediator in tumorigenesis (Lin and Karin, 2007).
IL-6 is another NF-κB-regulated pleiotropic pro-inflammatory mediator that enables tumor growth and inhibits apoptosis in a variety of human tumors (Rose-John and Schooltink, 2007). In contrast, IL-6 has also been reported as playing a crucial role in terminating inflammation (Hudson et al., 2008). IL-6 signaling via the membrane-bound receptor IL-6Ra is linked to the JAK/STAT pathway (predominantly through activation of STAT-3) and leads to the expression of genes encoding for anti-apoptotic cell cycle progression molecules (Lin and Karin, 2007). In contrast, a soluble form of the IL-6R can bind IL-6 with the same affinity as the membrane-bound form and the complex of IL-6 and the soluble IL-6R (sIL6R) can induce signaling in a process called IL-6 trans-signaling (Peters et al., 1998). Because the IL-6R is only sparely expressed, IL-6 trans-signaling dramatically increases the number of potential IL-6 target cells (Rose-John et al., 2006). Animal models of inflammatory colon cancer suggest that IL-6 trans-signaling serves as the major pro-inflammatory paradigm of IL-6 signaling under pathophysiologic conditions (Becker et al., 2004). It turns out that regenerative or anti-inflammatory activities of IL-6 are mediated by classic signaling whereas pro-inflammatory responses of interleukin-6 are rather mediated by trans-signaling (Rabe et al., 2008). This is important since therapeutic blockade of IL-6 by the neutralizing anti-IL-6 receptor monoclonal antibody tocilizumab™ has recently been approved for the treatment of inflammatory diseases. A recently performed clinical trial revealed that IL-6 inhibition by tocilizumab retards joint damage progression in patients with rheumatoid arthritis (Smolen et al., 2011). Interestingly, inhibition of IL-6R-mediated signaling using tocilizumab in a xenograft model of oral squamous cell carcinoma (OSCC) suppressed tumor growth and angiogenesis by down-regulating VEGF mRNA expression (Shinriki et al., 2009). Clinical studies inclusive those mentioned above have shown that inhibition of IL-6 signaling by tocilizumab is therapeutically effective not only in chronic inflammatory diseases such as rheumatoid arthritis (Nishimoto et al., 2004), juvenile idiopathic arthritis (Yokota et al., 2004), and Crohn’s disease (Ito et al., 2004) but also in Castleman’s disease (Nishimoto et al., 2005). In all of these diseases, tocilizumab ameliorates inflammatory manifestations, and normalizes acute phase protein levels. Given its success in treating these diseases, tocilizumab may also prove useful in treating IL-6-related cancers.
Elevated IL-6 levels are found in numerous tumors such as multiple myeloma (Klein et al., 1992), colorectal cancer (Chung and Chang, 2003), gastric carcinoma (Kai et al., 2005), and Hodgkin lymphoma (Cozen et al., 2004). Moreover, malignant ascites from patients with epithelial ovarian cancer was found to contain high levels of IL-6 (Offner et al., 1995). In breast cancer patients high IL-6 concentrations induced by an IL6 gene polymorphism correlate with poor prognosis (Berger, 2004). A comparison of non-metastasizing pancreatic cancer, benign prostatic hyperplasia, and metastasized pancreatic cancer revealed elevated levels of IL-6 in the latter, more aggressive tumor (Weiss et al., 2011). In this study it was shown that IL-6 leads to an increased expression of uPA and VEGF which implies a crucial role of IL-6 in angiogenesis of pancreatic tumors. In OSCC lysophosphatidic acid (LPA), a bioactive lipid with a growth factor-like activity induces the secretion of IL-6 and IL-8 in an NF-κB- and AP-1-dependent manner (Hwang et al., 2011). Direct stimulation of human osteoblasts with IL-6 and IL-8 induced the expression of RANKL and thereby promotes osteoclast formation. From these findings it can be concluded that IL-6 and IL-8 derived from LPA-stimulated OSCC play a crucial role in osteogenesis and bone resorption. Inhibition of IL-6 signaling in colon cancer resulted in a reduced tumor growth in mice (Becker et al., 2004; Greten et al., 2004). Incubation of cholangiocarcinoma cells with an anti-IL-6-neutralizing antiserum reduced AKT phosphorylation and down-regulated the expression of Mcl-1. This indicates a contribution of IL-6 in the AKT-mediated survival mechanisms (Kobayashi et al., 2005).
PGHS-2, formerly termed as COX-2, has emerged as another pro-inflammatory mediator in tumorigenesis whose expression is mediated by NF-κB. The expression of PGHS-2 is inducible in response to stimuli such as mitogens, cytokines, growth factors, or hormones. PGHS-2 is the rate-limiting enzyme involved in the conversion of arachidonic acid to prostanoids acting as key mediators of inflammation. Aberrant or increased expression of PGHS-2 has been shown to be involved in the pathogenesis of breast, gastric, colorectal, lung, prostate, head/neck, and pancreatic cancer. PGHS-2 affects cell proliferation, apoptosis, angiogenesis, and metastasis (Lu et al., 2006; Aggarwal and Gehlot, 2009). Over-expression of PGHS-2 results in the secretion of large amounts of VEGF and therefore, is associated with increased tumor cell invasion and poor prognosis (Raut et al., 2004; Ladetto et al., 2005). In human basal cell carcinoma cells elevated levels of PGHS-2 led to an up-regulated expression of the anti-apoptotic molecules Mcl-1 and Bcl-2, VEGF and basic fibroblast growth factor (bFGF; Tjiu et al., 2006). In chronic inflammation, endothelial cells express both, acute phase genes and adhesion molecules that enable recruitment of leukocytes to the site of tissue damage. Moreover, an enhanced production of prostaglandins mediated by PGHS-2 augments vasopermeability leading to a more pronounced recruitment of leukocytes (Jura et al., 2005). Leukocytes are the main source of RNS and ROS acting as chemical effectors in inflammation-driven carcinogenesis (Kundu and Surh, 2008). We identified a constitutively enhanced expression of PGHS-2 in human pancreatic adenocarcinoma cells that is further increased in the presence of IL-1 (Bauer et al., 2009; Hoffmann et al., 2011). The constitutive production of PGHS-2 and its key product PGE2 in the microenvironment of pancreatic carcinomas accounts for an enhanced malignancy of pancreatic tumor cells which is caused by inhibition of apoptosis, increase in cell proliferation, induction of angiogenesis, and invasion of malignant cells into surrounding tissue (Merati et al., 2001; Kong et al., 2002; Garcea et al., 2005). PGHS-2 also induces the expression of MMP-2 (Surh et al., 2001; Sansone et al., 2009; Wang et al., 2009b) and pro-inflammatory IL-6 and IL-1 via PGE2 (Takahashi et al., 2008). PGHS-2-mediated effects on growth, angiogenesis, invasiveness, and metastasis are augmented by the IL-1-induced up-regulation of the enzyme by forcing the progression of a positive amplification loop triggered by PGE2 and IL-6. Of note, it was demonstrated that PGE2 contributes to cancer progression by inhibiting DC differentiation and function, acting paradoxically as an immunosuppressive factor (Muthuswamy et al., 2010; Stock et al., 2011). In cervical cancer, PGE2 was found to induce a cytokine production profile and phenotypical features of tolerogenic DC suggesting that the altered expression of PGE2 might promote carcinogenesis by favoring (pre)cancer immunotolerance (Herfs et al., 2009). In addition, IL-6 derived from tumor cells or cells of the tumor microenvironment was shown to polarize DC toward immune tolerance through the induction of STAT-3 activation (Alshamsan, 2011). Therefore, tumor-induced p-STAT-3 in DC can be seen as a promising target for colon cancer immunotherapy. In this context, knocking-down the IL-6 receptor α-chain of DC vaccines significantly enhanced the frequency of tumor-specific CD8+ CTL-producing effector molecules such as IFN-γ, TNF, FasL, perforin, and granzyme B, and generated more CD8+ memory T cells, leading to the substantially prolonged survival of cytotoxic lymphocytes (Tc1) tumor-bearing mice (Hwang et al., 2010).
Most of the PGHS-2-induced effects are mediated through its product PGE2 (Yoshimatsu et al., 2001a,b). Thus, down-regulation of prostaglandins in tumor tissues by PGHS-2 inhibition blocks several neoplastic pathways leading to the suppression of tumor growth (Maier et al., 2004). In this context, PGHS-2 inhibitors hold promise for cancer chemoprevention. Among them, the non-steroidal anti-inflammatory drug (NSAID) celecoxib constitutes a potent and specific inhibitor of the inducible human PGHS-2. Celecoxib interferes with tumor initiation and tumor cell growth in vitro and in vivo. Preclinical studies demonstrate promising anti-cancer effects of celecoxib in colorectal, pancreatic as well as head and neck carcinomas. Additionally, celecoxib was found to increase tumor cell sensitivity toward radiochemotherapy (reviewed by Jendrossek, 2011). Celecoxib has also been found to impair tissue expression of VEGF, tumor angiogenesis, and metastasis in an experimental model of pancreatic cancer (Wei et al., 2004). Thus, modulation of PGHS-2 expression may be a promising approach in cancer therapy (Jimeno et al., 2006). However, due to the high toxicity of PGHS-2 inhibitors, the development of novel components is necessary (Spektor and Fuster, 2005; Lee et al., 2007). Randomized clinical trials and meta-analyses reported on an increased risk for cardiovascular diseases in patients receiving long-term treatment with PGHS-2 inhibitors. These cardiovascular adverse effects include myocardial infarction, stroke, and cardiovascular death/heart failure (summarized by Trelle et al., 2011). This increased rate of life-threatening cardiovascular side effects led to the withdrawal of the PGHS-2 inhibitors valdecoxib and rofecoxib from the market that had been approved by the United States Food and Drug Association. A patient-pooled analysis of adjudicated data from 7,950 patients in six placebo-controlled trials demonstrated a dose regimen-related increase in the risk of serious cardiovascular events after a daily administration of 400 and 800 mg celecoxib (Solomon et al., 2008). Although long-term treatment with high-dose celecoxib can enhance the risk for cardiovascular diseases, the drug is still used at lower doses in the United States since it is less toxic compared to other PGHS-2 inhibitors (Solomon et al., 2008; Trelle et al., 2011).
A novel approach to overcome the limitations associated with the toxicity of PGHS-2 inhibitors is to combine chemical PGHS-2 inhibitors at low doses with naturally occurring compounds such as the catechin EGCG which is a promising chemopreventive agent derived from green tea (summarized by Cerella et al., 2010). Our group investigated the effects of a combinatorial treatment with celecoxib and EGCG on the expression of IL-1-induced tumorigenic factors in human pancreatic adenocarcinoma cells. We found that the combined administration of celecoxib and EGCG can induce synergistic cancer preventive effects in pancreatic cancer cells by down-regulating tumorigenic factors and inducing apoptosis (Härdtner et al., 2009). Previous investigations of our group and others revealed anti-proliferative and apoptosis-inducing effects of EGCG and celecoxib in pancreatic cancer cells (Chen and Zhang, 2007; Inaba et al., 2008; Xu et al., 2008; Hoffmann et al., 2011). The anti-proliferative properties of NSAID such as celecoxib are related to effects on the cell cycle (Xiong, 2004) including changes in gene expression that favor cell cycle arrest (Yip-Schneider et al., 2001; Tseng et al., 2002). Interestingly, several studies revealed an anti-proliferative effect of PGHS-2-selective inhibitors not only in PGHS-2-positive but also in PGHS-2-negative pancreatic tumor cells implying that the inhibitory action of NSAID on cell proliferation can affect both, PGHS-2-dependent and -independent pathways (Molina et al., 1999; Yip-Schneider et al., 2001). Numerous investigations also documented an apoptosis-inducing potential of NSAID (Maier et al., 2004; Suganuma et al., 2011). Celecoxib targets several proteins distinct from PGHS-2 that are involved in the control of cell survival and cell death including the anti-apoptotic proteins survivin, Mcl-1, and Bcl-2 (Sakoguchi-Okada et al., 2007; Rudner et al., 2010). Further, PGHS-2-independent molecular targets of celecoxib comprise the survival kinase AKT/PKB and its up-stream regulator 3-phosphoinositide-dependent kinase-1 (PDK-1; Belham et al., 1999; Kulp et al., 2004), cyclin-dependent kinase inhibitors, and cyclins (Grosch et al., 2006), as well as the sarcoplasmic/endoplasmic reticulum calcium ATPase SERCA (Johnson et al., 2002). By counteracting these molecules celecoxib interferes with the activation status of caspases and finally induces apoptosis.
EGCG-mediated effects on apoptosis include caspase-3/-9 activation, PARP cleavage, Bax oligomerization, mitochondrial membrane depolarization, direct interaction with anti-apoptotic members of the Bcl-2 family as well as NF-κB inhibition (Lambert et al., 2005; Shimizu et al., 2005; Inaba et al., 2008). Celecoxib is reported to interfere, among others, with the NF-κB signaling pathway (Niederberger et al., 2001; Shirode and Sylvester, 2010) providing the basis for the synergism with EGCG. Based on these findings one can hypothesize that celecoxib in combination with EGCG may promote apoptosis directly or indirectly thus altering the cellular death threshold in tumor cells (Jendrossek, 2011). In previous studies EGCG has been shown to synergistically enhance the effects of TRAIL (Siddiqui et al., 2008) and PGHS-2 inhibitors NS-398 (Adhami et al., 2007) and celecoxib (Basu and Haldar, 2009). In a human lung (Suganuma et al., 2011) and prostate cancer (Adhami et al., 2007) model the combination of celecoxib and EGCG increased tumor cell apoptosis and decreased inflammation. Other natural compounds affecting the PGHS-2 expression include the non-flavonoid polyphenols curcumin from turmeric Curcuma longa, resveratrol from red wine, isoflavone genistein from lupin as well as omega-3 fatty acids from oily fish flaxseeds. Such a combinatory administration might have future clinical implications with respect to an adjuvant therapy in cancer patients, since it might reduce the adverse effects of high-dose celecoxib as a monotherapy.
Both, STAT-3 and NF-κB are crucial for cancer-related inflammation. NF-κB does not only mediate tumorigenesis but also exerts anti-tumorigenic effects in tumor cells and in the tumor microenvironment (for a review see Ben-Neriah and Karin, 2011). Evidence for a positive association of NF-κB activation with tumor-associated inflammation came from colitis-associated colon cancer (Greten et al., 2004; Pikarsky et al., 2004) and hepatitis-associated hepatocellular carcinoma (Pikarsky et al., 2004). Colitis-associated colon cancer represents a classical example for an inflammation-triggered malignancy. NF-κB activation in intestinal epithelial cells of this cancer model was found to enhance the survival of pre-malignant progenitor cells by inducing anti-apoptotic Bcl-XL (Greten et al., 2004; Pikarsky et al., 2004). NF-κB activation in cancer seems to be related, at least in part, by mutations in components of the signaling cascade or effects of inflammatory factors in the tumor microenvironment that accumulate after NF-κB activation (Karin et al., 2002). Transcriptional activation of NF-κB leads to the induction of pro-inflammatory cytokines (e.g., IL-1, IL-6, TNF), chemokines (IL-8), PGHS-2, MMP, and adhesion molecules (ICAM-1, VCAM-1). The presence of constitutively active NF-κB in most tumors correlate with a poor clinical outcome (Weichert et al., 2007). Moreover, most chemopreventive agents including nutraceuticals derived from different sources have the potential to suppress constitutive and inducible NF-κB activation pathways (Aggarwal and Gehlot, 2009) in order to block chronic inflammation.
Whilst NF-κB signaling contributes to both, inflammation-driven carcinogenesis and anti-tumor immunity, STAT-3 induces cancer-promoting inflammation and restrains anti-tumor immune responses by counteracting NF-κB-induced expression of anti-tumor Th1 cytokines (IL-12, IFN-γ; Kortylewski et al., 2005; Yu et al., 2007). Furthermore, STAT-3 contributes to the expansion and development of Treg and Th17 cells (Wang et al., 2009a; Wu et al., 2009). STAT-3 also induces the expression of tumorigenic mediators (cytokines, pro-angiogenic, and growth factors) and their corresponding receptors that in turn activate a STAT-3 mediated immunoregulatory circuit in the tumor microenvironment (Yu et al., 2007). Thus, the constitutive activation of STAT-3, does not only promote cancer-related inflammation but also suppresses anti-tumor immune responses (Yu et al., 2009).
As already mentioned, the association of cancer with chronic inflammation is related to intrinsic and extrinsic pathways, both leading to activation of NF-κB and STAT-3. Similarly to NF-κB, constitutively active STAT-3 is found in breast, ovarian, prostate, and brain tumors, leukemia, lymphoma, and multiple myeloma. STAT-3 activation results in the modulation of the expression of numerous genes that are crucial for maintaining/amplifying tumor-associated inflammation and promoting tumor growth and progression (Yu et al., 2009). The NF-κB family comprises homo- and heterodimeric transcription factors consisting of RelA, c-Rel, RelB, NF-κB1 (p50 and its precursor p105), and NF-κB2 (p52 and its precursor p100) with RelA-p50 as being the most prominent NF-κB transcription factor (Vallabhapurapu and Karin, 2009). Physiologically, NF-κB is sequestered in the cytosol by its inhibitory component, IκBα. Upon phosphorylation by the IκB kinase complex (IKK), IκBα is degraded in an ubiquitin-dependent manner in the proteasome. Then NF-κB translocates into the nucleus where predominantly RelA-p50 up-regulates the expression of Th1 immunostimulatory genes (IL-12, CD40, CD80) that are important for the control of microbial infections and tumor cells (Yu et al., 2007, 2009). STAT-3 opposes the anti-tumor immune responses mediated by NF-κB within a cell. On the one hand, STAT-3 is able to inhibit IKK during acute inflammation and thus attenuates Th1 immune responses (Lee et al., 2009). On the other hand, STAT-3 prolongs the nuclear retention of RelA during oncogenic and chronic inflammation by acting as a co-transcription factor for RelA thus contributing to the persistent activation of NF-κB during chronic inflammation and the malignant process (Yu et al., 2009). It has been shown previously that STAT-3 promotes nuclear localization of RelA by acetyltransferase p300-mediated acetylation affecting the NF-κB/IκBα interaction and avoiding its nuclear export (Chen and Greene, 2004). Since STAT-3 is a prerequisite for p300-mediated acetylation of RelA, constitutive activity of RelA in tumors requires continuous STAT-3 signaling (Lee et al., 2009). Accordingly, increased STAT-3 activity found in tumors preferentially leads to an association of NF-κB with STAT-3 via p300. Due to the NF-κB activating capacity of STAT-3 in malignancies constitutive activity of STAT-3 found in tumors preferentially requires RelA (Yu et al., 2009). As stated by the authors, this reciprocal relationship is related to the fact that numerous RelA-encoded target gene products function as STAT-3 activators (e.g., IL-6, IL-11, IL-17, IL-21, IL-23, PGHS-2). Remarkably, expression of IL-6, IL-17, IL-23, and PGHS-2 (all of them activating STAT-3) depends on STAT-3 as co-transcription factor for NF-κB. As a consequence, STAT-3 and NF-κB interact at multiple levels and thereby boost tumor-associated inflammation.
Heat shock proteins (HSP) are highly conserved proteins expressed in a wide range of species where they inhabit nearly all cellular and subcellular compartments. Environmental stress (e.g., heat, hypoxia, bacterial infections, heavy metals, oxidative stress, inflammation) as well as physiological processes (differentiation, proliferation, maturation) result in an increased HSP synthesis (Lindquist and Craig, 1988; DeNagel and Pierce, 1992). Intracellular residing HSP protect cells against lethal damage induced by environmental stress, and support folding and transport of newly produced polypeptides and aberrant proteins (Hartl, 1996). Depending on their intra-/extracellular localization HSP mediate different functions. On the one hand, up-regulated intracellular HSP levels protect tumor cells from lethal damage induced by environmental stress. On the other hand, membrane-bound and extracellular residing HSP with molecular weights of 70 and 90 kDa were identified as key regulators of the host’s immune system.
A variety of HSP were found on the plasma membrane of tumor cell lines as determined by selective cell surface protein profiling (Shin et al., 2003). These findings were confirmed by a broad screening program of human tumor biopsies in our laboratory using cmHsp70.1 mAb (Stangl et al., 2011). Phenotypic analyses revealed that Hsp70, the major stress-inducible member of the HSP70 group, is found on the plasma membrane in 50–70% of colon, lung, pancreas, mammary, head and neck, lung, and urogenital carcinomas (Multhoff et al., 1995a,b; Chen et al., 2002). Metastases exhibit an elevated Hsp70 membrane density compared to primary tumors in humans (unpublished observation). These data were confirmed in a xenograft tumor mouse model. After orthotopic injection of human tumor cells into immunodeficient animals the cell surface density of Hsp70 was greater on metastases than on primary tumors (Multhoff et al., 2000; Stangl et al., 2006). Interestingly, the corresponding normal tissue of the mice was always found to be membrane Hsp70-negative (Stangl et al., 2011). These findings might be explained by the fact that membrane Hsp70 might facilitate metastases, support adherence of tumor cells to endothelial cells and organs, or might confer resistance to an unfavorable milieu during metastasis. In line with these findings we could show that overall survival of patients with membrane Hsp70-positive squamous cell carcinomas of the lung and lower rectal carcinomas was significantly reduced compared to those patients with membrane Hsp70-negative tumors (Pfister et al., 2007). Apart from solid tumors also bone marrow samples of patients suffering from acute (AML) and chronic (CML) myeloid leukemia are frequently membrane Hsp70-positive (Gehrmann et al., 2003). Quantitative analysis revealed that ∼15–20% of the total Hsp70 is present in tumor cell membranes (Gehrmann et al., 2008). The anchorage of Hsp70 within the plasma membrane is most likely mediated by the tumor-specific glycosphingolipid Gb3 (Gehrmann et al., 2008). This led us to the hypothesis that membrane Hsp70 might provide an ideal tumor-specific molecule for a targeted immunotherapeutic approach.
Even in the absence of immunogenic peptides, Hsp70, or a peptide derived thereof in combination with pro-inflammatory cytokines such as IL-2 and IL-15 has the capacity to stimulate the cytolytic activity of NK cells against membrane Hsp70-positive tumor cells (Multhoff et al., 1997, 2001). The mechanism of tumor cell killing has been identified as perforin-independent granzyme B-mediated apoptosis (Gross et al., 2003b). Granzyme B derived from activated NK cells specifically binds to membrane Hsp70 on tumor cells and following Hsp70-mediated endocytosis, apoptosis is induced (Gross et al., 2003a,b). Hsp70 also has been detected on tumor-derived exosomes of membrane Hsp70-positive tumors (Gastpar et al., 2005). These data suggest that NK cells might be attracted to membrane Hsp70-positive tumors in vivo via the secretion of Hsp70 surface-positive exosomes. Incubation of NK cells with Hsp70 protein or a 14mer-peptide derived from the C-terminus of Hsp70 is accompanied by an up-regulation of activating receptors on NK cells such as CD94/NKG2C, NKG2D, NKp30, NKp44, and NKp46 (Gross et al., 2003b,c). Hsp70 membrane-positive tumors are thus efficiently eliminated by NK cells that had been pre-stimulated with low dose IL-2 plus Hsp70 peptide (Multhoff et al., 1999). Adoptive transfer of these TKD-stimulated NK cells in tumor-bearing mice revealed identical results in vivo (Botzler et al., 1998; Multhoff et al., 2000; Moser et al., 2002). It is known that IL-2-activated NK cells are able to induce regression of established lung and liver tumors (Schwarz et al., 1989; Yasumura et al., 1994; Vujanovic et al., 1995; Whiteside et al., 1998). Our group identified a specific migratory capacity of NK cells toward Hsp70-postive tumor cells and supernatants derived thereof. The same effect could be observed for the Hsp70 peptide TKD (Gastpar et al., 2004). From these results we speculated that killing of Hsp70-positive tumors in vivo might be related to an enhanced migratory and cytolytic capacity of pre-activated NK cells.
The rapid induction of HSP in response to environmental stress is based on a variety of genetic and biochemical processes referred to as the heat shock response (HSR; Shamovsky and Nudler, 2008). The link between HSR and cancer development has been emerging since more than 20 years. HSR is regulated mainly at the transcription level by heat shock factors (HSF). Among them, HSF-1 is considered as being the key transcription factor of stress-inducible HSP (Pirkkala et al., 2001; Akerfelt et al., 2010). As a consequence, HSP are over-expressed in a wide spectrum of human malignancies contributing to tumor growth, differentiation, invasiveness, and metastasis and being associated with poor prognosis in certain cancer types (Ciocca and Calderwood, 2005). HSP over-expression in tumor cells plays a pivotal role in tumorigenesis by inhibiting apoptosis and senescence. In breast cancer, transformation-induced activation of HSF-1 results in an up-regulated expression of Hsp27 and Hsp70 which in turn results in protection against apoptosis (Calderwood, 2010). HSF-1 also triggers expression of Hsp90, an essential factor in tumor growth due to its ability to chaperone a variety of oncogenic signaling proteins including Her-2/neu and c-Src (Kamal et al., 2003; Neckers and Lee, 2003; Calderwood, 2010). Several studies have shown that Her-2/neu (c-ErbB-2) is amplified and over-expressed in many tumors such as breast, ovarian, and gastric adenocarcinoma (Hynes and Stern, 1994). Since HSP over-expression also protects from drug-related apoptosis (Khaleque et al., 2005), these mechanisms highlight the role of HSP in tumor progression and therapy resistance.
Recent studies indicate an involvement of HSP such as Hsp70/Hsp72 and Hsp90 in the recognition of PAMP by binding to TLR-4 within lipid rafts (Triantafilou and Triantafilou, 2004; Wheeler et al., 2009). Since extracellular residing Hsp70 acts as a danger signal for the immune system (Matzinger, 1998), this stress protein has been added to the list of “alarmins.” Endogenous alarmins and exogenous PAMP both comprise the group of danger-associated molecular patterns (DAMP; Bianchi, 2007). Hsp70, added exogenously to cells stimulates the production of pro-inflammatory cytokines TNF, IL-1β, and IL-6 by antigen presenting cells (Asea et al., 2000a,b, 2002). Extracellular Hsp70 has also been found to induce IL-8 production in human bronchial epithelial cells (Chase et al., 2007). In vitro co-culturing of colon tumor cell spheroids with normal cells caused a significant tumor grade-dependent increase in IL-6 production thereby altering Hsp70 expression (Paduch et al., 2009). From these observations it can be concluded that Hsp70 may enhance the impact of tumorigenic mediators in the tumor microenvironment.
Cancer-related inflammation has emerged as one of the hallmarks of cancer (Hanahan and Weinberg, 2011). In the last two decades, several tumorigenic factors have been identified as being implicated in inflammation-associated carcinogenesis. These factors are released by tumor cells or cells of the tumor microenvironment such as stromal cells, endothelial cells, or host infiltrating cells, respectively, and include pro-inflammatory cytokines, pro-angiogenic and growth-promoting factors, anti-apoptotic and invasion-promoting factors, inflammatory enzymes, prostaglandins, iNOS as well as chemokines. Among them, IL-1, TNF, and IL-6 act as crucial mediators of inflammation-driven tumorigenesis forming an inflammatory network in cancer as outlined in Figure 2. These mediators activate the key transcription factors in tumor-associated inflammation: NF-κB, STAT-3, and HIF-1 impacting any stage of tumorigenesis such as initiation, promotion as well as progression, and metastasis. It is noteworthy that NF-κB might be the central player in tumorigenesis. NF-κB is activated by a great variety of lifestyle-related factors including infectious agents, irradiation, environmental stimuli, tobacco, stress, dietary agents, obesity, and alcohol accounting for almost 95% of all cancers (Aggarwal and Gehlot, 2009). Modern anti-tumor therapies thus aim to suppress NF-κB activation. Most of the chemopreventive agents have been found as being able to suppress NF-κB activation like the selective PGHS-2 inhibitor celecoxib. Moreover, lifestyle-related agents derived from different sources including fruits, legumes, vegetables, grains, spices, and exercise are also able to inhibit NF-κB leading to suppression of the inflammatory network (Aggarwal and Gehlot, 2009). Clinical and preclinical studies are conducted to suppress the inflammatory network by the use of, e.g., steroids (dexamethasone, prednisolone), TNF inhibitors (thalidomide = thalomid™, anti-TNF antibodies such as infliximab = remicade™, etanercept = enbrel™, adalimumab = humira™), IL-1 inhibitors (anakinra™ = IL-1 receptor antagonist), PGHS-2 inhibitors (celecoxib), NF-κB inhibitors (curcumin, EGCG, piperazine), and RANKL inhibitors (denosumab™ = fully human monoclonal anti-RANKL antibody). A promising approach in cancer therapy also might be targeting HSP, because up-regulated Hsp90 and Hsp70 in cancer cells have been recognized as important drug targets and are under intensive studies in recent years. HSP are currently being targeted in the therapy of breast cancer and other carcinomas and effective drugs for Hsp90 (e.g., geldanamycin and its analogs) have been synthesized and evaluated in clinical trials (Calderwood and Gong, 2011; Kim and Kim, 2011). HSP vaccines have been intensively studied in the preceding two decades, proving to be safe and effective in treating a number of malignancies (Murshid et al., 2011). However, therapeutical approaches that completely block one tumorigenic factor should be avoided, since it might also interfere with the physiological anti-tumor immune response and could therefore prove to be harmful. Instead, partial inhibition of numerous factors is preferred resulting in an enhanced efficacy thereby being less toxic to the patient.
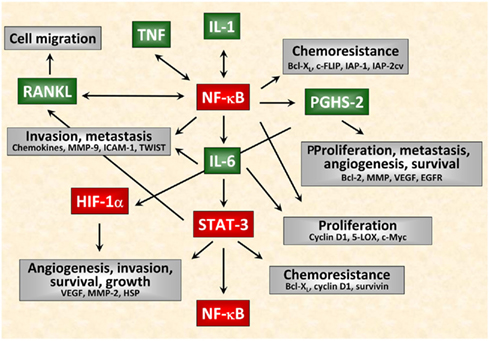
Figure 2. Interactions of inflammatory mediators in tumor cells. Schematical simplified representation of the complex intracellular signaling network in cancer. Among the tumorigenic factors produced by tumor cells or cells of the tumor microenvironment, IL-1, TNF, and IL-6 act as crucial mediators of inflammation-driven tumorigenesis. In particular IL-1 and TNF are major pleiotropic cytokines involved in tumor/host interactions. Nevertheless, these cytokines function as autocrine growth factors and modulate the expression of several tumorigenic factors at any stage of tumorigenesis not only affecting proliferation, migration, and survival of tumor cells but also angiogenesis, invasiveness, metastasis as well as chemoresistance of tumors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors want to thank Anett Lange for excellent editorial assistance. The work was supported by the Deutsche Forschungsgemeinschaft (DFG) SFB-824, DFG-Cluster of Excellence: Munich Advanced Photonics, Bundesministerium für Bildung und Forschung (BMBF) MOBITUM (01EZ0826) Innovative Therapies (01GU0823), m4 – Personalized Medicine and Targeted Therapies (01EX1021C), Kompetenzverbund Strahlenforschung (03NUK007E), and multimmune GmbH.
Aaltoma, S. H., Lipponen, P. K., and Kosma, V. M. (2001). Inducible nitric oxide synthase (iNOS) expression and its prognostic value in prostate cancer. Anticancer Res. 21, 3101–3106.
Adhami, V. M., Malik, A., Zaman, N., Sarfaraz, S., Siddiqui, I. A., Syed, D. N., Afaq, F., Pasha, F. S., Saleem, M., and Mukhtar, H. (2007). Combined inhibitory effects of green tea polyphenols and selective cyclooxygenase-2 inhibitors on the growth of human prostate cancer cells both in vitro and in vivo. Clin. Cancer Res. 13, 1611–1619.
Aggarwal, B. B., and Gehlot, P. (2009). Inflammation and cancer: how friendly is the relationship for cancer patients? Curr. Opin. Pharmacol. 9, 351–369.
Akerfelt, M., Morimoto, R. I., and Sistonen, L. (2010). Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Allen, I. C., TeKippe, E. M., Woodford, R. M., Uronis, J. M., Holl, E. K., Rogers, A. B., Herfarth, H. H., Jobin, C., and Ting, J. P. (2010). The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056.
Alshamsan, A. (2011). Induction of tolerogenic dendritic cells by IL-6-secreting CT26 colon carcinoma. Immunopharmacol. Immunotoxicol. doi:10.3109/08923973.2011.625034. [Epub ahead of print].
Ambs, S., Merriam, W. G., Ogunfusika, M. O., Bennett, W. P., Ishibe, N., Hussain, S. P., Tzeng, E. E., Geller, D. A., Billiar, T. R., and Harris, C. C. (1998). p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat. Med. 4, 1371–1376.
Ancrile, B., Lim, K. H., and Counter, C. M. (2007). Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 21, 1714–1719.
Anderson, D. M., Maraskovsky, E., Billingsley, W. L., Dougall, W. C., Tometsko, M. E., Roux, E. R., Teepe, M. C., DuBose, R. F., Cosman, D., and Galibert, L. (1997). A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390, 175–179.
Angst, E., Reber, H. A., Hines, O. J., and Eibl, G. (2008). Mononuclear cell-derived interleukin-1 beta confers chemoresistance in pancreatic cancer cells by upregulation of cyclooxygenase-2. Surgery 144, 57–65.
Apte, R. N., Krelin, Y., Song, X., Dotan, S., Recih, E., Elkabets, M., Carmi, Y., Dvorkin, T., White, R. M., Gayvoronsky, L., Segal, S., and Voronov, E. (2006). Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur. J. Cancer 42, 751–759.
Arnott, C. H., Scott, K. A., Moore, R. J., Robinson, S. C., Thompson, R. G., and Balkwill, F. R. (2004). Expression of both TNF-alpha receptor subtypes is essential for optimal skin tumour development. Oncogene 23, 1902–1910.
Asea, A., Kabingu, E., Stevenson, M. A., and Calderwood, S. K. (2000a). HSP70 peptidembearing and peptide-negative preparations act as chaperokines. Cell Stress Chaperones 5, 425–431.
Asea, A., Kraeft, S. K., Kurt-Jones, E. A., Stevenson, M. A., Chen, L. B., Finberg, R. W., Koo, G. C., and Calderwood, S. K. (2000b). HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 6, 435–442.
Asea, A., Rehli, M., Kabingu, E., Boch, J. A., Bare, O., Auron, P. E., Stevenson, M. A., and Calderwood, S. K. (2002). Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 277, 15028–15034.
Balkwill, F., and Mantovani, A. (2001). Inflammation and cancer: back to Virchow? Lancet 357, 539–545.
Barbie, D. A., Tamayo, P., Boehm, J. S., Kim, S. Y., Moody, S. E., Dunn, I. F., Schinzel, A. C., Sandy, P., Meylan, E., Scholl, C., Frohling, S., Chan, E. M., Sos, M. L., Michel, K., Mermel, C., Silver, S. J., Weir, B. A., Reiling, J. H., Sheng, Q., Gupta, P. B., Wadlow, R. C., Le, H., Hoersch, S., Wittner, B. S., Ramaswamy, S., Livingston, D. M., Sabatini, D. M., Meyerson, M., Thomas, R. K., Lander, E. S., Mesirov, J. P., Root, D. E., Gilliland, D. G., Jacks, T., and Hahn, W. C. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112.
Basu, A., and Haldar, S. (2009). Combinatorial effect of epigallocatechin-3-gallate and TRAIL on pancreatic cancer cell death. Int. J. Oncol. 34, 281–286.
Bauer, L., Venz, S., Junker, H., Brandt, R., and Radons, J. (2009). Nicotinamide phosphoribosyltransferase and prostaglandin H2 synthase 2 are up-regulated in human pancreatic adenocarcinoma cells after stimulation with interleukin-1. Int. J. Oncol. 35, 97–107.
Becker, C., Fantini, M. C., Schramm, C., Lehr, H. A., Wirtz, S., Nikolaev, A., Burg, J., Strand, S., Kiesslich, R., Huber, S., Ito, H., Nishimoto, N., Yoshizaki, K., Kishimoto, T., Galle, P. R., Blessing, M., Rose-John, S., and Neurath, M. F. (2004). TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21, 491–501.
Belham, C., Wu, S., and Avruch, J. (1999). Intracellular signalling: PDK1 – a kinase at the hub of things. Curr. Biol. 9, R93–R96.
Ben-Neriah, Y., and Karin, M. (2011). Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 12, 715–723.
Berger, F. G. (2004). The interleukin-6 gene: a susceptibility factor that may contribute to racial and ethnic disparities in breast cancer mortality. Breast Cancer Res. Treat. 88, 281–285.
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5.
Blanco, S., Sanz-Garcia, M., Santos, C. R., and Lazo, P. A. (2008). Modulation of interleukin-1 transcriptional response by the interaction between VRK2 and the JIP1 scaffold protein. PLoS ONE 3, e1660. doi:10.1371/journal.pone.0001660
Born, T. L., Thomassen, E., Bird, T. A., and Sims, J. E. (1998). Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J. Biol. Chem. 273, 29445–29450.
Botzler, C., Schmidt, J., Luz, A., Jennen, L., Issels, R., and Multhoff, G. (1998). Differential Hsp70 plasma-membrane expression on primary human tumors and metastases in mice with severe combined immunodeficiency. Int. J. Cancer 77, 942–948.
Brown, J. M., Corey, E., Lee, Z. D., True, L. D., Yun, T. J., Tondravi, M., and Vessella, R. L. (2001). Osteoprotegerin and rank ligand expression in prostate cancer. Urology 57, 611–616.
Cahill, C. M., and Rogers, J. T. (2008). Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1. J. Biol. Chem. 283, 25900–25912.
Calderwood, S. K. (2010). Heat shock proteins in breast cancer progression - a suitable case for treatment? Int. J. Hyperthermia 26, 681–685.
Calderwood, S. K., and Gong, J. (2011). Molecular chaperones in mammary cancer growth and breast tumor therapy. J. Cell Biochem. doi:10.1002/jcb.23461. [Epub ahead of print].
Cao, Z., Xiong, J., Takeuchi, M., Kurama, T., and Goeddel, D. V. (1996). TRAF6 is a signal transducer for interleukin-1. Nature 383, 443–446.
Cerella, C., Sobolewski, C., Dicato, M., and Diederich, M. (2010). Targeting COX-2 expression by natural compounds: a promising alternative strategy to synthetic COX-2 inhibitors for cancer chemoprevention and therapy. Biochem. Pharmacol. 80, 1801–1815.
Chang, L., Kamata, H., Solinas, G., Luo, J. L., Maeda, S., Venuprasad, K., Liu, Y. C., and Karin, M. (2006). The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 124, 601–613.
Chase, M. A., Wheeler, D. S., Lierl, K. M., Hughes, V. S., Wong, H. R., and Page, K. (2007). Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-kappaB-dependent mechanism. J. Immunol. 179, 6318–6324.
Chen, G., Sircar, K., Aprikian, A., Potti, A., Goltzman, D., and Rabbani, S. A. (2006). Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer 107, 289–298.
Chen, L., and Zhang, H. Y. (2007). Cancer preventive mechanisms of the green tea polyphenol (-)-epigallocatechin-3-gallate. Molecules 12, 946–957.
Chen, L. F., and Greene, W. C. (2004). Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5, 392–401.
Chen, X., Tao, Q., Yu, H., Zhang, L., and Cao, X. (2002). Tumor cell membrane-bound heat shock protein 70 elicits antitumor immunity. Immunol. Lett. 84, 81–87.
Chien, Y., Kim, S., Bumeister, R., Loo, Y. M., Kwon, S. W., Johnson, C. L., Balakireva, M. G., Romeo, Y., Kopelovich, L., Gale, M. Jr., Yeaman, C., Camonis, J. H., Zhao, Y., and White, M. A. (2006). RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127, 157–170.
Chung, Y. C., and Chang, Y. F. (2003). Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 83, 222–226.
Ciocca, D. R., and Calderwood, S. K. (2005). Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10, 86–103.
Colotta, F., Allavena, P., Sica, A., Garlanda, C., and Mantovani, A. (2009). Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081.
Cozen, W., Gill, P. S., Ingles, S. A., Masood, R., Martinez-Maza, O., Cockburn, M. G., Gauderman, W. J., Pike, M. C., Bernstein, L., Nathwani, B. N., Salam, M. T., Danley, K. L., Wang, W., Gage, J., Gundell-Miller, S., and Mack, T. M. (2004). IL-6 levels and genotype are associated with risk of young adult Hodgkin lymphoma. Blood 103, 3216–:3221.
Cross, S. S., Harrison, R. F., Balasubramanian, S. P., Lippitt, J. M., Evans, C. A., Reed, M. W., and Holen, I. (2006). Expression of receptor activator of nuclear factor kappabeta ligand (RANKL) and tumour necrosis factor related, apoptosis inducing ligand (TRAIL) in breast cancer, and their relations with osteoprotegerin, oestrogen receptor, and clinicopathological variables. J. Clin. Pathol. 59, 716–720.
De Paepe, B., Verstraeten, V. M., De Potter, C. R., and Bullock, G. R. (2002). Increased angiotensin II type-2 receptor density in hyperplasia, DCIS and invasive carcinoma of the breast is paralleled with increased iNOS expression. Histochem. Cell Biol. 117, 13–19.
DeNagel, D. C., and Pierce, S. K. (1992). A case for chaperones in antigen processing. Immunol. Today 13, 86–89.
DeNardo, D. G., Johansson, M., and Coussens, L. M. (2008). Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. 27, 11–18.
Dinarello, C. A. (2010). Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 29, 317–329.
Dostert, C., Petrilli, V., Van, B. R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677.
Dower, S. K., and Qwarnstrom, E. E. (2003). Signalling networks, inflammation and innate immunity. Biochem. Soc. Trans. 31, 1462–1471.
Dunne, A., and O’Neill, L. A. (2003). The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE 2003, re3.
Elgert, K. D., Alleva, D. G., and Mullins, D. W. (1998). Tumor-induced immune dysfunction: the macrophage connection. J. Leukoc. Biol. 64, 275–290.
El-Osta, H., Janku, F., and Kurzrock, R. (2010). Successful treatment of Castleman’s disease with interleukin-1 receptor antagonist (Anakinra). Mol. Cancer Ther. 9, 1485–1488.
Fahy, B. N., Schlieman, M. G., Virudachalam, S., and Bold, R. J. (2004). Inhibition of AKT abrogates chemotherapy-induced NF-kappaB survival mechanisms: implications for therapy in pancreatic cancer. J. Am. Coll. Surg. 198, 591–599.
Fernandez, M., Pino, A. M., Figueroa, P., and Rodriguez, J. P. (2010). The increased expression of receptor activator of nuclear-kappaB ligand (RANKL) of multiple myeloma bone marrow stromal cells is inhibited by the bisphosphonate ibandronate. J. Cell. Biochem. 111, 130–137.
Fernandez-Medarde, A., and Santos, E. (2011). Ras in cancer and developmental diseases. Genes Cancer 2, 344–358.
Forbes, S. A., Bindal, N., Bamford, S., Cole, C., Kok, C. Y., Beare, D., Jia, M., Shepherd, R., Leung, K., Menzies, A., Teague, J. W., Campbell, P. J., Stratton, M. R., and Futreal, P. A. (2011). COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950.
Franchi, L., Eigenbrod, T., Munoz-Planillo, R., and Nunez, G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247.
Garcea, G., Dennison, A. R., Steward, W. P., and Berry, D. P. (2005). Role of inflammation in pancreatic carcinogenesis and the implications for future therapy. Pancreatology 5, 514–529.
Gastpar, R., Gehrmann, M., Bausero, M. A., Asea, A., Gross, C., Schroeder, J. A., and Multhoff, G. (2005). Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 65, 5238–5247.
Gastpar, R., Gross, C., Rossbacher, L., Ellwart, J., Riegger, J., and Multhoff, G. (2004). The cell surface-localized heat shock protein 70 epitope TKD induces migration and cytolytic activity selectively in human NK cells. J. Immunol. 172, 972–980.
Gay, N. J., Gangloff, M., and O’Neill, L. A. (2011). What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 32, 104–109.
Gehrmann, M., Liebisch, G., Schmitz, G., Anderson, R., Steinem, C., De Maio, A., Pockley, G., and Multhoff, G. (2008). Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS ONE 3, e1925. doi:10.1371/journal.pone.0001925
Gehrmann, M., Schmetzer, H., Eissner, G., Haferlach, T., Hiddemann, W., and Multhoff, G. (2003). Membrane-bound heat shock protein 70 (Hsp70) in acute myeloid leukemia: a tumor specific recognition structure for the cytolytic activity of autologous NK cells. Haematologica 88, 474–476.
Ghiringhelli, F., Apetoh, L., Tesniere, A., Aymeric, L., Ma, Y., Ortiz, C., Vermaelen, K., Panaretakis, T., Mignot, G., Ullrich, E., Perfettini, J. L., Schlemmer, F., Tasdemir, E., Uhl, M., Genin, P., Civas, A., Ryffel, B., Kanellopoulos, J., Tschopp, J., Andre, F., Lidereau, R., McLaughlin, N. M., Haynes, N. M., Smyth, M. J., Kroemer, G., and Zitvogel, L. (2009). Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178.
Goldbach-Mansky, R., Dailey, N. J., Canna, S. W., Gelabert, A., Jones, J., Rubin, B. I., Kim, H. J., Brewer, C., Zalewski, C., Wiggs, E., Hill, S., Turner, M. L., Karp, B. I., Aksentijevich, I., Pucino, F., Penzak, S. R., Haverkamp, M. H., Stein, L., Adams, B. S., Moore, T. L., Fuhlbrigge, R. C., Shaham, B., Jarvis, J. N., O’Neil, K., Vehe, R. K., Beitz, L. O., Gardner, G., Hannan, W. P., Warren, R. W., Horn, W., Cole, J. L., Paul, S. M., Hawkins, P. N., Pham, T. H., Snyder, C., Wesley, R. A., Hoffmann, S. C., Holland, S. M., Butman, J. A., and Kastner, D. L. (2006). Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 355, 581–592.
Greten, F. R., Eckmann, L., Greten, T. F., Park, J. M., Li, Z. W., Egan, L. J., Kagnoff, M. F., and Karin, M. (2004). IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118, 285–296.
Grivennikov, S. I., and Karin, M. (2010). Inflammation and oncogenesis: a vicious connection. Curr. Opin. Genet. Dev. 20, 65–71.
Grosch, S., Maier, T. J., Schiffmann, S., and Geisslinger, G. (2006). Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J. Natl. Cancer Inst. 98, 736–747.
Gross, C., Hansch, D., Gastpar, R., and Multhoff, G. (2003a). Interaction of heat shock protein 70 peptide with NK cells involves the NK receptor CD94. Biol. Chem. 384, 267–279.
Gross, C., Koelch, W., DeMaio, A., Arispe, N., and Multhoff, G. (2003b). Cell surface-bound heat shock protein 70 (Hsp70) mediates perforin-independent apoptosis by specific binding and uptake of granzyme B. J. Biol. Chem. 278, 41173–41181.
Gross, C., Schmidt-Wolf, I. G., Nagaraj, S., Gastpar, R., Ellwart, J., Kunz-Schughart, L. A., and Multhoff, G. (2003c). Heat shock protein 70-reactivity is associated with increased cell surface density of CD94/CD56 on primary natural killer cells. Cell Stress Chaperones 8, 348–360.
Hacker, H., and Karin, M. (2006). Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Härdtner, C., Brandt, R., Multhoff, G., and Radons, J. (2009). (-)-Epigallocatechin-3-gallate compensates for celecoxib-mediated up-regulation of PGHS-2 expression in human pancreatic adenocarcinoma cells under tumor-associated inflammatory conditions. Eur. J. Immunol. 39(Suppl. 1/09), 530–531.
Hedrich, C. M., Bruck, N., Fiebig, B., and Gahr, M. (2011). Anakinra: a safe and effective first-line treatment in systemic onset juvenile idiopathic arthritis (SoJIA). Rheumatol. Int. doi: 10.1007/s00296-011-2249-4. [Epub ahead of print].
Hefler, L. A., Grimm, C., Lantzsch, T., Lampe, D., Leodolter, S., Koelbl, H., Heinze, G., Reinthaller, A., Tong-Cacsire, D., Tempfer, C., and Zeillinger, R. (2005). Interleukin-1 and interleukin-6 gene polymorphisms and the risk of breast cancer in caucasian women. Clin. Cancer Res. 11, 5718–5721.
Herfs, M., Herman, L., Hubert, P., Minner, F., Arafa, M., Roncarati, P., Henrotin, Y., Boniver, J., and Delvenne, P. (2009). High expression of PGE2 enzymatic pathways in cervical (pre)neoplastic lesions and functional consequences for antigen-presenting cells. Cancer Immunol. Immunother. 58, 603–614.
Hofbauer, L. C., and Schoppet, M. (2004). Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA 292, 490–495.
Hoffman, H. M., Throne, M. L., Amar, N. J., Sebai, M., Kivitz, A. J., Kavanaugh, A., Weinstein, S. P., Belomestnov, P., Yancopoulos, G. D., Stahl, N., and Mellis, S. J. (2008). Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 58, 2443–2452.
Hoffmann, J., Junker, H., Schmieder, A., Venz, S., Brandt, R., Multhoff, G., Falk, W., and Radons, J. (2011). EGCG downregulates IL-1RI expression and suppresses IL-1-induced tumorigenic factors in human pancreatic adenocarcinoma cells. Biochem. Pharmacol. 82, 1153–1162.
Hudson, B. I., Carter, A. M., Harja, E., Kalea, A. Z., Arriero, M., Yang, H., Grant, P. J., and Schmidt, A. M. (2008). Identification, classification, and expression of RAGE gene splice variants. FASEB J. 22, 1572–1580.
Hussain, S. P., and Harris, C. C. (2007). Inflammation and cancer: an ancient link with novel potentials. Int. J. Cancer 121, 2373–2380.
Hwang, W., Jung, K., Jeon, Y., Yun, S., Kim, T. W., and Choi, I. (2010). Knockdown of the interleukin-6 receptor alpha chain of dendritic cell vaccines enhances the therapeutic potential against IL-6 producing tumors. Vaccine 29, 34–44.
Hwang, Y. S., Lee, S. K., Park, K. K., and Chung, W. Y. (2011). Secretion of IL-6 and IL-8 from lysophosphatidic acid-stimulated oral squamous cell carcinoma promotes osteoclastogenesis and bone resorption. Oral Oncol. doi:10.1016/j.oraloncology.2011.08.022. [Epub ahead of print].
Hynes, N. E., and Stern, D. F. (1994). The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim. Biophys. Acta 1198, 165–184.
Inaba, H., Nagaoka, Y., Kushima, Y., Kumagai, A., Matsumoto, Y., Sakaguchi, M., Baba, K., and Uesato, S. (2008). Comparative examination of anti-proliferative activities of (-)-epigallocatechin gallate and (-)-epigallocatechin against HCT116 colorectal carcinoma cells. Biol. Pharm. Bull. 31, 79–84.
Ito, H., Takazoe, M., Fukuda, Y., Hibi, T., Kusugami, K., Andoh, A., Matsumoto, T., Yamamura, T., Azuma, J., Nishimoto, N., Yoshizaki, K., Shimoyama, T., and Kishimoto, T. (2004). A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 126, 989–996.
Jaiswal, M., LaRusso, N. F., and Gores, G. J. (2001). Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G626–G634.
Jefferies, C. A., and O’Neill, L. A. (2000). Rac1 regulates interleukin 1-induced nuclear factor kappaB activation in an inhibitory protein kappaBalpha-independent manner by enhancing the ability of the p65 subunit to transactivate gene expression. J. Biol. Chem. 275, 3114–3120.
Jendrossek, V. (2011). Targeting apoptosis pathways by celecoxib in cancer. Cancer Lett. doi:10.1016/j.canlet.2011.01.012. [Epub ahead of print].
Jimeno, A., Amador, M. L., Kulesza, P., Wang, X., Rubio-Viqueira, B., Zhang, X., Chan, A., Wheelhouse, J., Kuramochi, H., Tanaka, K., Danenberg, K., Messersmith, W. A., Almuete, V., Hruban, R. H., Maitra, A., Yeo, C. J., and Hidalgo, M. (2006). Assessment of celecoxib pharmacodynamics in pancreatic cancer. Mol. Cancer Ther. 5, 3240–3247.
Johnson, A. J., Hsu, A. L., Lin, H. P., Song, X., and Chen, C. S. (2002). The cyclo-oxygenase-2 inhibitor celecoxib perturbs intracellular calcium by inhibiting endoplasmic reticulum Ca2+-ATPases: a plausible link with its anti-tumour effect and cardiovascular risks. Biochem. J. 366, 831–837.
Jura, N., Archer, H., and Bar-Sagi, D. (2005). Chronic pancreatitis, pancreatic adenocarcinoma and the black box in-between. Cell Res. 15, 72–77.
Jurado, S., Garcia-Giralt, N., Diez-Perez, A., Esbrit, P., Yoskovitz, G., Agueda, L., Urreizti, R., Perez-Edo, L., Salo, G., Mellibovsky, L., Balcells, S., Grinberg, D., and Nogues, X. (2010). Effect of IL-1beta, PGE(2), and TGF-beta1 on the expression of OPG and RANKL in normal and osteoporotic primary human osteoblasts. J. Cell. Biochem. 110, 304–310.
Kai, H., Kitadai, Y., Kodama, M., Cho, S., Kuroda, T., Ito, M., Tanaka, S., Ohmoto, Y., and Chayama, K. (2005). Involvement of proinflammatory cytokines IL-1beta and IL-6 in progression of human gastric carcinoma. Anticancer Res. 25, 709–713.
Kamal, A., Thao, L., Sensintaffar, J., Zhang, L., Boehm, M. F., Fritz, L. C., and Burrows, F. J. (2003). A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, 407–410.
Kamata, H., Honda, S., Maeda, S., Chang, L., Hirata, H., and Karin, M. (2005). Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120, 649–661.
Karin, M., Cao, Y., Greten, F. R., and Li, Z. W. (2002). NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2, 301–310.
Karin, M., Lawrence, T., and Nizet, V. (2006). Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 124, 823–835.
Kawai, T., and Akira, S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650.
Kawata, M., Koinuma, D., Ogami, T., Umezawa, K., Iwata, C., Watabe, T., and Miyazono, K. (2011). TGF-beta-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by proinflammatory cytokines derived from RAW 264.7 macrophage cells. J. Biochem. doi: 10.1093/jb/mvr136. [Epub ahead of print].
Keating, S. E., Maloney, G. M., Moran, E. M., and Bowie, A. G. (2007). IRAK-2 participates in multiple toll-like receptor signaling pathways to NFkappaB via activation of TRAF6 ubiquitination. J. Biol. Chem. 282, 33435–33443.
Khaleque, M. A., Bharti, A., Sawyer, D., Gong, J., Benjamin, I. J., Stevenson, M. A., and Calderwood, S. K. (2005). Induction of heat shock proteins by heregulin beta1 leads to protection from apoptosis and anchorage-independent growth. Oncogene 24, 6564–6573.
Kim, L. S., and Kim, J. H. (2011). Heat shock protein as molecular targets for breast cancer therapeutics. J. Breast Cancer 14, 167–174.
Kishimoto, K., Matsumoto, K., and Ninomiya-Tsuji, J. (2000). TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J. Biol. Chem. 275, 7359–7364.
Klein, B., Lu, Z. Y., Gaillard, J. P., Harousseau, J. L., and Bataille, R. (1992). Inhibiting IL-6 in human multiple myeloma. Curr. Top. Microbiol. Immunol. 182, 237–244.
Kobayashi, S., Werneburg, N. W., Bronk, S. F., Kaufmann, S. H., and Gores, G. J. (2005). Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology 128, 2054–2065.
Kong, G., Kim, E. K., Kim, W. S., Lee, K. T., Lee, Y. W., Lee, J. K., Paik, S. W., and Rhee, J. C. (2002). Role of cyclooxygenase-2 and inducible nitric oxide synthase in pancreatic cancer. J. Gastroenterol. Hepatol. 17, 914–921.
Kortylewski, M., Kujawski, M., Wang, T., Wei, S., Zhang, S., Pilon-Thomas, S., Niu, G., Kay, H., Mule, J., Kerr, W. G., Jove, R., Pardoll, D., and Yu, H. (2005). Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11, 1314–1321.
Kulp, S. K., Yang, Y. T., Hung, C. C., Chen, K. F., Lai, J. P., Tseng, P. H., Fowble, J. W., Ward, P. J., and Chen, C. S. (2004). 3-phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res. 64, 1444–1451.
Kumar, A., Xu, J., Brady, S., Gao, H., Yu, D., Reuben, J., and Mehta, K. (2010). Tissue transglutaminase promotes drug resistance and invasion by inducing mesenchymal transition in mammary epithelial cells. PLoS ONE 5, e13390. doi:10.1371/journal.pone.0013390
Kundu, J. K., and Surh, Y. J. (2008). Inflammation: gearing the journey to cancer. Mutat. Res. 659, 15–30.
Lachmann, H. J., Kone-Paut, I., Kuemmerle-Deschner, J. B., Leslie, K. S., Hachulla, E., Quartier, P., Gitton, X., Widmer, A., Patel, N., and Hawkins, P. N. (2009a). Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 360, 2416–2425.
Lachmann, H. J., Lowe, P., Felix, S. D., Rordorf, C., Leslie, K., Madhoo, S., Wittkowski, H., Bek, S., Hartmann, N., Bosset, S., Hawkins, P. N., and Jung, T. (2009b). In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J. Exp. Med. 206, 1029–1036.
Ladetto, M., Vallet, S., Trojan, A., Dell’Aquila, M., Monitillo, L., Rosato, R., Santo, L., Drandi, D., Bertola, A., Falco, P., Cavallo, F., Ricca, I., De, M. F., Mantoan, B., Bode-Lesniewska, B., Pagliano, G., Francese, R., Rocci, A., Astolfi, M., Compagno, M., Mariani, S., Godio, L., Marino, L., Ruggeri, M., Omede, P., Palumbo, A., and Boccadoro, M. (2005). Cyclooxygenase-2 (COX-2) is frequently expressed in multiple myeloma and is an independent predictor of poor outcome. Blood 105, 4784–4791.
Lambert, J. D., Hong, J., Yang, G. Y., Liao, J., and Yang, C. S. (2005). Inhibition of carcinogenesis by polyphenols: evidence from laboratory investigations. Am. J. Clin. Nutr. 81, 284S–291S.
Lee, H., Herrmann, A., Deng, J. H., Kujawski, M., Niu, G., Li, Z., Forman, S., Jove, R., Pardoll, D. M., and Yu, H. (2009). Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 15, 283–293.
Lee, Y. H., Ji, J. D., and Song, G. G. (2007). Adjusted indirect comparison of celecoxib versus rofecoxib on cardiovascular risk. Rheumatol. Int. 27, 477–482.
Lewis, A. M., Varghese, S., Xu, H., and Alexander, H. R. (2006). Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med. 4, 48.
Li, J., Sarosi, I., Yan, X. Q., Morony, S., Capparelli, C., Tan, H. L., McCabe, S., Elliott, R., Scully, S., Van, G., Kaufman, S., Juan, S. C., Sun, Y., Tarpley, J., Martin, L., Christensen, K., McCabe, J., Kostenuik, P., Hsu, H., Fletcher, F., Dunstan, C. R., Lacey, D. L., and Boyle, W. J. (2000). RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. U.S.A. 97, 1566–1571.
Li, X., Kim, K. W., Cho, M. L., Ju, J. H., Kang, C. M., Oh, H. J., Min, J. K., Lee, S. H., Park, S. H., and Kim, H. Y. (2010). IL-23 induces receptor activator of NF-kappaB ligand expression in fibroblast-like synoviocytes via STAT3 and NF-kappaB signal pathways. Immunol. Lett. 127, 100–107.
Lin, W. J., and Yeh, W. C. (2005). Implication of Toll-like receptor and tumor necrosis factor alpha signaling in septic shock. Shock 24, 206–209.
Lin, W. W., and Karin, M. (2007). A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 117, 1175–1183.
Lu, H., Ouyang, W., and Huang, C. (2006). Inflammation, a key event in cancer development. Mol. Cancer Res. 4, 221–233.
Lust, J. A., Lacy, M. Q., Zeldenrust, S. R., Dispenzieri, A., Gertz, M. A., Witzig, T. E., Kumar, S., Hayman, S. R., Russell, S. J., Buadi, F. K., Geyer, S. M., Campbell, M. E., Kyle, R. A., Rajkumar, S. V., Greipp, P. R., Kline, M. P., Xiong, Y., Moon-Tasson, L. L., and Donovan, K. A. (2009). Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 84, 114–122.
Maier, T. J., Schilling, K., Schmidt, R., Geisslinger, G., and Grosch, S. (2004). Cyclooxygenase-2 (COX-2)-dependent and -independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. Biochem. Pharmacol. 67, 1469–1478.
Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. (2008). Cancer-related inflammation. Nature 454, 436–444.
Martin, M. U., and Wesche, H. (2002). Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim. Biophys. Acta 1592, 265–280.
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Menu, P., and Vince, J. E. (2011). The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin. Exp. Immunol. 166, 1–15.
Merati, K., said, S. M., Andea, A., Sarkar, F., Ben-Josef, E., Mohammad, R., Philip, P., Shields, A. F., Vaitkevicius, V., Grignon, D. J., and Adsay, N. V. (2001). Expression of inflammatory modulator COX-2 in pancreatic ductal adenocarcinoma and its relationship to pathologic and clinical parameters. Am. J. Clin. Oncol. 24, 447–452.
Meylan, E., Dooley, A. L., Feldser, D. M., Shen, L., Turk, E., Ouyang, C., and Jacks, T. (2009). Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 462, 104–107.
Mocellin, S., Rossi, C. R., Pilati, P., and Nitti, D. (2005). Tumor necrosis factor, cancer and anticancer therapy. Cytokine Growth Factor Rev. 16, 35–53.
Molina, M. A., Sitja-Arnau, M., Lemoine, M. G., Frazier, M. L., and Sinicrope, F. A. (1999). Increased cyclooxygenase-2 expression in human pancreatic carcinomas and cell lines: growth inhibition by nonsteroidal anti-inflammatory drugs. Cancer Res. 59, 4356–4362.
Mori, K., Berreur, M., Blanchard, F., Chevalier, C., Guisle-Marsollier, I., Masson, M., Redini, F., and Heymann, D. (2007a). Receptor activator of nuclear factor-kappaB ligand (RANKL) directly modulates the gene expression profile of RANK-positive Saos-2 human osteosarcoma cells. Oncol. Rep. 18, 1365–1371.
Mori, K., Le Goff, B., Berreur, M., Riet, A., Moreau, A., Blanchard, F., Chevalier, C., Guisle-Marsollier, I., Leger, J., Guicheux, J., Masson, M., Gouin, F., Redini, F., and Heymann, D. (2007b). Human osteosarcoma cells express functional receptor activator of nuclear factor-kappa B. J. Pathol. 211, 555–562.
Mori, T., Miyamoto, T., Yoshida, H., Asakawa, M., Kawasumi, M., Kobayashi, T., Morioka, H., Chiba, K., Toyama, Y., and Yoshimura, A. (2011). IL-1β and TNFα-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int. Immunol. 23, 701–712.
Moser, C., Schmidbauer, C., Gurtler, U., Gross, C., Gehrmann, M., Thonigs, G., Pfister, K., and Multhoff, G. (2002). Inhibition of tumor growth in mice with severe combined immunodeficiency is mediated by heat shock protein 70 (Hsp70)-peptide-activated, CD94 positive natural killer cells. Cell Stress Chaperones 7, 365–373.
Multhoff, G., Botzler, C., Jennen, L., Schmidt, J., Ellwart, J., and Issels, R. (1997). Heat shock protein 72 on tumor cells: a recognition structure for natural killer cells. J. Immunol. 158, 4341–4350.
Multhoff, G., Botzler, C., Wiesnet, M., Eissner, G., and Issels, R. (1995a). CD3- large granular lymphocytes recognize a heat-inducible immunogenic determinant associated with the 72-kD heat shock protein on human sarcoma cells. Blood 86, 1374–1382.
Multhoff, G., Botzler, C., Wiesnet, M., Muller, E., Meier, T., Wilmanns, W., and Issels, R. D. (1995b). A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int. J. Cancer 61, 272–279.
Multhoff, G., Mizzen, L., Winchester, C. C., Milner, C. M., Wenk, S., Eissner, G., Kampinga, H. H., Laumbacher, B., and Johnson, J. (1999). Heat shock protein 70 (Hsp70) stimulates proliferation and cytolytic activity of natural killer cells. Exp. Hematol. 27, 1627–1636.
Multhoff, G., Pfister, K., Botzler, C., Jordan, A., Scholz, R., Schmetzer, H., Burgstahler, R., and Hiddemann, W. (2000). Adoptive transfer of human natural killer cells in mice with severe combined immunodeficiency inhibits growth of Hsp70-expressing tumors. Int. J. Cancer 88, 791–797.
Multhoff, G., Pfister, K., Gehrmann, M., Hantschel, M., Gross, C., Hafner, M., and Hiddemann, W. (2001). A 14-mer Hsp70 peptide stimulates natural killer (NK) cell activity. Cell Stress Chaperones 6, 337–344.
Murshid, A., Gong, J., Stevenson, M. A., and Calderwood, S. K. (2011). Heat shock proteins and cancer vaccines: developments in the past decade and chaperoning in the decade to come. Expert Rev. Vaccines 10, 1553–1568.
Muthuswamy, R., Mueller-Berghaus, J., Haberkorn, U., Reinhart, T. A., Schadendorf, D., and Kalinski, P. (2010). PGE(2) transiently enhances DC expression of CCR7 but inhibits the ability of DCs to produce CCL19 and attract naive T cells. Blood 116, 1454–1459.
Muzio, M., Ni, J., Feng, P., and Dixit, V. M. (1997). IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278, 1612–1615.
Nakata, Y., Matsuda, K., Uzawa, A., Nomura, M., Akashi, M., and Suzuki, G. (1995). Administration of recombinant human IL-1 by Staphylococcus enterotoxin B prevents tolerance induction in vivo. J. Immunol. 155, 4231–4235.
Ng, P. K., Tsui, S. K., Lau, C. P., Wong, C. H., Wong, W. H., Huang, L., and Kumta, S. M. (2010). CCAAT/enhancer binding protein beta is up-regulated in giant cell tumor of bone and regulates RANKL expression. J. Cell. Biochem. 110, 438–446.
Niederberger, E., Tegeder, I., Vetter, G., Schmidtko, A., Schmidt, H., Euchenhofer, C., Brautigam, L., Grosch, S., and Geisslinger, G. (2001). Celecoxib loses its anti-inflammatory efficacy at high doses through activation of NF-kappaB. FASEB J. 15, 1622–1624.
Ninomiya-Tsuji, J., Kishimoto, K., Hiyama, A., Inoue, J., Cao, Z., and Matsumoto, K. (1999). The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398, 252–256.
Nishimoto, N., Kanakura, Y., Aozasa, K., Johkoh, T., Nakamura, M., Nakano, S., Nakano, N., Ikeda, Y., Sasaki, T., Nishioka, K., Hara, M., Taguchi, H., Kimura, Y., Kato, Y., Asaoku, H., Kumagai, S., Kodama, F., Nakahara, H., Hagihara, K., Yoshizaki, K., and Kishimoto, T. (2005). Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 106, 2627–2632.
Nishimoto, N., Yoshizaki, K., Miyasaka, N., Yamamoto, K., Kawai, S., Takeuchi, T., Hashimoto, J., Azuma, J., and Kishimoto, T. (2004). Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 50, 1761–1769.
Offner, F. A., Obrist, P., Stadlmann, S., Feichtinger, H., Klingler, P., Herold, M., Zwierzina, H., Hittmair, A., Mikuz, G., and Abendstein, B. (1995). IL-6 secretion by human peritoneal mesothelial and ovarian cancer cells. Cytokine 7, 542–547.
Okamoto, M., Liu, W., Luo, Y., Tanaka, A., Cai, X., Norris, D. A., Dinarello, C. A., and Fujita, M. (2010). Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 285, 6477–6488.
O’Neill, L. A. (2000). The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE 2000, re1.
O’Sullivan, B. J., Thomas, H. E., Pai, S., Santamaria, P., Iwakura, Y., Steptoe, R. J., Kay, T. W., and Thomas, R. (2006). IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J. Immunol. 176, 7278–7287.
Paduch, R., Jakubowicz-Gil, J., and Kandefer-Szerszen, M. (2009). Expression of HSP27, HSP72 and MRP proteins in in vitro co-culture of colon tumour cell spheroids with normal cells after incubation with rhTGF- beta1 and/or CPT-11. J. Biosci. 34, 927–940.
Park, H. R., Min, S. K., Cho, H. D., Kim, D. H., Shin, H. S., and Park, Y. E. (2003). Expression of osteoprotegerin and RANK ligand in breast cancer bone metastasis. J. Korean Med. Sci. 18, 541–546.
Peters, M., Muller, A. M., and Rose-John, S. (1998). Interleukin-6 and soluble interleukin-6 receptor: direct stimulation of gp130 and hematopoiesis. Blood 92, 3495–3504.
Pfister, K., Radons, J., Busch, R., Tidball, J. G., Pfeifer, M., Freitag, L., Feldmann, H. J., Milani, V., Issels, R., and Multhoff, G. (2007). Patient survival by Hsp70 membrane phenotype: association with different routes of metastasis. Cancer 110, 926–935.
Pidgeon, G. P., Harmey, J. H., Kay, E., Da Costa, M., Redmond, H. P., and Bouchier-Hayes, D. J. (1999). The role of endotoxin/lipopolysaccharide in surgically induced tumour growth in a murine model of metastatic disease. Br. J. Cancer 81, 1311–1317.
Pikarsky, E., Porat, R. M., Stein, I., Abramovitch, R., Amit, S., Kasem, S., Gutkovich-Pyest, E., Urieli-Shoval, S., Galun, E., and Ben-Neriah, Y. (2004). NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431, 461–466.
Pirkkala, L., Nykanen, P., and Sistonen, L. (2001). Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 15, 1118–1131.
Rabe, B., Chalaris, A., May, U., Waetzig, G. H., Seegert, D., Williams, A. S., Jones, S. A., Rose-John, S., and Scheller, J. (2008). Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood 111, 1021–1028.
Radons, J., Dove, S., Neumann, D., Altmann, R., Botzki, A., Martin, M. U., and Falk, W. (2003). The interleukin 1 (IL-1) receptor accessory protein Toll/IL-1 receptor domain: analysis of putative interaction sites in vitro mutagenesis and molecular modeling. J. Biol. Chem. 278, 49145–49153.
Radons, J., Gabler, S., Wesche, H., Korherr, C., Hofmeister, R., and Falk, W. (2002). Identification of essential regions in the cytoplasmic tail of interleukin-1 receptor accessory protein critical for interleukin-1 signaling. J. Biol. Chem. 277, 16456–16463.
Raut, C. P., Nawrocki, S., Lashinger, L. M., Davis, D. W., Khanbolooki, S., Xiong, H., Ellis, L. M., and McConkey, D. J. (2004). Celecoxib inhibits angiogenesis by inducing endothelial cell apoptosis in human pancreatic tumor xenografts. Cancer Biol. Ther. 3, 1217–1224.
Ravi, R., Mookerjee, B., Bhujwalla, Z. M., Sutter, C. H., Artemov, D., Zeng, Q., Dillehay, L. E., Madan, A., Semenza, G. L., and Bedi, A. (2000). Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 14, 34–44.
Reddy, S. A., Huang, J. H., and Liao, W. S. (1997). Phosphatidylinositol 3-kinase in interleukin 1 signaling. Physical interaction with the interleukin 1 receptor and requirement in NFkappaB and AP-1 activation. J. Biol. Chem. 272, 29167–29173.
Rose-John, S., Scheller, J., Elson, G., and Jones, S. A. (2006). Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J. Leukoc. Biol. 80, 227–236.
Rose-John, S., and Schooltink, H. (2007). Cytokines are a therapeutic target for the prevention of inflammation-induced cancers. Recent Results Cancer Res. 174, 57–66.
Rudner, J., Elsaesser, S. J., Muller, A. C., Belka, C., and Jendrossek, V. (2010). Differential effects of anti-apoptotic Bcl-2 family members Mcl-1, Bcl-2, and Bcl-xL on celecoxib-induced apoptosis. Biochem. Pharmacol. 79, 10–20.
Sakoguchi-Okada, N., Takahashi-Yanaga, F., Fukada, K., Shiraishi, F., Taba, Y., Miwa, Y., Morimoto, S., Iida, M., and Sasaguri, T. (2007). Celecoxib inhibits the expression of survivin via the suppression of promoter activity in human colon cancer cells. Biochem. Pharmacol. 73, 1318–1329.
Sandau, K. B., Faus, H. G., and Brune, B. (2000). Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI 3K pathway. Biochem. Biophys. Res. Commun. 278, 263–267.
Sansone, P., Piazzi, G., Paterini, P., Strillacci, A., Ceccarelli, C., Minni, F., Biasco, G., Chieco, P., and Bonafe, M. (2009). Cyclooxygenase-2/carbonic anhydrase-IX up-regulation promotes invasive potential and hypoxia survival in colorectal cancer cells. J. Cell. Mol. Med. 13, 3876–3887.
Sawa, T., and Ohshima, H. (2006). Nitrative DNA damage in inflammation and its possible role in carcinogenesis. Nitric Oxide 14, 91–100.
Schulze, J., Albers, J., Baranowsky, A., Keller, J., Spiro, A., Streichert, T., Zustin, J., Amling, M., and Schinke, T. (2010). Osteolytic prostate cancer cells induce the expression of specific cytokines in bone-forming osteoblasts through a Stat3/5-dependent mechanism. Bone 46, 524–533.
Schwarz, R. E., Vujanovic, N. L., and Hiserodt, J. C. (1989). Enhanced antimetastatic activity of lymphokine-activated killer cells purified and expanded by their adherence to plastic. Cancer Res. 49, 1441–1446.
Shamovsky, I., and Nudler, E. (2008). New insights into the mechanism of heat shock response activation. Cell. Mol. Life Sci. 65, 855–861.
Shchors, K., Shchors, E., Rostker, F., Lawlor, E. R., Brown-Swigart, L., and Evan, G. I. (2006). The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 20, 2527–2538.
Shimizu, M., Deguchi, A., Lim, J. T., Moriwaki, H., Kopelovich, L., and Weinstein, I. B. (2005). (-)-Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor-2 signaling pathways in human colon cancer cells. Clin. Cancer Res. 11, 2735–2746.
Shin, B. K., Wang, H., Yim, A. M., Le Naour, F., Brichory, F., Jang, J. H., Zhao, R., Puravs, E., Tra, J., Michael, C. W., Misek, D. E., and Hanash, S. M. (2003). Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 278, 7607–7616.
Shinriki, S., Jono, H., Ota, K., Ueda, M., Kudo, M., Ota, T., Oike, Y., Endo, M., Ibusuki, M., Hiraki, A., Nakayama, H., Yoshitake, Y., Shinohara, M., and Ando, Y. (2009). Humanized anti-interleukin-6 receptor antibody suppresses tumor angiogenesis and in vivo growth of human oral squamous cell carcinoma. Clin. Cancer Res. 15, 5426–5434.
Shirode, A. B., and Sylvester, P. W. (2010). Synergistic anticancer effects of combined gamma-tocotrienol and celecoxib treatment are associated with suppression in Akt and NFkappaB signaling. Biomed. Pharmacother. 64, 327–332.
Siddiqui, I. A., Malik, A., Adhami, V. M., Asim, M., Hafeez, B. B., Sarfaraz, S., and Mukhtar, H. (2008). Green tea polyphenol EGCG sensitizes human prostate carcinoma LNCaP cells to TRAIL-mediated apoptosis and synergistically inhibits biomarkers associated with angiogenesis and metastasis. Oncogene 27, 2055–2063.
Singh, R., Wang, B., Shirvaikar, A., Khan, S., Kamat, S., Schelling, J. R., Konieczkowski, M., and Sedor, J. R. (1999). The IL-1 receptor and Rho directly associate to drive cell activation in inflammation. J. Clin. Invest. 103, 1561–1570.
Smolen, J. S., Avila, J. C., and Aletaha, D. (2011). Tocilizumab inhibits progression of joint damage in rheumatoid arthritis irrespective of its anti-inflammatory effects: disassociation of the link between inflammation and destruction. Ann. Rheum. Dis. doi: 10.1136/annrheumdis-2011-200395. [Epub ahead of print].
Solomon, S. D., Wittes, J., Finn, P. V., Fowler, R., Viner, J., Bertagnolli, M. M., Arber, N., Levin, B., Meinert, C. L., Martin, B., Pater, J. L., Goss, P. E., Lance, P., Obara, S., Chew, E. Y., Kim, J., Arndt, G., and Hawk, E. (2008). Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation 117, 2104–2113.
Sparmann, A., and Bar-Sagi, D. (2004). Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 6, 447–458.
Spektor, G., and Fuster, V. (2005). Drug insight: cyclo-oxygenase 2 inhibitors and cardiovascular risk-where are we now? Nat. Clin. Pract. Cardiovasc. Med. 2, 290–300.
Stangl, S., Gehrmann, M., Riegger, J., Kuhs, K., Riederer, I., Sievert, W., Hube, K., Mocikat, R., Dressel, R., Kremmer, E., Pockley, A.G., Friedrich, L., Vigh, L., Skerra, A., and Multhoff, G. (2011). Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. U.S.A. 108, 733–738.
Stangl, S., Wortmann, A., Guertler, U., and Multhoff, G. (2006). Control of metastasized pancreatic carcinomas in SCID/beige mice with human IL-2/TKD-activated NK cells. J. Immunol. 176, 6270–6276.
Stock, A., Booth, S., and Cerundolo, V. (2011). Prostaglandin E2 suppresses the differentiation of retinoic acid-producing dendritic cells in mice and humans. J. Exp. Med. 208, 761–773.
Suganuma, M., Saha, A., and Fujiki, H. (2011). New cancer treatment strategy using combination of green tea catechins and anticancer drugs. Cancer Sci. 102, 317–323.
Sun, S. G., Lau, Y. S., Itonaga, I., Sabokbar, A., and Athanasou, N. A. (2006). Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J. Pathol. 209, 114–120.
Surh, Y. J., Chun, K. S., Cha, H. H., Han, S. S., Keum, Y. S., Park, K. K., and Lee, S. S. (2001). Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat. Res. 480–481, 243–268.
Sutton, C., Brereton, C., Keogh, B., Mills, K. H., and Lavelle, E. C. (2006). A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 203, 1685–1691.
Takahashi, T., Uehara, H., Bando, Y., and Izumi, K. (2008). Soluble EP2 neutralizes prostaglandin E2-induced cell signaling and inhibits osteolytic tumor growth. Mol. Cancer Ther. 7, 2807–2816.
Taketomi, A., Takenaka, K., Matsumata, T., Shimada, M., Higashi, H., Shirabe, K., Itasaka, H., Adachi, E., Maeda, T., and Sugimachi, K. (1997). Circulating intercellular adhesion molecule-1 in patients with hepatocellular carcinoma before and after hepatic resection. Hepatogastroenterology 44, 477–483.
Tang, Z. N., Zhang, F., Tang, P., Qi, X. W., and Jiang, J. (2011). Hypoxia induces RANK and RANKL expression by activating HIF-1alpha in breast cancer cells. Biochem. Biophys. Res. Commun. 408, 411–416.
Thiery, J. P., Acloque, H., Huang, R. Y., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890.
Tjiu, J. W., Liao, Y. H., Lin, S. J., Huang, Y. L., Tsai, W. L., Chu, C. Y., Kuo, M. L., and Jee, S. H. (2006). Cyclooxygenase-2 overexpression in human basal cell carcinoma cell line increases antiapoptosis, angiogenesis, and tumorigenesis. J. Invest. Dermatol. 126, 1143–1151.
Trelle, S., Reichenbach, S., Wandel, S., Hildebrand, P., Tschannen, B., Villiger, P. M., Egger, M., and Juni, P. (2011). Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ 342, c7086.
Triantafilou, M., and Triantafilou, K. (2004). Heat-shock protein 70 and heat-shock protein 90 associate with Toll-like receptor 4 in response to bacterial lipopolysaccharide. Biochem. Soc. Trans. 32, 636–639.
Tseng, W. W., Deganutti, A., Chen, M. N., Saxton, R. E., and Liu, C. D. (2002). Selective cyclooxygenase-2 inhibitor rofecoxib (Vioxx) induces expression of cell cycle arrest genes and slows tumor growth in human pancreatic cancer. J. Gastrointest. Surg. 6, 838–843.
Vallabhapurapu, S., and Karin, M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733.
Voronov, E., Carmi, Y., and Apte, R. N. (2007). Role of IL-1-mediated inflammation in tumor angiogenesis. Adv. Exp. Med. Biol. 601, 265–270.
Vujanovic, N. L., Yasumura, S., Hirabayashi, H., Lin, W. C., Watkins, S., Herberman, R. B., and Whiteside, T. L. (1995). Antitumor activities of subsets of human IL-2-activated natural killer cells in solid tissues. J. Immunol. 154, 281–289.
Wang, C., Deng, L., Hong, M., Akkaraju, G. R., Inoue, J., and Chen, Z. J. (2001). TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351.
Wang, L., Yi, T., Kortylewski, M., Pardoll, D. M., Zeng, D., and Yu, H. (2009a). IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 206, 1457–1464.
Wang, S., Liu, Q., Zhang, Y., Liu, K., Yu, P., Liu, K., Luan, J., Duan, H., Lu, Z., Wang, F., Wu, E., Yagasaki, K., and Zhang, G. (2009b). Suppression of growth, migration and invasion of highly-metastatic human breast cancer cells by berbamine and its molecular mechanisms of action. Mol. Cancer 8, 81.
Watters, T. M., Kenny, E. F., and O’Neill, L. A. (2007). Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol. Cell Biol. 85, 411–419.
Weaver, C. T., Hawrylowicz, C. M., and Unanue, E. R. (1988). T helper cell subsets require the expression of distinct costimulatory signals by antigen-presenting cells. Proc. Natl. Acad. Sci. U.S.A. 85, 8181–8185.
Wei, D., Wang, L., He, Y., Xiong, H. Q., Abbruzzese, J. L., and Xie, K. (2004). Celecoxib inhibits vascular endothelial growth factor expression in and reduces angiogenesis and metastasis of human pancreatic cancer via suppression of Sp1 transcription factor activity. Cancer Res. 64, 2030–2038.
Weichert, W., Boehm, M., Gekeler, V., Bahra, M., Langrehr, J., Neuhaus, P., Denkert, C., Imre, G., Weller, C., Hofmann, H. P., Niesporek, S., Jacob, J., Dietel, M., Scheidereit, C., and Kristiansen, G. (2007). High expression of RelA/p65 is associated with activation of nuclear factor-kappaB-dependent signaling in pancreatic cancer and marks a patient population with poor prognosis. Br. J. Cancer 97, 523–530.
Weiss, T. W., Simak, R., Kaun, C., Rega, G., Pfluger, H., Maurer, G., Huber, K., and Wojta, J. (2011). Oncostatin M and IL-6 induce u-PA and VEGF in prostate cancer cells and correlate in vivo. Anticancer Res. 31, 3273–3278.
Wesche, H., Henzel, W. J., Shillinglaw, W., Li, S., and Cao, Z. (1997a). MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity 7, 837–847.
Wesche, H., Korherr, C., Kracht, M., Falk, W., Resch, K., and Martin, M. U. (1997b). The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP kinases). J. Biol. Chem. 272, 7727–7731.
Wheeler, D. S., Chase, M. A., Senft, A. P., Poynter, S. E., Wong, H. R., and Page, K. (2009). Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir. Res. 10, 31.
Whiteside, T. L., Vujanovic, N. L., and Herberman, R. B. (1998). Natural killer cells and tumor therapy. Curr. Top. Microbiol. Immunol. 230, 221–244.
Wilson, K. T., Fu, S., Ramanujam, K. S., and Meltzer, S. J. (1998). Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in Barrett’s esophagus and associated adenocarcinomas. Cancer Res. 58, 2929–2934.
Wink, D. A., Vodovotz, Y., Laval, J., Laval, F., Dewhirst, M. W., and Mitchell, J. B. (1998). The multifaceted roles of nitric oxide in cancer. Carcinogenesis 19, 711–721.
Wu, S., Rhee, K. J., Albesiano, E., Rabizadeh, S., Wu, X., Yen, H. R., Huso, D. L., Brancati, F. L., Wick, E., McAllister, F., Housseau, F., Pardoll, D. M., and Sears, C. L. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022.
Xiong, H. Q. (2004). Molecular targeting therapy for pancreatic cancer. Cancer Chemother. Pharmacol. 54(Suppl. 1), S69–S77.
Xu, X. F., Xie, C. G., Wang, X. P., Liu, J., Yu, Y. C., Hu, H. L., and Guo, C. Y. (2008). Selective inhibition of cyclooxygenase-2 suppresses the growth of pancreatic cancer cells in vitro and in vivo. Tohoku J. Exp. Med. 215, 149–157.
Yamada, T., Tsuda, M., Takahashi, T., Totsuka, Y., Shindoh, M., and Ohba, Y. (2011). RANKL expression specifically observed in vivo promotes epithelial mesenchymal transition and tumor progression. Am. J. Pathol. 178, 2845–2856.
Yamashita, M., Kurokawa, K., Sato, Y., Yamagata, A., Mimura, H., Yoshikawa, A., Sato, K., Nakano, A., and Fukai, S. (2010). Structural basis for the Rho- and phosphoinositide-dependent localization of the exocyst subunit Sec3. Nat. Struct. Mol. Biol. 17, 180–186.
Yang, J., and Weinberg, R. A. (2008). Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev. Cell 14, 818–829.
Yasumura, S., Lin, W. C., Hirabayashi, H., Vujanovic, N. L., Herberman, R. B., and Whiteside, T. L. (1994). Immunotherapy of liver metastases of human gastric carcinoma with interleukin 2-activated natural killer cells. Cancer Res. 54, 3808–3816.
Yip-Schneider, M. T., Sweeney, C. J., Jung, S. H., Crowell, P. L., and Marshall, M. S. (2001). Cell cycle effects of nonsteroidal anti-inflammatory drugs and enhanced growth inhibition in combination with gemcitabine in pancreatic carcinoma cells. J. Pharmacol. Exp. Ther. 298, 976–985.
Yokota, S., Miyamae, T., Imagawa, T., Iwata, N., Katakura, S., and Mori, M. (2004). Inflammatory cytokines and systemic-onset juvenile idiopathic arthritis. Mod. Rheumatol. 14, 12–17.
Yoshimatsu, K., Altorki, N. K., Golijanin, D., Zhang, F., Jakobsson, P. J., Dannenberg, A. J., and Subbaramaiah, K. (2001a). Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin. Cancer Res. 7, 2669–2674.
Yoshimatsu, K., Golijanin, D., Paty, P. B., Soslow, R. A., Jakobsson, P. J., DeLellis, R. A., Subbaramaiah, K., and Dannenberg, A. J. (2001b). Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 7, 3971–3976.
Yu, H., Kortylewski, M., and Pardoll, D. (2007). Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7, 41–51.
Yu, H., Pardoll, D., and Jove, R. (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809.
Zeisberg, M., and Neilson, E. G. (2009). Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 119, 1429–1437.
Keywords: inflammation, carcinogenesis, tumorigenic factors, heat shock proteins, NF-κB, STAT-3, IL-1
Citation: Multhoff G, Molls M and Radons J (2012) Chronic inflammation in cancer development. Front. Immun. 2:98. doi: 10.3389/fimmu.2011.00098
Received: 25 October 2011; Accepted: 28 December 2011;
Published online: 12 January 2012.
Edited by:
Cristina Bonorino, Pontificia Universidade Catolica do Rio Grande do Sul, BrazilReviewed by:
Lidija Klampfer, Montefiore Medical Center and Albert Einstein Cancer Center, USACopyright: © 2012 Multhoff, Molls and Radons. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Gabriele Multhoff, Department of Radiation Oncology, Klinikum rechts der Isar, Technische Universität München, Ismaningerstr. 22, D-81675 Munich, Germany. e-mail:gabriele.multhoff@lrz.tu-muenchen.de
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.