
- Department of Clinical Microbiology and Immunology, Tel Aviv University, Tel Aviv, Israel
Therapy for non-Hodgkin’s lymphoma has progressed significantly over the last decades. However, the majority of patients remain incurable, and novel therapies are needed. Because immunotherapy ideally offers target selectivity, an ever increasing number of immunotherapies, both passive and active, are undergoing development. The champion of passive immunotherapy to date is the anti-CD20 monoclonal antibody rituximab that revolutionized the standard of care for lymphoma. The great success of rituximab catalyzed the development of new passive immunotherapy strategies that are currently undergoing clinical evaluation. These include improvement of rituximab efficacy, newer generation anti-CD20 antibodies, drug-conjugated and radio labeled anti-CD20 antibodies, monoclonal antibodies targeting non-CD20 lymphoma antigens, and bispecific antibodies. Active immunotherapy aims at inducing long-lasting antitumor immunity, thereby limiting the likelihood of relapse. Current clinical studies of active immunotherapy for lymphoma consist largely of vaccination and immune checkpoint blockade. A variety of protein- and cell-based vaccines are being tested in ongoing clinical studies. Recently completed phase III clinical trials of an idiotype protein vaccine suggest that the vaccine may have clinical activity in a subset of patients. Efforts to enhance the efficacy of active immunotherapy are ongoing with an emphasis on optimization of antigen delivery and presentation of vaccines and modulation of the immune system toward counteracting immunosuppression, using antibodies against immune regulatory checkpoints. This article discusses results of the various immunotherapy approaches applied to date for B-cell lymphoma and the ongoing trials to improve their effect.
Introduction
Indolent non-Hodgkin’s lymphomas (NHL), the most common of which is follicular lymphoma (FL), remain incurable. Therefore, new therapeutic modalities that target tumor cells more specifically and with less toxicity are needed. Targeted immunotherapy ideally fits these two requirements, hence both passive and active immunotherapies are widely studied.
Passive immunotherapy based on monoclonal antibodies (mAbs) has already improved the clinical outcome in lymphoma. The anti-CD20 mAb rituximab is presently utilized as a standard of care for lymphoma. However, although rituximab significantly improves duration of remissions and overall survival (Czuczman et al., 2004; Fisher et al., 2005a; Hiddemann et al., 2005), patients eventually relapse from residual tumor cells and die of the disease. Newer generations of improved antibodies are therefore awaited.
Active immunotherapy trials so far consist largely on vaccines that use the immunoglobulin idiotype (Id) as a tumor-specific antigen. Each B lymphocyte expresses on its surface an immunoglobulin molecule, capable of recognizing and binding to a unique antigen. The Id refers to the unique amino acid sequences within the complementarity determining regions (CDR) in the variable regions of the immunoglobulin heavy and light chains. The entire immunoglobulin with its unique variable regions is commonly referred to as the Id protein. Since B-cell lymphomas are clonal in nature, arising from a single B-cell, their immunoglobulin Id is distinct from the Id expressed by their non-malignant counterparts. Therefore, the Id is regarded as a tumor-specific antigen that can be targeted by immunotherapy. Unlike passive immunotherapy, regulatory approval has not yet been granted to any regimen of active immunotherapy for lymphoma, despite evidence of biological and clinical efficacy as well as clinical benefit of Id vaccination in lymphoma patients (Bendandi et al., 1999; Inoges et al., 2006; Schuster et al., 2011). Further trials of vaccines as well as immune checkpoint blockade are ongoing.
This review describes current immunotherapy approaches for B-cell lymphoma and discusses the ongoing efforts and prospects for their improvement.
Passive Immunotherapy by Antibodies
The first antibody-based therapy for B-cell lymphoma utilized anti-Id mAbs. Although these antibodies were eventually abandoned because they need to be tailored for each patient, they provided evidence that antibody-based therapy can be safe and highly effective (Miller et al., 1982), paving the way for subsequent development of the anti-CD20 mAb rituximab.
Anti-CD20 Antibodies
Rituximab, a human–mouse chimeric IgG1 antibody that targets CD20 revolutionized the treatment of FL and is now a standard component of first-line therapy. As a single agent or in combination with chemotherapy it improves the overall response rate (ORR), the duration of response and overall survival (Czuczman et al., 2004; Fisher et al., 2005a; Hiddemann et al., 2005).
Mechanisms of action of rituximab
Rituximab depletes both normal and malignant B-cells. Four major mechanisms have been proposed for the action of rituximab: antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis, complement-dependent cytotoxicity (CDC), direct induction of apoptosis, and “vaccination effect” resulting from cross-priming (Glennie et al., 2007; Rezvani and Maloney, 2011). These mechanisms have been demonstrated in vitro and in mouse models, and clinical observations support their activity in patients. However, their relative contribution to the overall clinical effect remains unclear. The controversy regarding the relative contribution of each mechanism originates at least in part from the use of different anti-CD20 mAbs and different experimental models. Anti-CD20 mAbs are divided into two subtypes based on their functional activity upon antigen ligation. Type I anti-CD20 mAbs, such as rituximab, redistribute CD20 into membrane lipid rafts and potently activate complement, whereas type II anti-CD20 mAbs that do not redistribute CD20 are weak complement activators but potent inducers of programmed cell death (PCD). Both subtypes are equal in their ability to activate Fcγ receptor (FcγR)-bearing effector cells (Chan et al., 2003; Cragg et al., 2003; Glennie et al., 2007).
Growing evidence indicates that recruitment of innate effector cells via Fc/FcγR interaction is critical to the therapeutic efficacy of rituximab. In mouse models, depletion of both normal and malignant B-cells by anti-CD20 mAbs was dependent on active FcγRs (Uchida et al., 2004; Minard-Colin et al., 2008). In the clinical setting, FL patients who carry the genetic polymorphism 158 V/V that codes for a high affinity FcγRIIIa show higher response rates to rituximab as compared to patients with low affinity polymorphisms (158 V/F or 158 F/F; Koene et al., 1997; Cartron et al., 2002; Weng and Levy, 2003), supporting an important role for ADCC. In chronic lymphocytic leukemia (CLL) however, FcγR polymorphisms failed to predict the response to rituximab (Farag et al., 2004), suggesting that mechanisms of tumor clearance independent of Fc/FcγR interactions may be more important in CLL. It should be noted that although natural killer (NK) cells, macrophages, and neutrophils have been implicated in the elimination of B-cells by anti-CD20 antibodies, the nature of the critical effector cells responsible for the therapeutic effect remains disputed.
Complement-dependent cytotoxicity may represent another effector mechanism, although the role of complement remains controversial. In some murine models of lymphoma, rituximab effectively destroyed tumor cells in mice with a functioning complement system but its activity was ablated in complement-deficient mice (Di Gaetano et al., 2003; Golay et al., 2006). However, other studies showed no requirement for complement in the depletion of normal B-cells using C1q-, C3-, and C4-deficient mice (Uchida et al., 2004; Hamaguchi et al., 2005). A number of factors may account for the controversy regarding the involvement of CDC in anti-CD20 antibody immunotherapy. One major factor is the type of tumor, as many tumors are protected from CDC by complement defense molecules. In addition, the microenvironment may play a role in the sensitivity of cells to CDC. Thus, it has been demonstrated that unlike circulating cells, B-cells in the marginal zone compartment exhibit dependency on complement for anti-CD20 antibody killing (Gong et al., 2005). Tumor burden is another factor that may determine the mechanism of action of anti-CD20 mAbs. It has been recently reported that while low tumor load can be eliminated by complement alone, elimination of high tumor load requires multiple effector mechanisms (Boross et al., 2011). In humans, there is evidence that complement is activated by rituximab. Thus, rituximab infusion results in rapid depletion of complement components due to their consumption (Kennedy et al., 2004). However, some studies point to deleterious rather than therapeutic effects of complement activation. In fact, some of the side effects of rituximab treatment have been ascribed to complement activation (van der Kolk et al., 2001). In addition, blockade of ADCC by deposited C3b complement component has been demonstrated (Wang et al., 2008). Moreover, FL patients with a C1qA polymorphism associated with low C1q levels showed correlation with prolonged response to rituximab (Racila et al., 2008). Therefore, the involvement of complement in anti-CD20 therapy, particularly its suggested detrimental role, has to be further substantiated.
A third proposed mechanism of action of rituximab is direct cell killing through signaling. Binding of rituximab has been shown to induce PCD in vitro in several B-cell lines (Hofmeister et al., 2000; Shan et al., 2000). The PCD was dependent on crosslinking of rituximab, which is believed to be mediated in vivo through FcγR-bearing cells. The in vitro findings were supported by the demonstration that rituximab induces caspase activation in CLL patients, which correlates with tumor cell depletion (Byrd et al., 2002). In contrast, many other B-cell lines appear insensitive to direct killing in vitro, and de Haij et al. (2010) showed in a mouse model that Fc-dependent crosslinking of antigen–antibody immune complexes is insufficient for the therapeutic activity in vivo. Hence, the ability of anti-CD20 mAbs to induce cell death through signaling remains controversial.
“Vaccination” by cross-priming has more recently been suggested as an additional mechanism of action. The concept of cross-priming implies that following antibody-induced apoptosis, cell debris are taken up by antigen presenting cells and then presented to cytotoxic T lymphocytes specific for lymphoma-associated antigens. Such an adaptive immune induction by CD20 antibodies may underlie the durable remissions experienced by some rituximab-treated patients (Hainsworth et al., 2002). Although cross-priming induced by rituximab has been demonstrated in vitro (Selenko et al., 2001), its occurrence in vivo remains uncertain. However, several studies support a role for a “vaccine effect.” Thus, treatment of immunocompetent mice with anti-CD20 mAb induces T-cell-mediated protection against human CD20-expressing tumor cells (Abes et al., 2010), and anti-CD20 mAb treatment synergizes with dendritic cell (DC) vaccination in the therapy of established murine lymphoma (Gadri et al., 2009). Moreover, a clinical study demonstrated that rituximab therapy induces an Id-specific T-cell response in FL patients (Hilchey et al., 2009). It is hoped that future studies will offer a decisive answer as to the importance of this mechanism.
Rituximab resistance
Despite being the most successful anti-cancer mAb to date, the effectiveness of rituximab is ultimately limited due to development of resistance with relapse of the disease. Rituximab resistance is mediated by both tumor and host factors. Lymphoma cells may develop resistance because of increased expression of complement regulatory proteins that impair CDC (Takei et al., 2006), blockade of ADCC by deposited C3b complement component (Wang et al., 2008), overexpression of anti-apoptotic proteins or down-regulation of pro-apoptotic proteins (Olejniczak et al., 2008), and down-regulation of CD20 either through loss of expression (Davis et al., 1999), shaving of rituximab/CD20 complexes by phagocytic cells (Beum et al., 2006), or antigenic modulation through internalization that has been previously believed to be of little significance (Beers et al., 2010). Host-related resistance is mediated by exhaustion of complement components (Kennedy et al., 2004; Klepfish et al., 2008) or effector cells (Berdeja et al., 2007). The relative contribution of these mechanisms to rituximab resistance remains to be determined in further clinical studies.
Prospective improvement of anti-CD20 antibody therapy
Understanding the mechanisms of rituximab action and resistance are critical for the development of strategies to improve the clinical outcome of anti-CD20 antibody therapy. Based on present knowledge, two strategies are being investigated: improvement of rituximab efficacy and development of newer generation anti-CD20 mAbs.
Improvement of rituximab efficacy. Enhancing sensitivity of lymphoma cells to rituximab is one approach to improve its efficacy. Rituximab-resistant lymphoma cells exhibit up-regulation of components of the ubiquitin–proteasome system. Of several proteasome inhibitors that have been shown in vitro to sensitize lymphoma cells to rituximab, bortezomib has entered clinical trials. Following a phase II trial showing that the combination of bortezomib with rituximab is feasible as a salvage treatment in relapsed or refractory indolent lymphomas (de Vos et al., 2009), a phase III trial is ongoing. Similarly, since anti-apoptotic proteins of the Bcl-2 family have been implicated in rituximab resistance, Bcl-2 inhibitors such as the antisense oligonucleotide oblimersen are investigated. A phase II study combining oblimersen with rituximab resulted in a promising response rate of 60% in FL patients, some of which were refractory to prior treatment with rituximab (Pro et al., 2008). Results of larger randomized trials in rituximab-resistant patients are awaited.
Enhancement of ADCC is another potential approach to improve rituximab efficacy. Along this line, several cytokines have been combined with rituximab to boost ADCC. Disappointingly, response rates to the combination treatment were not significantly different from those obtained with rituximab alone. For instance, in a phase II study of rituximab combined with IL-2 in rituximab-refractory indolent NHL, no clinical responses could be demonstrated although the IL-2 expanded FcR-bearing cells in vivo and enhanced in vitro ADCC (Khan et al., 2006). When G-CSF was combined with rituximab in CLL, ORR was comparable to that seen with rituximab monotherapy. Interestingly, remission duration was remarkably long (van der Kolk et al., 2003; Gruber et al., 2011). This finding should be substantiated in further trials. Agents other than cytokines have been also investigated as adjuncts to rituximab to enhance ADCC. The toll-like receptor 9 (TLR9) agonist CpG oligonucleotide (ODN) has been combined with rituximab in a phase II study in patients with relapsed or refractory FL (Friedberg et al., 2009a). Clinical responses were observed in 48% of patients despite adverse prognostic factors, warranting further studies. Noteworthy, a recent preclinical study suggests that TLR3 agonists may be preferred agents for enhancing CD20 mAb efficacy (Horikawa et al., 2011). According to this study, a rare IL-10 producing regulatory B-cell subset that is not entirely depleted by anti-CD20 mAbs (B10 cells) inhibits lymphoma depletion during immunotherapy. In contrast to TLR9 agonists, TLR3 agonists do not activate B10 cells hence enhance lymphoma depletion more effectively. Although poly(I:C) is not used clinically because of its toxicity, poly(I:C)-like agents are available for clinical use (Ambrus et al., 2006).
While the contribution of complement to the therapeutic efficacy of rituximab remains controversial, strategies to overcome complement resistance and enhance CDC are being investigated. Since rituximab resistance is associated with up-regulation of membrane complement regulatory proteins, their blocking may enhance rituximab efficacy. Neutralizing antibodies abrogating the function of CD55 and CD59 markedly enhanced the antitumor activity of rituximab in vitro and in vivo in immunodeficient mice (Macor et al., 2007). Similarly, knockdown of CD55 expression using siRNA attenuated the resistance of NHL cells (clinical samples) to CDC activated by rituximab (Terui et al., 2006). Another target for siRNA is mortalin, the mitochondrial hsp70 involved in complement resistance. In vitro experiments showed that mortalin silencing in human B-cell lymphoma sensitized the cells to complement attack by rituximab (Masarwa et al., 2008). It is therefore assumed that combination of rituximab with agents that block complement regulatory pathways may enhance tumor cell killing. None of these approaches has yet reached clinical trials. However, the in vivo application of siRNA and antisense oligonucleotides is a rapidly developing and promising field.
Additionally, repletion of depleted complement components by infusion of fresh frozen plasma has been tested in CLL patients as a means to overcome rituximab resistance (Klepfish et al., 2008; Xu et al., 2010). These results remain to be confirmed in controlled trials. It should however be kept in mind that complement activation by rituximab may act as a double edged sword, hence extensive complement supply may not be desirable.
New generation anti-CD20 mAbs. Several new generation anti-CD20 mAbs are currently undergoing clinical investigation (Table 1). These mAbs are humanized or fully human, unlike the chimeric rituximab. Their design was based on current insights into the mechanisms of rituximab action and resistance aiming at improving their functions, including greater antibody-target binding, enhanced FcRγ binding, enhanced CDC and PCD. It remains to be determined whether they are doing better compared to rituximab in naïve patients and whether they show therapeutic activity in rituximab-refractory patients.

Table 1. New generation anti-CD20 mAbs in clinical development.
Among the new generation mAbs, ofatumumab is at the most advanced-stage of clinical development. It is a fully human type I mAb that binds a unique epitope in the small extracellular loop of the CD20 molecule, which is in close proximity of the cell membrane. Antibody binding to this epitope results in a slow off-rate and an exceptionally high ability to recruit C1q and lyse cells through complement activation (Teeling et al., 2006). Ofatumumab induces CDC in vitro in a range of rituximab-resistant cells (Teeling et al., 2004). Importantly, ofatumumab showed toxicity profile similar to that of rituximab, indicating that the increased ability of ofatumumab to activate complement does not further increase toxicity (Coiffier et al., 2008; Hagenbeek et al., 2008). A phase I/II clinical trial with ofatumumab in relapsed/refractory FL patients showed encouraging ORR of 42% (Hagenbeek et al., 2008). However, a subsequent study on heavily pre-treated patients with rituximab-resistant FL showed disappointing ORR of 11% (Hagenbeek et al., 2009), suggesting that ofatumumab monotherapy is ineffective in rituximab-refractory FL. The mAb may however be of benefit in combination with chemotherapy. Studies of ofatumumab combined with CHOP chemotherapy in patients with FL are ongoing (NCT00494780). According to an early report, ofatumumab was highly effective in combination with CHOP chemotherapy in previously untreated FL patients (van Meerten and Hagenbeek, 2011). The mAb appears to be more effective in CLL than in FL (Wierda et al., 2010a). Ofatumumab monotherapy resulted in high response rates in patients with fludarabine–alemtuzumab refractory CLL (58% ORR) and patients with bulky fludarabine refractory CLL (47% ORR). Retrospective analysis of the treated patients revealed that ofatumumab was active in fludarabine refractory CLL irrespective of prior rituximab. ORR for rituximab-treated, rituximab-refractory, and rituximab-naïve patients was 43, 44, and 53% (Wierda et al., 2011). In 2009 ofatumumab received FDA approval for use in fludarabine and alemtuzumab refractory CLL. Yet, it is important to bear in mind that much higher doses of ofatumumab were administered than previously used for rituximab.
Veltuzumab is a humanized type I mAb with CDRs identical to rituximab except for a single amino acid substitution in CDR3-VH, which translates into a slower dissociation rate. When compared with rituximab, veltuzumab showed greater antitumor activity in murine models and greater B-cell depletion in cynomolgus monkeys (Goldenberg et al., 2009). In a phase I/II clinical trial, veltuzumab appeared safe and the ORR to single agent veltuzumab was 44% in patients with relapsed/recurrent FL, which is comparable with that of rituximab in a similar patient population (Morschhauser et al., 2009). Importantly, veltuzumab was active when injected subcutaneously and at doses lower than the standard dose of rituximab, properties that warrant further studies and are currently being investigated in NHL and CLL (NCT00546793).
Ocrelizumab is a humanized type I anti-CD20 mAb that differs from rituximab at several positions within the CDRs. Compared with rituximab, ocrelizumab demonstrated superior binding to low affinity FcγRIIIa variants, increased ADCC, and lower CDC. In a phase I/II trial in patients with relapsed/refractory FL after prior rituximab therapy, ORR was 38% with apparently milder adverse events, probably due to decreased complement activation (Morschhauser et al., 2010a). These results are in the range of rituximab re-treatment. The clinical development of ocrelizumab was suspended in 2010 following excess death rates of opportunistic infections in lupus erythematosis and rheumatoid arthritis, although studies are ongoing in multiple sclerosis. As for CD20-positive malignancies, there has been little further development.
Third-generation type I humanized anti-CD20 mAbs, such as PRO131921 and AME-133v, are mAbs in which the Fc fragment was modified to increase its affinity to FcγRIIIa and thus enhance ADCC. In preclinical studies, these mAbs had superior activity compared to rituximab (Bowels et al., 2006). In phase I trials, PRO131921 and AME-133v showed clinical activity in rituximab-pre-treated relapsed FL (Friedberg et al., 2009b; Tobinai et al., 2011). AME-133v is currently undergoing a phase I/II trial in relapsed/refractory FL (NCT00354926).
Unlike other ani-CD20 mAbs in clinical development, the third-generation GA101 is a type II antibody. It has a glyco-engineered Fc fragment with non-fucosylated oligosaccharides to enhance the affinity to FcγR, particularly FcγRIIIa, therefore enhance ADCC (Mössner et al., 2010). Being a type II anti-CD20 mAb, GA101 induces effective direct and immune cell-mediated cell killing. In preclinical models, GA101 was significantly more effective than rituximab in B-cell depletion and in tumor rejection. Preliminary results of a phase I/II study showed that GA101 as a single agent has a similar safety profile to rituximab and a promising efficacy (ORR of 58%) in patients with relapsed/refractory B-NHL (mainly FL) who previously received rituximab (Salles et al., 2008). Phase III trials of GA101 combined with chemotherapy have been initiated in previously untreated diffuse large B-cell lymphoma (DLBCL; NCT01287741) and rituximab-refractory indolent NHL (NCT01059630). Clinical data of this promising agent are awaited.
CD20 antibody radio conjugates. Radio immunotherapy has emerged as a promising treatment option for lymphoma. Two radio conjugates of anti-CD20 mAbs are currently approved by the FDA: 131I-tositumomab (Bexxar) and 90Y-ibritumomab tiutexan (Zevalin). Both of them have demonstrated high clinical efficacy in heavily pre-treated patients, including patients with chemotherapy- and rituximab-refractory disease with poor prognostic characteristics. Thus, in five clinical trials, patients with indolent NHL that were treated with one course of 131I-tositumomab demonstrated ORR ranging from 47 to 68% with complete response (CR) rates ranging between 20 and 38% (Fisher et al., 2005b). The 5-year progression-free survival (PFS) was 17% with a median follow-up of 5.3 years. Eighty-one (32%) of 250 patients had a long-term durable response (PFS greater than 1 year), and the median duration of response for those patients was 45.8 months. Similar durable responses were observed in 90Y-ibritumomab tiuxetan-treated patients with relapsed/refractory NHL (Witzig et al., 2007). In four clinical trials, 59 (39%) of 153 patients with indolent FL had a long-term response (PFS of more than 1 year), and the median time to progression was 30.9 months.
Radioimmunotherapy has also demonstrated excellent response rates in the front-line treatment of previously untreated advanced-stage indolent FL, either as a single agent or in combination with chemotherapy. Kaminski et al. (2005) reported that treatment with 131I-tositumomab as single agent in 76 patients with stage III or IV FL resulted in ORR of 95%, with a CR rate of 74%. The 5-year PFS for all patients was 59%, and the median PFS was 6.1 years with a median follow-up of 5.1 years. In a phase II trial of CHOP chemotherapy followed by 131I-tositumomab for previously untreated FL, the ORR was 91% including a 69% CR rate. After a median follow-up time of 5.1 years, the 5-year ORR was 87%, and the PFS rate was 67% (Press et al., 2006). This outcome was superior to historical figures of patients treated with CHOP alone, which led to phase III randomized trial of CHOP plus rituximab versus CHOP plus 131I-tositumomab in newly diagnosed NHL (NCT00006721). The study is ongoing and results are eagerly awaited. Another randomized phase III trial to evaluate the efficacy of consolidation with 90Y-ibritumomab tiuxetan in patients with advanced-stage FL in first remission was already completed and published (Morschhauser et al., 2008). 90Y-ibritumomab tiuxetan consolidation significantly prolonged the median PFS by 2 years in all patients regardless of whether patients achieved partial response (PR) or CR after induction treatment. After 90Y-ibritumomab tiuxetan consolidation, 77% of patients in PR after induction converted to CR, resulting in a final CR rate of 87%.
Altogether, there is substantial evidence demonstrating that radioimmunotherapy is extremely effective. However, this treatment modality is not yet widely applied. Favorable awaited results of ongoing trials in patients with indolent NHL and in patients with more aggressive lymphomas may speed its adoption in the clinic.
Targeting Other Lymphoma Antigens
Although mAbs to CD20 dominate clinical studies in lymphoma, mAbs to other antigens are being developed (Table 2). Antibodies with activity that warrants further development are briefly reviewed.

Table 2. Clinical development of mAbs against antigens other than CD20.
CD22
CD22 is a B-cell restricted surface molecule involved in regulation of B-cell activation. Epratuzumab is a humanized anti-CD22 mAb that has shown only modest single agent activity in pre-treated FL (Leonard et al., 2003). However, the combination of epratuzumab with rituximab demonstrated enhanced anti-lymphoma activity compared with either mAb alone. A phase II trial, evaluating the combination of epratuzumab and rituximab in patients with different types of refractory/relapsed NHL showed ORR of 47%, with the best responses being observed in patients with FL and DLBCL (64%; Strauss et al., 2006). In another phase II study, the combination of epratuzumab, rituximab, and CHOP chemotherapy in previously untreated DLBCL provided ORR of 96% (Micallef et al., 2011). These results led to the production of anti-CD20/anti-CD22 bispecific mAbs (Rossi et al., 2009). The rationale was that crosslinking of the two target molecules by bispecific antibodies may have therapeutic advantage over the combination of the two parental mAbs, an assumption that needs to be verified in clinical studies.
Epratuzumab has been also applied as an antibody radio conjugate. In a phase I/II study in patients with relapsed/refractory NHL, treatment with 90Y-epratuzumab tetraxetan demonstrated ORR and median PFS of 62% (CR48%) and 9.5 months, respectively (Morschhauser et al., 2010b).
Inotuzumab ozogamicin (CMC-544) is a mAb-drug conjugate composed of a humanized anti-CD22 mAb linked to the potent cytotoxic agent calicheamicin. In two studies of patients with relapsed/refractory FL and DLBCL who were treated with inotuzumab ozogamicin alone or in combination with rituximab, the response rates and PFS results indicated promising efficacy, albeit not as impressive in rituximab-refractory patients (Dang et al., 2009; Advani et al., 2010). These results support continued clinical development of this regimen.
CD30
CD30 is a pro-survival receptor expressed on Reed Sternberg cells of Hodgkin’s lymphoma (HL) and on anaplastic large-cell lymphoma. Several mAbs have been developed against CD30 including SGN-30, MDX-060, MDX-1401, and XmAb2513. Despite promising results in preclinical studies, early clinical studies with naked anti-CD30 mAbs demonstrated minimal activity. In contrast, the drug conjugate Brentuximab vedotin (SGN-35), a chimeric anti-CD30 mAb combined with the antimitotic agent vedotin, has shown highly promising clinical efficacy. In a pilot phase I study, brentuximab vedotin was administered to 45 patients with relapsed or refractory CD30-positive malignancies, primarily HL (Younes et al., 2010). Objective responses, including 11 complete remissions, were observed in 17 patients. The median duration of response was at least 9.7 months. Tumor regression was observed in 36 of 42 patients who could be evaluated (86%). This is an excellent result for a single agent treatment in heavily pre-treated refractory HL. Based on these results, a phase II trial of brentuximab vedotin in patients with relapsed/refractory HL is ongoing (NCT00848926) and results are eagerly awaited.
CD40
Agonistic mAbs to CD40 may be particularly effective due to stimulation of host antitumor immune responses. Two anti-CD40 mAbs have been investigated. Dacetuzumab (SGN-40), a humanized mAb, failed to demonstrate clinical efficacy in phase I/II trials and its development was therefore discontinued. Lucatumumab (HCD122), a fully human mAb, is currently investigated in a phase I/II study in progressive NHL and HL after at least two prior therapies (NCT00670592). In addition, a phase I study of lucatumumab and bendamustine combination in rituximab-refractory FL is ongoing (NCT01275209). In regard to the limited clinical efficacy of agonistic CD40 mAbs, it has been recently demonstrated that co-engagement of the Fc domain of agonistic CD40 mAbs with the inhibitory FcγRIIB is required for CD40-mediated immune activation and antitumor activity (Fubin and Ravetch, 2011). This unexpected finding has direct implication for immunotherapy. Thus, the design of agonistic CD40 mAbs with increased binding affinity to FcγRIIB may enhance their clinical efficacy.
CD52
CD52 is expressed on most lymphoid malignancies. Alemtuzumab (Campath-1H), a humanized anti-CD52 mAb, is approved by the FDA for the treatment of fludarabine-resistant CLL. Also, as demonstrated in a phase III study in CLL patients, first-line treatment with alemtuzumab resulted in significantly improved PFS, ORR, and CR compared with chlorambucil (Hillmen et al., 2007). A phase II trial of alemtuzumab and dose-adjusted EPOCH–Rituximab in relapsed or refractory DLBCL is currently ongoing (NCT01030900).
CD19
CD19, a B-cell restricted membrane glycoprotein involved in regulation of B-cell activation, was one of the first targets for immunotherapy by naked mAbs and by immunotoxins. However, because clinical efficacy of anti-CD19 mAbs was modest and because anti-CD20 mAbs emerged as more promising agents, the development of anti-CD19 mAb therapy has been halted. Nonetheless, promising results have been obtained with the bispecific T-cell engaging (BiTE) blinatumomab (MT103), a CD19 × CD3 bispecific single chain antibody that recruits effector T-cells to lymphoma cells, resulting in effective tumor cell killing (Bargou et al., 2008). Doses as low as 0.005 mg/m2/day in NHL patients led to elimination of target cells in blood. Partial and complete tumor regressions were first observed at a dose level of 0.015 mg, and all seven patients treated at a dose level of 0.06 mg experienced tumor regression. Ongoing studies (Viardot et al., 2010) suggest that T-cell engaging antibodies appear to have a promising therapeutic potential that warrants further development.
Active Immunotherapy
As opposed to passive immunotherapy with mAbs, active immunotherapy may induce a polyclonal response directed against multiple epitopes, hence limiting tumor escape that gives rise to relapse. In addition, active immunotherapy can induce long-lasting antitumor immunity, thereby limiting the likelihood of relapse. On the other hand, the disadvantage of active immunotherapy is its reliance on the patient’s immune system, which may be dysfunctional. Current clinical studies of active immunotherapy for lymphoma consist largely of vaccination and immune checkpoint blockade that will be herein reviewed.
Vaccination
The most thoroughly studied target for vaccination in lymphoma is the immunoglobulin Id. A major obstacle in production of Id vaccines derives from its patient-specific nature that requires the generation of a custom-made product. However, the manufacturing issues were overcome by advances in hybridoma and recombinant DNA technology. The Id may be used as either protein or DNA in therapeutic vaccines. In the traditional rescue hybridization technique, the Id protein is produced by fusing the lymphoma cells with mouse myeloma cells to generate Id-secreting hybridomas (Levy and Dilley, 1978). For recombinant Id protein production, genes encoding the tumor-specific immunoglobulin variable regions are cloned by polymerase chain reaction, ligated into an expression vector and transfected into bacterial, plant, insect, or mammalian cells that then produce the Id protein (Park and Neelapu, 2008). For Id DNA vaccination, the immunoglobulin heavy and light chains are cloned and inserted into a plasmid vector for naked DNA injection.
Most clinical trials of Id vaccines for lymphoma have been performed on patients with FL. One reason for this is the indolent nature of FL that gives time for the vaccine to be produced and for the patient’s immune system to recover after aggressive chemotherapy. In addition, FL is regarded as particularly immune responsive based on occurrence of spontaneous regressions, high response rates to mAb treatment, occasional responsiveness to interferon-α and IL-2, and correlation of survival time with gene expression profile of tumor infiltrating immune cells (Hollander, 2009).
Id protein vaccines
The native Id is a weak immunogen. However, it can be made immunogenic by coupling it to a carrier protein and by administering it with an immunologic adjuvant. Based on preclinical and early clinical studies, the most widely used Id protein vaccine formulation consists of the Id protein coupled to the powerful immunogenic keyhole limpet hemocyanin (KLH) with the addition of granulocyte–macrophage colony-stimulating factor (GM-CSF), which facilitates induction of tumor-specific CD8 T-cells (Bendandi et al., 1999).
Phase I/II clinical trials. Several early phase trials of Id–KLH plus GM-CSF vaccines in FL have demonstrated promising clinical responses and clinical benefit. Bendandi et al. (1999) conducted a phase I/II clinical trial in which 20 FL patients in first chemotherapy-induced complete remission received an Id–KLH + GM-CSF vaccine. Despite being in complete remission, all 11 patients with detectable t(14; 18) chromosomal translocations in their primary tumors had cells from the malignant clone detectable in the blood by PCR. However, 8 of 11 patients showed complete elimination of residual tumor cells (detectable by PCR) immediately after completing vaccination and sustained their molecular remissions for a median of 18 months. Tumor-specific CD8 and CD4 T-cells were found in 19 out of 20 patients (95%), whereas anti-Id antibodies were detected in 15 out of 20 patients (75%). Vaccination was thus associated with clearance of residual tumor cells from blood and long-term disease-free survival. In another phase I/II study, 22 patients in first remission after chemotherapy were immunized with Id protein conjugated to KLH and administered with GM-CSF. Among 21 evaluable patients, 62% mounted Id-specific immune responses. At median follow-up of more than 6 years, median PFS was 38 months (Timmerman et al., 2009).
Clinical trials were performed not only in patients in first chemotherapy-induced remission. In a phase II study, 25 patients received an Id–KLH plus GM-CSF vaccine following second chemotherapy-induced complete remission (Inoges et al., 2006). Twenty patients (80%) had either humoral or cellular Id-specific immune responses. The median duration of the second CR among the 20 immune responders was significantly longer than the median duration of their first CR or the chemotherapy-induced second response. This trial confirmed that Id vaccination induced a specific immune response in the majority of patients with FL and that specific immune responses were associated with a highly significant increase in disease-free survival.
Although the majority of early phase vaccination studies were conducted in patients who were in complete remission or very good partial remission after cytoreductive therapy, some studies evaluated vaccination of patients with previously treated measurable disease. One such phase II trial in patients with measurable disease demonstrated that Id–KLH plus GM-CSF vaccine alone can induce tumor regression and durable clinical responses (Redfern et al., 2006).
Phase III clinical trials. The encouraging results of the early phase trials prompted initiation of three double-blind, randomized phase III clinical trials of Id vaccination after initial cytoreductive therapy (Table 3). Disappointingly, two of these studies, sponsored by Genitope Inc., and Favrille Inc., found no statistically significant differences in PFS between the Id–KLH plus GM-CSF vaccine and a control vaccine containing KLH plus GM-CSF, although the vaccine was generally safe (Levy et al., 2008; Freedman et al., 2009). Interestingly, the Genitope study demonstrated a correlation of immune and clinical response with a significant difference in PFS among Id-vaccinated patients who developed an anti-Id immune response compared to Id-vaccinated patients who did not mount an immune response (40 versus 16 months). This result suggests that a subgroup of patients may benefit from Id vaccination. The third phase III study, conducted by NCI/Biovest International, demonstrated an improved disease-free survival of 44.2 months for patients in the experimental arm versus 30.6 months in the control arm (p = 0.045; Schuster et al., 2011). This study is criticized because it was terminated before completion of the planned accrual, resulting in a smaller sample size than originally planned and a limited statistical power with an inability to achieve the originally stipulated threshold of p < 0.01.
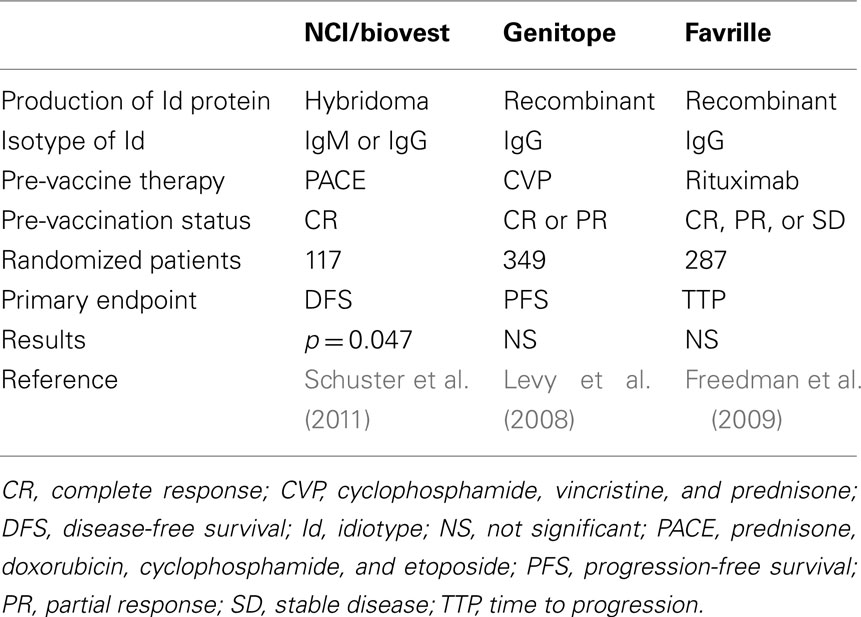
Table 3. Phase III clinical trials of Id vaccination in follicular lymphoma.
Several differences in trial design and vaccine formulation may be responsible for the different clinical outcomes of the three phase III trials. First, differences in patient eligibility seem to be important. While the Biovest trial was limited to patients who were in complete remission at the time of vaccination, the Genitope and Favrille trials also included patients who achieved only PR or even stable disease after chemotherapy. If these data are confirmed in future studies, it would strongly support the value of vaccination in the minimal disease state. With this regard, a number of trials have already shown that Id vaccination can induce immune responses after high-dose cytotoxic therapy and even after myeloablative conditioning and bone marrow transplantation (Davis et al., 2001). Second, the Id protein in the Biovest study was a hybridoma product, whereas the other two studies used a recombinant Id, suggesting that manufacturing procedures may be important. Third, the pre-vaccine therapy was different in the three trials. Finally, the Id isotype may be of importance. While the Id isotype in the Biovest study was either IgM or IgG, lymphomas in the other two studies were exclusively of the IgG isotype. An intriguing finding in the Biovest study was that among patients receiving an IgM-Id vaccine, median time to relapse was 52.9 versus 28.7 months in IgM isotype control-treated patients (p = 0.001), while among patients receiving IgG-Id vaccine, median time to relapse was 35.1 versus 32.4 months in IgG tumor isotype control-treated patients (p = 0.87; Schuster et al., 2011). If these data are validated, determination of Id isotype may facilitate early identification of patients who are more likely to benefit from vaccination.
Combination of Id vaccines and rituximab. The standard of care for FL changed during the course of the three phase III clinical trials and now includes the mAb rituximab. Because rituximab depletes both normal and malignant B-cells, it is expected to impair or delay the anti-Id humoral response in vaccinated patients. Hence, it is crucial to determine its effects on Id-specific T-cell responses. Neelapu et al. (2005) reported that vaccination by Id–KLH plus GM-CSF in patients with mantle cell lymphoma following rituximab-containing chemotherapy induced vigorous CD4 and CD8 antitumor type I cytokine responses in the absence of circulating B-cells. These results show that severe B-cell depletion does not impair T-cell priming in humans, suggesting that Id vaccines may be used in combination with rituximab. However, vaccine boosts after B-cell recovery may be necessary for optimal humoral responses.
Prospective improvement of Id vaccines. Increasing Id immunogenicity, delivery, and presentation. Increasing antigen immunogenicity, delivery, and presentation may improve the clinical outcome of Id vaccines. Methods to improve the immunogenicity of Id vaccines are being sought. The Id is currently chemically conjugated to KLH using glutaraldehyde. However, the extensive protein crosslinking via glutaraldehyde could destroy immunogenic epitopes and inhibit proteolytic processing. Therefore, to increase immunogenicity, milder chemical reagents are tested. It has been demonstrated that a sulfhydryl-based Id-carrier protein conjugation system using maleimide chemistry enhanced the efficacy of Id–KLH vaccines (Betting et al., 2008). In addition, carriers more potent than KLH may be introduced. Several bacterial products that have high immune-enhancing activity and that can be linked to Id by chemical conjugation or by genetic fusion have already been suggested (Neelson et al., 2008; Chen et al., 2009).
Methods are also being sought for targeting antigen to antigen presenting cells in order to enhance antigen uptake and processing. Several cell surface receptors have been suggested as potential targets. Targeting can be achieved for instance by linking Id to the B subunit of E. coli heat labile toxin, which binds to GM1 ganglioside receptors and facilitates antigen uptake through receptor-mediated endocytosis (Chen et al., 2009). Antigen can be also targeted to “scavenger” receptors such as the mannose receptor, which binds to carbohydrate structures rich in terminal mannose residues. In this regard, it is interesting to note that recombinant proteins produced in insect cells are characterized by terminal mannose residues. It has been reported that recombinant insect cell-derived Id showed higher binding to and activation of DCs as well as enhanced immune stimulation, which involved mannose receptor targeting (Betting et al., 2009).
Liposomes are widely accepted as effective carriers for vaccines. Hence, incorporation of Id into cytokine-carrying liposomes has been proposed as a means to enhance Id delivery. An early study in FL patients demonstrated that delivery by a liposomal Id/IL-2 vaccine is safe, induces sustained tumor-specific CD4 and CD8 T-cell responses, and may therefore serve as a model for vaccine development (Neelapu et al., 2004).
DNA vaccines. Idiotype DNA vaccines consist of a plasmid encoding the immunoglobulin single chain variable fragment. The DNA vaccine is injected into the skin or muscle, resulting in production of the protein by the patient’s cells and presentation to the immune system. Since this approach does not require in vitro production of a purified protein, it is faster and less expensive. This is particularly important in the case of a customized vaccine. Preclinical studies showed that the Id DNA vaccine is not immunogenic unless linked to a xenogeneic carrier (Syrengelas et al., 1996; King et al., 1998). An early phase I/II study in FL patients immunized with such a chimeric DNA vaccine was quite disappointing: only 1 out of 12 patients mounted an Id-specific immune response and no clinical response was observed. To enhance the immunogenicity of Id DNA vaccines, the Id was genetically fused to proinflammatory chemokines such as MCP-3 and MIP-3α that target the Id to DCs. These fusion vaccines enhance antigen uptake and presentation through chemokine receptor-mediated endocytosis and exert a chemotaxis effect on DCs, thus enhancing anti-Id immune responses (Biragyn et al., 1999). Preclinical studies in lymphoma models demonstrated superior antitumor effects compared to an Id–KLH protein vaccine.
The failure to translate the success of DNA vaccination in preclinical models to the clinic is believed to be due to failure in delivery of DNA. This failure reduced the initial enthusiasm. However, novel delivery systems such as electroporation dramatically improve performance of DNA vaccination and are now in clinical trials for several types of cancer (Stevenson et al., 2011).
DC vaccines. Dendritic cells loaded in vitro with antigen are attractive vehicles for therapeutic cancer vaccines. In a pilot clinical study, vaccination with autologous DCs pulsed ex vivo with tumor-specific Id protein was examined in four patients with FL. All patients developed antitumor cellular immune responses. In addition, clinical responses have been observed with one patient experiencing complete tumor regression, a second patient having partial tumor regression, and a third patient resolving all evidence of disease as detected by molecular analysis (Hsu et al., 1996). In an extended trial with 35 patients, among 28 patients with residual disease at the time of vaccination, 10 experienced tumor regression (Timmerman et al., 2002). Randomized controlled clinical trials are required to definitively answer the question of clinical benefit induced by DC-based Id vaccination in lymphoma. This field has not progressed as anticipated, partly due to skepticism regarding the feasibility and prospects of DC vaccines to gain regulatory approval. The recent FDA approval of sipuleucel-T, a DC vaccine against prostate cancer, provided a formal proof for the clinical efficacy of DC vaccines, and is likely to prompt development of this therapeutic modality.
Combining Id vaccines with new adjuvants. Increasing evidence argues that the success of an anti-cancer vaccine may rely on immunoadjuvant stimulating effects. Hence, co-administration of vaccines with various adjuvants is being evaluated for improving vaccine performance. One promising class of adjuvants with potent immunostimulatory properties consists of CpG ODNs that are capable of activating antigen presenting cells, B-cells, and NK cells by engaging TLR9. B-cell malignancies are uniquely sensitive to CpG ODNs in that the malignant cells themselves express TLR9, thus respond to CpG ODNs by up-regulation of MHC and costimulatory molecules, and by proliferation that can lead to activation induced cell death (Weiner, 2009). Preclinical studies in animal models showed that CpG ODN is a potent adjuvant when combined with vaccines (Liu et al., 1998). As discussed elsewhere in this review, CpG ODN also demonstrates potent immunostimulatory activity when combined with other therapeutic agents.
Tumor cell vaccines
One drawback of Id vaccination is the restriction of the antitumor response to a single antigen. Therefore, vaccine formulations based on autologous neoplastic cells have been investigated. These formulations might potentially induce autoimmunity, but have the advantage of widening the spectrum of target tumor-associated antigens.
Unlike other malignant cells, B-lymphoma cells can be activated to express MHC class II and costimulatory cells, thus present their own tumor-associated antigens and elicit T-cell responses. Lymphoma cells are therefore excellent candidates for tumor cell-based vaccination. One approach to activate malignant B-cells is by ligation of CD40 via CD40–CD40 ligand (CD40L) interaction. This can be achieved either by mixing lymphoma cells with CD40L expressing cells or by engineering lymphoma cells to express CD40L. Following successful studies in animal models, this concept has been tested in patients with CLL. In phase I trials, in which ex vivo CD40L-transduced autologous tumor cells were reinfused to patients, reductions in leukemia cell counts and lymph node size were seen (Wierda et al., 2000, 2010b). In another phase I trial in CLL, in which patients received subcutaneous injections of autologous tumor cells transduced with both CD40L and IL-2, three patients (30%) had >50% reduction in the size of affected lymph nodes (Biagi et al., 2005), suggesting that CD40-based immunotherapy may have clinical benefit.
A vaccine formulation based on DCs loaded with killed tumor cells rather than Id protein has also been studied. In a pilot study, 18 patients with relapsed and measurable indolent NHL were injected subcutaneously with DCs loaded with tumor cells. The vaccination achieved significant objective clinical responses, and was associated with significant immune modulation (Di Nicola et al., 2009). Six patients had objective clinical responses including three continuous CR and three PR, with a median follow-up of 50.5 months. Eight patients had stable disease, whereas four had progressive disease. Clinical responses were significantly associated with a reduction in regulatory T-cells (Treg) and maturation of lymphocytes to the effector memory stage in either peripheral blood or tumor specimen samples. These results indicate that immunization of patients with tumor cell-loaded DCs represents a potentially effective strategy for the treatment of indolent NHL patients with measurable disease.
In situ vaccination
To circumvent the logistical difficulty of ex vivo manufacture of a customized vaccine, an alternate approach of in situ vaccination has been developed. It consists of low-dose irradiation to a single lymphoma site, followed by intratumoral injection of CpG at the same site. This maneuver kills some tumor cells at the treated site, and the CpG activates nearby DCs, enhancing presentation of the released tumor antigens by the DCs. Residual viable lymphoma cells can also be activated by CpG and present the released antigens to T-cells. B-cell NHL are considered highly suited to this maneuver because they are sensitive to radiotherapy and because they express TLR9. Preclinical studies in a lymphoma model indicated that the combination of local immunostimulation and cytotoxic therapy was required to elicit powerful systemic antitumor responses and tumor rejection (Li et al., 2007). In a recent phase I/II study, 15 patients with low grade lymphoma were treated by in situ vaccination (Brody et al., 2010). One patient had a CR, three others had PR, and two patients had stable but continually regressing disease for periods significantly longer than that achieved with prior therapies. In situ vaccination induced tumor-reactive memory CD8 T-cells. However, some patients’ tumors were able to induce a suppressive, regulatory phenotype in autologous T-cells in vitro. These patients tended to have a shorter time to disease progression. One clinically responding patient received a second course of vaccination after relapse resulting in a second, more rapid clinical response. These encouraging preliminary results warrant further study of CpG-based in situ vaccination. The induction of tumor-reactive CD8 T-cells and inferior outcome of patients with Treg-inducing tumors suggest that enhancing T-effector cells and inhibiting Treg might further improve this regimen of vaccination.
Immune Checkpoint Intervention
There is accumulating evidence that therapeutic vaccination is enhanced by disruption of immunoregulatory pathways that limit the magnitude and duration of the immune response. Although most of the evidence comes from preclinical studies, several approaches for immune checkpoint blockade have already been tested in clinical trials for lymphoma patients.
Blockade of T-cell inhibitory signals
Anti-CTLA-4 mAb. Cytotoxic T-lymphocyte antigen 4 (CTLA-4), a member of the CD28 family, is a key negative regulator of T-cell activation. It is inducibly expressed in conventional T-cells after activation and constitutively expressed in Treg. Blockade of CTLA-4 by mAbs enhanced T-cell activation, improved immune responsiveness to anti-cancer vaccines in preclinical studies, and had clinical activity against several types of cancer (Peggs et al., 2008). Following a phase III trial, demonstrating overall survival benefit in metastatic melanoma, the anti-CTLA-4 human mAb ipilimumab was approved by the FDA in 2011 to treat patients with late-stage melanoma. Ipilimumab has also been evaluated for treatment of B-cell lymphoma. In a pilot clinical trial with four NHL patients after failure of Id vaccination, administration of ipilimumab showed tumor regression in two patients, one of which obtained a PR of 14-month duration (O’Mahony et al., 2007). In a more recent phase I study, 18 patients with relapsed/refractory B-cell NHL were treated with ipilimumab (Ansell et al., 2009). Two patients showed clinical responses; one patient had an ongoing CR (>31 months), and one had a PR lasting 19 months. In 5 of 16 cases tested (31%), T-cell proliferation to recall antigens was significantly increased after ipilimumab therapy. Although response rates have been low in these early trials, the overall survival benefit observed with ipilimumab in melanoma prompts further studies. Evaluation of ipilimumab is ongoing, both as monotherapy and in combination with other therapies.
Anti-PD-1 mAb. Programmed death-1 (PD-1) is another T-cell inhibitory receptor that is inducibly expressed after activation of T-cells. Ligation of PD-1 by its ligands PD-L1 and PD-L2 inhibits T-cell activation. It has been demonstrated that PD-L1 is expressed by NHL and inhibits the activity of tumor-associated T-cells (Andorsky et al., 2011), suggesting PD-L1 blockade as a potentially useful strategy for lymphoma immunotherapy. CT-011 is a humanized anti-PD-1 mAb that has been demonstrated to induce regression of several tumor types in mice. A phase I trial using CT-011 in advanced hematologic malignancies demonstrated some clinical responses including one CR in FL (Berger et al., 2008). A phase II trial with CT-011 in DLBCL following autologous stem cell transplantation has been completed and publication of data is awaited. According to media releases, CT-011 met the primary endpoint of improved PFS, and statistically significant results have also been achieved in the secondary endpoint of overall survival. In addition, a phase II trial of CT-011 combined with rituximab in patients with relapsed FL is ongoing (NCT00904722).
Depletion of Treg
Treg are implicated in the dampening of antitumor T-cell responses. Accumulating data indicate that the presence of Treg at the tumor site or in peripheral blood correlates with poor prognosis (Wilke et al., 2010). In B-cell NHL, Treg at the tumor site have been shown to suppress activity of infiltrating CD4 and CD8 T-cells (Yang et al., 2006a,b), suggesting that Treg depletion may enhance clinically beneficial antitumor responses. Denileukin diftitox (Ontak) is a diphtheria toxin-IL-2 fusion protein shown to deplete Treg in several tumors. In a phase II study of denileukin diftitox in 29 patients with recurrent indolent B-cell NHL, three PR were obtained (Kuzel et al., 2007). Despite the modest clinical response, the prospective introduction of additional Treg-depleting or Treg-inhibiting reagents may improve the outcome.
Conclusion
Rituximab improved tremendously the management of NHL, prolonging remission, and survival. However, resistance to rituximab leads to eventual relapse in most patients. Although several new generation anti-CD20 mAbs show improved mode of action compared to rituximab, their clinical efficacy in rituximab-refractory patients is disappointingly modest. It appears that type I anti-CD20 mAbs, which comprise the majority of newly developed anti-CD20 mAbs, do not provide significant improvement over rituximab. In contrast, the third-generation type II mAb GA101 has already shown promising efficacy in rituximab-refractory patients, and results of phase III trials are awaited. As it seems that the direct PCD induced by type II anti-CD20 mAbs may be required for elimination of rituximab-resistant lymphoma cells, efforts should be directed toward rigorous clinical studies of these mAbs, particularly in rituximab-refractory patients.
Radioimmunotherapy with anti-CD20 mAbs demonstrated excellent clinical efficacy in chemotherapy and rituximab-refractory FL. Yet, this treatment is under-utilized, perhaps due to the complexity of delivery compared to naked antibodies, concerns about late toxicity or uncertainty regarding their integration with standard therapies. In view of compelling data that the two approved drugs 131I-tositumomab and 90Y-ibritumomab tiutexan can significantly improve the therapeutic outcome in refractory disease, their integration in current treatment schemes needs to be re-examined. If radioimmunotherapy is not adopted by the clinical community as a more widely used therapeutic tool, some patients may lose a chance to benefit from this effective treatment.
Antibodies targeting antigens other than CD20 showed disappointing clinical efficacy. However, drug conjugates such as inotuzumab ozogamicin (anti-CD22) or brentuximab vedotin (anti-CD30) and bispecific antibodies such as blinatumomab (anti-CD19/CD3) have demonstrated impressive clinical efficacy. It is hoped that these mAbs may improve the outcome in rituximab-refractory lymphoma, but phase III trials are needed to prove their superiority.
In comparison with the success of anti-CD20 mAbs, lymphoma vaccines appear disappointing. Patients with CR following induction chemotherapy, in particular patients with an IgM vaccine isotype, may benefit from Id vaccine therapy. However, a more effective vaccine is needed for patients who do not achieve minimal residual disease. Thus, next generation vaccines are being developed. These include DNA-based and DC-based Id vaccines as well as tumor cell vaccines. Optimal vaccine activity will most probably require combination with immune adjuvants that potentiate vaccine-induced antitumor immunity, or with antibodies that block immune regulatory checkpoints. Whatever strategies for vaccine development are selected, lymphoma vaccines will have to be optimally integrated with standard therapies, particularly with rituximab.
Finally, in the forthcoming era of personalized medicine, it will become extremely important to determine which lymphoma patients are most likely to benefit from a particular therapy out of the existing therapeutic arsenal. This will likely include tumor and microenvironment profiling by genomic and biomarker analysis.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abes, R., Gelize, E., Fridman, W. H., and Teillaud, J. L. (2010). Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 116, 926–934.
Advani, A., Coiffier, B., Czuczman, M. S., Dreyling, M., Foran, J., Gine, E., Gisselbrecht, C., Ketterer, N., Nasta, S., Rohatiner, A., Schmidt-Wolf, I. G., Schuler, M., Sierra, J., Smith, M. R., Verhoef, G., Winter, J. N., Boni, J., Vandendries, E., Shapiro, M., and Fayad, L. (2010). Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: results of a phase I study. J. Clin. Oncol. 28, 2085–2093.
Ambrus, J. L. Sr., Chadha, K. C., Islam, A., Akhter, S., and Ambrus, J. L. Jr. (2006). Treatment of viral and neoplastic diseases with double-stranded RNA derivatives and other new agents. Exp. Biol. Med. (Maywood) 231, 1283–1286.
Andorsky, D. J., Yamada, R. E., Said, J., Pinkus, G. S., Betting, D. J., and Timmerman, J. M. (2011). Programmed death ligand 1 is expressed by non-Hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin. Cancer Res. 17, 4232–4244.
Ansell, S. M., Hurvitz, S. A., Koenig, P. A., LaPlant, B. R., Kabat, B. F., Fernando, D., Habermann, T. M., Inwards, D., Verma, M., Yamada, R., Erlichman, C., Lowy, I., and Timmerman, J. M. (2009). Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 15, 6446–6453.
Bargou, R., Leo, E., Zugmaier, G., Klinger, M., Goebeler, M., Knop, S., Noppeney, R., Viardot, A., Hess, G., Schuler, M., Einsele, H., Brandl, C., Wolf, A., Kirchinger, P., Klappers, P., Schmidt, M., Riethmüller, G., Reinhardt, C., Baeuerle, P. A., and Kufer, P. (2008). Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321, 974–977.
Beers, S. A., French, R. R., Chan, H. T., Lim, S. H., Jarrett, T. C., Vidal, R. M., Wijayaweera, S. S., Dixon, S. V., Kim, H., Cox, K. L., Kerr, J. P., Johnston, D. A., Johnson, P. W., Verbeek, J. S., Glennie, M. J., and Cragg, M. S. (2010). Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115, 5191–5201.
Bendandi, M., Gocke, C. D., Kobrin, C. B., Benko, F. A., Sternas, L. A., Pennington, R., Watson, T. M., Reynolds, C. W., Gause, B. L., Duffey, P. L., Jaffe, E. S., Creekmore, S. P., Longo, D. L., and Kwak, L. W. (1999). Complete molecular remissions induced by patient-specific vaccination plus granulocyte-monocyte colony-stimulating factor against lymphoma. Nat. Med. 5, 1171–1177.
Berdeja, J. G., Hess, A., Lucas, D. M., O’Donnell, P., Ambinder, R. F., Diehl, L. F., Carter-Brookins, D., Newton, S., and Flinn, I. W. (2007). Systemic interleukin-2 and adoptive transfer of lymphokine-activated killer cells improves antibody-dependent cellular cytotoxicity in patients with relapsed B-cell lymphoma treated with rituximab. Clin. Cancer Res. 13, 2392–2399.
Berger, R., Rotem-Yehudar, R., Slama, G., Landes, S., Kneller, A., Leiba, M., Koren-Michowitz, M., Shimoni, A., and Nagler, A. (2008). Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin. Cancer Res. 14, 3044–3051.
Betting, D. J., Kafi, K., Abdollahi-Fard, A., Hurvitz, S. A., and Timmerman, J. M. (2008). Sulfhydryl-based tumor antigen-carrier protein conjugates stimulate superior antitumor immunity against B cell lymphomas. J. Immunol. 181, 4131–4140.
Betting, D. J., Mu, X. Y., Kafi, K., McDonnel, D., Rosas, F., Gold, D. P., and Timmerman, J. M. (2009). Enhanced immune stimulation by a therapeutic lymphoma tumor antigen vaccine produced in insect cells involves mannose receptor targeting to antigen presenting cells. Vaccine 27, 250–259.
Beum, P. V., Kennedy, A. D., Williams, M. E., Lindorfer, M. A., and Taylor, R. P. (2006). The shaving reaction: rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia by THP-1 monocytes. J. Immunol. 176, 2600–2609.
Biagi, E., Rousseau, R., Yvon, E., Schwartz, M., Dotti, G., Foster, A., Havlik-Cooper, D., Grilley, B., Gee, A., Baker, K., Carrum, G., Rice, L., Andreef, M., Popat, U., and Brenner, M. (2005). Responses to human CD40 ligand/human interleukin-2 autologous cell vaccine in patients with B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 11, 6916–6923.
Biragyn, A., Tani, K., Grimm, M. C., Weeks, S., and Kwak, L. W. (1999). Genetic fusion of chemokines to a self tumor antigen induces protective, T-cell dependent antitumor immunity. Nat. Biotechnol. 17, 253–258.
Boross, P., Jansen, J. H., de Haij, S., Beurskens, F. J., van der Poel, C. E., Bevaart, L., Nederend, M., Golay, J., van de Winkel, J. G., Parren, P. W., and Leusen, J. H. (2011). The in vivo mechanism of action of CD20 monoclonal antibodies depends on local tumor burden. Haematologica 96, 1822–1830.
Bowels, J. A., Wang, S. Y., Link, B. K., Allan, B., Beuerlein, G., Campbell, M. A., Marquis, D., Ondek, B., Wooldridge, J. E., Smith, B. J., Breitmeyer, J. B., and Weiner, G. J. (2006). Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood 108, 2548–2654.
Brody, J. D., Ai, W. Z., Czerwinski, D. K., Torchia, J. A., Levy, M., Advani, R. H., Kim, Y. H., Hoppe, R. T., Knox, S. J., Shin, L. K., Wapnir, I., Tibshirani, R. J., and Levy, R. (2010). In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J. Clin. Oncol. 28, 4324–4332.
Byrd, J. C., Kitada, S., Flinn, I. W., Aron, J. L., Pearson, M., Lucas, D., and Reed, J. C. (2002). The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood 99, 1038–1043.
Cartron, G., Dacheux, L., Salles, G., Solal-Celigny, P., Bardos, P., Colombat, P., and Waiter, H. (2002). Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99, 754–758.
Chan, H. T., Hughes, D., French, R. R., Tutt, A. L., Walshe, C. A., Teeling, J. L., Glennie, M. J., and Cragg, M. S. (2003). CD20-induced lymphoma cell death is independent of both caspase activity and its redistribution into TritonX-100 insoluble membrane rafts. Cancer Res. 63, 5480–5489.
Chen, C. G., Lu, Y. T., Savelyeva, N., Stevenson, F. K., and Zhu, D. (2009). Amplification of immune responses against a DNA-delivered idiotype lymphoma antigen by fusion to the B subunit of E. coli heat labile toxin. Vaccine 27, 4289–4296.
Coiffier, B., Lepretre, S., Pedersen, L. M., Gadeberg, O., Fredriksen, H., van Oers, M. H., Wooldridge, J., Kloczko, J., Holowiecki, J., Hellmann, A., Walewsk, J., Flensburg, M., Petersen, J., and Robak, T. (2008). Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell lymphocytic leukemia: a phase 1-2 study. Blood 111, 1094–1100.
Cragg, M. S., Morgan, S. M., Chan, H. T., Morgan, B. P., Filatov, A. V., Johnson, P. W., French, R. R., and Glennie, M. J. (2003). Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood 101, 1045–1052.
Czuczman, M. S., Weaver, R., Alkuzweny, B., Berlfein, J., and Grillo-López, A. J. (2004). Prolonged clinical and molecular remission in patients with low-grade or follicular non-Hodgkin’s lymphoma treated with rituximab plus CHOP chemotherapy: 9-year follow-up. J. Clin. Oncol. 22, 4711–4716.
Dang, N. H., Smith, M. R., Offner, F., Verhoef, G., Johnson, P., Rohatiner, A. Z., Kaufman, J. L., Gine, E., Coiffier, B., Czuczman, M. S., Foran, J., Durrant, S., Hua, S., Vandendries, E. R., and Fayad, L. (2009). Anti-CD22 immunoconjugate inotuzumab ozogamicin (CMC-544)+rituximab: clinical activity including survival in patients with recurrent/refractory follicular or “aggressive” lymphoma. Blood 114, abstr. 584.
Davis, T. A., Czerwinski, D. K., and Levy, R. (1999). Therapy of B-cell lymphoma with anti-CD20 antibodies can result in the loss of CD20 antigen expression. Clin. Cancer Res. 5, 611–615.
Davis, T. A., Hsu, F. J., Caspar, C. B., van Beckhoven, A., Czerwinsk, D. K., Liles, T. M., Taidi, B., Benike, C. J., Engleman, E. G., and Levy, R. (2001). Idiotype vaccination following ABMT can stimulate specific anti-idiotype immune responses in patients with B-cell lymphoma. Biol. Blood Marrow Transplant. 7, 517–522.
de Haij, S., Jansen, J. H., Boross, P., Beurskens, F. J., Bakema, J. E., Bos, D. L., Martens, A., Verbeek, J. S., Parren, P. W., van de Winkel, J. G., and Leusen, J. H. (2010). In vivo cytotoxicity of type I CD20 antibodies critically depends on Fc receptor ITAM signaling. Cancer Res. 70, 3209–3217.
de Vos, S., Goy, A., Dakhil, S. R., Saleh, M. N., McLaughlin, P., Belt, R., Flowers, C. R., Knapp, M., Hart, L., Patel-Donnelly, D., Glenn, M., Gregory, S. A., Holladay, C., Zhang, T., and Boral, A. L. (2009). Multicenter randomized phase II study of weekly or twice-weekly bortezomib plus rituximab in patients with relapsed or refractory follicular or marginal-zone B-cell lymphoma. J. Clin. Oncol. 27, 5023–5030.
Di Gaetano, N., Cittera, E., Nota, R., Vecchi, A., Grieco, V., Scanziani, E., Botto, M., Introna, M., and Golay, J. (2003). Complement activation determines the therapeutic activity of rituximab in vivo. J. Immunol. 171, 1581–1587.
Di Nicola, M., Zappasodi, R., Carlo-Stella, C., Mortarini, R., Pupa, S. M., Magn, M., Devizzi, L., Matteucc, P., Baldassari, P., Ravagnani, F., Cabras, A., Anichini, A., and Gianni, A. M. (2009). Vaccination with autologous tumor-loaded dendritic cells induces clinical and immunologic responses in indolent B-cell lymphoma patients with relapsed and measurable disease: a pilot study. Blood 113, 18–27.
Farag, S. S., Flinn, I. W., Modali, R., Lehman, T. A., Young, D., and Byrd, J. C. (2004). Fc gamma RIIIa and Fc gamma RIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood 103, 1472–1474.
Fisher, R. I., LeBlanc, M., Press, O. W., Maloney, D. G., Unger, J. M., and Miller, T. P. (2005a). New treatment options have changed the survival of patients with follicular lymphoma. J. Clin. Oncol. 23, 8447–8452.
Fisher, R. I., Kaminski, M. S., Wahl, R. L., Knox, S. J., Zelenetz, A. D., Vose, J. M., Leonard, J. P., Kroll, S., Goldsmith, S. J., and Coleman, M. (2005b). Tositumomab and iodine-131 tositumomab produces durable complete remissions in a subset of heavily pretreated patients with low-grade and transformed non-Hodgkin’s lymphomas. J. Clin. Oncol. 23, 7565–7573.
Freedman, A., Neelapu, S. S., Nichols, C., Robertson, M. J., Djulbegovic, B., Winter, J. N., Bender, J. F., Gold, D. P., Ghalie, R. G., Stewart, M. E., Esquibel, V., and Hamlin, P. (2009). Placebo-controlled phase III trial of patient-specific immunotherapy with mitumprotimut-T and granulocyte-macrophage colony-stimulating factor after rituximab in patients with follicular lymphoma. J. Clin. Oncol. 27, 3036–3043.
Friedberg, J. W., Kelly, J. L., Neuberg, D., Peterson, D. R., Kutok, J. L., Salloum, R., Brenn, T., Fisher, D. C., Ronan, E., Dalton, V., Rich, L., Marquis, D., Sims, P., Rothberg, P. G., Liesveld, J., Fisher, R. I., Coffman, R., Mosmann, T., and Freedman, A. S. (2009a). Phase II study of a TLR-9 agonist (1018 ISS) with rituximab in patients with relapsed or refractory follicular lymphoma. Br. J. Haematol. 146, 282–291.
Friedberg, J. W., Vose, J. M., Kahl, B. S., Brunvand, M. W., Goy, A., Kasamon, I. L., Burington, B., Li, J., Ho, W., and Cheson, B. D. (2009b). A phase I study of PRO131921, a novel anti-CD20 monoclonal antibody in patients with relapsed/refractory CD20+indolent NHL: correlation between clinical responses and AUC pharmacokinetics. Blood 114, abstr. 3742.
Fubin, L., and Ravetch, J. V. (2011). Inhibitory Fcγ receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 333, 1030–1034.
Gadri, Z., Kukulansky, T., Bar-Or, E., Haimovich, H., and Hollander, N. (2009). Synergistic effect of dendritic cell vaccination and anti-CD20 antibody treatment in the therapy of murine lymphoma. J. Immunother. 32, 333–340.
Glennie, M. J., French, R. R., Cragg, M. S., and Taylor, R. P. (2007). Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol. Immunol. 44, 3823–3837.
Golay, J., Cittera, E., Di Gaetano, N., Manganini, M., Mosca, M., Nebuloni, M., van Rooijen, N., Vago, L., and Introna, M. (2006). The role of complement in the therapeutic activity of rituximab in a murine B lymphoma model homing in lymph nodes. Haematologica 91, 176–183.
Goldenberg, D. M., Rossi, E. A., Stein, R., Cardillo, T. M., Czuczman, M. S., Hernandez-Ilazaliturri, F. J., Hansen, H. J., and Chang, C. H. (2009). Properties and structure-function relationships of veltuzumab (hA20), a humanized anti-CD20 monoclonal antibody. Blood 113, 1062–1070.
Gong, Q., Ou, Q., Ye, S., Lee, W. P., Cornelius, J., Diehl, L., Lin, W. Y., Hu, Z., Lu, Y., Chen, Y., Wu, Y., Meng, Y. G., Gribling, P., Lin, Z., Nguyen, K., Tran, T., Zhang, Y., Rosen, H., Martin, F., and Chan, A. C. (2005). Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 174, 817–826.
Gruber, M. M., Fleiss, K., Porpaczy, E., Skrabs, C., Hauswirth, A. W., Gaiger, A., Vanura, K., Heintel, D., Shehata, M., Einberger, C., Thalhamme, R., Fonatsch, C., and Jäger, U. (2011). Prolonged progression-free survival in patients with chronic lymphocytic leukemia receiving granulocyte colony-stimulating factor during treatment with fludarabine, cyclophosphamide, and rituximab. Ann. Hematol. 90, 1131–1136.
Hagenbeek, A., Fayad, L., Delwail, V., Rossi, J. F., Jacobson, E., Kuliczkowski, K., Link, B. K., Pinter-Brown, L., Radford, J., Hellmann, A., Gallop-Evans, E., Gupta, I., Arning, M., Begtrup, K., Schultz, M., Bang, B., Russell, C. A., and Czuczman, M. S. (2009). Evaluation of ofatumumab, a novel human CD20 monoclonal antibody, as single agent therapy in rituximab-refractory follicular lymphoma. Blood 114, abstr. 935.
Hagenbeek, A., Gadeberg, O., Johnson, P., Pedersen, L. M., Walewski, J., Hellmann, A., Link, B. K., Robak, T., Wojtukiewicz, M., Pfreundschuh, M., Kneba, M., Engert, A., Sonneveld, P., Flensburg, M., Petersen, J., Losic, N., and Radford, J. (2008). First clinical use of ofatumumab, a novel fully human anti-CD20 monoclonal antibody in relapsed or refractory follicular lymphoma: results of a phase 1/2 trial. Blood 111, 5486–5495.
Hainsworth, J. D., Litchy, S., Burris, H. A. III, Scullin, D. C. Jr., Corso, S. W., Yardley, D. A., Morrissey, L., and Greco, F. A. (2002). Rituximab as first-line and maintenance therapy for patients with indolent non-Hodgkin’s lymphoma. J. Clin. Oncol. 20, 4261–4267.
Hamaguchi, Y., Uchida, J., Chain, D. W., Venture, G. M., Poe, J. C., Haas, K. M., and Tedder, T. F. (2005). The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J. Immunol. 174, 4389–4399.
Hiddemann, W., Kneba, M., Dreyling, M., Schmitz, N., Lengfelder, E., Schmits, R., Reiser, M., Metzner, B., Harder, H., Hegewisch-Becker, S., Fischer, T., Kropff, M., Reis, H. E., Freund, M., Wörmann, B., Fuchs, R., Planker, M., Schimke, J., Eimermacher, H., Trümper, L., Aldaoud, A., Parwaresch, R., and Unterhalt, M. (2005). Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 106, 3725–3732.
Hilchey, S. P., Hyrien, O., Mosmann, T. R., Livingstone, A. M., Friedberg, J. W., Young, F., Fisher, R. I., Kelleher, R. J. Jr., Bankert, R. B., and Bernstein, S. H. (2009). Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for “vaccine effect” of rituximab. Blood 113, 3809–3812.
Hillmen, P., Skotnicki, A. B., Robak, T., Jaksic, B., Dmoszynska, A., Wu, J., Sirard, C., and Mayer, J. (2007). Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J. Clin. Oncol. 25, 5616–5623.
Hofmeister, J. K., Cooney, D., and Coggeshall, K. M. (2000). Clustered CD20 induced apoptosis: src-family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3-dependent apoptosis. Blood Cells Mol. Dis. 26, 133–143.
Hollander, N. (2009). Current vaccination strategies for the treatment of B-cell lymphoma and multiple myeloma. Crit. Rev. Immunol. 29, 399–418.
Horikawa, M., Minard-Colin, V., Matsushita, T., and Tedder, T. F. (2011). Regulatory B-cell production of IL-10 inhibits lymphoma depletion during CD20 immunotherapy in mice. J. Clin. Invest. 121, 4268–4280.
Hsu, F. J., Benike, C., Fagnoni, F., Liles, T. M., Czerwinski, D., Taidi, B., Engleman, E. G., and Levy, R. (1996). Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat. Med. 2, 52–58.
Inoges, S., Rodriguez-Calvillo, M., Zabalegui, N., Lopez-Díaz de Cerio, A., Villanueva, H., Soria, E., Suarez, L., Rodríguez-Caballero, A., Pastor, F., García-Munoz, R., Panizo, C., Perez-Calvo, J., Melero, I., Rocha, E., Orfao, A., and Bendandi, M. (2006). Clinical benefit associated with idiotypic vaccination in patients with follicular lymphoma. J. Natl. Cancer Inst. 98, 1292–1301.
Kaminski, M. S., Tuck, M., Estes, J., Kolstad, A., Ross, C. W., Zasadny, K., Regan, D., Kison, P., Fisher, S., Kroll, S., and Wahl, R. L. (2005). 131I-tositumomab therapy as initial treatment for follicular lymphoma. N. Engl. J. Med. 352, 441–449.
Kennedy, A. D., Beum, P. V., Solga, M. D., DiLillo, D. J., Lindorfer, M. A., Hess, C. E., Densmore, J. J., Williams, M. E., and Taylor, R. P. (2004). Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J. Immunol. 172, 3280–3288.
Khan, K. D., Emmanouilides, C., Benson, D. M. Jr., Hurst, D., Garcia, P., Michelson, G., Milan, S., Ferketich, A. K., Piro, L., Leonard, J. P., Porcu, P., Eisenbeis, C. F., Banks, A. L., Chen, L., Byrd, J. C., and Caligiuri, M. A. (2006). A phase 2 study of rituximab in combination with recombinant interleukin-2 for rituximab-refractory indolent non-Hodgkin’s lymphoma. Clin. Cancer Res. 12, 7046–7053.
King, C. A., Spellerberg, M. B., Zhu, D., Rice, J., Sahota, S. S., Thompsett, A. R., Hamblin, T. J., Radl, J., and Stevenson, F. K. (1998). DNA vaccines with single-chain Fv fused to fragment C of tetanus toxin induce protective immunity against lymphoma and myeloma. Nat. Med. 4, 1281–1286.
Klepfish, A., Rachmilewitz, E. A., Kotsianidis, I., Patchenko, P., and Schattner, A. (2008). Adding fresh frozen plasma to rituximab for the treatment of patients with refractory advanced CLL. QJM 101, 737–740.
Koene, H. R., Kleijer, M., Algra, J., Roos, D., von Dem Borne, A. E., and de Haas, M. (1997). Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood 90, 1109–1114.
Kuzel, T. M., Li, S., Eklund, J., Foss, F., Gascoyne, R., Abramson, N., Schwerkoske, J. F., Weller, E., and Horning, S. J. (2007). Phase II study of denileukin diftitox for previously treated indolent non-Hodgkin lymphoma: final results of E1497. Leuk. Lymphoma 48, 2397–2402.
Leonard, J. P., Coleman, M., Ketas, J. C., Chadburn, A., Ely, S., Furman, R. R., Wegener, W. A., Hansen, H. J., Ziccardi, H., Eschenberg, M., Gayko, U., Cesano, A., and Goldenberg, D. M. (2003). Phase I/II trial of epratuzumab (humanized anti-CD22 antibody) in indolent non-Hodgkin’s lymphoma. J. Clin. Oncol. 21, 3051–3059.
Levy, R., and Dilley, J. (1978). Rescue of immunoglobulin secretion from human neoplastic lymphoid cells by somatic cell hybridization. Proc. Natl. Acad. Sci. U.S.A. 75, 2411–2415.
Levy, R., Robertson, M. J., Ganjoo, K., Leonard, J., Vose, J., and Denney, D. (2008). Results of a phase 3 trial evaluating safety and efficacy of specific immunotherapy, recombinant idiotype (Id) conjugated to KLH (Id-KLH) with GM-CSF, compared to non-specific immunotherapy, KLH with GM-CSF, in patients with follicular non-Hodgkin’s lymphoma. American Association for Cancer Research Meeting Abstract, San Diego, LB-204.
Li, J., Song, W., Czerwinski, D. K., Varghese, B., Uematsu, S., Akira, S., Krieg, A. M., and Levy, R. (2007). Lymphoma immunotherapy with CpG oligodeoxynucleotides requires TLR9 either in the host or in the tumor itself. J. Immunol. 179, 2493–2500.
Liu, H. M., Newbrough, S. E., Bhatia, S. K., Dahle, C. E., Krieg, A. M., and Weiner, G. J. (1998). Immunostimulatory CpG oligodeoxynucleotides enhance the immune response to vaccine strategies involving granulocyte-macrophage colony-stimulating factor. Blood 92, 3730–3736.
Macor, P., Tripodo, C., Zorzet, S., Piovan, E., Bossi, F., Marzari, R., Amadori, A., and Tedesco, F. (2007). In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increase the antitumor activity of rituximab. Cancer Res. 67, 10556–19563.
Masarwa, M., Donin, N., Ziporen, L., and Fishelson, Z. (2008). Silencing of the mitochondrial hsp70 mortalin as an adjuvant cancer therapy. Mol. Immunol. 45, 4104–4105.
Micallef, I. N., Maurer, M. J., Wiseman, G. A., Nikcevich, D. A., Kurtin, P. J., Cannon, M. W., Perez, D. G., Soori, G. S., Link, B. K., Habermann, T. M., and Witzig, T. E. (2011). Epratuzumab with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy in patients with previously untreated diffuse large B-cell lymphoma. Blood 118, 4053–4061.
Miller, R. A., Maloney, D. G., Warnke, R., and Levy, R. (1982). Treatment of B-cell lymphoma with monoclonal anti-idiotype antibody. N. Engl. J. Med. 306, 517–522.
Minard-Colin, V., Xiu, Y., Poe, J. C., Horikawa, M., Magro, C. M., Hamaguchi, Y., Haas, K. M., and Tedder, T. F. (2008). Lymphocyte depletion during CD20 immunotherapy in mice is mediated by macrophage FcgammaRI, FcgammaRIII, and FcgammaRIV. Blood 112, 1205–1213.
Morschhauser, F., Leonard, J. P., Fayad, L., Coiffier, B., Petillon, M. O., Coleman, M., Schuster, S. J., Dyer, M. J., Horne, H., Teoh, N., Wegener, W. A., and Goldenberg, D. M. (2009). Humanized anti-CD20 antibody veltuzumab in refractory/recurrent non-Hodgkin’s lymphoma: phase I/II results. J. Clin. Oncol. 27, 3346–3353.
Morschhauser, F., Marlton, P., Vitolo, U., Lindén, O., Seymour, J. F., Crump, M., Coiffier, B., Foà, R., Wassne, E., Burger, H. U., Brennan, B., and Mendila, M. (2010a). Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann. Oncol. 21, 1870–1876.
Morschhauser, F., Kraeber-Bodéré, F., Wegener, W. A., Harousseau, J. L., Petillon, M. O., Huglo, D., Trümper, L. H., Meller, J., Pfreundschuh, M., Kirsch, C. M., Naumann, R., Kropp, J., Horne, H., Teoh, N., Le Gouill, S., Bodet-Milin, C., Chatal, J. F., and Goldenberg, D. M. (2010b). High rates of durable responses with anti-CD22 fractionated radioimmunotherapy: results of a multicenter, phase I/II study in non-Hodgkin’s lymphoma. J. Clin. Oncol. 28, 3709–3716.
Morschhauser, F., Radford, J., Van Hoof, A., Vitolo, U., Soubeyran, P., Tilly, H., Huijgens, P. C., Kolstad, A., d’Amore, F., Gonzalez Diaz, M., Petrini, M., Sebban, C., Zinzani, P. L., van Oers, M. H., van Putten, W., Bischof-Delaloye, A., Rohatiner, A., Salles, G., Kuhlmann, J., and Hagenbeek, A. (2008). Phase III trial of consolidation therapy with yttrium-90-ibritumomab tiuxetan compared with no additional therapy after first remission in advanced follicular lymphoma. J. Clin. Oncol. 26, 5156–5164.
Mössner, E., Brünker, P., Moser, S., Püntener, U., Schmidt, C., Herter, S., Grau, R., Gerdes, C., Nopora, A., van Puijenbroek, E., Ferrara, C., Sondermann, P., Jäger, C., Strein, P., Fertig, G., Friess, T., Schüll, C., Bauer, S., Dal Porto, J., Del Nagro, C., Dabbagh, K., Dyer, M. J., Poppema, S., Klein, C., and Umaña, P. (2010). Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 115, 4393–4402.
Neelapu, S. S., Baskar, S., Gause, B. L., Kobrin, C. B., Watson, T. M., Frye, A. R., Pennington, R., Harvey, L., Jaffe, E. S., Robb, R. J., Popescu, M. C., and Kwak, L. W. (2004). Human autologous tumor-specific T-cell responses induced by liposomal delivery of a lymphoma antigen. Clin. Cancer Res. 10, 8309–8317.
Neelapu, S. S., Kwak, L. W., Kobrin, C. B., Reynolds, C. W., Janik, J. E., Dunleavy, K., Whit, T., Harvey, L., Pennington, R., Stetler-Stevenson, M., Jaffe, E. S., Steinberg, S. M., Gress, R., Hakim, F., and Wilson, W. H. (2005). Vaccine induced tumor-specific immunity despite severe B-cell depletion in mantle cell lymphoma. Nat. Med. 11, 986–991.
Neelson, P., Pan, Z. K., and Paterson, Y. (2008). Listeriolysin O is an improved protein carrier for lymphoma immunoglobulin idiotype and provides systemic protection against 38C13 lymphoma. Cancer Immunol. Immunother. 57, 493–505.
Olejniczak, S. H., Hernandez-Ilizaliturri, F. H., Clements, J. L., and Czuczman, M. S. (2008). Acquired resistance to rituximab is associated with chemotherapy resistance resulting from decreased Bax and Bak expression. Clin. Cancer Res. 14, 1550–1560.
O’Mahony, D., Morris, J. C., Quinn, C., Gao, W., Wilson, W. H., Gause, B., Pittaluga, S., Neelapu, S., Brown, M., Fleisher, T. A., Gulley, J. L., Schlom, J., Nussenblatt, R., Albert, P., Davis, T. A., Lowy, I., Petrus, M., Waldmann, T. A., and Janik, J. E. (2007). A pilot study of CTLA-4 blockade after cancer vaccine failure in patients with advanced malignancy. Clin. Cancer Res. 13, 958–964.
Park, H. J., and Neelapu, S. S. (2008). Developing idiotype vaccines for lymphoma: from preclinical studies to phase III clinical trials. Br. J. Haematol. 142, 179–191.
Peggs, K. S., Quezada, S. A., and Allison, J. P. (2008). Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol. Rev. 224, 141–165.
Press, O. W., Unger, J. M., Braziel, R. M., Maloney, D. G., Miller, T. P., Leblanc, M., Fisher, R. I., and Southwest Oncology Group. (2006). Phase II trial of CHOP chemotherapy followed by tositumomab/iodine I-131tositumomabfor previously untreated follicular non-Hodgkin’s lymphoma: five year follow-up of Southwest Oncology Group Protocol S9911. J. Clin. Oncol. 24, 4143–4149.
Pro, B., Leber, B., Smith, M., Fayad, L., Romaguera, J. E., Hagemeister, F. B., Rodriguez, M. A., McLaughlin, P., Samaniego, F., Zwiebel, J., Lopez, A., Kwak, L. W., and Younes, A. (2008). Phase II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in combination with rituximab in patients with recurrent B-cell non-Hodgkin lymphoma. Br. J. Haematol. 143, 355–360.
Racila, E., Link, B. K., Weng, W. K., Witzig, T. E., Ansell, S., Maurer, M. J., Huang, J., Dahle, C., Halwani, A., Levy, R., and Weiner, G. J. (2008). A polymorphism in the complement component C1q correlates with prolonged response following rituximab therapy of follicular lymphoma. Clin. Cancer Res. 14, 6697–6703.
Redfern, C. H., Guthrie, T. H., Bessudo, A., Densmor, J. J., Holman, P. R., Janakiraman, N., Leonard, J. P., Levy, R. L., Just, R. G., Smith, M. R., Rosenfelt, F. P., Wiernik, P. H., Carter, W. D., Gold, D. P., Melink, T. J., Gutheil, J. C., and Bender, J. F. (2006). Phase II clinical trial of idiotype vaccination in previously treated patients with indolent non-Hodgkin’s lymphoma resulting in durable clinical responses. J. Clin. Oncol. 24, 3107–3112.
Rezvani, A. R., and Maloney, D. G. (2011). Rituximab resistance. Best Pract. Res. Clin. Haematol. 24, 203–216.
Rossi, E. A., Goldenberg, D. M., Cardillo, T. M., Stein, R., and Chang, C. H. (2009). Hexavalent bispecific antibodies represent a new class of anticancer therapeutics: 1. Properties of anti-CD20/CD22 antibodies in lymphoma. Blood 113, 6161–6171.
Salles, G. A., Morschhauser, F., Cartron, G., Lamy, T., Milpied, N. J., Thieblemont, C., Tilly, H., Birkett, J., and Burgess, M. (2008). A phase I/II study of RO5072759 (GA101) in patients with relapsed/refractory CD20+ malignant disease. Blood 112, abstr. 234.
Schuster, S. J., Neelapu, S. S., Gause, B. L., Janik, J. E., Muggia, F. M., Gockerman, J. P., Winter, J. N., Flowers, C. R., Nikcevich, D. A., Sotomayor, E. M., McGaughey, D. S., Jaffe, E. S., Chong, E. A., Reynolds, C. W., Berry, D. A., Santos, C. F., Popa, M. A., McCord, A. M., and Kwak, L. W. (2011). Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J. Clin. Oncol. 29, 2787–2794.
Selenko, N., Majdic, O., Draxier, S., Berer, A., Jager, U., Knapp, W., and Stockl, J. (2001). CD20 antibody (C2B8)-induced apoptosis of lymphoma cells promote phagocytosis by dendritic cells and cross-priming of CD8+ cytotoxic T cells. Leukemia 15, 1619–1629.
Shan, D., Ledbetter, J. A., and Press, O. W. (2000). Signaling events involved in anti-CD20-induced apoptosis of malignant human B cells. Cancer Immunol. Immunother. 48, 673–683.
Stevenson, F. K., Mander, A., Chudley, L., and Ottensmeier, C. H. (2011). DNA fusion vaccines enter the clinic. Cancer Immunol. Immunother. 60, 1147–1151.
Strauss, S. J., Morschhauser, F., Rech, J., Repp, R., Solal-Celigny, P., Zinzani, P. L., Engert, A., Coiffier, B., Hoelzer, D. F., Wegener, W. A., Teoh, N. K., Goldenberg, D. M., and Lister, T. A. (2006). Multicenter phase II trial of immunotherapy with humanized anti-CD22 antibody, epratuzumab, in combination with rituximab, in refractory or recurrent non-Hodgkin’s lymphoma. J. Clin. Oncol. 24, 3880–3886.
Syrengelas, A. D., Chen, T. T., and Levy, R. (1996). DNA immunization induces protective immunity against B-cell lymphoma. Nat. Med. 2, 1038–1041.
Takei, K., Tamazaki, T., Sawada, U., Ishizuka, H., and Aizawa, S. (2006). Analysis of changes in CD20, CD55, and CD59 expression on established rituximab-resistant B-lymphoma cell line. Leuk. Res. 30, 625–631.
Teeling, J. L., French, R. R., Cragg, M. S., van den Brake, J., Pluyter, M., Huang, H., Chan, C., Parren, P. W., Hack, C. E., Dechant, M., Valerius, T., van de Winkel, J. G., and Glennie, M. J. (2004). Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood 104, 1793–1800.
Teeling, J. L., Mackus, W. J., Wiegman, L. J., van den Brakel, J. H., Beers, S. A., French, R. R., van Meerten, T., Ebeling, S., Vink, T., Slootstra, J. W., Parren, P. W., Glennie, M. J., and van de Winkel, J. G. (2006). The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J. Immunol. 177, 362–371.
Terui, Y., Sakurai, T., Mishima, Y., Mishima, Y., Sugimura, N., Sasaoka, C., Kojima, K., Yokoyama, M., Mizunuma, N., Takahashi, S., Ito, Y., and Hatake, K. (2006). Blockade of bulky lymphoma-associated CD55 expression by RNA interference overcomes resistance to complement-dependent cytotoxicity with rituximab. Cancer Sci. 97, 72–79.
Timmerman, J. M., Czerwinski, D. K., Davis, T. A., Hsu, F. J., Benike, C., Hao, Z. M., Taidi, B., Rajapaksa, R., Caspar, C. B., Okada, C. Y., van Beckhoven, A., Liles, T. M., Engleman, E. G., and Levy, R. (2002). Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood 99, 1517–1526.
Timmerman, J. M., Vose, J. M., Czerwinski, D. K., Weng, W. K., Ingolia, D., Mayo, M., Denney, D. W., and Levy, R. (2009). Tumor-specific recombinant idiotype immunization after chemotherapy as initial treatment for follicular non-Hodgkin lymphoma. Leuk. Lymphoma 50, 37–46.
Tobinai, K., Ogura, M., Kobayashi, Y., Uchida, T., Watanabe, T., Oyama, T., Maruyama, D., Suzuki, T., Mori, M., Kasai, M., Cronier, D., Wooldridge, J. E., and Koshiji, M. (2011). Phase I study of LY2469298, an Fc-engineered humanized anti-CD20 antibody, in patients with relapsed or refractory follicular lymphoma. Cancer Sci. 102, 432–438.
Uchida, J., Hamaguchi, Y., Oliver, J. A., Ravetch, J. V., Poe, J. C., Haas, K. M., and Tedder, T. F. (2004). The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 199, 1659–1669.
van der Kolk, L. E., Grillo-Lopez, A. J., Baars, J. W., Hack, C. E., and van Oers, M. H. (2001). Complement activation plays a key role in the side-effects of rituximab treatment. Br. J. Haematol. 115, 807–811.
van der Kolk, L. E., Grillo-Lopez, A. J., Baars, J. W., and van Oers, M. H. (2003). Treatment of relapsed non-Hodgkin’s lymphoma with a combination of chimeric anti-CD20 monoclonal antibodies (rituximab) and G-CSF: final report of safety and efficacy. Leukemia 17, 1658–1664.
van Meerten, T., and Hagenbeek, A. (2011). Novel antibodies against follicular non-Hodgkin’s lymphoma. Best Pract. Res. Clin. Haematol. 24, 231–256.
Viardot, A., Goebeler, M., Scheele, J. S., Zugmaier, G., Noppeney, R., Knop, S., Topp, M. S., Nagorsen, D., Klinger, M., Schmidt, M., Klappers, P., Kufer, P., Einsele, H., and Bargou, R. (2010). Treatment of patients with non-Hodgkin lymphoma (NHL) with CD19/CD3 bispecific antibody Blinatumomab (MT103): double-step dose increase to continuous infusion of 60 μg/m2/d is tolerable and highly effective. Blood 116, abstr. 2880.
Wang, S. Y., Racila, E., Taylor, R. P., and Weiner, G. J. (2008). NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood 111, 1456–1463.
Weiner, G. J. (2009). CpG oligodeoxynucleotide-based therapy of lymphoid malignancies. Adv. Drug Deliv. Rev. 61, 263–267.
Weng, W. K., and Levy, R. (2003). Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 21, 3940–3947.
Wierda, W. G., Cantwell, M. J., Woods, S. J., Rassenti, L. Z., Prussak, C. E., and Kipps, T. J. (2000). CD40-ligand (CD154) gene therapy for chronic lymphocytic leukemia. Blood 96, 2917–2924.
Wierda, W. G., Kipps, T. J., Mayer, J., Stilgenbauer, S., Williams, C. D., Hellmann, A., Robak, T., Furman, R. R., Hillmen, P., Trneny, M., Dyer, M. J., Padmanabhan, S., Piotrowska, M., Kozak, T., Chan, G., Davis, R., Losic, N., Wilms, J., Russell, C. A., and Osterborg, A. (2010a). Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J. Clin. Oncol. 28, 1749–1755.
Wierda, W. G., Castro, J. E., Aguillon, R., Sampath, D., Jalayer, A., McMannis, J., Prussak, C. E., Keating, M., and Kipps, T. J. (2010b). A phase I study of immune gene therapy for patients with CLL using a membrane-stable, humanized CD154. Leukemia 24, 1893–1900.
Wierda, W. G., Padmanabhan, S., Chan, G. W., Gupta, I. V., Lisby, S., and Osterborg, A. (2011). Ofatumumab is active in patients with fludarabine-refractory CLL irrespective of prior rituximab: results from the phase II international study. Blood 118, 5126–5129.
Wilke, C. M., Wu, K., Zhao, E., Wang, G., and Zou, W. (2010). Prognostic significance of regulatory T cells in tumor. Int. J. Cancer 127, 748–758.
Witzig, T. E., Molina, A., Gordon, L. I., Emmanouilides, C., Schilder, R. J., Flinn, I. W., Darif, M., Macklis, R., Vo, K., and Wiseman, G. A. (2007). Long-term responses in patients with recurring or refractory B-cell non-Hodgkin lymphoma treated with yttrium 90 ibritumomab tiuxetan. Cancer 109, 1804–1810.
Xu, W., Miao, K. R., Zhu, D. X., Fang, C., Zhu, H. Y., Dong, H. J., Wang, D. M., Wu, Y. J., Qiao, C., and Li, J. Y. (2010). Enhancing the action of rituximab by adding fresh frozen plasma for the treatment of fludarabine refractoy chronic lymphocytic leukemia. Int. J. Cancer 128, 2192–2201.
Yang, Z. Z., Novak, A. J., Stenson, M. J., Witzig, T. E., and Ansell, S. M. (2006a). Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood 107, 3639–3646.
Yang, Z. Z., Novak, A. J., Ziesmer, S. C., Witzig, T. E., and Ansell, S. M. (2006b). Attenuation of CD8 T-cell function by CD4CD25 regulatory T cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 66, 10145–10152.
Keywords: anti-CD20, clinical trials, idiotype, immunotherapy, lymphoma, monoclonal antibodies, vaccines
Citation: Hollander N (2012) Immunotherapy for B-Cell lymphoma: current status and prospective advances. Front. Immun. 3:3. doi: 10.3389/fimmu.2012.00003
Received: 24 November 2011;
Paper pending published: 19 December 2011;
Accepted: 05 January 2012;
Published online: 24 January 2012.
Edited by:
Lee Mark Wetzler, Boston University School of Medicine, USACopyright: © 2012 Hollander. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nurit Hollander, Department of Clinical Microbiology and Immunology, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel. e-mail: hollandn@post.tau.ac.il