 Martina M. McGrath Nader Najafian*
Martina M. McGrath Nader Najafian*- Transplantation Research Center, Brigham and Women’s Hospital and Children’s Hospital, Harvard Medical School, Boston, MA, USA
Negative costimulatory molecules, acting through so-called inhibitory pathways, play a crucial role in the control of T cell responses. This negative “second signal” opposes T cell receptor activation and leads to downregulation of T cell proliferation and promotes antigen specific tolerance. Much interest has focused upon these pathways in recent years as a method to control detrimental alloresponses and promote allograft tolerance. However, recent experimental data highlights the complexity of negative costimulatory pathways in alloimmunity. Varying effects are observed from molecules expressed on donor and recipient tissues and also depending upon the activation status of immune cells involved. There appears to be significant overlap and redundancy within these systems, rendering this a challenging area to understand and exploit therapeutically. In this article, we will review the literature at the current time regarding the major negative costimulation pathways including CTLA-4:B7, PD-1:PD-L1/PD-L2 and PD-L1:B7-1, B7-H3, B7-H4, HVEM:BTLA/CD160, and TIM-3:Galectin-9. We aim to outline the role of these pathways in alloimmunity and discuss their potential applications for tolerance induction in transplantation.
Introduction
CD4+ T cells are initially stimulated through the T cell receptor (TCR), by the recognition of antigen presented with major histocompatibility complex (MHC) Class II molecules. Full T cell activation requires a “second signal” or binding of a costimulatory molecule at the time of TCR ligation. The most important costimulatory molecule is CD28 and mice deficient in CD28 or one of its ligands (B7-1 or B7-2), display severely impaired CD4+ T cell proliferation. The absence of a “second signal” at the time of TCR ligation leads to the development of T cell anergy, a state characterized by decreased proliferative capacity, the inability to secrete IL-2 and reduced ability to undergo activation. While positive costimulatory signals promote T cell proliferation and differentiation into effector phenotypes, negative signals lead to arrest of T cell responses and promote regulation and tolerance. Therefore, T cell activation involves a delicate balance between positive and negative costimulatory signals.
Solid organ transplantation is a lifesaving therapy for patients with end-stage organ failure and while short-term allograft survival has improved dramatically, long-term outcomes remain disappointingly poor. Current immunosuppressive protocols are highly effective at suppressing acute rejection but are associated with significant morbidity with long-term use. To date, induction of donor specific tolerance has remained an elusive goal for all but the most aggressive therapeutic strategies, suitable only for a small subset of patients. Innate mechanisms of regulation and coinhibition are as yet incompletely understood but remain the focus of intensive research. In targeting our goal of inducing allospecific tolerance without global immunosuppression, the most promising strategies would appear to be those aimed at limiting positive costimulation, in combination with selective signaling through inhibitory pathways. However, there are a number of coinhibitory molecules and substantial overlap exists between these pathways. Therefore, successful induction of tolerance may require manipulation of more than one to overcome the inherent redundancy between them. To this end, a detailed understanding of the relative importance of each negative costimulatory pathway, their expression patterns, receptors and ligands, and the interactions between them is crucial to develop coherent strategies to control aggressive alloimmune responses.
Below, we discuss the current state of knowledge of the most widely studied negative costimulatory pathways (Figure 1) and their potential applications in alloimmunity. To provide some pathophysiological context and highlight the overlap between pathways observed in disease pathogenesis, we have provided examples of disease states in which each of these molecules has been implicated. The examples provided are intended to be illustrative only, as a complete description is beyond the scope of this text.
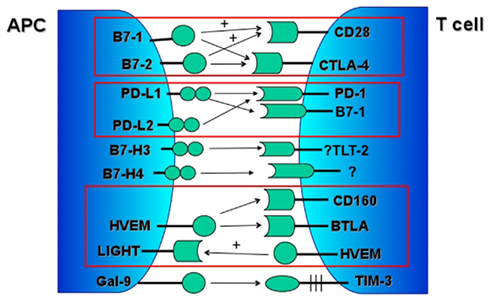
Figure 1. Major negative costimulatory pathways. Associated positive signaling pathways marked with “+.”
CTLA-4:B7-1/B7-2
Cytolytic T lymphocyte-associated antigen 4 (CTLA-4) is a member of the Ig superfamily and is closely structurally related to CD28, with which it shares approximately 30% homology. CTLA-4 is not expressed by naïve T cells but is rapidly upregulated on T cell activation and CD28 engagement (Walunas et al., 1994). It is highly expressed on regulatory T (Tregs) cells and is central to their suppressive function and the maintenance of peripheral tolerance (Wing et al., 2008).
Cytolytic T lymphocyte-associated antigen 4 is mainly located within intracellular vesicles (Mead et al., 2005). Upon TCR ligation, it is trafficked to the cell surface where it forms a homodimer. However, it appears that CTLA-4 continuously undergoes endocytosis, where cell surface CTLA-4 is rapidly re-internalized independent of ligand binding (Linsley et al., 1996). Its fate after being internalized remains incompletely understood, but it appears that some molecules are degraded in lysosomes and some are trafficked back to the cell surface (Linsley et al., 1996; Egen and Allison, 2002). CTLA-4 shares the same ligands as CD28; B7-1 (CD80) and B7-2 (CD86), but binds with 10- to 50-fold greater binding affinity.
B7-1 and B7-2 are both widely expressed on B cells, macrophages, dendritic cells (DCs) and T cells and are rarely found on non-lymphoid tissues. B7-2 is constitutively expressed on antigen presenting cells (APCs) at low levels and is rapidly upregulated, whereas B7-1 expression is induced, later than B7-2. While their function on APCs has been extensively studied, their role on T cells is incompletely understood. Recent studies have suggested that B7 on T cells may serve to down-regulate responses and deliver negative signals to T cells, through CTLA-4 ligation via T-T interaction (Taylor et al., 2004). B7 deficiency on T cells resulted in accelerated graft versus host disease (GVHD) in a model of allogeneic bone marrow transplantation (Taylor et al., 2004). Furthermore, B7 deficient T cells demonstrate resistance to suppression by Tregs in an in vivo colitis model (Paust et al., 2004). As discussed below, B7-1 has also been demonstrated to interact with PD-L1, producing a coinhibitory signal.
More recent data, using receptor array techniques, indicates that ICOS ligand, B7-H2, is also a costimulatory ligand for CD28, with a distinct binding site from ICOS. B7-H2 binds both CD28 and CTLA-4, albeit at a lower affinity than B7-1 or B7-2 (Yao et al., 2011). Furthermore, CD28 binds B7-H2 and B7-1/B7-2 through different interfaces, potentially allowing simultaneous binding of these ligands. Interestingly however, Abatacept, (CTLA-4-Ig), binds B7-H2 and also blocks the interaction between B7-H2-Ig and CD28, suggesting that CTLA-4 may have a greater affinity for B7-H2 than CD28 (Yao et al., 2011). In terms of function, B7-H2 binding to CD28 costimulates T cell proliferation and appears to play a central role in IFNγ production from memory T cells. While B7-H2 may act synergistically with B7-1 and B7-2 to deliver CD28-mediated costimulatory signals, the impact of B7-H2:CTLA-4 interaction remains largely unstudied. This link between the ICOS:B7-H2 positive costimulatory pathway and CTLA-4 is interesting as this could also potentially represent a regulatory mechanism to control ICOS-induced T cell activation, However, these data were acquired in vitro and therefore the true significance of these observations in vivo remains unknown.
Cytolytic T lymphocyte-associated antigen 4 ligation blocks T cell activation, inhibits CD28-dependent IL-2 production and inhibits cell cycle progression (Walunas et al., 1994, 1996). Despite a large body of literature, there remains considerable ongoing investigation into its exact mechanism of action. CTLA-4 mediated inhibition of T cell activation is currently thought to arise through both “cell intrinsic” and “cell extrinsic” mechanisms. Firstly, CTLA-4 acts as an antagonist of CD28 by competitively binding B7-1 and B7-2, thereby blocking positive costimulatory signaling. This hypothesis is consistent with the known greater affinity and avidity of CTLA-4 for these ligands. More recently it has been proposed that CTLA-4 expression may also increase T cell mobility and oppose the TCR induced “stop signal” needed for contact between T cells and APCs, thereby limiting the potential for T cell activation (Schneider et al., 2006). In addition, through binding B7-1 and B7-2, CTLA-4 blocks transmission of signals from the TCR by inhibiting the formation of ZAP-70 containing microclusters, leading to reduced calcium mobilization, which then limits T cell capacity for proliferation (Schneider et al., 2008). A splice variant of CTLA-4 has also been described. This variant lacks the extra-cellular ligand-binding domain and is proposed to constitutively generate a ligand-independent inhibitory signal (Vijayakrishnan et al., 2004). The importance of this splice variant in control of T effector cell responses is suggested by its increased expression in disease-resistant strains of NOD mice when compared to diabetes-susceptible congenic strains (Vijayakrishnan et al., 2004; Araki et al., 2009). However, this splice variant does not appear to be present in humans and therefore appears unlikely to represent a central mode of action of CTLA-4 in immunity.
In addition, it has been suggested that CTLA-4 exerts its effect through “cell extrinsic” mechanisms of immune suppression. A recent paper elegantly demonstrates the capacity of CTLA-4 to capture B7-2 and internalize it for degradation; leading to impaired T cell activation (Qureshi et al., 2011). This process was diminished through deletion of the cytoplasmic tail of CTLA-4 and through the use of blocking antibodies such as anti-CTLA-4, but not by blockade of CD28, demonstrating that this mechanism is specific to CTLA-4. Furthermore, while transendocytosis of B7-2 by CTLA-4 occurs constitutively, it is upregulated after TCR activation, providing an explanation for the increased Treg suppressive activity observed after T cell activation (Qureshi et al., 2011). Other cell extrinsic mechanisms of action for CTLA-4 have been proposed including induction of indoleamine 2,3-dioxygenase (IDO) activity, (thereby leading to localized tryptophan depletion and decreased T cell proliferation; Munn et al., 1999) and the presence of a soluble form of CTLA-4, which could locally affect T cell activation (Magistrelli et al., 1999). However, conflicting data exist for both of these mechanisms and their true significance and role in immunity remains unclear.
CTLA-4 in Disease States
Cytolytic T lymphocyte-associated antigen 4 holds a central role in the control of T cell responses, as evidenced by the fact that CTLA-4 deficient mice die at 3–4 weeks of age of uncontrolled lymphoproliferative disease (Tivol et al., 1995). In humans, CTLA-4 gene polymorphisms have been implicated in many autoimmune diseases including thyroid disease, type I diabetes mellitus, rheumatoid arthritis (RA) and systemic lupus erythematosis (Ueda et al., 2003). Dysregulation of intracellular expression of CTLA-4 has been proposed as a cause of the lymphoproliferative disorder, Chediak–Higashi syndrome (Barrat et al., 1999).
Impact of CTLA-4 on Treg Function
The central role of CTLA-4 on Treg function has been demonstrated in studies where administration of anti-CTLA-4 led to the development of organ specific autoimmunity in immune competent mice, without reducing total Treg numbers (Takahashi et al., 2000). Furthermore, Tregs from CTLA-4−/− mice exhibit weak suppressive activity in vitro. This function is independent of CD28 expression as Tregs from CD28 deficient mice show normal suppressive activity (Takahashi et al., 2000). Finally, other studies suggest that engagement of CTLA-4 may lead to the development of antigen specific Tregs, leading to inhibition of T cell responses in certain experimental models (Vasu et al., 2004).
Recent studies outline the role of reverse signaling through B7-1 and B7-2 in control of immune responses. Engagement of B7 on DCs by CTLA-4 initially leads to increased IFNγ, which acts in a paracrine fashion to increase indoleamine 2,3-dioxygenase (IDO) thereby inhibiting T cell proliferation (Grohmann et al., 2002; Fallarino et al., 2003). As Tregs highly express CTLA-4, a mechanism of Treg suppression has been proposed where IDO production by B7 expressing DCs appears to act as an important bridge between Tregs and naïve responder cells (Mellor et al., 2004). The importance of this pathway in vivo is underpinned by data from a murine model of islet transplantation, where inhibition of IDO abrogated the graft prolonging effects of CTLA-4-Ig (Grohmann et al., 2002).
CTLA-4 in Models of Transplantation
Cytolytic T lymphocyte-associated antigen 4-Ig is a fusion protein, which avidly binds B7, thereby competitively inhibiting CD28 costimulation. This antibody has been used to study the effects of costimulation blockade in experimental models of skin, heart, and kidney transplantation. Administration of CTLA-4-Ig on day 2 after renal or cardiac transplantation lead to improved allograft survival, decreased pro-inflammatory cytokine production and decreased histological evidence of chronic allograft vasculopathy when compared with treatment with cyclosporine alone (Azuma et al., 1996; Russell et al., 1996). When donor specific transfusion at the time of transplantation was combined with CTLA-4-Ig, long-term graft survival with donor specific tolerance was seen (Lin et al., 1993). Treatment of murine recipients of xenogeneic pancreatic islets with CTLA-4-Ig at the time of transplant similarly led to prolonged allograft survival and donor specific tolerance (Lenschow et al., 1992). Finally, combined with CD40 blockade, treatment with CTLA-4-Ig has been demonstrated to lead to long-term allograft survival in both cardiac and a highly immunogenic skin transplant model (Larsen et al., 1996).
Interestingly, delayed treatment with CTLA-4-Ig, administered 8 weeks after transplantation, lead to decreased progression of chronic allograft rejection with attenuation of cardiac allograft vasculopathy (Chandraker et al., 1998). This suggests that ongoing T cell recognition of foreign antigen is an important mediator of late allograft injury and costimulation blockade, even given late after transplantation, may delay progression. Similar results were seen in a study where an anti-CTLA-4 blocking antibody was given at various timepoints, up to 30 days after cardiac transplantation. Immediate rejection was not seen but instead treatment led to delayed severe acute cellular rejection at approximately day 45 post transplant, again indicating the active role of CTLA-4 in the maintenance of tolerance post transplantation (Chandraker et al., 2005).
Administration of CTLA-4-Ig in CD28 deficient transplant recipients leads to accelerated allograft rejection compared to wild type. In this model of cardiac transplantation, similar acceleration of rejection was observed after treatment with a blocking anti-CTLA-4 antibody, demonstrating that the negative regulatory function of CTLA-4 extends beyond its ability to competitively inhibit CD28 signals (Lin et al., 1998).
More recent studies have examined the link between CTLA-4 expression and CD45 mediated allograft tolerance. Anti-CD45RB is a potent immunomodulatory agent and has been shown to induce donor specific tolerance in models of renal and islet transplantation (Lazarovits et al., 1996). Interestingly, administration of anti-CD45RB is associated with rapid upregulation of CTLA-4 expression in vivo. Blockade of CTLA-4 at the time of islet transplantation abrogated the graft prolonging effect of anti-CD45RB, demonstrating the dependence of this pathway on CTLA-4 induction to promote allograft survival (Fecteau et al., 2001).
CTLA-4 in Human Studies of Transplantation
Genetic studies suggest that polymorphisms in CTLA-4 are associated with differing incidence of acute allograft rejection. In a cohort of over 200 liver transplant patients, single nucleotide polymorphisms (SNPs) previously linked with decreased CTLA-4 function were found to be more common in patients with shorter allograft survival (Marder et al., 2003). Similarly, a study of Korean kidney transplant recipients linked the same SNP with an increased risk of late acute allograft rejection (Kim et al., 2010).
Belatacept in Kidney Transplantation
Initial attempts to generate an antibody to block CD28 were unsuccessful, as the majority of antibodies generated lead to signaling rather than blockade of this pathway. As described above, CTLA-4-Ig, the first antibody developed to successfully block CD28:B7 interactions, is a recombinant fusion protein comprising the Fc domain of human IgG1 linked to the extra-cellular portion of CTLA-4 (Vincenti et al., 2011). CTLA-4-Ig, known commercially as Abatacept, was licensed for clinically for the treatment of RA. However, it proved ineffective in non-human primate studies of transplantation and the molecule was re-engineered, leading to the generation of Belatacept. Belatacept differs from Abatacept by two amino acid substitutions at the ligand-binding domain and binds B7-1 and B7-2 with greater avidity, thereby producing greater immunosuppressive effects (Vincenti et al., 2011).
Phase III human trials in kidney transplant recipients have demonstrated similar graft survival in those treated with Belatacept versus cyclosporine, despite increased rates of acute rejection in the early post transplant period (Vincenti et al., 2010). At 3 year follow up, patients treated with Belatacept showed better renal function, with higher mean eGFR and lower rates of donor specific antibody formation (Vincenti et al., 2012). On the basis of these studies, Belatacept was approved by the FDA in June 2011, for the prevention of acute rejection post renal transplant.
However, despite the early success of Belatacept in transplantation, there remains some concern as to the overall impact of blocking CD28 signaling through competitive antibody binding to B7. This strategy also blocks the ability of CTLA-4 to bind B7-1 and -2 and in particular, this may negatively impact on the development of antigen specific Tregs (Vasu et al., 2004) and on the function of existing Tregs, possibly blocking their suppressive activity. Indeed, a recently published study reported that treatment with CTLA-4-Ig prolonged survival in a fully mismatched model of cardiac rejection but precipitated accelerated rejection in a partially mismatched model, where engraftment is dependent upon Treg function (Riella et al., 2012). Furthermore, intact CTLA-4 function has also previously been shown to be critical for the maintenance of graft tolerance in a model of induced tolerance in skin transplantation (Markees et al., 1998). These issues may provide some explanation as to why CTLA-4-Ig does not reproducibly induce tolerance in certain models of transplantation. Alternative agents currently under investigation include anti-CD28 antibodies, which selectively block CD28 without impacting CTLA-4 function. Targeting the CD28/CTLA-4: B7-H1/2 pathway in this manner may instead promote Treg function and has generated some promising data in non-human primate studies of renal transplantation (Poirier et al., 2010).
Conclusions
CD28 is the most important T cell costimulatory molecule and CTLA-4 holds a central role in the control of CD28-mediated immune responses, including alloimmune responses. Data from human studies indicate that blockade of this pathway using Belatacept should prove to be a useful addition to treatment protocols post kidney transplantation. It is hoped that the advent of costimulation blockade will reduce the severity of chronic allograft rejection both through control of immune mediated injury, and through decreased exposure to calcineurin inhibitors, both of which contribute to reduced long-term renal allograft survival.
However, several important issues remain unanswered. Increased rates of early acute rejection were observed in clinical trials of Belatacept. While these episodes were responsive to therapy, the long-term impact of these episodes of early acute rejection on allograft survival are as yet unclear. Furthermore, the nature of the T cell response and whether it relates to blockade of CTLA-4 mediated suppression of T effector cells remain unknown. It is also unclear if CTLA-4-Ig blocks the recently described coinhibitory interaction between B7 and PD-L1 (described below) and how biologically significant this will prove to be in complex models of alloimmunity. Finally, some data suggests that CTLA-4:B7 interaction may inhibit differentiation of pro-inflammatory Th17 cells and that blockade of CTLA-4 exacerbates Th17-driven experimental autoimmune encephalitis (EAE; Ying et al., 2010). Further studies are needed to explore these issues in detail, to understand the complex interactions between CD28 and CTLA-4 and their links with other costimulatory and coinhibitory pathways in vivo.
PD-1: PD-L1/PD-L2; PD-L1: B7-1
PD-1 is a type I transmembrane protein and is a member of the immunoglobulin superfamily. Its intracellular domain contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) as well as an immunoreceptor tyrosine-based switch motif (ITSM; Okazaki et al., 2001). In humans and mice, it is expressed on activated T and B cells and myeloid cells. Its two ligands, PD-L1 and PD-L2, show differential expression patterns. PD-L1 is constitutively expressed on all haematopoietic cells and many non-haematopoietic tissues and is upregulated after activation whereas PD-L2 is inducibly expressed on DCs and macrophages only. Upon ligand engagement, PD-1 is phosphorylated and recruits SHP-1 and -2 to the antigen-receptor complex (Okazaki et al., 2001).
PD-1 signaling inhibits PI3k activity (Parry et al., 2005), leading to decreased T cell proliferation, reduced IFNγ and IL-2 production and increased T cell apoptosis (Sandner et al., 2005). Using multi-photon laser-scanning microscopy techniques to study the movement of T cells in vivo, Fife et al. described the mechanism of inhibition of T cell activation induced by PD-1:PD-L1 signaling. In this model, T cell activation was decreased by blocking the TCR-induced stop signal, thereby maintaining T cell mobility within lymph nodes and decreasing interactions between T cells and antigen bearing DCs (Fife et al., 2009). Of note, PD-1 mediated inhibition depends upon the strength of TCR ligation, with greatest inhibition delivered at low levels of TCR stimulation. Furthermore, this inhibition can be overcome by stronger costimulatory signals, such as those delivered through CD28 (Freeman et al., 2000).
PD-1 in Disease States
PD-1 plays an important role in the maintenance of self-tolerance and its deficiency leads to varying disease states depending upon the genetic background of the animals studied. PD-1−/− on a Balb/c background suffer from autoimmune cardiomyopathy while C57BL/6 PD-1−/− develop progressive arthritis and a lupus-like glomerulonephritis (Fife and Bluestone, 2008). Furthermore, PD-1 blockade and deficiency in NOD mice leads to accelerated autoimmune diabetes (Ansari et al., 2003; Wang et al., 2005a) and PD-1 blockade leads to the development of accelerated and clinically more severe EAE in mouse models (Salama et al., 2003). Of note, PD-L1 has been implicated in the development of T cell exhaustion (Urbani et al., 2006; further discussed below) and its tissue expression also plays a crucial role in the maintenance of peripheral tolerance (Keir et al., 2006). Similar to other coinhibitory molecules, polymorphisms in PD-1 have been linked with a range of autoimmune conditions in humans, including MS, SLE, Type I DM, and Grave’s disease (Keir et al., 2008).
Impact of PD-1 on Treg Function
Regulatory T cells upregulate PD-1 expression on activation (Raimondi et al., 2006) and blockade of PD-1 appears to decrease the suppressive activity of Tregs in vivo (Kitazawa et al., 2007). Furthermore, in the setting of transplantation, PD-L1 blockade appears to alter the balance of Tregs/Teffector cells and promote aggressive alloresponses (Sandner et al., 2005).
PD-1:PD-L1/PD-L2 in Models of Transplantation
In keeping with its role as a negative costimulatory pathway, blockade of PD-1 leads to accelerated allograft rejection in fully mismatched cardiac allografts. However, partially mismatched cardiac transplants survive long-term in PD-1 deficient recipients (Ito et al., 2005; Tao et al., 2005). This discrepancy suggests that PD-1 signals play a greater role in disease models where CD4+ and CD8+ T cell-dependent responses are prominent. More detailed studies have demonstrated that blockade of PD-L1 specifically, but not PD-L2, was associated with accelerated rejection in skin and cardiac allograft models, characterized by increased proliferation and decreased apoptosis of allospecific CD4+ T cells (Barrat et al., 1999; Sandner et al., 2005; Yang et al., 2008).
Administration of a fusion protein which is agonistic for PD-1, PD-L1-Ig, has been demonstrated to prolong cardiac allograft survival in both CD28−/− recipients and, when given with cyclosporine, WT recipients of full mismatched cardiac allografts. This survival was associated with decreased IFNγ expression and reduction in pro-inflammatory cytokine production (Ozkaynak et al., 2002). Furthermore, PD-L1-Ig synergizes with anti-CD154mAb to promote long-term islet allograft survival (Gao et al., 2003). Finally, intact PD-1:PD-L1 signaling has been reported to be central to the spontaneous tolerance in murine liver allografts (Morita et al., 2010).
In a further layer of complexity, tissue expression of PD-1 ligands appears to play an important role in allograft outcome. Donor, but not recipient, PD-L1 deficiency is associated with an increased tempo of cardiac allograft rejection; characterized by accelerated chronic allograft vasculopathy, greater frequency of effector T cells and increased IFNγ production (Yang et al., 2008). Studies using bone marrow chimeras have demonstrated that tolerance induced by CTLA-4-Ig is dependent upon the presence of PD-L1 on donor endothelium (Riella et al., 2011). Mechanistic studies showed increased pro-inflammatory cytokine production and increased CD8+ effector/memory T cell frequency in recipients of grafts lacking PD-L1 endothelial expression (Riella et al., 2011). Furthermore, the effect of PD-1 pathway appears to differ between cell types. In a GVHD model, blockade of PD-L2 but not PD-L1 lead to increased proliferation of allospecific CD8+ T cells with little effect of CD4+ T cells (Habicht et al., 2007). Differing PD-1 ligand expression in a lymphoreplete versus a lymphopenic model may also influence these observations.
Recently, B7-1 has been recognized as an alternative binding partner for PD-L1, albeit at lower affinity than for PD-1 (Butte et al., 2007). This interaction also transmits a coinhibitory signal leading to decreased T cell proliferation, reduced cytokine production and reduction in activation marker expression. In a MHC Class II mismatched model of chronic cardiac allograft rejection, PD-L1 blockade accelerated rejection in B7-2, but not B7-1, deficient recipients (Yang et al., 2011). Moreover, administration of an antibody which specifically blocked the B7-1:PD-L1 interaction led to more severe chronic allograft vasculopathy with upregulation of pro-inflammatory cytokines and reduction in splenic Tregs (Yang et al., 2011). These data demonstrate the relevance of this interaction using in vivo models of transplantation and are of particular relevance in light of the recent availability of the costimulation blocker, Belatacept, in clinical practice. The impact of Belatacept on the interaction between B7-1 and PD-L1 is unknown and may be an important area of future investigation.
PD-1 Pathway in Human Studies of Transplantation
Preliminary studies have looked at the predictive value of urinary levels of PD-1, PD-L1, and PD-L2 mRNA in the diagnosis of acute allograft rejection in human recipients. Elevated urinary PD-1 mRNA, was associated with biopsy proven acute rejection and when measured in combination with urinary mRNA for OX40, OX40L, and Foxp3, was a strong predictor of acute allograft rejection (Afaneh et al., 2010).
In human transplant recipients, the role of allograft tissue expression of PD-1 and its ligands have also been examined. PD-L1 is expressed on human renal tubular epithelial cells and has been reported to suppress alloreactive T cell responses (Starke et al., 2010). Furthermore, while these molecules are not seen on native kidneys, immunohistochemical staining of renal allograft samples revealed induction of PD-1, PD-L1, and PD-L2 in rejecting allografts. No correlation was seen between staining and serum creatinine or proteinuria. However, the expression of PD-L1 was significantly higher in those grafts showing vascular (Banff 2a and 2b) versus tubulointerstitial rejection (Banff 1a and 1b; Starke et al., 2010).
Conclusions
The differential expression of ligands for CTLA-4 and PD-1 has lead to hypotheses that these negative costimulatory pathways occupy non-redundant roles in control of immune responses. CTLA-4 may control early T cell activation, whereas PD-1 may be more important in control of later stages and in particular T cell activation within the transplanted organ. Therefore, these pathways appear to play complementary roles in alloimmunity and robust strategies to promote tolerance may involve targeting both.
Data from human studies suggest that PD-1 and PD-L1 may be useful biomarkers for non-invasive diagnosis of allograft rejection, most likely in combination with a panel of other molecules. More importantly, animal models have demonstrated that tissue expression of PD-1 and its ligands play an important role in modulating the alloresponse to the graft. Furthermore, upregulation of these molecules is seen in rejecting grafts, in murine and human studies, perhaps in response to local tissue injury. Whether this is associated with limitation of the alloresponse or possibly a marker of greater responsiveness to therapy, is as yet unknown, and will form an important area of future investigation. Finally, an exciting prospect exists that manipulation of graft expression of these molecules, either through targeted gene therapy or the use of drugs to upregulate expression, may promote a tolerogenic environment and influence graft survival.
B7-H3
B7-H3 is a member of the B7 family of costimulatory molecules. It is a type-1 transmembrane protein with a short cytoplasmic tail and no known signaling domain. It is broadly expressed in human tissues and in the immune system, its expression is induced on monocytes, DCs, and T cells upon activation (Chapoval et al., 2001). This wide pattern of expression suggests B7-H3 may have diverse immunological and non-immunological functions. In keeping with this, its exact role in immunity remains unclear, with positive and negative costimulatory functions being reported. Initial studies of human B7-H3 using a B7-H3-Ig fusion protein, suggested it was a positive costimulatory molecule, demonstrating increased T cell proliferation, IFNγ production and CTL generation (Chapoval et al., 2001). Its receptor is found on activated T cells and is distinct from CD28, CTLA-4, ICOS, and PD-1 (Chapoval et al., 2001).
Triggering receptor expressed on myeloid cells (TREM)-like transcript 2 (TLT-2) has been proposed as a receptor for B7-H3 (Hashiguchi et al., 2008). TLT-2 is constitutively expressed on CD8+ T cells and is induced on CD4+ T cells after activation. In this study, B7-H3:TLT-2 was reported to enhance T cell responses in vitro, with modest increases IL-2 and IFN-γ production on CD4+ and CD8+ T cells seen after TLT-2 and B7-H3 interaction (Hashiguchi et al., 2008). However, a more recent paper has strongly refuted these findings, and reports no interaction between B7-H3 and TLT-2 on murine or human cells (Leitner et al., 2009).
In mice, B7-H3 is constitutively expressed on professional APCs and a small population of splenic T cells. Its expression is increased on DCs after incubation with LPS (Prasad et al., 2004). Contrary to the human study, data from murine studies using a blocking antibody against B7-H3 demonstrate its role as a negative regulator of T cell activation (Prasad et al., 2004). An agonistic fusion protein, B7-H3-Ig, was found to markedly decrease proliferation, IL-2 and IFNγ production by T cells incubated with aCD3 (Suh et al., 2003; Prasad et al., 2004; Leitner et al., 2009). Furthermore, when T cells were activated with aCD3 in the presence of B7-H3-Ig, reduced activity of NF-κB, NFAT, and inhibition of AP-1 activation were observed (Prasad et al., 2004). The effect of B7-H3 signaling was overcome with CD28-mediated costimulation, suggesting B7-H3 may exert greatest effect in situations where CD28 costimulation is limited. In addition, use of a blocking mAb, anti-B7-H3, lead to greatly enhanced T cell proliferation (Prasad et al., 2004). Unlike the human data, murine studies did not confirm a role for B7-H3 in CTL responses in vitro or in vivo (Suh et al., 2003).
B7-H3 in Disease States
In vivo, treatment with an antagonistic mAb, anti-B7-H3, lead to earlier onset of EAE, more clinically severe disease and greater numbers of brain-infiltrating CD4+ T cells in treated than controls (Prasad et al., 2004). Finally, B7-H3 deficient APCs show greater costimulatory capacity than wild type (Suh et al., 2003).
In a murine model of airway inflammation, B7-H3 deficient mice developed more severe airway inflammation and increased T cell infiltration than controls in Th1 but not Th2 conditions (Suh et al., 2003). Furthermore, B7-H3 expression on DCs was enhanced by IFNγ but suppressed by IL-4. Taken together, these data suggest a role for B7-H3 in negative regulation of T cell responses occurring under Th1 polarizing conditions.
Extensive studies of B7-H3 function in cancer biology have also revealed contradictory data. In several mouse cancer models, ectopic overexpression of B7-H3 leads to activation of tumor specific CTLs and slowed growth or tumor eradication (Hofmeyer et al., 2008). However, human studies in non-small cell, prostate and ovarian cancers have shown increased disease severity where malignant cells showed increased B7-H3 expression, suggesting B7-H3 may be acting as an immune evasion pathway in this setting (Hofmeyer et al., 2008).
B7-H3 in Models of Transplantation
The role of B7-H3 in transplantation has not yet been extensively studied but again, conflicting data exists. Using a complete MHC mismatched cardiac allograft model, prolonged allograft survival was seen in B7-H3−/− recipients treated with Cyclosporine or Rapamycin (Wang et al., 2005b). Analysis of graft infiltrating cells revealed decreased expression of both cytokines IL-2 and IFN-γ and chemoattractant proteins MCP-1 and IP-10 in treated B7-H3−/− recipients compared with control animals. No differences were seen in expression of regulatory cytokines (Wang et al., 2005b). Chronic allograft rejection was investigated using two models; a complete MHC mismatched allograft combined with anti-CD154 treatment and a Class II mismatched cardiac transplant. Decreased chronic allograft vasculopathy with similar cytokine expression profiles were observed in B7-H3−/− recipients in both models.
In contrast however, our group has extensive preliminary data demonstrating the role of B7-H3 as a negative regulator of alloimmunity. B7-H3−/− recipients show attenuated allograft survival and treatment with an agonistic fusion protein, B7-H3-Ig prolongs allograft survival in a complete MHC mismatch model (Ueno, Yeung, personal communication).
The differing effects observed between these transplantation studies may possibly relate to specifics of the individual B7-H3 knockouts described and also the binding affinity/functional activity of fusion proteins studied. Furthermore, the existence of separate “costimulatory” and “coinhibitory” ligands for B7-H3 has not yet been excluded. The contradictory findings suggest the role played by B7-H3 varies, dependent upon the system being studied. Furthermore, B7-H3 has wide tissue expression and little is known as to its local activity in regulating immunity. Clearly, further detailed studies are required to elucidate the exact role of B7-H3 in alloimmunity and its relative importance as a potential therapeutic target.
B7-H4
B7-H4 is a type I transmembrane protein and is a recently described member of the B7 family. While B7-H4 mRNA is found in a variety of tissues, it is not expressed on naïve T cells, B cells or DCs. Its expression is upregulated after in vitro stimulation of human T cells, B cells, monocytes, and DCs (Prasad et al., 2003; Sica et al., 2003). Its receptor, as yet unidentified, is thought to be expressed on activated T cells and appears to be distinct from known CD28 family members. Finally, it has also been shown to suppress neutrophil-mediated immune processes, thereby playing a role in innate immunity (Zhu et al., 2009).
In studies using an agonistic fusion protein, B7-H4 was demonstrated to inhibit T cell proliferation both in vitro and in vivo (Prasad et al., 2003; Sica et al., 2003). Signaling through B7-H4 lead to suppression of IL-2 and IFN-γ production in vitro. In addition, there appears to be an interaction between B7-H4 and Treg function. In a model of GVHD, treatment with B7-H4-Ig suppressed the generation of CTLs both in vitro and in vivo (Sica et al., 2003). In vitro studies suggest that suppressive function of APCs is associated with increased B7-H4 expression. This appears to be mediated through Treg production of IL-10 (Kryczek et al., 2006). Finally, blockade of B7-H4 lead to accelerated onset and worsening severity of murine EAE, further indicating its important role in control of T cell responses (Prasad et al., 2003).
B7-H4 in Disease States
Early treatment with B7-H4-Ig reduced the incidence of diabetes in NOD mice, associated with reduced insulitis scores and increased pancreatic infiltration with Foxp3+ CD4+ T cells in treated mice (Wang et al., 2011). In a similar vein, B7-H4 was overexpressed on an insulinoma cell line transplanted into C57BL/6 mice with Streptozotocin-induced diabetes. Improved survival was observed in the B7-H4 group; this was associated with an increased splenic Treg population and reciprocally reduced IFN-γ-producing and increased IL-4 producing splenocytes (Yuan et al., 2009). Another group found that B7-H4 overexpression promoted islet allograft survival and induced donor specific tolerance (Wang et al., 2009a).
Similar to studies of B7-H3 in cancer biology, B7-H4 appears to play a role in tumor immune evasion. It appears to be highly expressed on human tumors including breast, lung, prostate, and ovarian malignancies. In keeping with its known role as a negative regulator of immune responses, the presence of B7-H4 positive cells has been repeatedly shown to correlate with reduced numbers of tumor infiltrating lymphocytes and worse outcomes (Yi and Chen, 2009).
B7-H4 in Models of Transplantation
A small number of studies have addressed the role of B7-H4 in alloimmunity. Our group has recently described the effect of B7-H4 blockade in murine model of cardiac transplantation. In a fully mismatched cardiac allograft model, blockade of B7-H4 did not affect allograft survival. However B7-H4 blockade significantly accelerated allograft rejection in CD28−/− recipients and in CD80/CD86DKO recipients, in whom fully mismatched allografts usually survive long-term (Yamaura et al., 2010). This rejection was accompanied by increased frequency of IFN-γ and IL-4 producing splenocytes and increased levels of donor specific antibodies. Furthermore, blockade of this pathway with anti-B7-H4 precipitated acute rejection in mice treated with CTL-A4-Ig where long-term allograft acceptance is the norm (Yamaura et al., 2010).
These data suggest that B7-H4 plays a dominant negative regulatory role in controlling alloreactive T cells in the absence of CD28/CTLA-4:B7 signaling. In this fashion, it appears to play a non-redundant role in fine-tuning the immune response in vivo. Although not yet fully elucidated, this role may prove of increasing relevance going forward; particularly where costimulatory blockade using agents such as Belatacept becomes increasingly common in clinical practice.
BTLA/CD160/LIGHT:HVEM
This complex pathway involves a number of players, the most widely studied of which are B and T cell lymphocyte attenuator (BTLA), herpesvirus entry mediator (HVEM), and LIGHT. These three molecules interact directly with each other but it appears that HVEM occupies a central role and can direct either positive or negative costimulatory signals depending upon its receptor binding. Central to understanding the role of this pathway in immunity is an analysis of the complex interactions and relative roles of each of the molecules involved.
B and T cell lymphocyte attenuator is a member of the Ig superfamily and shares structural homology with PD-1 and CTLA-4. BTLA is a type I membrane glycoprotein with a single C-type Ig domain and three conserved tyrosine motifs in its cytoplasmic tail; two of which a found within (ITIMs; Watanabe et al., 2003). As predicted by its structure, BTLA acts as a negative costimulatory molecule. Binding to its ligand, HVEM, induces phosphorylation of its ITIM domain. This is followed by association with SH2 domain-containing protein tyrosine phosphatase 1 (SHP-1) and SHP-2, leading to attenuation of signals promoting cellular activation and growth (Sedy et al., 2005). It is broadly expressed across cells of both innate and adaptive immunity and is found on CD4+ and CD8+ T cells, B lymphocytes, DCs, NK cells, and also endothelial cells.
B and T cell lymphocyte attenuator expression is low on naïve CD4+ T cells (10–15%; Tao et al., 2008) but increases with antigen specific stimulation and peaks on day 2 (Hurchla et al., 2005). This declines by day 7 but secondary reactivation of T cells leads to rapid BTLA induction (Hurchla et al., 2005). In terms of T cell subsets; greater expression is seen on Th1 than Th2 cells (Hurchla et al., 2005) and little is expressed on Tregs. In addition, BTLA is highly expressed on anergic CD4+ T cells in vivo (Hurchla et al., 2005). The highest levels of BTLA expression on T cells were seen with anergy induction (Hurchla et al., 2005). BTLA deficient T cells proliferate more vigorously than wild type when stimulated with aCD3 or APCs. This appears to be primarily due to increased CD8+ T cell proliferation. Furthermore, BTLA−/− CD8+ T cells are more efficient at differentiating into memory T cells than WT cells (Krieg et al., 2005). This is thought to explain the higher proportion of memory T cells observed in both BTLA−/− and HVEM−/− animals (Krieg et al., 2005).
B and T cell lymphocyte attenuator is unique among coinhibitory members of the Ig superfamily, in that it binds to HVEM, which is a member of the TNF receptor superfamily. Other ligands for HVEM have been described, including CD160, which also transduces a negative costimulatory signal (Cai et al., 2008). HVEM also binds the canonical TNF ligands, LTα (Murphy and Murphy, 2010; lymphotoxin alpha), and LIGHT (Mauri et al., 1998; lymphotoxin-like, exhibits inducible expression, and competes with herpes simplex virus glycoprotein D for HVEM, a receptor expressed by T lymphocytes) both of which are positive costimulatory molecules. HVEM engagement with BTLA and CD160 both activate inhibitory signaling in lymphocytes (Sedy et al., 2005; Murphy et al., 2006; Cai et al., 2008). BTLA and CD160 both bind to the N-terminal membrane distal cysteine-rich domain-1 (CRD-1) of HVEM (Sedy et al., 2005), whereas the positive costimulatory molecules, LIGHT and LTα, bind to CRD-2 and CRD-3 (Rooney et al., 2000). LTα has not yet been studied in alloimmunity and will not be discussed further here.
Herpesvirus entry mediator is widely expressed across cells of innate and adaptive immunity and is also found on endothelial cells. However, its relative role in differing cell subsets remains unclear. Signaling through HVEM activates NF-kB, and in this manner it costimulates B cell proliferation and immunoglobulin secretion (Duhen et al., 2004) and on DCs (Salio et al., 1999), NK cells (Kwon et al., 1997) and non-haematopoietic cells, it promotes activation of effector functions (Marsters et al., 1997). During T cell activation, HVEM shows a reciprocal expression pattern to BTLA, with high HVEM expression seen on naive CD4+ which markedly decreases with T cell activation (Morel et al., 2000). This returns to high levels as CD4+ cells become quiescent.
LIGHT is expressed on monocytes, granulocytes, and immature DCs (Tamada et al., 2000). LIGHT expression on peripheral blood lymphocytes is induced with TCR activation but it is constitutively expressed on CD4+ cells and NK cells in mucosal tissues. (Cohavy et al., 2005) LIGHT does not itself appear to have signaling motifs but it shows potent CD28 independent costimulatory activity. Its interaction with HVEM provides a positive costimulatory signal (Sedy et al., 2005) leading to increased MAP kinase activity, T cell activation (Shi et al., 2002), and increased inflammatory responses (Ware, 2008).
LIGHT exists in both membrane-bound and soluble forms, cleaved by matrix metalloproteases. Interestingly, binding of LIGHT appears to modulate the interaction between BTLA and HVEM and its effect on this interaction varies greatly dependent upon the form. As described above, LIGHT has a separate binding site to BTLA on HVEM (Rooney et al., 2000) and its binding, in soluble or membrane-bound forms to HVEM, modulates the signal produced when BTLA binds, providing a tight feedback loop. Both membrane-bound and soluble LIGHT can induce NF-κB activation through HVEM but membrane-bound LIGHT shows much more robust activity than the soluble form (Cheung et al., 2009). Conversely, membrane-bound LIGHT non-competitively disrupts binding between BTLA and HVEM (Cheung et al., 2005). The regulation of LIGHT cleavage in vivo has not yet been elucidated but it appears to play an important role in determining the downstream effect of BTLA:HVEM interaction.
CD160 is the second coinhibitory molecule involved in this pathway. It binds HVEM and inhibits CD4+ proliferation and cytokine production (Cai et al., 2008). CD160 is highly expressed on human NK cell subsets, CD8+ T cells, NKT cells, γδ T cells, and on all intestinal intra-epithelial T lymphocytes (IELs; Maeda et al., 2005). However, functional studies of CD160 have not been reported to date and little is known about its role in auto- or allo-immunity in vivo. Although both act to control T cell proliferation, CD160 and BTLA appear to show distinct expression patterns, suggesting their inhibitory functions may be non-overlapping in vivo.
BTLA Promotes T Cell Survival
Aside from its role as coinhibitory molecule, there are data to suggest BTLA also promotes T cell survival. In vitro data using an NF-κB reporter shows that BTLA binding to HVEM leads to NF-κB activation and promotes cell survival (Cheung et al., 2009). Furthermore, the presence of soluble LIGHT increased the avidity of BTLA binding to HVEM and augmented the observed NF-κB activation.
BTLA−/− T cells fail to sustain GVHD in a non-irradiated GVHD model. BTLA−/− T cells show normal expansion initially after adoptive transfer, but failed to sustain inflammation (Hurchla et al., 2007). After the first week, BTLA−/− T cells show a sharp decline in effector cell numbers, which is accompanied by resolution of GVHD (Hurchla et al., 2007). Furthermore, BTLA−/− T cells fail to elicit a strong anti-host response as demonstrated by lack of depletion of host B cells. Similar findings were seen with antibody blockade of BTLA on wild type cells. Mechanistic studies showed that these T cells show decreased proliferation and decreased cytolytic activity (Hurchla et al., 2007).
Impact of BTLA:HVEM on Treg Function
The expression of BTLA is low on Tregs and BTLA−/− Tregs demonstrate normal suppressive activity (Tao et al., 2008). Upon activation, T effector cells express increased BTLA but markedly down-regulate HVEM. In contrast, activated Tregs strongly upregulate HVEM but not BTLA. The importance of this pathway in Treg function is emphasized by findings that HVEM−/− Tregs show decreased suppressive activity and WT Tregs are unable to suppress BTLA−/− T effector cells (Tao et al., 2008). In addition, studies using fully mismatched cardiac allografts suggest that long-term allograft survival could not be achieved in the absence of HVEM on Tregs (Tao et al., 2008). While HVEM does not appear to be a dominant functional pathway on Tregs (as HVEM−/− mice do not display spontaneous autoimmunity observed in Foxp3 deficient mice), these data suggest a potentially important role in alloimmunity, which requires further detailed evaluation.
BTLA:HVEM in Disease States
BTLA−/− mice are more prone to develop autoantibodies and develop a hepatitis-like syndrome with advancing age (Oya et al., 2008). Expansion of CD4+ and NKT cell populations in the liver parenchyma with associated endothelialitis and portal inflammation has also been described (Oya et al., 2008). BTLA−/− also show greater allergic airway inflammation (Deppong et al., 2006) and greater susceptibility to EAE (Greenwald et al., 2005). HVEM−/− mice show increased susceptibility to Con A mitogen induced, autoimmune hepatitis (Wang et al., 2005c). LIGHT−/− mice show impaired T cell responses (Tamada et al., 2000).
In a murine model of inflammatory bowel disease, Rag−/− mice developed more severe colitis upon adoptive transfer of HVEM−/− T cells compared to transfer of wild type T cells. Transfer for BTLA−/− cells did not accelerate colitis in the same fashion (Steinberg et al., 2008). It has been proposed that this may be due to lack of T cell survival as observed in the GVHD model described above (Hurchla et al., 2007). Treatment of HVEM−/− Rag−/− recipients with a BTLA agonist reversed the observed colitis (Steinberg et al., 2008). These data suggest BTLA:HVEM plays an important role in control of effector T cell responses in vivo. Furthermore, data from Rag−/− studies suggests the negative costimulatory effect of BTLA signaling may be the more dominant pathway regulating in vivo immune responses, rather than positive costimulation mediated through LIGHT.
A Taiwanese study of 94 patients with RA reported an association between a SNP [C(+800)] in BTLA and increased risk of RA. This SNP is located in the region between the two ITIMs in the cytoplasmic tail of BTLA (Lin et al., 2006). In addition, a small Japanese study showed an association between a different BTLA SNP (590C) and RA, but not with SLE or Sjogren’s syndrome. In this study, patients bearing this SNP were reported to have presented with disease earlier than those without (Oki et al., 2011). The functional consequences of either of these SNPs remain unknown.
BTLA in Models of Transplantation
Several studies have described prolonged islet cell allograft survival after treatment with anti-BTLA antibodies. One such antibody, PJ196, given in combination with CTLA-4-Ig, has been reported to lead to long-term islet cell allograft survival (Truong et al., 2007a). The mechanism of action of this antibody is unclear. BTLA expressing cells were not depleted, but BTLA surface expression was markedly down-regulated after treatment with PJ196. Furthermore, earlier data had suggested this antibody does not simply act as a BTLA agonist as it did not decrease in vitro T cell proliferation. After combination treatment with CTLA-4-Ig, islet graft histology demonstrated massive cellular infiltration, primarily consisting of CD4+ Foxp3+ Tregs. Such infiltrate was not observed after treatment with anti-BTLA alone, leading the authors to speculate that combined treatment may have enhanced the local milieu for Treg generation, possibly through down-regulating coinhibitory signals on Tregs and thereby enhancing their function (Truong et al., 2007a).
In a related publication from the same group, treatment with a depleting anti-BTLA antibody, 6F7, alone led to modest non-significant prolongation of islet allograft survival, when compared with control. No graft prolongation was seen in BTLA−/− recipients and addition of CTLA-4-Ig did not significantly prolong allograft survival in BTLA−/− mice. Combination of anti-BTLA (6F7) with CTLA-4-Ig again lead to long-term islet allograft survival in this model (Truong et al., 2007b). Treatment was associated with a reduction in total T cell, CD4+ cell and B cell number. Furthermore, the percentage of CD4+ cells expressing PD-1 was significantly increased in the anti-BTLA treatment group.
As seen in the earlier study, allograft histology demonstrated local accumulation of CD4+ Foxp3+ cells in tolerant animals. In vivo donor specific tolerance was present and third party grafts were appropriately rejected. However, in vitro studies showed no difference in T cell proliferation or cytolytic killing in response to allogeneic stimulus in tolerant mice. In vivo CFSE proliferation studies confirmed that 6F7 does not show significant impact on proliferation of allogeneic CD4+ or CD8+ T cells (Truong et al., 2007b). The function of this mAb in vivo is unclear, but the lack of effect on proliferation of activated T cells suggests that similar to PJ196, 6F7 is not simply an agonistic antibody. Through an unclear mechanism, increased expression of PD-1 was observed along with increased local Treg accumulation in the treated animals. It is unclear if anti-BTLA mAb acts to increase PD-1 expression either directly or if this occurs through indirect mechanisms by blocking BTLA signaling.
Further studies have examined the role of BTLA in the survival of Class I or Class II mismatched cardiac allograft survival. In contrast to wild type recipients, where partially mismatched cardiac allografts survive long-term, both BTLA−/− and HVEM−/− recipients rejected their grafts within 4 weeks of transplantation. Treatment with a blocking anti-BTLA mAb (6A6) led to accelerated rejection, at a similar tempo (Tao et al., 2005).
While targeting PD-1 did not induce acute rejection in this model, recipients deficient in both BTLA and PD-1 rejected their allografts, at a slightly faster tempo than BTLA−/− alone. Moreover, targeting BTLA via any one of either BTLA−/−, HVEM−/− recipients, or administration of a blocking anti-BTLA mAb, all lead to accelerated allograft rejection in a mismatched model (Tao et al., 2005). Similarly, T cell alloreactivity was greatly enhanced in the absence of BTLA or HVEM. While BTLA was induced on alloreactive T cells post partially mismatched cardiac transplantation, PD-1 was not.
Interestingly, and unexpectedly, in the setting of transplantation across a full MHC mismatch, T cells from BTLA−/− mice display decreased proliferation and cytokine production and these recipients show slightly prolonged cardiac allograft survival. As discussed previously (in PD-1 section), following fully mismatched cardiac transplantation; PD-1 expression is greatly increased on CD4+ and CD8+ alloreactive T cells. Unexpectedly, this increase in expression is more marked in BTLA−/− recipients. Treatment with anti-PD-1 blockade or transplantation into a BTLA/PD-1 double knockout recipient abrogated this effect and led to acute allograft rejection, similar to controls (Tao et al., 2005).
Based on the above data, it appears that BTLA signaling plays an important role in the maintenance of tolerance to partially mismatched cardiac allografts, a well-studied model of chronic rejection. However, in the setting of stronger alloimmune stimulus, as seen with fully mismatched transplantation, the impact of BTLA on allograft tolerance is much less. In these circumstances, the pro-tolerogenic effect of PD-1 appears to be dominant. Again, the effect of targeting BTLA on induction of PD-1 expression remains unclear, but these data suggest an important link between these two negative costimulatory pathways.
The differences in survival in the cardiac and islet transplant data possibly relate to the antibodies used. The cardiac transplants were carried out using a non-depleting, blocking anti-BTLA mAb (6A6) whereas the islet cell transplants used a depleting antibody (6F7) or one that down-regulated BTLA expression (PJ196).
Several groups have studied the role of BTLA using GVHD models. Using a parent into non-irradiated F1 murine model of GVH, treatment with an antagonistic anti-BTLA mAb (4G12b) was shown to decrease anti-host response in terms of infiltration of bone marrow and thymus along with a marked reduction in the cytotoxic T cell activity (Del Rio et al., 2011). Further studies have shown that anti-BTLA mAb (6A6), administered at the time of allogeneic hematopoietic stem cell transplantation (HSCT) prevented the development of GVHD (Albring et al., 2010). However, once GVHD was established, treatment with 6A6 could not reverse the disease. In this model, lymphocyte depletion was not seen and the effect was dependent on the presence of BTLA on donor cells. Overall, treatment at the time of transplantation appeared to rebalance the T cell expansion in these recipients, with decreased effector T cell populations, greater proportions of Tregs with increased Treg/Teff ratio (Albring et al., 2010).
Finally, a recently published study used an agonistic BTLA mAb (BYK-1), which does not block the interaction of BTLA and HVEM. Administration of this mAb profoundly inhibited the generation of donor anti-host CTLs (Sakoda et al., 2011). Similar to the study using 6A6 mAb, treatment at the time of transplantation prevented the development of GVHD in recipient mice and led to long-term survival and successful engraftment (Sakoda et al., 2011). These effects were shown to be dependent on donor T cells and additionally, T cell-expressed BTLA was demonstrated to deliver a pro-survival signal, through HVEM (Sakoda et al., 2011). Taken together, these data suggest BTLA blockade may be a potential therapeutic target in HSCT to prevent the development of GVHD while allowing survival and engraftment of donor cells.
Conclusions
Attempts to dissect and understand this pathway are complicated by several major issues. These include the number of binding partners for HVEM, their varied functions, and the fact that most lymphoid cells can express any of these receptors/ligands (BTLA, HVEM, LIGHT, and CD160) at differing stages of their development and activation. However, active regulation of HVEM expression appears to be a central tenet to controlling its interactions with BTLA, CD160, and LIGHT. As such, HVEM appears to occupy a central role, acting as a molecular switch, to direct T cell activation in an inhibitory or stimulatory direction.
The observed bidirectional signaling through BTLA:HVEM, the effect of LIGHT on their interaction and the relative expression of these molecules are all issues that need to be considered when interpreting disease or transplantation models involving this pathway. Studies using differing models suggest that the dominance of one signal over the other varies according to the type of immune response involved. Some studies suggest that the positive costimulatory signal through HVEM may predominate in pathogenesis of GVHD, where blockade of BTLA has led to impaired allogeneic responses and HVEM−/− mice suffer less severe GVHD. Meanwhile, in models of autoimmunity and inflammatory responses, BTLA negative costimulatory signal appears dominant, as BTLA−/− mice show greater susceptibility to these conditions. However, the impact of LIGHT modulating the BTLA–HVEM interaction has not yet been adequately investigated in vivo and remains an important area of future investigation. Furthermore, the link between this pathway and PD-1 remains to be elucidated.
Further research in this area is crucial to clarify the roles of these molecules and understand their hierarchy of importance in models of transplantation. Theoretically, blockade LIGHT:HVEM costimulation is desirable, as is promotion of coinhibition through BTLA:HVEM signaling. However, recent data highlighting the costimulatory effect of BTLA binding to HVEM, leading to increased NF-κB activation and pro-survival signals, sound a note of caution. Therefore, the development of antibodies binding BTLA to promote its signaling rather than HVEM may prove the most promising target. In vivo data from models of transplantation appear to indicate that those antibodies that deplete or down-regulate BTLA expression are more effective in controlling alloresponses. However, much work remains to be done to develop our understanding of this pathway as a potential therapeutic target.
TIM-3:Galectin-9
The T cell immunoglobulin mucin (TIM) family members are a novel group of costimulatory molecules, expressed on a wide variety of innate and adaptive immune cells. While TIM-3 is present on predominantly on Th1 cells, it is found on Th17 cells at lower levels and is also expressed on CD8+ T cells, DCs, macrophages, and mast cells (Freeman et al. (2010)). In studies using an antagonistic TIM-3Ig fusion protein, TIM-3 blockade in naïve CD4+ cells was demonstrated to increase proliferation and increase IFNγ production (Sabatos et al., 2003). Galectin-9, an S type lectin, is the ligand for TIM-3. It is expressed on Tregs, B cells, and mast cells and is also found on non-immune cells such as endothelial cells and fibroblasts (Zhu et al., 2005).
Upon binding galectin-9, the intracellular tail of TIM-3 is phosphorylated by the interleukin inducible T cell kinase (ITK; van de Weyer et al., 2006), leading to intracellular calcium influx and apoptosis. TIM-3 signaling is thought to be a critical inhibitory mechanism whereby Th1 responses are controlled. Furthermore, IFNγ upregulates galectin-9 expression, leading to an elegant feedback loop (Kashio et al., 2003; Zhu et al., 2005). Similarly, administration of soluble galectin-9 decreases Th17 differentiation (Seki et al., 2008) while blockade of TIM-3 increases IL-17 production (Hastings et al., 2009; Boenisch et al., 2010), demonstrating the role of this pathway in controlling Th17 responses.
Data on the role of TIM-3 signaling in innate immunity are conflicting. Interruption of signaling during induction of EAE leads to macrophage expansion and activation resulting in a more severe clinical phenotype (Monney et al., 2002). Similarly, TIM-3 blockade during liver ischemia–reperfusion injury increases neutrophil infiltration, cytokine release, and hepatocyte apoptosis (Uchida et al., 2010). In contrast, ex vivo studies suggest TIM-3 acts synergistically with TLR stimuli to increase pro-inflammatory TNF-α secretion from DCs, which may in turn promote T effector responses (Anderson et al., 2007).
TIM-3 in Models of Transplantation
Galectin-9 is expressed on Tregs and studies demonstrate that blockade of TIM-3:galectin-9 reduces the suppressive activity of Tregs in vitro. In vivo TIM-3 blockade at the time of transplantation abrogates the tolerizing effect of donor derived Treg transfusion, leading to accelerated graft loss when compared with controls (Wang et al., 2009b). Similarly, in a model of islet transplant tolerance induced with DST and anti-CD154, TIM-3 blockade was associated with loss of tolerance and rapid allograft rejection (Sanchez-Fueyo et al., 2003) and TIM-3 interactions have been demonstrated to be essential for the generation of donor specific Tregs (Sanchez-Fueyo et al., 2003).
TIM-3:galectin-9 interactions appear to be central to Treg function and tolerance induction. Galectin-9 expression on T cells is limited solely to Tregs (Sabatos et al., 2003; Wang et al., 2009b). TIM-3 blockade abrogates induced peripheral tolerance, causes significant attenuation of suppressive activity of natural Tregs and increased auto- and allo-immune responses including autoantibody production (Sabatos et al., 2003; Muthukumarana et al., 2008; Wang et al., 2009b). Furthermore, TIM-3 signaling appears to facilitate the acquisition of donor specific tolerance; TIM-3 deficient mice are refractory to tolerance induction (Sabatos et al., 2003) and the generation of allospecific Tregs during tolerance induction has been demonstrated to be dependent upon intact TIM-3:galectin-9 pathway (Sabatos et al., 2003; Sanchez-Fueyo et al., 2003). In a murine model of islet cell transplantation, tolerant allografts displayed blunted intra-graft expression of TIM-3 and IFN-γ as compared with rejected grafts (Sabatos et al., 2003).
In fact, TIM-3 broadly modulates the alloresponse in vivo. In a fully mismatched model of chronic cardiac allograft rejection, accelerated graft loss is observed with TIM-3 blockade (Boenisch et al., 2010). This rejection is characterized by increased donor specific alloantibody production, increased Th1 and Th17 polarization, and suppression of adaptive Treg induction (Boenisch et al., 2010). Administration of exogenous stable galectin-9 has been used to promote TIM-3 signaling in experimental models. Treatment has been associated with prolonged allograft survival of both skin and cardiac allografts (Wang et al., 2008; He et al., 2009). These studies used models of both acute and chronic allograft rejection, and all consistently demonstrated that TIM-3:galectin-9 interaction leads to Th1 and Th17 suppression while promoting Treg differentiation (Wang et al., 2008; He et al., 2009).
TIM-3 in Human Transplant Recipients
In human transplant recipients, TIM-3 may be helpful as a biomarker of Th1 activation and rejection. Small studies indicate that TIM-3 mRNA levels are significantly higher within rejecting allografts and that there is a strong correlation between intra-graft TIM-3 and IFN-γ levels. Interestingly, treatment-refractory rejection episodes showed relatively lower levels of TIM-3, suggesting a link between lack of negative costimulatory signaling and poorer allograft outcomes (Ponciano et al., 2007). In addition, measurement of TIM-3 mRNA in urine and blood have proved accurate in the differentiation of delayed graft function (DGF) with acute tubular necrosis versus DGF with acute rejection (Manfro et al., 2008). Patients with acute rejection showed much higher urine TIM-3 mRNA levels than those with other causes of allograft dysfunction or non-rejecting controls (Renesto et al., 2007; Manfro et al., 2008). While larger studies will be needed to validate these findings, these data suggest potential utility of TIM-3 mRNA measurement as a non-invasive tool in the diagnosis of allograft dysfunction.
Other Functions of TIM-3
TIM-3 also functions as a phosphatidylserine receptor and mediates phagocytosis of apoptotic cells (Nakayama et al., 2009). Blockade of this phagocytic capacity has been shown to promote autoantibody production and impair cross presentation by CD8+ DCs (Nakayama et al., 2009). As discussed in greater detail below, increased TIM-3 expression on CD8+ T cells has been associated with an exhausted phenotype, with decreased viral clearance and reduced anti-tumor immunity (Golden-Mason et al., 2009; Mengshol et al., 2010; Sehrawat et al., 2010). The mechanisms involved, including the role of galectin-9 in these conditions, remain unclear at this time.
Conclusions
Taken together, experimental data indicate that TIM-3:galectin-9 plays a central role in regulation of Th1 responses including alloimmune responses. Through signaling on Tregs, TIM-3 is involved in the generation of antigen specific tolerance. Administration of galectin-9 has promoted allograft survival in both a stringent skin graft model and a vascularized model of transplantation. Human studies correlate TIM-3 levels with alloimmune activation and risk of rejection. These data indicate that targeting this pathway pharmacologically may hold significant promise in the management of detrimental alloresponses (Wang et al., 2008; He et al., 2009). However, several important issues remain to be clarified. In particular, the impact of targeting TIM-3 on phagocytosis of apoptotic bodies and maintenance of peripheral tolerance, and the relative importance of galectin-9 expression on donor and recipient tissues in modulating the immune response, are unclear. Furthermore, galectin-9 may also function through other, as yet unidentified, receptors on T cells, which may contribute to the observed effects (Zhu et al., 2005; Bi et al., 2008).
T Cell Exhaustion, Negative Costimulation, and Transplantation
T cell exhaustion is a state of T cell dysfunction characterized by progressive loss of proliferative and effector functions, culminating in clonal deletion (Virgin et al., 2009). It is most commonly seen in chronic viral infections and malignancies and is thought to relate to chronic antigenic stimulation (Wherry et al., 2003). There appears to be a link between the strength of antigenic stimulation and degree of T cell exhaustion, where larger amounts of epitopes lead to more severe degrees of exhaustion, even with similar viral loads. To date, it has been most widely studied in CD8+ T cells but is also thought to affect CD4+ T cells. Immune regulation is central to T cell exhaustion and expression of coinhibitory molecules appears to be a crucial feature. Exhausted T cells display high expression of PD-1 (Barber et al., 2006) but increased CD160, TIM-3, and CTLA-4 expression have all been described on virus-specific CD8+ T cells in chronic infection (Crawford and Wherry, 2009). Furthermore, studies from the cancer literature have reported reversal of T cell exhaustion through blockade of TIM-3 and PD-1 signaling (Sakuishi et al., 2010).
The role of T cell exhaustion in solid organ transplantation has not yet been widely studied. Several groups have reported on the impact of alloantigen load on alloreactive CD8+ T cells and demonstrated evidence of T cell exhaustion with decreased proliferative capacity and reduced effector cytokine production (Quezada et al., 2003; Steger et al., 2008). However, the expression of coinhibitory molecules was not examined in these studies. The true impact of T cell exhaustion on allograft outcomes remains unclear and whether manipulation of negative costimulatory pathways could lead to exhaustion of specific alloreactive T cell clones is unknown but is a fascinating potential research avenue.
Summary and Future Directions
With widespread expression across immune cells and tissues, negative costimulatory molecules are an appealing target for allospecific tolerance induction. An increasing body of literature outlines the complexity of these pathways, involving variable expression of receptors and ligands but also overlapping functions and interactions of different pathways.
In terms of hierarchy of costimulatory molecules, CD28 occupies a central role in T cell activation and therefore in opposing this, CTLA-4 is considered the most important coinhibitory molecule. CTLA-4-Ig blocks CD28-mediated costimulation through competitive binding to B7, but does not consistently induce allograft tolerance across varied transplant models. This observation is thought to be due to the presence of other positive costimulatory pathways unaffected by CTLA-4-Ig, but also may represent inadvertent blockade of tolerogenic CTLA-4 signals. To begin to address this issue, several groups are currently working on antibodies which can block CD28 specifically, without affecting CTLA-4 function. Recent non-human primate studies of transplantation using a novel anti-CD28 appear to hold some promise in this area. However, there is likely to be a requirement for concomitant negative costimulatory signals to induce robust allospecific tolerance. A more complete understanding of the mechanisms of action of CTLA-4, as provided by recent landmark studies, will help to inform and guide further investigation in this area.
Studies of the PD-1 pathways have greatly advanced our understanding of costimulation and the importance of tissue-expressed ligands in T cell activation and directing the alloresponse. Antibody blockade of both PD-1 and PD-L1 has been shown to accelerate allograft rejection, associated with increased proliferation of allospecific CD4+ T cells. Prolonged allograft survival is seen after administration of a PD-1 agonist in the setting of CD28 deficiency; suggesting that this may be a promising therapy to combine with blockade of CD28. Tissue expression of PD-L1 also appears to play an important role in directing the alloresponse, as indicated by data showing donor PD-L1 deficiency leads to accelerated allograft rejection and tolerance induced by CTLA-4-Ig appears to be dependent on PD-L1 on donor endothelium.
B7-H3 and B7-H4 have not been extensively investigated in alloimmunity but current data suggests these may play an important role in fine-tuning the immune responses in the setting of limited CD28-mediated stimulation. In particular, B7-H3 appears to negatively regulate T cell responses under Th1 polarizing conditions and manipulation of this pathway may prove useful to prevent rejection, a predominantly Th1 mediated process. However, as described above, the current data is conflicting and these findings need to be resolved to fully assess the role of B7-H3 in alloimmunity. In addition to negative costimulation, B7-H4 also appears to promote Treg function by increasing IL-10 production. B7-H4 may also prove a useful pathway to target, as adjunctive therapy with CD28 blockade.
Furthermore, there is broad similarity in the expression of B7-H3, B7-H4, PDL-1, and PDL-2 in lymphoid and non-lymphoid tissues, and their receptors are all expressed on activated T cells. These features suggest a role for these molecules in regulating activation of naïve T cells in lymphoid tissue and effector T cell function in the periphery. Signals transmitted by these B7 ligands may be tissue specific and regulated by inflammatory cytokines; thereby influencing the nature and extent of T cell function. Therapeutic strategies aimed at manipulating expression of or signaling through these molecules locally either within the graft or draining lymph nodes may be important future avenues to promote allospecific tolerance without systemic immunosuppression.
Through limiting the development of GVHD and promoting engraftment, manipulation of BTLA signaling may prove an important strategy in the management of stem cell transplantation. The data from islet cell transplantation is also quite promising but these models do not address the complex interactions with other ligands such as LIGHT. Of particular interest, there appears to be interaction between BTLA and PD-1 in the induction of tolerance. BTLA−/− mice show greater induction of PD-1 expression and improved allograft survival, in a PD-1 dependent manner. This relationship warrants further exploration and may prove a useful link between these coinhibitory pathways. However, the complex interactions between the different molecules involved in BTLA:HVEM signaling and the potential for bidirectional signaling, render this a challenging pathway to exploit. At this time, much remains to be understood before any potential agent could be brought closer to clinical applications.
TIM-3 and its ligand, galectin-9, are exciting new players in the field of coinhibition. Recent data demonstrates the ability of TIM-3 to broadly modulate the immune response. Administration of galectin-9 has been demonstrated to prolong allograft survival with decreases in Th1 and Th17 alloimmunity and increased allospecific Treg generation. Furthermore, TIM-3 blockade has been shown to prevent tolerance induction. These data suggest a central role for TIM-3 in regulation of tolerance and that promotion of TIM-3 signaling, perhaps through administration of exogenous stable galectin-9, may prove an additional important tool in the quest to induce tolerance in transplant recipients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grant funding from the National Institutes of Health (R01AI070820 and R56 AI089777-01A1; Nader Najafian) and the American Society of Transplantation (Basic Science Fellowship grant; Martina M. McGrath).
References
Afaneh, C., Muthukumar, T., Lubetzky, M., Ding, R., Snopkowski, C., Sharma, V. K., Seshan, S., Dadhania, D., Schwartz, J. E., and Suthanthiran, M. (2010). Urinary cell levels of mRNA for OX40, OX40L, PD-1, PD-L1, or PD-L2 and acute rejection of human renal allografts. Transplantation 90, 1381–1387.
Albring, J. C., Sandau, M. M., Rapaport, A. S., Edelson, B. T., Satpathy, A., Mashayekhi, M., Lathrop, S. K., Hsieh, C. S., Stelljes, M., Colonna, M., Murphy, T. L., and Murphy, K. M. (2010). Targeting of B and T lymphocyte associated (BTLA) prevents graft-versus-host disease without global immunosuppression. J. Exp. Med. 207, 2551–2559.
Anderson, A. C., Anderson, D. E., Bregoli, L., Hastings, W. D., Kassam, N., Lei, C., Chandwaskar, R., Karman, J., Su, E. W., Hirashima, M., Bruce, J. N., Kane, L. P., Kuchroo, V. K., and Hafler, D. A. (2007). Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318, 1141–1143.
Ansari, M. J., Salama, A. D., Chitnis, T., Smith, R. N., Yagita, H., Akiba, H., Yamazaki, T., Azuma, M., Iwai, H., Khoury, S. J., Auchincloss, H. Jr., and Sayegh, M. H. (2003). The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 198, 63–69.
Araki, M., Chung, D., Liu, S., Rainbow, D. B., Chamberlain, G., Garner, V., Hunter, K. M., Vijayakrishnan, L., Peterson, L. B., Oukka, M., Sharpe, A. H., Sobel, R., Kuchroo, V. K., and Wicker, L. S. (2009). Genetic evidence that the differential expression of the ligand-independent isoform of CTLA-4 is the molecular basis of the Idd5.1 type 1 diabetes region in nonobese diabetic mice. J. Immunol. 183, 5146–5157.
Azuma, H., Chandraker, A., Nadeau, K., Hancock, W. W., Carpenter, C. B., Tilney, N. L., and Sayegh, M. H. (1996). Blockade of T-cell costimulation prevents development of experimental chronic renal allograft rejection. Proc. Natl. Acad. Sci. U.S.A. 93, 12439–12444.
Barber, D. L., Wherry, E. J., Masopust, D., Zhu, B., Allison, J. P., Sharpe, A. H., Freeman, G. J., and Ahmed, R. (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687.
Barrat, F. J., Le Deist, F., Benkerrou, M., Bousso, P., Feldmann, J., Fischer, A., and de Saint Basile, G. (1999). Defective CTLA-4 cycling pathway in Chediak-Higashi syndrome: a possible mechanism for deregulation of T lymphocyte activation. Proc. Natl. Acad. Sci. U.S.A. 96, 8645–8650.
Bi, S., Earl, L. A., Jacobs, L., and Baum, L. G. (2008). Structural features of galectin-9 and galectin-1 that determine distinct T cell death pathways. J. Biol. Chem. 283, 12248–12258.
Boenisch, O., D’Addio, F., Watanabe, T., Elyaman, W., Magee, C. N., Yeung, M. Y., Padera, R. F., Rodig, S. J., Murayama, T., Tanaka, K., Yuan, X., Ueno, T., Jurisch, A., Mfarrej, B., Akiba, H., Yagita, H., and Najafian, N. (2010). TIM-3: a novel regulatory molecule of alloimmune activation. J. Immunol. 185, 5806–5819.
Butte, M. J., Keir, M. E., Phamduy, T. B., Sharpe, A. H., and Freeman, G. J. (2007). Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122.
Cai, G., Anumanthan, A., Brown, J. A., Greenfield, E. A., Zhu, B., and Freeman, G. J. (2008). CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat. Immunol. 9, 176–185.
Chandraker, A., Azuma, H., Nadeau, K., Carpenter, C. B., Tilney, N. L., Hancock, W. W., and Sayegh, M. H. (1998). Late blockade of T cell costimulation interrupts progression of experimental chronic allograft rejection. J. Clin. Invest. 101, 2309–2318.
Chandraker, A., Huurman, V., Hallett, K., Yuan, X., Tector, A.J., Park, C. H., Lu, E., Zavazava, N., and Oaks, M. (2005). CTLA-4 is important in maintaining long-term survival of cardiac allografts. Transplantation 79, 897–903.
Chapoval, A. I., Ni, J., Lau, J. S., Wilcox, R. A., Flies, D. B., Liu, D., Dong, H., Sica, G. L., Zhu, G., Tamada, K., and Chen, L. (2001). B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2, 269–274.
Cheung, T. C., Humphreys, I. R., Potter, K. G., Norris, P. S., Shumway, H. M., Tran, B. R., Patterson, G., Jean-Jacques, R., Yoon, M., Spear, P. G., Murphy, K. M., Lurain, N. S., Benedict, C. A., and Ware, C. F. (2005). Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc. Natl. Acad. Sci. U.S.A. 102, 13218–13223.
Cheung, T. C., Steinberg, M. W., Oborne, L. M., Macauley, M. G., Fukuyama, S., Sanjo, H., D’Souza, C., Norris, P. S., Pfeffer, K., Murphy, K. M., Kronenberg, M., Spear, P. G., and Ware, C. F. (2009). Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc. Natl. Acad. Sci. U.S.A. 106, 6244–6249.
Cohavy, O., Zhou, J., Ware, C. F., and Targan, S. R. (2005). LIGHT is constitutively expressed on T and NK cells in the human gut and can be induced by CD2-mediated signaling. J. Immunol. 174, 646–653.
Crawford, A., and Wherry, E. J. (2009). The diversity of costimulatory and inhibitory receptor pathways and the regulation of antiviral T cell responses. Curr. Opin. Immunol. 21, 179–186.
Del Rio, M. L., Kurtz, J., Perez-Martinez, C., Ghosh, A., Perez-Simon, J. A., and Rodriguez-Barbosa, J. I. (2011). B- and T lymphocyte attenuator targeting protects against the acute phase of graft versus host reaction by inhibiting donor anti-host cytotoxicity. Transplantation 92, 1085–1093.
Deppong, C., Juehne, T. I., Hurchla, M., Friend, L. D., Shah, D. D., Rose, C. M., Bricker, T. L., Shornick, L. P., Crouch, E. C., Murphy, T. L., Holtzman, M. J., Murphy, K. M., and Green, J. M. (2006). Cutting edge: B and T lymphocyte attenuator and programmed death receptor-1 inhibitory receptors are required for termination of acute allergic airway inflammation. J. Immunol. 176, 3909–3913.
Duhen, T., Pasero, C., Mallet, F., Barbarat, B., Olive, D., and Costello, R. T. (2004). LIGHT costimulates CD40 triggering and induces immunoglobulin secretion; a novel key partner in T cell-dependent B cell terminal differentiation. Eur. J. Immunol. 34, 3534–3541.
Egen, J. G., and Allison, J. P. (2002). Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 16, 23–35.
Fallarino, F., Grohmann, U., Hwang, K. W., Orabona, C., Vacca, C., Bianchi, R., Belladonna, M. L., Fioretti, M. C., Alegre, M. L., and Puccetti, P. (2003). Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 4, 1206–1212.
Fecteau, S., Basadonna, G. P., Freitas, A., Ariyan, C., Sayegh, M. H., and Rothstein, D. M. (2001). CTLA-4 up-regulation plays a role in tolerance mediated by CD45. Nat. Immunol. 2, 58–63.
Fife, B. T., and Bluestone, J. A. (2008). Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 224, 166–182.
Fife, B. T., Pauken, K. E., Eagar, T. N., Obu, T., Wu, J., Tang, Q., Azuma, M., Krummel, M. F., and Bluestone, J. A. (2009). Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 10, 1185–1192.
Freeman, G. J., Casasnovas, J. M., Umetsu, D. T., and DeKruyff, R. H. (2010). TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 235, 172–189.
Freeman, G. J., Long, A. J., Iwai, Y., Bourque, K., Chernova, T., Nishimura, H., Fitz, L. J., Malenkovich, N., Okazaki, T., Byrne, M. C., Horton, H. F., Fouser, L., Carter, L., Ling, V., Bowman, M. R., Carreno, B. M., Collins, M., Wood, C. R., and Honjo, T. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034.
Gao, W., Demirci, G., Strom, T. B., and Li, X. C. (2003). Stimulating PD-1-negative signals concurrent with blocking CD154 co-stimulation induces long-term islet allograft survival. Transplantation 76, 994–999.
Golden-Mason, L., Palmer, B. E., Kassam, N., Townshend-Bulson, L., Livingston, S., McMahon, B. J., Castelblanco, N., Kuchroo, V., Gretch, D. R., and Rosen, H. R. (2009). Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 83, 9122–9130.
Greenwald, R. J., Freeman, G. J., and Sharpe, A. H. (2005) The B7 family revisited. Annu. Rev. Immunol. 23, 515–548.
Grohmann, U., Orabona, C., Fallarino, F., Vacca, C., Calcinaro, F., Falorni, A., Candeloro, P., Belladonna, M. L., Bianchi, R., Fioretti, M. C., and Puccetti, P. (2002). CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3, 1097–1101.
Habicht, A., Kewalaramani, R., Vu, M. D., Demirci, G., Blazar, B. R., Sayegh, M. H., and Li, X. C. (2007). Striking dichotomy of PD-L1 and PD-L2 pathways in regulating alloreactive CD4(+) and CD8(+) T cells in vivo. Am. J. Transplant. 7, 2683–2692.
Hashiguchi, M., Kobori, H., Ritprajak, P., Kamimura, Y., Kozono, H., and Azuma, M. (2008). Triggering receptor expressed on myeloid cell-like transcript 2 (TLT-2) is a counter-receptor for B7-H3 and enhances T cell responses. Proc. Natl. Acad. Sci. U.S.A. 105, 10495–10500.
Hastings, W. D., Anderson, D. E., Kassam, N., Koguchi, K., Greenfield, E. A., Kent, S. C., Zheng, X. X., Strom, T. B., Hafler, D. A., and Kuchroo, V. K. (2009). TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur. J. Immunol. 39, 2492–2501.
He, W., Fang, Z., Wang, F., Wu, K., Xu, Y., Zhou, H., Du, D., Gao, Y., Zhang, W. N., Niki, T., Hirashima, M., Yuan, J., and Chen, Z. K. (2009). Galectin-9 significantly prolongs the survival of fully mismatched cardiac allografts in mice. Transplantation 88, 782–790.
Hofmeyer, K. A., Ray, A., and Zang, X. (2008). The contrasting role of B7-H3. Proc. Natl. Acad. Sci. U.S.A. 105, 10277–10278.
Hurchla, M. A., Sedy, J. R., Gavrieli, M., Drake, C. G., Murphy, T. L., and Murphy, K. M. (2005). B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly induced in anergic CD4+ T cells. J. Immunol. 174, 3377–3385.
Hurchla, M. A., Sedy, J. R., and Murphy, K. M. (2007). Unexpected role of B and T lymphocyte attenuator in sustaining cell survival during chronic allostimulation. J. Immunol. 178, 6073–6082.
Ito, T., Ueno, T., Clarkson, M. R., Yuan, X., Jurewicz, M. M., Yagita, H., Azuma, M., Sharpe, A. H., Auchincloss, H. Jr., Sayegh, M. H., and Najafian, N. (2005). Analysis of the role of negative T cell costimulatory pathways in CD4 and CD8 T cell-mediated alloimmune responses in vivo. J. Immunol. 174, 6648–6656.
Kashio, Y., Nakamura, K., Abedin, M. J., Seki, M., Nishi, N., Yoshida, N., Nakamura, T., and Hirashima, M. (2003). Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J. Immunol. 170, 3631–3636.
Keir, M. E., Butte, M. J., Freeman, G. J., and Sharpe, A. H. (2008). PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704.
Keir, M. E., Liang, S. C., Guleria, I., Latchman, Y. E., Qipo, A., Albacker, L. A., Koulmanda, M., Freeman, G. J., Sayegh, M. H., and Sharpe, A. H. (2006). Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 203, 883–895.
Kim, H. J., Jeong, K. H., Lee, S. H., Moon, J. Y., Lee, T. W., Kang, S. W., Park, S. J., Kim, Y. H., and Chung, J. H. (2010). Polymorphisms of the CTLA4 gene and kidney transplant rejection in Korean patients. Transpl. Immunol. 24, 40–44.
Kitazawa, Y., Fujino, M., Wang, Q., Kimura, H., Azuma, M., Kubo, M., Abe, R., and Li, X. K. (2007). Involvement of the programmed death-1/programmed death-1 ligand pathway in CD4+CD25+ regulatory T-cell activity to suppress alloimmune responses. Transplantation 83, 774–782.
Krieg, C., Han, P., Stone, R., Goularte, O. D., and Kaye, J. (2005). Functional analysis of B and T lymphocyte attenuator engagement on CD4+ and CD8+ T cells. J. Immunol. 175, 6420–6427.
Kryczek, I., Wei, S., Zou, L., Zhu, G., Mottram, P., Xu, H., Chen, L., and Zou, W. (2006). Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. J. Immunol. 177, 40–44.
Kwon, B. S., Tan, K. B., Ni, J., Oh, K. O., Lee, Z. H., Kim, K. K., Kim, Y. J., Wang, S., Gentz, R., Yu, G. L., Harrop, J., Lyn, S. D., Silverman, C., Porter, T. G., Truneh, A., and Young, P. R. (1997). A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J. Biol. Chem. 272, 14272–14276.
Larsen, C. P., Elwood, E. T., Alexander, D. Z., Ritchie, S. C., Hendrix, R., Tucker-Burden, C., Cho, H. R., Aruffo, A., Hollenbaugh, D., Linsley, P. S., Winn, K. J., and Pearson, T. C. (1996). Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature 381, 434–438.
Lazarovits, A. I., Poppema, S., Zhang, Z., Khandaker, M., Le Feuvre, C. E., Singhal, S.K., Garcia, B. M., Ogasa, N., Jevnikar, A. M., White, M. H., Singh, G., Stiller, C. R., and Zhong, R. Z. (1996). Prevention and reversal of renal allograft rejection by antibody against CD45RB. Nature 380, 717–720.
Leitner, J., Klauser, C., Pickl, W. F., Stöckl, J., Majdic, O., Bardet, A. F., Kreil, D. P., Dong, C., Yamazaki, T., Zlabinger, G., Pfistershammer, K., and Steinberger, P. (2009). B7-H3 is a potent inhibitor of human T-cell activation: no evidence for B7-H3 and TREML2 interaction. Eur. J. Immunol. 39, 1754–1764.
Lenschow, D. J., Zeng, Y., Thistlethwaite, J. R., Montag, A., Brady, W., Gibson, M. G., Linsley, P. S., and Bluestone, J. A. (1992). Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science 257, 789–792.
Lin, H., Bolling, S. F., Linsley, P. S., Wei, R. Q., Gordon, D., Thompson, C. B., and Turka, L. A. (1993). Long-term acceptance of major histocompatibility complex mismatched cardiac allografts induced by CTLA4Ig plus donor-specific transfusion. J. Exp. Med. 178, 1801–1806.
Lin, H., Rathmell, J. C., Gray, G. S., Thompson, C. B., Leiden, J. M., and Alegre, M. L. (1998). Cytotoxic T lymphocyte antigen 4 (CTLA4) blockade accelerates the acute rejection of cardiac allografts in CD28-deficient mice: CTLA4 can function independently of CD28. J. Exp. Med. 188, 199–204.
Lin, S. C., Kuo, C. C., and Chan, C. H. (2006). Association of a BTLA gene polymorphism with the risk of rheumatoid arthritis. J. Biomed. Sci. 13, 853–860.
Linsley, P. S., Bradshaw, J., Greene, J., Peach, R., Bennett, K. L., and Mittler, R. S. (1996). Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 4, 535–543.
Maeda, M., Carpenito, C., Russell, R. C., Dasanjh, J., Veinotte, L. L., Ohta, H., Yamamura, T., Tan, R., and Takei, F. (2005). Murine CD160, Ig-like receptor on NK cells and NKT cells, recognizes classical and nonclassical MHC class I and regulates NK cell activation. J. Immunol. 175, 4426–4432.
Magistrelli, G., Jeannin, P., Herbault, N., Benoit De Coignac, A., Gauchat, J. F., Bonnefoy, J. Y., and Delneste, Y. (1999). A soluble form of CTLA-4 generated by alternative splicing is expressed by nonstimulated human T cells. Eur. J. Immunol. 29, 3596–3602.
Manfro, R. C., Aquino-Dias, E. C., Joelsons, G., Nogare, A. L., Carpio, V. N., and Goncalves, L. F. (2008). Noninvasive Tim-3 messenger RNA evaluation in renal transplant recipients with graft dysfunction. Transplantation 86, 1869–1874.
Marder, B. A., Schroppel, B., Lin, M., Schiano, T., Parekh, R., Tomer, Y., and Murphy, B. (2003). The impact of costimulatory molecule gene polymorphisms on clinical outcomes in liver transplantation. Am. J. Transplant. 3, 424–431.
Markees, T. G., Phillips, N. E., Gordon, E. J., Noelle, R. J., Shultz, L. D., Mordes, J. P., Greiner, D. L., and Rossini, A. A. (1998). Long-term survival of skin allografts induced by donor splenocytes and anti-CD154 antibody in thymectomized mice requires CD4(+) T cells, interferon-gamma, and CTLA4. J. Clin. Invest. 101, 2446–2455.
Marsters, S. A., Ayres, T. M., Skubatch, M., Gray, C. L., Rothe, M., and Ashkenazi, A. (1997). Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-kappaB and AP-1. J. Biol. Chem. 272, 14029–14032.
Mauri, D. N., Ebner, R., Montgomery, R. I., Kochel, K. D., Cheung, T. C., Yu, G. L., Ruben, S., Murphy, M., Eisenberg, R. J., Cohen, G. H., Spear, P. G., and Ware, C. F. (1998). LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8, 21–30.
Mead, K. I., Zheng, Y., Manzotti, C. N., Perry, L. C., Liu, M. K., Burke, F., Powner, D. J., Wakelam, M. J., and Sansom, D. M. (2005). Exocytosis of CTLA-4 is dependent on phospholipase D and ADP ribosylation factor-1 and stimulated during activation of regulatory T cells. J. Immunol. 174, 4803–4811.
Mellor, A. L., Chandler, P., Baban, B., Hansen, A. M., Marshall, B., Pihkala, J., Waldmann, H., Cobbold, S., Adams, E., and Munn, D. H. (2004). Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2,3 dioxygenase. Int. Immunol. 16, 1391–1401.
Mengshol, J. A., Golden-Mason, L., Arikawa, T., Smith, M., Niki, T., McWilliams, R., Randall, J. A., McMahan, R., Zimmerman, M. A., Rangachari, M., Dobrinskikh, E., Busson, P., Polyak, S. J., Hirashima, M., and Rosen, H. R. (2010). A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS ONE 5, e9504. doi: 10.1371/journal.pone.0009504
Monney, L., Sabatos, C. A., Gaglia, J. L., Ryu, A., Waldner, H., Chernova, T., Manning, S., Greenfield, E. A., Coyle, A. J., Sobel, R. A., Freeman, G. J., and Kuchroo, V. K. (2002). Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541.
Morel, Y., Schiano de Colella, J. M., Harrop, J., Deen, K. C., Holmes, S. D., Wattam, T. A., Khandekar, S. S., Truneh, A., Sweet, R. W., Gastaut, J. A., Olive, D., and Costello, R. T. (2000). Reciprocal expression of the TNF family receptor herpes virus entry mediator and its ligand LIGHT on activated T cells: LIGHT down-regulates its own receptor. J. Immunol. 165, 4397–4404.
Morita, M., Fujino, M., Jiang, G., Kitazawa, Y., Xie, L., Azuma, M., Yagita, H., Nagao, S., Sugioka, A., Kurosawa, Y., Takahara, S., Fung, J., Qian, S., Lu, L., and Li, X. K. (2010). PD-1/B7-H1 interaction contribute to the spontaneous acceptance of mouse liver allograft. Am. J. Transplant. 10, 40–46.
Munn, D. H., Shafizadeh, E., Attwood, J. T., Bondarev, I., Pashine, A., and Mellor, A. L. (1999). Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 189, 1363–1372.
Murphy, K. M., Nelson, C. A., and Sedy, J. R. (2006). Balancing co-stimulation and inhibition with BTLA and HVEM. Nat. Rev. Immunol. 6, 671–681.
Murphy, T. L., and Murphy, K. M. (2010). Slow down and survive: enigmatic immunoregulation by BTLA and HVEM. Annu. Rev. Immunol. 28, 389–411.
Muthukumarana, P. A., Zheng, X. X., Rosengard, B. R., Strom, T. B., and Metcalfe, S. M. (2008). In primed allo-tolerance, TIM-3-Ig rapidly suppresses TGFbeta, but has no immediate effect on Foxp3. Transpl. Int. 21, 593–597.
Nakayama, M., Akiba, H., Takeda, K., Kojima, Y., Hashiguchi, M., Azuma, M., Yagita, H., and Okumura, K. (2009). Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113, 3821–3830.
Okazaki, T., Maeda, A., Nishimura, H., Kurosaki, T., and Honjo, T. (2001). PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. U.S.A. 98, 13866–13871.
Oki, M., Watanabe, N., Owada, T., Oya, Y., Ikeda, K., Saito, Y., Matsumura, R., Seto, Y., Iwamoto, I., and Nakajima, H. (2011). A functional polymorphism in B and T lymphocyte attenuator is associated with susceptibility to rheumatoid arthritis. Clin. Dev. Immunol. 305656.
Oya, Y., Watanabe, N., Owada, T., Oki, M., Hirose, K., Suto, A., Kagami, S., Nakajima, H., Kishimoto, T., Iwamoto, I., Murphy, T. L., Murphy, K. M., and Saito, Y. (2008). Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum. 58, 2498–2510.
Ozkaynak, E., Wang, L., Goodearl, A., McDonald, K., Qin, S., O’Keefe, T., Duong, T., Smith, T., Gutierrez-Ramos, J. C., Rottman, J. B., Coyle, A. J., and Hancock, W. W. (2002). Programmed death-1 targeting can promote allograft survival. J. Immunol. 169, 6546–6553.
Parry, R. V., Chemnitz, J. M., Frauwirth, K. A., Lanfranco, A. R., Braunstein, I., Kobayashi, S. V., Linsley, P. S., Thompson, C. B., and Riley, J. L. (2005). CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 25, 9543–9553.
Paust, S., Lu, L., McCarty, N., and Cantor, H. (2004). Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc. Natl. Acad. Sci. U.S.A. 101, 10398–10403.
Poirier, N., Azimzadeh, A. M., Zhang, T., Dilek, N., Mary, C., Nguyen, B., Tillou, X., Wu, G., Reneaudin, K., Hervouet, J., Martinet, B., Coulon, F., Allain-Launay, E., Karam, G., Soulillou, J. P., Pierson, R. N. III, Blancho, G., and Vanhove, B. (2010). Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci. Transl. Med. 2, 17ra0.
Ponciano, V. C., Renesto, P. G., Nogueira, E., Rangel, E. B., Cenedeze, M. A., Franco, M. F., Câmara, N. O., and Pacheco-Silva, A. (2007). Tim-3 expression in human kidney allografts. Transpl. Immunol. 17, 215–222.
Prasad, D. V., Nguyen, T., Li, Z., Yang, Y., Duong, J., Wang, Y., and Dong, C. (2004). Murine B7-H3 is a negative regulator of T cells. J. Immunol. 173, 2500–2506.
Prasad, D. V., Richards, S., Mai, X. M., and Dong, C. (2003). B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity 18, 863–873.
Quezada, S. A., Fuller, B., Jarvinen, L. Z., Gonzalez, M., Blazar, B. R., Rudensky, A. Y., Strom, T. B., and Noelle, R. J. (2003). Mechanisms of donor-specific transfusion tolerance: preemptive induction of clonal T-cell exhaustion via indirect presentation. Blood 102, 1920–1926.
Qureshi, O. S., Zheng, Y., Nakamura, K., Attridge, K., Manzotti, C., Schmidt, E. M., Baker, J., Jeffery, L. E., Kaur, S., Briggs, Z., Hou, T. Z., Futter, C. E., Anderson, G., Walker, L. S., and Sansom, D. M. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603.
Raimondi, G., Shufesky, W. J., Tokita, D., Morelli, A. E., and Thomson, A. W. (2006). Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J. Immunol. 176, 2808–2816.
Renesto, P. G., Ponciano, V. C., Cenedeze, M. A., Saraiva Camara, N. O., and Pacheco-Silva, A. (2007). High expression of Tim-3 mRNA in urinary cells from kidney transplant recipients with acute rejection. Am. J. Transplant. 7, 1661–1665.
Riella, L. V., Liu, T., Yang, J., Chock, S., Shimizu, T., Mfarrej, B., Batal, I., Xiao, X., Sayegh, M. H., and Chandraker, A. (2012). Deleterious effect of CTLA4-Ig on a treg-dependent transplant model. Am. J. Transplant. doi: 10.1111/j.1600-6143.2011.03929.x. [Epub ahead of print].
Riella, L. V., Watanabe, T., Sage, P. T., Yang, J., Yeung, M., Azzi, J., Vanguri, V., Chandraker, A., Sharpe, A. H., Sayegh, M. H., and Najafian, N. (2011). Essential role of PDL1 expression on nonhematopoietic donor cells in acquired tolerance to vascularized cardiac allografts. Am. J. Transplant. 11, 832–840.
Rooney, I. A., Butrovich, K. D., Glass, A. A., Borboroglu, S., Benedict, C. A., Whitbeck, J. C., Cohen, G. H., Eisenberg, R. J., and Ware, C. F. (2000). The lymphotoxin-beta receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. J. Biol. Chem. 275, 14307–14315.
Russell, M. E., Hancock, W. W., Akalin, E., Wallace, A. F., Glysing-Jensen, T., Willett, T. A., and Sayegh, M. H. (1996). Chronic cardiac rejection in the LEW to F344 rat model. Blockade of CD28-B7 costimulation by CTLA4Ig modulates T cell and macrophage activation and attenuates arteriosclerosis. J. Clin. Invest. 97, 833–838.
Sabatos, C. A., Chakravarti, S., Cha, E., Schubart, A., Sánchez-Fueyo, A., Zheng, X. X., Coyle, A. J., Strom, T. B., Freeman, G. J., and Kuchroo, V. K. (2003). Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 4, 1102–1110.
Sakoda, Y., Park, J. J., Zhao, Y., Kuramasu, A., Geng, D., Liu, Y., Davila, E., and Tamada, K. (2011). Dichotomous regulation of GVHD through bidirectional functions of the BTLA-HVEM pathway. Blood 117, 2506–2514.
Sakuishi, K., Apetoh, L., Sullivan, J. M., Blazar, B. R., Kuchroo, V. K., and Anderson, A. C. (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194.
Salama, A. D., Chitnis, T., Imitola, J., Ansari, M. J., Akiba, H., Tushima, F., Azuma, M., Yagita, H., Sayegh, M. H., and Khoury, S. J. (2003). Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 198, 71–78.
Salio, M., Cella, M., Suter, M., and Lanzavecchia, A. (1999). Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 29, 3245–3253.
Sanchez-Fueyo, A., Tian, J., Picarella, D., Domenig, C., Zheng, X. X., Sabatos, C. A., Manlongat, N., Bender, O., Kamradt, T., Kuchroo, V. K., Gutiérrez-Ramos, J. C., Coyle, A. J., and Strom, T. B. (2003). Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 4, 1093–1101.
Sandner, S. E., Clarkson, M. R., Salama, A. D., Sanchez-Fueyo, A., Domenig, C., Habicht, A., Najafian, N., Yagita, H., Azuma, M., Turka, L. A., and Sayegh, M. H. (2005). Role of the programmed death-1 pathway in regulation of alloimmune responses in vivo. J. Immunol. 174, 3408–3415.
Schneider, H., Downey, J., Smith, A., Zinselmeyer, B. H., Rush, C., Brewer, J. M., Wei, B., Hogg, N., Garside, P., and Rudd, C. E. (2006). Reversal of the TCR stop signal by CTLA-4. Science 313, 1972–1975.
Schneider, H., Smith, X., Liu, H., Bismuth, G., and Rudd, C. E. (2008). CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur. J. Immunol. 38, 40–47.
Sedy, J. R., Gavrieli, M., Potter, K. G., Hurchla, M. A., Lindsley, R. C., Hildner, K., Scheu, S., Pfeffer, K., Ware, C. F., Murphy, T. L., and Murphy, K. M. (2005). B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 6, 90–98.
Sehrawat, S., Reddy, P. B., Rajasagi, N., Suryawanshi, A., Hirashima, M., and Rouse, B. T. (2010). Galectin-9/TIM-3 interaction regulates virus-specific primary and memory CD8 T cell response. PLoS Pathog. 6, e1000882. doi: 10.1371/journal.ppat.1000882
Seki, M., Oomizu, S., Sakata, K. M., Sakata, A., Arikawa, T., Watanabe, K., Ito, K., Takeshita, K., Niki, T., Saita, N., Nishi, N., Yamauchi, A., Katoh, S., Matsukawa, A., Kuchroo, V., and Hirashima, M. (2008). Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin. Immunol. 127, 78–88.
Shi, G., Luo, H., Wan, X., Salcedo, T. W., Zhang, J., and Wu, J. (2002). Mouse T cells receive costimulatory signals from LIGHT, a TNF family member. Blood 100, 3279–3286.
Sica, G. L., Choi, I. H., Zhu, G., Tamada, K., Wang, S. D., Tamura, H., Chapoval, A. I., Flies, D. B., Bajorath, J., and Chen, L. (2003). B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity 18, 849–861.
Starke, A., Lindenmeyer, M. T., Segerer, S., Neusser, M. A., Rüsi, B., Schmid, D. M., Cohen, C. D., Wüthrich, R. P., Fehr, T., and Waeckerle-Men, Y. (2010). Renal tubular PD-L1 (CD274) suppresses alloreactive human T-cell responses. Kidney Int. 78, 38–47.
Steger, U., Denecke, C., Sawitzki, B., Karim, M., Jones, N. D., and Wood, K. J. (2008). Exhaustive differentiation of alloreactive CD8+ T cells: critical for determination of graft acceptance or rejection. Transplantation 85, 1339–1347.
Steinberg, M. W., Turovskaya, O., Shaikh, R. B., Kim, G., McCole, D. F., Pfeffer, K., Murphy, K. M., Ware, C. F., and Kronenberg, M. (2008). A crucial role for HVEM and BTLA in preventing intestinal inflammation. J. Exp. Med. 205, 1463–1476.
Suh, W. K., Gajewska, B. U., Okada, H., Gronski, M. A., Bertram, E. M., Dawicki, W., Duncan, G. S., Bukczynski, J., Plyte, S., Elia, A., Wakeham, A., Itie, A., Chung, S., Da Costa, J., Arya, S., Horan, T., Campbell, P., Gaida, K., Ohashi, P. S., Watts, T. H., Yoshinaga, S. K., Bray, M. R., Jordana, M., and Mak, T. W. (2003). The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 4, 899–906.
Takahashi, T., Tagami, T., Yamazaki, S., Uede, T., Shimizu, J., Sakaguchi, N., Mak, T. W., and Sakaguchi, S. (2000). Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192, 303–310.
Tamada, K., Shimozaki, K., Chapoval, A. I., Zhai, Y., Su, J., Chen, S. F., Hsieh, S. L., Nagata, S., Ni, J., and Chen, L. (2000). LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J. Immunol. 164, 4105–4110.
Tao, R., Wang, L., Han, R., Wang, T., Ye, Q., Honjo, T., Murphy, T. L., Murphy, K. M., and Hancock, W. W. (2005). Differential effects of B and T lymphocyte attenuator and programmed death-1 on acceptance of partially versus fully MHC-mismatched cardiac allografts. J. Immunol. 175, 5774–5782.
Tao, R., Wang, L., Murphy, K. M., Fraser, C. C., and Hancock, W. W. (2008). Regulatory T cell expression of herpesvirus entry mediator suppresses the function of B and T lymphocyte attenuator-positive effector T cells. J. Immunol. 180, 6649–6655.
Taylor, P. A., Lees, C. J., Fournier, S., Allison, J. P., Sharpe, A. H., and Blazar, B. R. (2004). B7 expression on T cells down-regulates immune responses through CTLA-4 ligation via T-T interactions [corrections]. J. Immunol. 172, 34–39.
Tivol, E. A., Borriello, F., Schweitzer, A. N., Lynch, W. P., Bluestone, J. A., and Sharpe, A. H. (1995). Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547.
Truong, W., Plester, J. C., Hancock, W. W., Kaye, J., Merani, S., Murphy, K. M., Murphy, T. L., Anderson, C. C., and Shapiro, A. M. (2007a). Negative and positive co-signaling with anti-BTLA (PJ196) and CTLA4Ig prolongs islet allograft survival. Transplantation 84, 1368–1372.
Truong, W., Plester, J. C., Hancock, W. W., Merani, S., Murphy, T. L., Murphy, K. M., Kaye, J., Anderson, C. C., and Shapiro, A. M. (2007b). Combined coinhibitory and costimulatory modulation with anti-BTLA and CTLA4Ig facilitates tolerance in murine islet allografts. Am. J. Transplant. 7, 2663–2674.
Uchida, Y., Ke, B., Freitas, M. C., Yagita, H., Akiba, H., Busuttil, R. W., Najafian, N., and Kupiec-Weglinski, J. W. (2010). T-cell immunoglobulin mucin-3 determines severity of liver ischemia/reperfusion injury in mice in a TLR4-dependent manner. Gastroenterology 139, 2195–2206.
Ueda, H., Howson, J. M., Esposito, L., Heward, J., Snook, H., Chamberlain, G., Rainbow, D. B., Hunter, K. M., Smith, A. N., Di Genova, G., Herr, M. H., Dahlman, I., Payne, F., Smyth, D., Lowe, C., Twells, R. C., Howlett, S., Healy, B., Nutland, S., Rance, H. E., Everett, V., Smink, L. J., Lam, A. C., Cordell, H. J., Walker, N. M., Bordin, C., Hulme, J., Motzo, C., Cucca, F., Hess, J. F., Metzker, M. L., Rogers, J., Gregory, S., Allahabadia, A., Nithiyananthan, R., Tuomilehto-Wolf, E., Tuomilehto, J., Bingley, P., Gillespie, K. M., Undlien, D. E., Rønningen, K. S., Guja, C., Ionescu-Tîrgoviste, C., Savage, D. A., Maxwell, A. P., Carson, D. J., Patterson, C. C., Franklyn, J. A., Clayton, D. G., Peterson, L. B., Wicker, L. S., Todd, J. A., and Gough, S. C. (2003). Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423, 506–511.
Urbani, S., Amadei, B., Tola, D., Massari, M., Schivazappa, S., Missale, G., and Ferrari, C. (2006). PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol. 80, 11398–11403.
van de Weyer, P. S., Muehlfeit, M., Klose, C., Bonventre, J. V., Walz, G., and Kuehn, E. W. (2006). A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem. Biophys. Res. Commun. 351, 571–576.
Vasu, C., Prabhakar, B. S., and Holterman, M. J. (2004). Targeted CTLA-4 engagement induces CD4+CD25+CTLA-4high T regulatory cells with target (allo)antigen specificity. J. Immunol. 173, 2866–2876.
Vijayakrishnan, L., Slavik, J. M., Illes, Z., Greenwald, R. J., Rainbow, D., Greve, B., Peterson, L. B., Hafler, D. A., Freeman, G. J., Sharpe, A. H., Wicker, L. S., and Kuchroo, V. K. (2004). An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity 20, 563–575.
Vincenti, F., Charpentier, B., Vanrenterghem, Y., Rostaing, L., Bresnahan, B., Darji, P., Massari, P., Mondragon-Ramirez, G. A., Agarwal, M., Di Russo, G., Lin, C. S., Garg, P., and Larsen, C. P. (2010). A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study). Am. J. Transplant. 10, 535–546.
Vincenti, F., Dritselis, A., and Kirkpatrick, P. (2011). Belatacept. Nat. Rev. Drug Discov. 10, 655–656.
Vincenti, F., Larsen, C. P., Alberu, J., Bresnahan, B., Garcia, V. D., Kothari, J., Lang, P., Urrea, E. M., Massari, P., Mondragon-Ramirez, G., Reyes-Acevedo, R., Rice, K., Rostaing, L., Steinberg, S., Xing, J., Agarwal, M., Harler, M. B., and Charpentier, B. (2012). Three-year outcomes from BENEFIT, a randomized, active-controlled, parallel-group study in adult kidney transplant recipients. Am. J. Transplant. 12, 210–217.
Virgin, H. W., Wherry, E. J., and Ahmed, R. (2009). Redefining chronic viral infection. Cell 138, 30–50.
Walunas, T. L., Bakker, C. Y., and Bluestone, J. A. (1996). CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 183, 2541–2550.
Walunas, T. L., Lenschow, D. J., Bakker, C. Y., Linsley, P. S., Freeman, G. J., Green, J. M., Thompson, C. B., and Bluestone, J. A. (1994). CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405–413.
Wang, F., He, W., Yuan, J., Wu, K., Zhou, H., Zhang, W., and Chen, Z. K. (2008). Activation of Tim-3-Galectin-9 pathway improves survival of fully allogeneic skin grafts. Transpl. Immunol. 19, 12–19.
Wang, J., Yoshida, T., Nakaki, F., Hiai, H., Okazaki, T., and Honjo, T. (2005a). Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. U.S.A. 102, 11823–11828.
Wang, L., Fraser, C. C., Kikly, K., Wells, A. D., Han, R., Coyle, A. J., Chen, L., and Hancock, W. W. (2005b). B7-H3 promotes acute and chronic allograft rejection. Eur. J. Immunol. 35, 428–438.
Wang, Y., Subudhi, S. K., Anders, R. A., Lo, J., Sun, Y., Blink, S., Wang, Y., Wang, J., Liu, X., Mink, K., Degrandi, D., Pfeffer, K., and Fu, Y. X. (2005c). The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. J. Clin. Invest. 115, 711–717.
Wang, X., Hao, J., Metzger, D. L., Mui, A., Ao, Z., Akhoundsadegh, N., Langermann, S., Liu, L., Chen, L., Ou, D., Verchere, C. B., and Warnock, G. L. (2011). Early treatment of NOD mice with B7-H4 reduces the incidence of autoimmune diabetes. Diabetes. 60, 3246–3255.
Wang, X., Hao, J., Metzger, D. L., Mui, A., Ao, Z., Verchere, C. B., Chen, L., Ou, D., and Warnock, G. L. (2009a). Local expression of B7-H4 by recombinant adenovirus transduction in mouse islets prolongs allograft survival. Transplantation 87, 482–490.
Wang, F., Wan, L., Zhang, C., Zheng, X., Li, J., and Chen, Z. K. (2009b). Tim-3-Galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology 214, 342–349.
Ware, C. F. (2008). Targeting lymphocyte activation through the lymphotoxin and LIGHT pathways. Immunol. Rev. 223, 186–201.
Watanabe, N., Gavrieli, M., Sedy, J. R., Yang, J., Fallarino, F., Loftin, S. K., Hurchla, M. A., Zimmerman, N., Sim, J., Zang, X., Murphy, T. L., Russell, J. H., Allison, J. P., and Murphy, K. M. (2003). BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat. Immunol. 4, 670–679.
Wherry, E. J., Blattman, J. N., Murali-Krishna, K., van der Most, R., and Ahmed, R. (2003). Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77, 4911–4927.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., Nomura, T., and Sakaguchi, S. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275.
Yamaura, K., Watanabe, T., Boenisch, O., Yeung, M., Yang, S., Magee, C. N., Padera, R., Datta, S., Schatton, T., Kamimura, Y., Azuma, M., and Najafian, N. (2010) In vivo function of immune inhibitory molecule B7-H4 in alloimmune responses. Am. J. Transplant. 10, 2355–2362.
Yang, J., Popoola, J., Khandwala, S., Vadivel, N., Vanguri, V., Yuan, X., Dada, S., Guleria, I., Tian, C., Ansari, M. J., Shin, T., Yagita, H., Azuma, M., Sayegh, M. H., and Chandraker, A. (2008). Critical role of donor tissue expression of programmed death ligand-1 in regulating cardiac allograft rejection and vasculopathy. Circulation 117, 660–669.
Yang, J., Riella, L. V., Chock, S., Liu, T., Zhao, X., Yuan, X., Paterson, A. M., Watanabe, T., Vanguri, V., Yagita, H., Azuma, M., Blazar, B. R., Freeman, G. J., Rodig, S. J., Sharpe, A. H., Chandraker, A., and Sayegh, M. H. (2011). The novel costimulatory programmed death ligand 1/B7.1 pathway is functional in inhibiting alloimmune responses in vivo. J. Immunol. 187, 1113–1119.
Yao, S., Zhu, Y., Zhu, G., Augustine, M., Zheng, L., Goode, D. J., Broadwater, M., Ruff, W., Flies, S., Xu, H., Flies, D., Luo, L., Wang, S., and Chen, L. (2011). B7-h2 is a costimulatory ligand for CD28 in human. Immunity 34, 729–740.
Yi, K. H., and Chen, L. (2009). Fine tuning the immune response through B7-H3 and B7-H4. Immunol. Rev. 229, 145–151.
Ying, H., Yang, L., Qiao, G., Li, Z., Zhang, L., Yin, F., Xie, D., and Zhang, J. (2010). Cutting edge: CTLA-4 – B7 interaction suppresses Th17 cell differentiation. J. Immunol. 185, 1375–1378.
Yuan, C. L., Xu, J. F., Tong, J., Yang, H., He, F. R., Gong, Q., Xiong, P., Duan, L., Fang, M., Tan, Z., Xu, Y., Chen, Y. F., Zheng, F., and Gong, F. L. (2009). B7-H4 transfection prolongs beta-cell graft survival. Transpl. Immunol. 21, 143–149.
Zhu, C., Anderson, A. C., Schubart, A., Xiong, H., Imitola, J., Khoury, S. J., Zheng, X. X., Strom, T. B., and Kuchroo, V. K. (2005). The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6, 1245–1252.
Keywords: Negative costimulation, Tolerance, CTLA-4, PD-1:PD-L1, B7-H3, B7-H4, BTLA, TIM-3
Citation: McGrath MM and Najafian N (2012) The role of coinhibitory signaling pathways in transplantation and tolerance. Front. Immun. 3:47. doi: 10.3389/fimmu.2012.00047
Received: 20 December 2011; Accepted: 28 February 2012;
Published online: 19 March 2012.
Edited by:
Shohei Hori, RIKEN, JapanReviewed by:
Taku Okazaki, The University of Tokushima, JapanNick David Jones, University of Birmingham, UK
Miyuki Azuma, Tokyo Medical and Dental University, Japan
Copyright: © 2012 McGrath and Najafian. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nader Najafian, Transplantation Research Center, Brigham and Women’s Hospital and Children’s Hospital, Harvard Medical School, Boston, MA 02115, USA. e-mail: nnajafian@rics.bwh.harvard.edu