 Pierre-Emmanuel Joubert Matthew L. Albert*
Pierre-Emmanuel Joubert Matthew L. Albert*- Laboratory of Dendritic Cell Immunobiology, Department of Immunology, INSERM U818, Institut Pasteur, Paris, France
Phagocytosis of dying cells constitutes an important mechanism of antigen capture for the cross-priming of CD8+ T cells. This process has been shown to be critical for achieving tumor and viral immunity. While most studies have focused on the mechanisms inherent in the dendritic cell that account for exogenous antigen accessing MHC I, several recent reports have highlighted the important contribution made by the antigen donor cell. Specifically, the cell stress and cell death pathways that precede antigen transfer are now known to impact cross-presentation and cross-priming. Herein, we review the current literature regarding a role for macroautophagy within the antigen donor cell. Further examination of this point of immune regulation is warranted and may contribute to a better understanding of how to optimize immunotherapy for treatment of cancer and chronic infectious disease.
Introduction
Significant evidence for an indirect pathway for the loading of MHC class I molecules has emerged over the last 35 years (Bevan, 1976; Rock et al., 1990). The most compelling data comes from in vivo experiments in mouse models demonstrating that viral, tumor, and histocompatibility antigens can be transferred from MHC-mismatched donor cells to host bone marrow derived conventional dendritic cells (cDCs), and elicit antigen-specific CTL responses that are restricted to self MHC molecules (Falo et al., 1995; Reis e Sousa and Germain, 1995; Sigal et al., 1999; Mellman and Steinman, 2001; Boon et al., 2006; Petersen et al., 2010). Bevan (1976) originally coined this phenomenon “cross-priming,” as antigen is “crossing the MHC barrier” that had initially been invoked in the generation of MHC class I peptide epitopes. As it is now understood that the activation of naïve T cells is a property restricted to cDCs, these in vivo observations offered a solution to the question of how CD8+ T cells are activated for the targeting of cells which express antigen that is not directly expressed by cDCs. Examples of such antigen include tumor-restricted proteins and viruses which do not infect professional antigen presenting cells (APCs; e.g., human papillomavirus; Fausch et al., 2003). While these observations indicate that the immune system possesses a natural mechanism by which exogenous antigens may access MHC I molecules of APCs, there remains much to be discovered regarding the mechanisms of antigen transfer.
Our in vitro studies and the in vivo work of others demonstrated that immature cDCs are capable of capturing antigen derived from internalized dying cells and cross-presenting donor antigen on MHC I molecules for engagement of CD8+ T cells (Albert et al., 1998, 2001; Kurts et al., 2010; Pang and Neefjes, 2010; Flinsenberg et al., 2011). cDC trafficking of tissue-restricted antigen derived from internalized dying cells has been demonstrated for models of gut-, skin-, and pancreas-restricted protein antigen (Huang et al., 2000; Belz et al., 2002; Scheinecker et al., 2002; Turley et al., 2003). In the latter model system, the use of transgenic mice expressing inhibitors of apoptosis in beta cells and the in vivo injection of biochemical modulators of death pathways have confirmed the critical role for cell death in both antigen transfer and T cell activation (Hugues et al., 2002; Turley et al., 2003; Giodini and Albert, 2010; Locher et al., 2010; Flinsenberg et al., 2011).
Over the last decade, there has been an explosion of information regarding cell stress and cell death. These death pathways may synergize and/or compete, each vying to deliver the fatal blow. Importantly, the mechanisms of cell stress and cell death are now recognized as critical determinants of the subsequent immune response – impacting trafficking of the APC, altering the antigenic repertoire that is transferred upon phagocytosis and influencing the cDC activation state (Albert, 2004). While most studies have focused on apoptotic cell death vs. necrotic cell death, there is increasing awareness that macroautophagy, within the antigen donor cell, influences the outcome of cross-presentation. Herein we focus on the ability of DCs to capture and cross-present cell-associated antigen, reviewing in detail the recent evidence for macroautophagy in the donor cell as an important mechanism for facilitating antigen delivery to cDCs.
Macroautophagy and Antigen Presentation
Macroautophagy (referred to herein as autophagy) has been defined as an “auto-digestive” process that promotes the delivery of intracytosolic components to lysosomal or vacuolar compartments for terminal degradation and recycling (Deretic and Levine, 2009; Figure 1). Autophagy has distinct roles in different cellular contexts and occurs at a basal level in all nucleated cells. Constitutive autophagy is important for the turnover of unfolded proteins or damaged organelles and maintains cellular homeostasis (Kroemer et al., 2010). For example, autophagy is important for reducing oxidative stress by selectively targeting damaged mitochondria (Wang and Klionsky, 2011; Mai et al., 2012). Similarly, endoplasmic reticulum (ER), peroxisomes, ribosomes, protein aggregates, and even intracellular pathogens may be eliminated via autophagy (Yu et al., 2008; Joubert et al., 2009; Komatsu and Ichimura, 2010; Mizushima and Komatsu, 2011). Autophagy is also considered as part of the host response to cellular stress, including nutrient deprivation or adenosine triphosphate (ATP) depletion. Under such conditions, autophagy protects cells by supplying recycled nutrients to support essential cellular process until restoration of homeostasis. Autophagy induced by cellular stress is generally considered to be a non-selective, bulk degradative process.
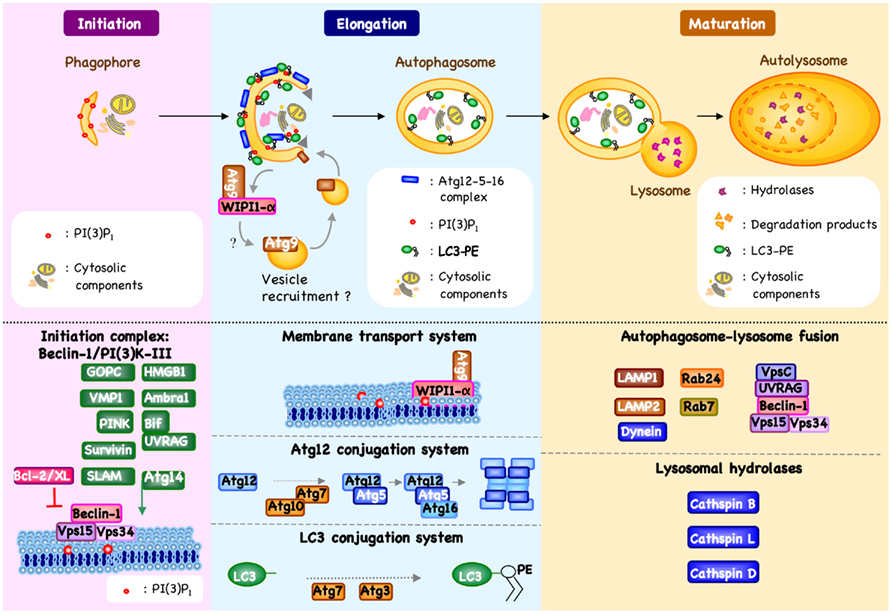
Figure 1. Machinery of autophagy. Beclin-1/PI3K-III complex activation, which is regulated by different mechanisms, results in the formation of phosphatidylinositol 3-phosphate [PI(3)P1] and the induction of an autophagic vesicle, which is characterized by a double-membrane, and termed an autophagosome (Crotzer and Blum, 2010; Mehrpour et al., 2010; Kang et al., 2011). Two ubiquitin-like systems have been shown essential for autophagosome formation. In the first, autophagy-related gene-12 (Atg12) is conjugated to Atg5, together forming a complex with Atg16L1, which decorate the outer membrane of the isolation membrane. Microtubule-associated protein 1 light chain-3 (LC3, also known as Atg8) constitutes the second ubiquitin-like system and conjugates phosphatidylethanolamine (PE) at the outer and inner autophagosomal membrane. Unlike the Atg12/Atg5/Atg16L1 complex that is recycled by the protease Atg4, the LC3-PE (referred to as LC3-II) remains associated with the inner membrane of autophagosome (Mehrpour et al., 2010). The incorporation of phospholipid into the autophagosome membrane is essential for its elongation, and regulates the membrane transport system. Autophagosome maturation is characterized by the formation of an autolysosome, the product of fusion with the lysosome.
More recently, autophagy has been recognized as an important modulator of host immunity, with a particular role for the processing of antigen for presentation by MHC and the initiation of adaptive immune responses (Dengjel et al., 2005; Deretic and Levine, 2009; Crotzer and Blum, 2010). Specifically, autophagy has been described to participate in the translocation of endogenous protein into the MHC class II loading compartment, which facilitates MHC class II presentation and CD4+ T cells activation (Dengjel et al., 2005). As one key example, it has been demonstrated that the constitutive degradation of cellular components through autophagy provides a critical source of self antigen in the thymus for education of CD4+ T cell precursors (Nedjic et al., 2008). In addition to CD4+ T cell activation, there is now evidence to suggest a role for autophagy – or at least autophagic genes – in the generation of MHC I/peptide complexes, within the APC. Based on early studies using silencing of Atg12 or Atg5, or pharmacological inhibitors, it was argued that autophagy does not have a major impact on MHC class I presentation of multiple endogenous or exogenous antigens (Nimmerjahn et al., 2003; Paludan et al., 2005; Lee et al., 2010). Moreover, conjugation of a viral epitope to LC3 did not alter MHC class I presentation of this epitope (Schmid et al., 2007). Recent data however, has challenged these conclusions, at least in the context of selected model systems. Interestingly, inhibition of autophagy in IFN-γ-treated B16 mouse melanoma cells diminished MHC class I protein surface expression and tumor cell cytolysis by CD8+ T lymphocytes (Li et al., 2010). MHC class I presentation by these tumors was proteasome dependent, suggesting a possible connection between autophagy and the conventional pathway for MHC class I presentation. In another study, the group of Desjardins has highlighted an unconventional autophagy pathway that modulates MHC class I presentation in macrophages infected by Herpes simplex virus (HSV; English et al., 2009). Early after infection (6–8 h), viral capsid antigen presentation in infected macrophages occurs via the conventional MHC class I pathway, but during the late stages of infection (8–12 h), viral capsid presentation is dependent on acidic organelles as well as Atg5 expression. This study also reported that HSV-infected cells contain specific and unconventional LC3+ vesicles that are closely associated with the nuclear envelope. This led the authors to suggest that virus infection induced a novel form of autophagy, which may contribute to the escape of antigen into the cytosol.
Connecting Autophagy and Cross-Priming
While basal or induced autophagy in APCs does not seem to be strongly implicated in their ability to cross-present exogenous antigen, some early evidence suggested a role for autophagy within the antigen donor cell. In 2002, a study from Vile and colleagues suggested for the first time that autophagic vacuoles within the donor cell could be correlated with efficient cross-priming of associated-antigens (Bateman et al., 2002). Viral fusogenic membrane glycoprotein (FMGs) had been known to kill solid tumor cells after the formation of large multinucleated syncytia. The authors observed that FMG-mediated killing of syncytia was not dependent on a classical apoptosis pathway. Instead, death of syncytia was associated with nuclear fusion and premature chromosome condensation as well as severe ATP depletion (Bateman et al., 2002). Interestingly, cytoplasmic vacuoles were demonstrated to be acidic, suggesting that the death of syncytia was correlated with autophagic activity. In the same study, the authors demonstrated that FMG-mediated death was accompanied by release of vesicles reminiscent of exosomes, which they called syncitiosomes. Importantly, dying syncytia produced significantly more syncitiosomes than normal cells or cells killed by pro-apoptotic or pro-necrotic signals, including irradiation, freeze thaw, or osmotic shock (Bateman et al., 2002). These syncitiosomes served as an efficient source of antigen for cDCs, out-performing classical exosomes, or dead cell corpses. Lacking from this study, however, was the mechanistic association of Atg proteins with death of syncytia and effective cross-priming.
An additional clue linked autophagy to the removal of apoptotic corpses in mouse embryoid bodies (EBs) and during chick retinal development (Qu et al., 2007; Mellen et al., 2008). In both studies, autophagy is required for dying cells to have sufficient energy to generate the engulfment signals necessary for the clearance of corpses by phagocytes. Indeed, EBs derived from cells lacking autophagy genes (e.g., atg5 or beclin-1), failed to express “eat-me” signal such as phosphatidylserine (PS) exposure, and secreted lower levels of “come-get-me” signals, such as lysophosphatidylcholine (LPC; Qu et al., 2007). These defects were associated with low levels of cellular ATP and could be reversed by treatment with the metabolic substrate, methyl pyruvate. Similar results were observed after treatment of retinas with 3-methyladenine (3-MA), a pharmacological inhibitor of autophagy (Mellen et al., 2008). Extrapolation of these studies might suggest another mechanism by which autophagy regulates cross-presentation. Indeed, the role of autophagy in metabolism may be generally relevant for phagocyte function, and aspects of innate and adaptive immune responses.
Autophagy within Tumor Cells Favors Cross-Priming of Tumor-Associated Antigen-Specific CD8+ T Cells
Seminal studies from Hu and colleagues illustrated a direct role for autophagy in the cross-priming of associated-antigen (Li et al., 2009). In their initial studies, HEK 293T cells expressing ovalbumin (V-TfR-GFP-OVA) or melanoma cells that endogenously express the gp100 tumor antigen were used as antigen donor cells (ADC). The authors showed that autophagy in ADC regulates the efficiency of cross-presentation both in vitro and in vivo (Li et al., 2009). Inhibition of autophagy with 3-MA or siRNA knockdown of the essential autophagic genes beclin-1 and Atg12, demonstrated that early steps of autophagy – including initiation and elongation of the double-membrane structure, sequestration of cytosolic antigens, and formation of autophagosomes – were required for efficient antigen cross-presentation. Providing additional support, drug, or stress-induced autophagy (i.e., treatment with rapamycin or starvation) resulted in enhanced cross-priming of OVA or gp100-specific CD8+ T cells. In these studies, it is important to note that late steps of autophagy, including late lysosomal fusion and degradation, seemed to have little impact on antigen cross-presentation (Li et al., 2009). Perhaps most interestingly, autophagosomes were isolated, purified, and exposed to DCs, demonstrating efficient delivery of antigen and cross-presentation (Li et al., 2009). Future studies will be required to confirm that contaminating microsomes or secreted exosomes did not contribute to antigen transfer in these experiments.
More recently, the same group observed that isolated autophagosomes from dying tumor cells expressed not only long-lived protein – well know to be sequestered in autophagosomes – but also short-lived proteins (SLiPs), including defective ribosomal initiation products (DRiPs; Yewdell et al., 1996; Li et al., 2011). Immunization with these autophagosomes – named “Dribbles” by the authors to refer to DRiPs-containing blebs – were effective in initiating tumor immunity and inducing the regression of 3LL Lewis lung tumors as well as delaying growth of B16F10 melanoma. Strikingly, DRibbles were more potent than GM-CSF gene modified tumor cells. Importantly, DRibble-derived antigen processing by cDCs was mediated by classical components of the MHC I processing machinery, including TAP1 and proteasome, and it did not involve the lysosomal pathway of the cDC (Li et al., 2011). These observations suggested that DRibble-derived antigen must be released into the cytosol after phagocytosis to be processed by the classical MHC class I pathway. Although mechanisms by which DRibbles could favor antigen cross-presentation remain to be confirmed, these studies were the first to identify capture of autophagosome-associated antigen as a mechanism for achieving tumor immunity.
Virus-Mediated Abortive Autophagy Enhances Antigen Cross-Priming
Studies from our own laboratory have also identified an important role for autophagy within the ADC, serving to enhance antigen cross-priming of CD8+ T cells (Uhl et al., 2009). The experimental system employed permitted direct comparison between two forms of programmed cell death (PCD): (i) the classical caspase-dependent apoptosis, which it occur in wild-type (WT) mouse embryonic fibroblast (MEFs); and (ii) caspase-independent cell death, which occurs with increased features of autophagy, and achieved experimentally through the use of Bax−/−/Bak−/− MEFs (Shimizu et al., 2004). Both cell types were infected with influenza A virus as a source of antigen, followed by UV irradiation – serving to both inhibit viral replication and induce genotoxic stress – and injected in vivo as a source of antigen for studying the efficiency of cross-priming (Uhl et al., 2009). Interestingly, mice immunized with cells undergoing enhanced autophagy showed a significantly higher CD8+ T cell response specific for both HA518–526 (in Balb/c hosts) and NP366–374 (in C57BL/6 hosts). Strikingly, silencing of the essential autophagic gene Atg5 in both WT and Bax−/−/Bak−/− MEFs inhibited antigen cross-priming. Careful evaluation of influenza infected WT MEFs indicated that viral infection induced accumulation of autophagosomes, now known to be a result of influenza M2-inhibition of autophagosome/lysosome fusion (Gannage et al., 2009). Notably, influenza infection is capable of inducing both autophagy and apoptosis; different from other triggers of cell stress and cell death, these two processes can be found simultaneously within the same cell (de la Calle et al., 2011). We argue that the finding of double positive cells – co-labeled with anti-caspase 3 antibodies and harboring LC3 punctae – is a result of abortive autophagy. This is supported by studies using chloroquine (de la Calle et al., 2011), which interestingly, has also been shown to enhance antigen cross-priming (Accapezzato et al., 2005), and is currently being tested in combination with rapamycin as a means of inducing in situ tumor immunity (Amaravadi et al., 2011). Additional work is required in order to establish abortive autophagosomes as the critical source of antigen for facilitating efficient transfer from donor cells to cDCs.
Autophagy may Favor Release of “Immunogenic” Proteins
The first established link between autophagy genes and pro-inflammatory responses was established in plasmacytoid DCs (pDCs), with the demonstration that Atg5 is involved in autophagy-mediated delivery of TLR7 agonists from the cytosol of infected cells into the lumen of the endosome, thus accounting for induction of type I interferon (IFN; Lee et al., 2007). Somewhat paradoxically, several studies have shown that the absent or hypomorphic expression of autophagic genes in certain cell types can result in enhanced production of type I interferon or other cytokines; including pro-inflammatory molecules such as IL1β and IL-18, as well as adipocytokines, such as leptin and adiponectin (Jounai et al., 2007; Cadwell et al., 2008; Saitoh et al., 2008; Tal et al., 2009). Thus, autophagy machinery could have a dual function in regulating cytokine production, acting not only to stimulate antiviral type I IFN responses in pDCs, but may also limit excess innate immune activation in other cell type, including fibroblasts. Alternatively, autophagy genes may differentially regulate distinct PRRs: enhancing TLR engagement through the delivery of ligands into the endosome; while inhibiting cytosolic sensors through direct or indirect mechanisms.
Defining how autophagy alters the inflammatory milieu is an important issue, as type I IFNs has been shown to regulate antigen cross-priming (Jounai et al., 2007; Uhl et al., 2009; Wei et al., 2010). Using the same model as previously described, our group has demonstrated that mice immunized with Bax−/−/Bak−/− dying cells – undergoing high level of autophagy – induced in cDCs a significantly higher type I IFN production in cDCs as compared to mice immunized with WT MEFs (Uhl et al., 2009). Supporting a role for type I IFN, it was shown that immunization of mice deficient for the IFNα/β receptor (IFNAR) resulted in a dramatically reduced cross-priming response (Uhl et al., 2009). The mechanism by which autophagy within dying cells favors the production of type I IFN by APCs remains to be defined; one possibility is that viral nucleic acids present in the autophagic corpse serves to engage sensors within the phagocytic cDC (Schulz et al., 2005).
In addition to pro-inflammatory cytokines, other “immunogenic proteins” may be released during autophagic processes, which in turn favor cross-priming. For example, the high-mobility group box 1 protein (HMGB1) provides an interesting connection between autophagy and cross-priming. HMGB1 is a highly conserved chromatin-binding protein that facilitates DNA bending and promotes transcription (Maruyama, 2011). In addition to its intra-nuclear role, HMGB1 also functions as an extracellular signaling molecule and can interact with at least three different surface receptors that are expressed on APC, namely the receptor for advanced glycosylation (RAGE), TLR2, and TLR4 (Nogueira-Machado et al., 2011). The binding of HMGB1 to TLR4 may not be a direct interaction as recent data indicates that HMGB1 chaperones LPS (Yang et al., 2012), as well as other PRR ligands (Yanai et al., 2009). Indeed, the role of HMGB1 as a co-factor for PAMPs may be central to its role in stimulating the processing and presentation of derived antigens. Importantly, secretion of HMGB1 by dying tumor cells has been shown to inhibit fusion of the APC’s phagosome with lysosomes, thereby preventing rapid degradation of tumor antigens and enabling processing and presentation onto MHC I (Apetoh et al., 2007). Furthermore, Scaffidi et al. (2002) showed that HMGB1 participates in the recruitment of phagocytes. Indeed, HMGB1−/− cells have a greatly reduced ability to promote inflammation, which indicates that the release of HMGB1 can signal the demise of a cell to its neighbors. While initial studies indicated that HMGB1 is released (passively) from necrotic cells and that during apoptosis HMGB1 becomes hypoacetylated and remains bound to the chromatin of the dying cell (Sims et al., 2010), the biology now seems a bit more complex. Recent work from Ferguson and colleagues indicate that the redox state, in addition to the acetylation state, may impact the bioactivity of HMGB1 (Kazama et al., 2008). ROS mediated oxidation of HMGB1 inhibited its pro-inflammatory potential. The authors went on to demonstrate that oxidation was caspase-dependant, acting via the cleavage of mitochondrial components of the electron transport system (Kazama et al., 2008). Based on the ability of autophagic processes to limit accumulation of ROS, it is interesting to consider that this may contribute to the pro-inflammatory effects of autophagy within dying ADC. This hypothesis is supported by evidence that HMGB1 released from autophagic tumor cells is immune stimulatory (Thorburn et al., 2009). Inhibition of autophagy resulted in HMGB1 retention, and in the induction of caspase-mediated cell death. The mechanism by which autophagy regulates secretion of HMGB1 remain unknown, but seems to be occurring in a manner similar to other leaderless cytokines. Indeed, exogenous HMGB1 can modulate the future of tumor cells and reduced HMGB1 induced pro-survival autophagy via the activation of RAGE receptor and beclin-1 whereas oxidized HMGB1 favored activation of caspase-9 and -3 that lead to apoptotic cell death (Tang et al., 2010). Thus, autophagy could favor the release of reduced HMGB1 by limiting cytoplasmic ROS level; in turn reduced HMGB1 may increase autophagy in neighboring cells as well as activate the recruitment of immune cells, stimulate cytokines secretion, and facilitate antigen cross-priming.
Other immunogenic signals may also be regulated by autophagy, including exposure of PS and secretion of LPC (Qu et al., 2007; Mellen et al., 2008). Recently, the groups of Zitvogel and Kroemer established a link between autophagy and ATP release during chemotherapy treatment (Michaud et al., 2011). This study showed that autophagy is dispensable for chemotherapy-induced cell death but required for its immunogenicity both in vitro and in vivo. In response to chemotherapy, autophagy-competent, but not Atg5 or Atg7-shRNA transfected tumors, were capable of recruiting cDCs and T cells into the tumor micro-environment, via ATP mediated chemoattraction. Although this work did not formally show T cell cross-priming, it helped establish a new concept related to autophagy regulation of immunity. Further investigation regarding other secreted and/or exposed proteins would help further clarify the mechanisms by which autophagy within ADC favors antigen cross-priming.
Autophagic Flux or Autophagosomes?
One critical unknown concerns the cell biology of autophagy within ADC as it relates to protection and delivery of antigen to APCs. Some evidence suggests that autophagosomes, but not necessarily autophagic flux, are required for efficient cross-priming. This is supported by the importance of early steps of autophagy – including initiation and elongation of the double-membrane structure, sequestration of cytosolic antigens, and formation of autophagosomes – for achieving efficient antigen cross-presentation of tumor cells (Li et al., 2009, 2011). Additionally, our own studies indicate that blocking autophagic flux within dying cells enhanced the cross-priming of viral antigens (Giodini and Albert, unpublished data). Accumulation of autophagosomes may also account for the seminal observations of Barnaba and colleagues, who shown that chloroquine enhance cross-priming (Accapezzato et al., 2005). In this context, it is possible that inhibition of lysosomal fusion serves to protect epitope within the autophagosome of dying cells and facilitates delivery of intact or partially processed antigen to APC.
One final consideration is the capture of free autophagic vesicles – passively released following cell death or actively secreted via a mechanism referred to as exophagy (Abrahamsen and Stenmark, 2010) – could be recognized by APCs as a source of antigen or immunogenic signals to enhance cross-priming. It has indeed been demonstrated in vitro that tumor cell-derived autophagosomes can be isolated from culture media; and may engage the C-type lectin receptor, CLEC9A (Li et al., 2011). These data may extend the previous role for CLEC9A as a receptor for necrotic cells and a mediator of efficient SYK-dependant cross-priming (Sancho et al., 2009).
Conclusion
In addition to cell death pathways, increasing evidence indicates an important role for cell stress, within ADC, as a regulator of adaptive immune responses. Autophagic processes have been shown to enhance the delivery of tumor and viral antigen to cDCs. Moreover, autophagy triggers release of immunostimulatory proteins (e.g., HMGB1) and bioactive molecules (e.g., LPC, ATP), which together favor recruitment and activation of the APC (Figure 2). Once within the phagosome, autophagic cells may serve as stimulators of PRRs, further enhancing the phagocyte’s cross-priming potential. Another interesting line of investigation will be the autophagosome itself, which may serve to protect antigen from degradation and facilitate delivery APCs. A detailed evaluation of how autophagy favors cross-priming may help launch new immunotherapy strategies for treating cancer or chronic disease. Moreover, it may provide a mechanistic understanding of currently used chemotherapies (e.g., cyclophosphamide or methotrexate) or experimental compounds (e.g., rapamycin given in combination with hydroxychloroquine).
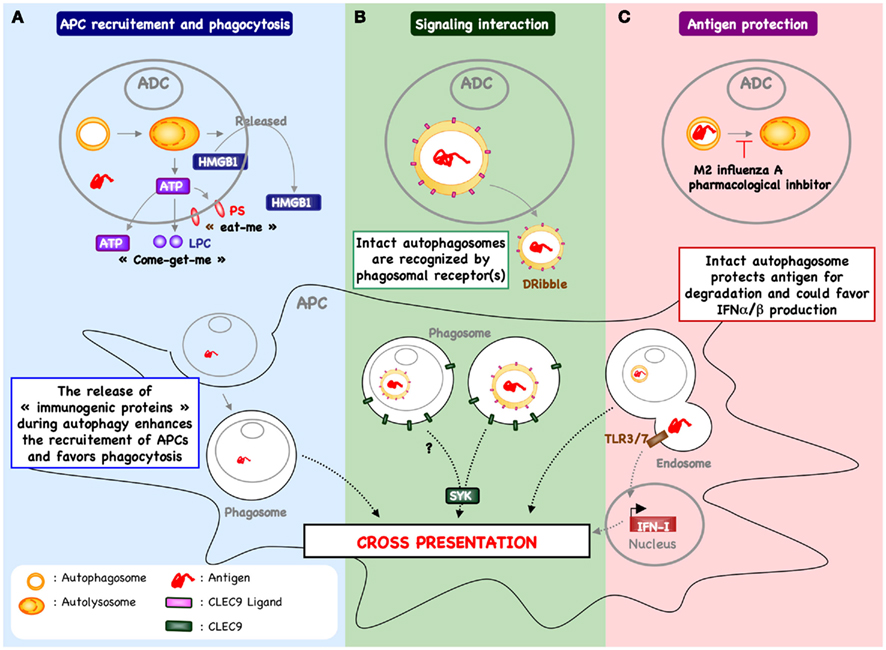
Figure 2. Autophagy within antigen donor cells and cross-priming. (A) The process of autophagy within stressed or dying cells has been demonstrated to enhance recruitment of antigen presenting cells (APCs) and phagocytosis. For example, in response to chemotherapy, autophagy within cancer cells favors the release of ATP and enhances the recruitment of cDC and T cells into the tumor micro-environment (Michaud et al., 2011). In addition, the release of ATP triggers the exposure of “eat-me” signal (e.g., PS) and the release of “come-get-me” signal (e.g., LPC), an essential feature of efficient capture of dying cells by APC (Qu et al., 2007; Mellen et al., 2008). Autophagy can also be involved in the release of other “immunogenic proteins” known to support the cross-priming (e.g., HMGB1). The mechanism by which autophagy regulates the release of HMGB1 remain to be determined. (B) Free autophagosome containing antigen – passively released following cell death or actively secreted – may be recognized directly by APC, inducing a signal transduction pathway that leads to increased cross-presentation. One example includes DRibbles-derived antigen cross-priming that is partially dependent of CLEC9A (Li et al., 2011). These data suggest that autophagosome express the ligand of CLEC9A, which is recognized by APC to enhance cross-priming. (C) Blocking autophagic flux in antigen donor cells – using pharmacological inhibitor or during influenza A infection – favors antigen cross-priming (Uhl et al., 2009). In this context, it is possible that inhibition of lysosomal fusion serves to protect epitope within the autophagosome of dying cells and facilitates delivery of intact or partially processed antigen to APC. In addition, the capture of intact autophagosome could enhance cytokine production (e.g., type I interferon) and cDC activation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Institut Pasteur, INSERM, ANRS, the European Research Council, and the French National Research Agency (ANR). The authors also thank Matt Buckwalter for his helpful edits to the text.
References
Abrahamsen, H., and Stenmark, H. (2010). Protein secretion: unconventional exit by exophagy. Curr. Biol. 20, R415–R418.
Accapezzato, D., Visco, V., Francavilla, V., Molette, C., Donato, T., Paroli, M., Mondelli, M. U., Doria, M., Torrisi, M. R., and Barnaba, V. (2005). Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J. Exp. Med. 202, 817–828.
Albert, M. L. (2004). Death-defying immunity: do apoptotic cells influence antigen processing and presentation? Nat. Rev. Immunol. 4, 223–231.
Albert, M. L., Jegathesan, M., and Darnell, R. B. (2001). Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat. Immunol. 2, 1010–1017.
Albert, M. L., Sauter, B., and Bhardwaj, N. (1998). Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 392, 86–89.
Amaravadi, R. K., Lippincott-Schwartz, J., Yin, X. M., Weiss, W. A., Takebe, N., Timmer, W., Dipaola, R. S., Lotze, M. T., and White, E. (2011). Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 17, 654–666.
Apetoh, L., Ghiringhelli, F., Tesniere, A., Criollo, A., Ortiz, C., Lidereau, R., Mariette, C., Chaput, N., Mira, J. P., Delaloge, S., Andre, F., Tursz, T., Kroemer, G., and Zitvogel, L. (2007). The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 220, 47–59.
Bateman, A. R., Harrington, K. J., Kottke, T., Ahmed, A., Melcher, A. A., Gough, M. J., Linardakis, E., Riddle, D., Dietz, A., Lohse, C. M., Strome, S., Peterson, T., Simari, R., and Vile, R. G. (2002). Viral fusogenic membrane glycoproteins kill solid tumor cells by nonapoptotic mechanisms that promote cross presentation of tumor antigens by dendritic cells. Cancer Res. 62, 6566–6578.
Belz, G. T., Behrens, G. M., Smith, C. M., Miller, J. F., Jones, C., Lejon, K., Fathman, C. G., Mueller, S. N., Shortman, K., Carbone, F. R., and Heath, W. R. (2002). The CD8alpha(+) dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J. Exp. Med. 196, 1099–1104.
Bevan, M. J. (1976). Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 143, 1283–1288.
Boon, T., Coulie, P. G., Van Den Eynde, B. J., and Van Der Bruggen, P. (2006). Human T cell responses against melanoma. Annu. Rev. Immunol. 24, 175–208.
Cadwell, K., Liu, J. Y., Brown, S. L., Miyoshi, H., Loh, J., Lennerz, J. K., Kishi, C., Kc, W., Carrero, J. A., Hunt, S., Stone, C. D., Brunt, E. M., Xavier, R. J., Sleckman, B. P., Li, E., Mizushima, N., Stappenbeck, T. S., and Virgin, H. W. T. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263.
de la Calle, C., Joubert, P. E., Law, K. W. H., Hasan, M., and Albert, M. L. (2011). Simultaneous assessment of autophagy and apoptosis using multispectral imaging cytometry. Autophagy 7, 1045–1051.
Dengjel, J., Schoor, O., Fischer, R., Reich, M., Kraus, M., Muller, M., Kreymborg, K., Altenberend, F., Brandenburg, J., Kalbacher, H., Brock, R., Driessen, C., Rammensee, H. G., and Stevanovic, S. (2005). Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 7922–7927.
Deretic, V., and Levine, B. (2009). Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5, 527–549.
English, L., Chemali, M., Duron, J., Rondeau, C., Laplante, A., Gingras, D., Alexander, D., Leib, D., Norbury, C., Lippe, R., and Desjardins, M. (2009). Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487.
Falo, L. D. Jr., Kovacsovics-Bankowski, M., Thompson, K., and Rock, K. L. (1995). Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. Nat. Med. 1, 649–653.
Fausch, S. C., Da Silva, D. M., and Kast, W. M. (2003). Differential uptake and cross-presentation of human papillomavirus virus-like particles by dendritic cells and Langerhans cells. Cancer Res. 63, 3478–3482.
Flinsenberg, T. W., Compeer, E. B., Boelens, J. J., and Boes, M. (2011). Antigen cross-presentation: extending recent laboratory findings to therapeutic intervention. Clin. Exp. Immunol. 165, 8–18.
Gannage, M., Dormann, D., Albrecht, R., Dengjel, J., Torossi, T., Ramer, P. C., Lee, M., Strowig, T., Arrey, F., Conenello, G., Pypaert, M., Andersen, J., Garcia-Sastre, A., and Munz, C. (2009). Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380.
Giodini, A., and Albert, M. L. (2010). A whodunit: an appointment with death. Curr. Opin. Immunol. 22, 94–108.
Huang, F. P., Platt, N., Wykes, M., Major, J. R., Powell, T. J., Jenkins, C. D., and Macpherson, G. G. (2000). A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 191, 435–444.
Hugues, S., Mougneau, E., Ferlin, W., Jeske, D., Hofman, P., Homann, D., Beaudoin, L., Schrike, C., Von Herrath, M., Lehuen, A., and Glaichenhaus, N. (2002). Tolerance to islet antigens and prevention from diabetes induced by limited apoptosis of pancreatic beta cells. Immunity 16, 169–181.
Joubert, P. E., Meiffren, G., Gregoire, I. P., Pontini, G., Richetta, C., Flacher, M., Azocar, O., Vidalain, P. O., Vidal, M., Lotteau, V., Codogno, P., Rabourdin-Combe, C., and Faure, M. (2009). Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 6, 354–366.
Jounai, N., Takeshita, F., Kobiyama, K., Sawano, A., Miyawaki, A., Xin, K. Q., Ishii, K. J., Kawai, T., Akira, S., Suzuki, K., and Okuda, K. (2007). The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A. 104, 14050–14055.
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580.
Kazama, H., Ricci, J. E., Herndon, J. M., Hoppe, G., Green, D. R., and Ferguson, T. A. (2008). Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29, 21–32.
Komatsu, M., and Ichimura, Y. (2010). Selective autophagy regulates various cellular functions. Genes Cells 15, 923–933.
Kroemer, G., Marino, G., and Levine, B. (2010). Autophagy and the integrated stress response. Mol. Cell 40, 280–293.
Kurts, C., Robinson, B. W., and Knolle, P. A. (2010). Cross-priming in health and disease. Nat. Rev. Immunol. 10, 403–414.
Lee, H. K., Lund, J. M., Ramanathan, B., Mizushima, N., and Iwasaki, A. (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401.
Lee, H. K., Mattei, L. M., Steinberg, B. E., Alberts, P., Lee, Y. H., Chervonsky, A., Mizushima, N., Grinstein, S., and Iwasaki, A. (2010). In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32, 227–239.
Li, B., Lei, Z., Lichty, B. D., Li, D., Zhang, G. M., Feng, Z. H., Wan, Y., and Huang, B. (2010). Autophagy facilitates major histocompatibility complex class I expression induced by IFN-gamma in B16 melanoma cells. Cancer Immunol. Immunother. 59, 313–321.
Li, Y., Wang, L. X., Pang, P., Cui, Z., Aung, S., Haley, D., Fox, B. A., Urba, W. J., and Hu, H. M. (2011). Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin. Cancer Res. 17, 7047–7057.
Li, Y., Wang, L. X., Pang, P., Twitty, C., Fox, B. A., Aung, S., Urba, W. J., and Hu, H. M. (2009). Cross-presentation of tumor associated antigens through tumor-derived autophagosomes. Autophagy 5, 576–577.
Locher, C., Conforti, R., Aymeric, L., Ma, Y., Yamazaki, T., Rusakiewicz, S., Tesniere, A., Ghiringhelli, F., Apetoh, L., Morel, Y., Girard, J. P., Kroemer, G., and Zitvogel, L. (2010). Desirable cell death during anticancer chemotherapy. Ann. N. Y. Acad. Sci. 1209, 99–108.
Mai, S., Muster, B., Bereiter-Hahn, J., and Jendrach, M. (2012). Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy 8.
Mehrpour, M., Esclatine, A., Beau, I., and Codogno, P. (2010). Overview of macroautophagy regulation in mammalian cells. Cell Res. 20, 748–762.
Mellen, M. A., De La Rosa, E. J., and Boya, P. (2008). The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 15, 1279–1290.
Mellman, I., and Steinman, R. M. (2001). Dendritic cells: specialized and regulated antigen processing machines. Cell 106, 255–258.
Michaud, M., Martins, I., Sukkurwala, A. Q., Adjemian, S., Ma, Y., Pellegatti, P., Shen, S., Kepp, O., Scoazec, M., Mignot, G., Rello-Varona, S., Tailler, M., Menger, L., Vacchelli, E., Galluzzi, L., Ghiringhelli, F., Di Virgilio, F., Zitvogel, L., and Kroemer, G. (2011). Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577.
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741.
Nedjic, J., Aichinger, M., Emmerich, J., Mizushima, N., and Klein, L. (2008). Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 455, 396–400.
Nimmerjahn, F., Milosevic, S., Behrends, U., Jaffee, E. M., Pardoll, D. M., Bornkamm, G. W., and Mautner, J. (2003). Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 33, 1250–1259.
Nogueira-Machado, J. A., Volpe, C. M., Veloso, C. A., and Chaves, M. M. (2011). HMGB1, TLR and RAGE: a functional tripod that leads to diabetic inflammation. Expert Opin. Ther. Targets 15, 1023–1035.
Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T., and Munz, C. (2005). Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596.
Pang, B., and Neefjes, J. (2010). Coupled for cross-presentation in tumor immunotherapy. Sci. Transl. Med. 2, 44ps40.
Petersen, T. R., Dickgreber, N., and Hermans, I. F. (2010). Tumor antigen presentation by dendritic cells. Crit. Rev. Immunol. 30, 345–386.
Qu, X., Zou, Z., Sun, Q., Luby-Phelps, K., Cheng, P., Hogan, R. N., Gilpin, C., and Levine, B. (2007). Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128, 931–946.
Reis e Sousa, C., and Germain, R. N. (1995). Major histocompatibility complex class I presentation of peptides derived from soluble exogenous antigen by a subset of cells engaged in phagocytosis. J. Exp. Med. 182, 841–851.
Rock, K. L., Gamble, S., and Rothstein, L. (1990). Presentation of exogenous antigen with class I major histocompatibility complex molecules. Science 249, 918–921.
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., Omori, H., Noda, T., Yamamoto, N., Komatsu, M., Tanaka, K., Kawai, T., Tsujimura, T., Takeuchi, O., Yoshimori, T., and Akira, S. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268.
Sancho, D., Joffre, O. P., Keller, A. M., Rogers, N. C., Martinez, D., Hernanz-Falcon, P., Rosewell, I., and Reis E Sousa, C. (2009). Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458, 899–903.
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195.
Scheinecker, C., Mchugh, R., Shevach, E. M., and Germain, R. N. (2002). Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J. Exp. Med. 196, 1079–1090.
Schmid, D., Pypaert, M., and Munz, C. (2007). Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26, 79–92.
Schulz, O., Diebold, S. S., Chen, M., Naslund, T. I., Nolte, M. A., Alexopoulou, L., Azuma, Y. T., Flavell, R. A., Liljestrom, P., and Reis E Sousa, C. (2005). Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 433, 887–892.
Shimizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thompson, C. B., and Tsujimoto, Y. (2004). Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 6, 1221–1228.
Sigal, L. J., Crotty, S., Andino, R., and Rock, K. L. (1999). Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature 398, 77–80.
Sims, G. P., Rowe, D. C., Rietdijk, S. T., Herbst, R., and Coyle, A. J. (2010). HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 28, 367–388.
Tal, M. C., Sasai, M., Lee, H. K., Yordy, B., Shadel, G. S., and Iwasaki, A. (2009). Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 2770–2775.
Tang, D., Kang, R., Cheh, C. W., Livesey, K. M., Liang, X., Schapiro, N. E., Benschop, R., Sparvero, L. J., Amoscato, A. A., Tracey, K. J., Zeh, H. J., and Lotze, M. T. (2010). HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310.
Thorburn, J., Horita, H., Redzic, J., Hansen, K., Frankel, A. E., and Thorburn, A. (2009). Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 16, 175–183.
Turley, S., Poirot, L., Hattori, M., Benoist, C., and Mathis, D. (2003). Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J. Exp. Med. 198, 1527–1537.
Uhl, M., Kepp, O., Jusforgues-Saklani, H., Vicencio, J. M., Kroemer, G., and Albert, M. L. (2009). Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 16, 991–1005.
Wei, J., Waithman, J., Lata, R., Mifsud, N. A., Cebon, J., Kay, T., Smyth, M. J., Sadler, A. J., and Chen, W. (2010). Influenza A infection enhances cross-priming of CD8+ T cells to cell-associated antigens in a TLR7- and type I IFN-dependent fashion. J. Immunol. 185, 6013–6022.
Yanai, H., Ban, T., Wang, Z., Choi, M. K., Kawamura, T., Negishi, H., Nakasato, M., Lu, Y., Hangai, S., Koshiba, R., Savitsky, D., Ronfani, L., Akira, S., Bianchi, M. E., Honda, K., Tamura, T., Kodama, T., and Taniguchi, T. (2009). HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462, 99–103.
Yang, D., Postnikov, Y. V., Li, Y., Tewary, P., De La Rosa, G., Wei, F., Klinman, D., Gioannini, T., Weiss, J. P., Furusawa, T., Bustin, M., and Oppenheim, J. J. (2012). High-mobility group nucleosome-binding protein 1 acts as an alarmin and is critical for lipopolysaccharide-induced immune responses. J. Exp. Med. 209, 157–171.
Yewdell, J. W., Anton, L. C., and Bennink, J. R. (1996). Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J. Immunol. 157, 1823–1826.
Keywords: autophagy, cross-priming, antigen presenting cells, dendritic cells, cell death, tumor immunity, viral immunity
Citation: Joubert P-E and Albert ML (2012) Antigen cross-priming of cell-associated proteins is enhanced by macroautophagy within the antigen donor cell. Front. Immun. 3:61. doi: 10.3389/fimmu.2012.00061
Received: 26 January 2012; Paper pending published: 07 February 2012;
Accepted: 09 March 2012; Published online: 28 March 2012.
Edited by:
Christian Munz, University of Zurich, SwitzerlandReviewed by:
Christian Kurts, Friedrich Wilhelms-Universität Bonn, GermanyPeter M. Van Endert, Université Paris Descartes/INSERM, France
Copyright: © 2012 Joubert and Albert. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Matthew L. Albert, Laboratory of Dendritic Cell Immunobiology, Department of Immunology, INSERM U818, Institut Pasteur, Paris, France. e-mail: albertm@pasteur.fr