 Melissa A. Brown* Julianne K. Hatfield
Melissa A. Brown* Julianne K. Hatfield
- Department of Microbiology and Immunology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA
There is abundant evidence that mast cells are active participants in events that mediate tissue damage in autoimmune disease. Disease-associated increases in mast cell numbers accompanied by mast cell degranulation and elaboration of numerous mast cell mediators at sites of inflammation are commonly observed in many human autoimmune diseases including multiple sclerosis, rheumatoid arthritis, and bullous pemphigoid. In animal models, treatment with mast cell stabilizing drugs or mast cell ablation can result in diminished disease. A variety of receptors including those engaged by antibody, complement, pathogens, and intrinsic danger signals are implicated in mast cell activation in disease. Similar to their role as first responders in infection settings, mast cells likely orchestrate early recruitment of immune cells, including neutrophils, to the sites of autoimmune destruction. This co-localization promotes cellular crosstalk and activation and results in the amplification of the local inflammatory response thereby promoting and sustaining tissue damage. Despite the evidence, there is still a debate regarding the relative role of mast cells in these processes. However, by definition, mast cells can only act as accessory cells to the self-reactive T and/or antibody driven autoimmune responses. Thus, when evaluating mast cell involvement using existing and somewhat imperfect animal models of disease, their importance is sometimes obscured. However, these potent immune cells are undoubtedly major contributors to autoimmunity and should be considered as important targets for therapeutic disease intervention.
Introduction
Autoimmune and allergic diseases share fundamentally important features in that both are the result of “hypersensitive” immune responses directed toward inherently harmless antigens. It is this overzealous response that causes the pathology. In fact, as late as the 1960s many disease models that we now know are autoimmune were labeled as “experimental allergic” neuritis, encephalomyelitis, orchitis, uveitis, and glomerulonephritis, reflecting this concept (Mackay and Anderson, 2010). But this nomenclature has been largely abandoned and the myriad of common mechanisms that link allergy and autoimmunity are sometimes forgotten. Mast cells, clearly implicated in both immediate- and delayed-type allergic responses, may underlie the relationship between various types of hypersensitive diseases, yet their role in autoimmunity is still often questioned. Here we review the data that implicates mast cells in a variety of autoimmune diseases and address some of the possible reasons for the controversy. We also discuss how many of the same mast cell mediated mechanisms that confer protection in infection settings or mediate allergic responses are pathologic in the face of a T- or B-cell directed response to a self antigen.
Autoimmunity: Directed by the Adaptive Immune System but Sustained by Innate Immune Cells
There is a complex interplay of susceptibility factors that must coalesce in an individual in order for autoimmune disease to develop. Genetics, hormonal influences and environment play important roles and some of these factors have been identified (for review see Kivity et al., 2009; Rubtsov et al., 2010; Rai and Wakeland, 2011; Pennell et al., 2012). However, many of the specific determinants that initiate an autoimmune response and allow it to be sustained and cause pathology are still enigmatic. By definition, the “directors” of autoimmune responses are cells of the adaptive immune system. Thus, an early event in the development of autoimmunity is the activation and expansion of T and/or antibody-producing B cells bearing self molecule-reactive receptors. Naïve autoreactive T or B cells first encounter antigen in secondary lymphoid organs where they undergo differentiation and acquire their effector function. These primed CD4+ T helper cells, CD8+ T cytolytic cells or secreted antibody molecules enter the blood stream and migrate to sites of inflamed tissues expressing relevant autoantigens (Figure 1). T cells, through the elaboration of cytotoxic mediators, and antibodies, through complement fixation or their ability to activate resident accessory cells such as macrophages and mast cells via Fc receptor engagement, can play direct roles in tissue destruction at these sites (Lohr et al., 2005).
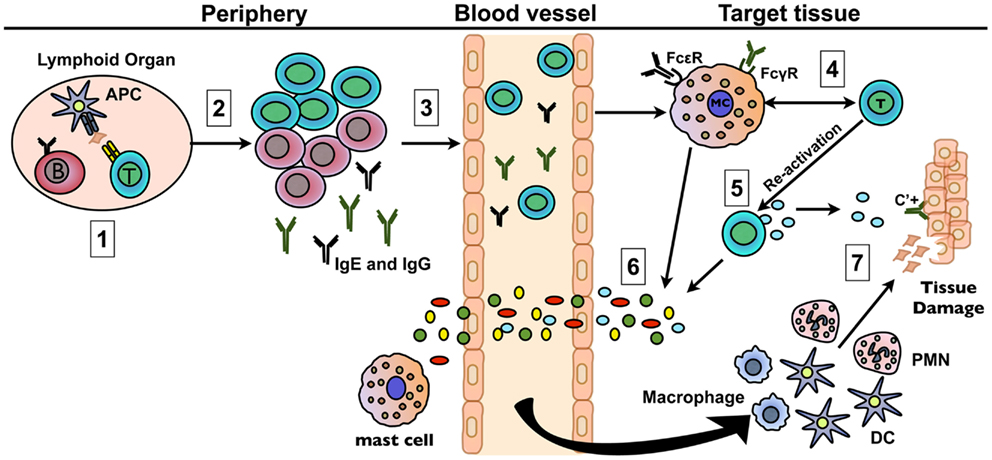
Figure 1. Mast cells amplify autoimmune responses through multiple mechanisms. (1) Naïve autoreactive B and T cells are activated in the secondary lymphoid organs via antigen presentation by APCs. (2) B and T cells undergo differentiation and proliferation leading to a large pool of autoreactive effector cells. Plasma cells secrete self-reactive IgG and/or IgE antibodies. (3) T cells and antibody direct the immune response to the target tissue by entering the bloodstream and trafficking to sites of relevant autoantigen expression. (4) Mast cells are activated early in disease either through TLR/NLR engagement or T cell-mast cell crosstalk via cell–cell contact or soluble mediators. FcR and C3aR/C5aR engagement on mast cells may also play a role. The possibility of activation through cross-talk with other cell types has not been verified. (5) T cells are reactivated through antigen encounter within the target tissue or through cross-talk with mast cells and other resident immune cells. (6) The subsequent release of mediators by mast cells and T cells results in increased vascular permeability and in the recruitment of other inflammatory cells, including neutrophils. (7) The resulting increasing cascade of mediators released by the cells that have co-localized at target tissue sites contribute to amplified inflammation that can directly or indirectly sustain tissue damage.
While the adaptive immune response initiates autoimmune inflammation, innate immune cells are critical for sustaining the response that leads to pathology (reviewed in Bach et al., 2004; Tenner, 2004; Marshak-Rothstein and Ohashi, 2007; Pisetsky, 2008; Chervonsky, 2009; Maciejewska Rodrigues et al., 2009). However, because autoimmunity is dependent on self-reactive T and/or B cells, innate immune cells can only play a modifying role at best. It is this secondary, albeit critical, role of innate cells that often makes it difficult to definitively determine their relative contribution because of the overlaps in immune cell function. The reliance on inherently imperfect animal models of human disease to define the interplay between adaptive and innate immune cells exacerbates this problem. The spontaneous or actively induced development of disease in animals is rarely physiologic and is often associated with an overly robust autoreactive B or T cell response, one that does not recapitulate the gradual inflammatory processes that characterize the development of most chronic human autoimmune illnesses. Thus, the contribution of modulatory cells can be masked, especially if they exert their critical effects early in disease. Such limitations have sometimes led to inconsistent results and controversy in the field about which cells make substantial contributions to disease pathogenesis. The challenge in convincingly implicating such accessory cells is perhaps best illustrated by the studies of mast cells in autoimmunity. Despite a wealth of supportive evidence, the contribution of mast cells to autoimmunity, particularly to rheumatoid arthritis (RA) and multiple sclerosis (MS), is still debated.
Mast Cells: Potent Immunomodulatory Cells
Mast cells are c-kithi, FcεRI+ granular cells of the innate immune system that reside in most tissues, but do not circulate in a mature form in the blood. They are most prevalent at sites that interface with the external environment and are often found in close association with blood vessels and nerves (reviewed in Rao and Brown, 2008). Depending on the mode of activation, mast cells can release unique arrays of immune modulating molecules that exist preformed in granules within minutes. Activation also induces the new synthesis of cell surface associated and soluble immune mediators that can exert prolonged effects on a response (Galli et al., 2005; Metz et al., 2007). Although cross-linking of the high affinity IgE receptor (FcεRI) is the best studied mode of mast cell stimulation, the last few years have been transformative in terms of identifying a range of other mast cell activating receptors that act in this capacity leading to either protective or pathologic outcomes. Indeed, there are few infection or disease states in which these potent immunomodulatory cells have not been directly or indirectly implicated (Rao and Brown, 2008) and thus it follows that mast cells are most certainly modifiers of autoimmune disease as well.
Mast Cells as Modifiers of Autoimmunity I: Correlative Evidence
The idea that mast cells are involved in the initiation and sustaining events of autoimmunity is not based on conventional wisdom, but rather on substantial data from studies of both human disease and animal models (Benoist and Mathis, 2002; Lee et al., 2002; Gregory and Brown, 2006; Rao and Brown, 2008; Sayed et al., 2008; Walker et al., 2011). Here we focus primarily on studies of RA, bullous pemphigoid (BP), type I diabetes and multiple sclerosis (MS), where the evidence for a mast cell contribution is particularly convincing.
In RA the primary targets of the autoantibody-directed immune response are the synovial joints (Lee and Weinblatt, 2001; Lee et al., 2002). Although mast cells are normal residents within the synovium, their numbers are increased in human disease and this is associated with the local production of several mast cell-derived mediators (reviewed in Nigrovic and Lee, 2007). Tryptase, a preformed mast cell-specific protease, is thought to initiate the inflammatory response by complexing with heparin to induce the release of neutrophil chemotactic factors such as TNF, IL-1β, and IL-17, as well as IL-33 by synovial fibroblasts (Shin et al., 2009; Hueber et al., 2010). IL-33, in turn, acts directly on mast cells to initiate the expression of additional inflammatory cytokines and chemokines (Xu et al., 2008, 2010; Verri et al., 2010). Tryptase can also directly activate synovial fibroblasts through its interaction with the protease-activated receptor 2 (PAR2) enhancing their ability to express proteases that degrade cartilage and bone (Palmer et al., 2007; Sawamukai et al., 2010). In human RA, a 6- to 25-fold expansion of mast cells has been observed in affected joints when compared to numbers in normal joints (Nigrovic and Lee, 2007). Mast cell numbers also increase more than threefold in multiple animal models of RA (Aloe et al., 1993; Kakizoe et al., 1999; Shin et al., 2006) and it has been postulated that they are essential for the joint-specific vascular leak phenomenon involving both histamine and serotonin (Binstadt et al., 2006). In collagen-induced arthritis, increased mast cell numbers and the associated neutrophil influx to the joints as well as exacerbated clinical severity are reduced by treatment with the mast cell stabilizer, sodium nedocromil (Pimentel et al., 2011).
Bullous pemphigoid is a skin blistering disease that can be induced in mice by injection of an antibody (anti-BP180) directed to hemidesmosomes, structures involved in cell–cell adherence (Kasperkiewicz and Zillikens, 2007). Early events in this model of disease include massive degranulation of skin mast cells that precede the characteristic neutrophil influx and blister formation. Cromolyn sodium, another mast cell stabilizing drug, blocks these events (Chen et al., 2001; Navi et al., 2007). In human disease, degranulated mast cells are prominent in lesions where histamine, tryptase, and several mast cell-associated chemokines are detected in blister fluid (Baba et al., 1976; Dvorak et al., 1982; Katayama et al., 1984; D’Auria et al., 2000).
In Type I diabetes, CD8+ T cells are thought to play a predominant role in the destruction of the insulin-producing islet cells of the pancreas although CD4+ T cells and antibodies are also implicated. The resulting loss of blood glucose regulation can lead to retinopathy, neuropathy, cardiovascular disease, and nephropathy (Bluestone et al., 2010). Gene expression profiling of the pancreatic lymph nodes in the spontaneous BB rat model revealed activated mast cell transcripts that correlate with disease and treatment with cromolyn sodium, results in a significant delay in disease onset (Geoffrey et al., 2006). Inhibitors of tyrosine kinase can also inhibit and reverse spontaneous disease in the NOD mouse model of Type I diabetes, however, specific inhibitors of c-kit, a tyrosine kinase-associated receptor required for mast cell development, have marginal effects on limiting disease progression (Louvet et al., 2008). Paradoxically, a protective role for mast cells in this disease has been proposed based on results of studies in a model of spontaneous diabetes in NOD mice. Treatment of NOD mice with anti-FcεRI antibodies, which activate mast cells and basophils to produce IL-4, delays diabetes (Hubner et al., 2011). It is postulated that in this model system IL-4 is exerting anti-inflammatory effects by skewing CD4+ T helper cells to a Th2 dominated response.
Multiple sclerosis is a CD4+ T cell-dependent disease in which myelinated nerves in the relatively immunologically sequestered CNS are targeted for destruction leading to variable neurologic dysfunction (Sospedra and Martin, 2005; Hauser and Oksenberg, 2006). Mast cells are observed in plaques of patients assessed post-mortem as are transcripts encoding tryptase, histamine, and FcεRI, identified by microarray analyses (Lock et al., 2002). Tryptase and histamine are also detected in the cerebral spinal fluid of some patients (Tuomisto et al., 1983; Rozniecki et al., 1995). In experimental autoimmune encephalomyelitis (EAE), a mouse model of MS induced by immunization with myelin peptides in CFA, treatment of mice with proxicromil an inhibitor of mast cell degranulation, cyproheptadine, a serotonin receptor antagonist or reserpine, which depletes mast cell granules, inhibits disease (Dietsch and Hinrichs, 1989; Dimitriadou et al., 2000).
Mast Cells as Modifiers of Autoimmunity II: Confounding Evidence from in vivo Studies of Mast Cell Deficient Mice
Although compelling, measures of increased mast cell numbers, mast cell activation profiles, or efficacy of mast cell-inhibitory drugs are not proof of mast cell involvement. The most definitive studies implicating mast cells have relied on the use of mast cell deficient mice, most commonly those with mutations in Kit, the gene encoding c-kit, the stem cell factor (SCF) receptor (W/Wv, W−sh mice) or in Kitlg, the gene encoding the c-kit ligand, SCF (Sl/Sld mice). The differentiation and long term survival of mast cells is dependent on sustained high level SCF/c-kit signaling and mice with W/Wv, W−sh/W−sh, or Sl/Sld mutations that compromise this signaling exhibit defects in mast cell development (Galli and Kitamura, 1987; Grimbaldeston et al., 2005). These mice are imperfect models because they also bear other hematologic and non-hematologic defects including neutropenia, anemia, pigment production defects, reduced intestinal mobility and sterility (W/Wv), or time-dependent loss of mast cells, cardiac abnormalities, and neutrophilia (W−sh/W−sh; Galli and Kitamura, 1987; Grimbaldeston et al., 2005; Nigrovic et al., 2008). Sl/Sld mice also develop lymphocytic leukemia at a high rate and have shortened life spans (Galli and Kitamura, 1987). Systemic or local reconstitution of Kit mutant mice with bone marrow derived mast cells (BMMC) selectively restores most mast cell populations, although not always to wild type levels, but does not correct other mutant Kit related abnormalities. If a wild type phenotype is re-established in Kit mutant mice by selective repopulation of tissue mast cells, it is generally accepted that mast cells are implicated.
Several recent reports have described the generation of unique strains of mast cell deficient mice that do not appear to have the accompanying profound hematologic defects of Kit mutant mice, although their characterization is still incomplete (Dudeck et al., 2011; Feyerabend et al., 2011; Lilla et al., 2011; Otsuka et al., 2011). These mice provide the promise that a more unequivocal assessment of mast cell contributions in vivo can be achieved. Of particular interest to this discussion of autoimmune disease are the Cpa3Cre/+ mice. Carboxypeptidase 3 (Cpa3) is a mast cell specific protease expressed early in development and Cpa3Cre/+ mice contain a transgene encoding cre-recombinase under the control of the carboxypeptidase 3 (Cpa3) promoter (Feyerabend et al., 2011). It is presumed that the high level expression of Cre-recombinase in mast cell precursors that express Cpa3 is toxic, causing genomic instability and the ultimate demise of this cell lineage at an early stage in development. With the exception of a small diminution in basophil numbers, no other cell lineages appear to be affected. Below, we review the controversial evidence that supports and refutes a role for mast cells in autoimmunity using mast cell deficient mice.
Evidence for Mast Cell Influence in Autoimmunity Using KitW/Wv Mice
Results in KitW/Wv mice have provided the most consistent supportive evidence for mast cell involvement in autoimmunity. Of note, in most autoimmune disease models examined using these mice, including RA, BP, and EAE, the mast cell-dependent neutrophil recruitment discussed above is an important pathogenic event, one that is absent in KitW/Wv mice (D’Auria et al., 2000; Chen et al., 2006; Kasperkiewicz and Zillikens, 2007; Monach et al., 2010; Sayed et al., 2010; Kaplan, 2011; Walker et al., 2011; Warde, 2011). Despite the inherent neutropenia of KitW/Wv mice, restoration of mast cells results in normal disease-related neutrophil influx to inflamed tissues indicating that the few neutrophils present in these mice are mobilized in a mast cell-dependent manner. The relationship of mast cells and neutrophils was first defined in infection settings where mast cells, acting as first responders, release multiple mediators including histamine, PAF, TNF, CXCL2, and IL-8 within minutes of activation and initiate the influx of neutrophils (Zhang et al., 1992; Malaviya et al., 1996a; Biedermann et al., 2000). As in infection, in autoimmune disease, neutrophils likely promote disease-associated changes in vascular permeability, but also contribute directly to self-tissue damage through the release of proteases, reactive oxygen intermediates, and chemoattractants that regulate the influx of other inflammatory cells, contributing to a cascade of damaging events (reviewed in Kumar and Sharma, 2010; Mantovani et al., 2011).
Mast cell dependence has been demonstrated in models of RA (K/BxN serum transfer-induced and antigen-induced RA) using (WB xC57BL/6)F1 (WBB6) KitW/Wv mice where, as in human RA, neutrophils are the dominant lineage in synovial effusions (Nigrovic and Lee, 2007). K/BxN mice exhibit an early and spontaneous onset of RA-like symptoms due to the presence of glucose-6-phosphate reactive autoantibodies and transfer of K/BxN serum into wild type but not WBB6 KitW/Wv mice, induces disease (Corr and Crain, 2002; Lee et al., 2002). Disease susceptibility is restored to Kit mutant mice by BMMC reconstitution. Mast cell reconstitution with IL-1β−/− BMMCs fails to restore disease susceptibility in the K/BxN serum transfer model, indicating that disease pathogenesis is mediated by mast cell-derived IL-1β (Nigrovic et al., 2007). Mast cells also directly modulate access of arthritogenic antibodies to the joint in the hyperacute phase of K/BxN serum transfer arthritis, a feature likely directly or indirectly related to the effects of mast cells on vascular permeability. Labeled antibodies were specifically deposited in the joint of wild type mice within minutes after transfer, while deposition was delayed and reduced in W/Wv animals (Wipke et al., 2002, 2004). In antigen-induced arthritis, a chronic arthritis model induced by the direct injection of methylated bovine serum albumen into the joint, there is little difference in early disease severity in wild type and KitW/Wv mice, but mast cell deficient mice develop significantly reduced cartilage damage over time, a phenotype reversed by BMMC reconstitution (van den Broek et al., 1988). However, in the collagen-induced arthritis model, there are no clinical disease differences leading the authors to suggest that either this model is mast cell-independent or that mast cells have a minor role (Pitman et al., 2011).
In the BP model, WBB6 KitW/Wv mice exhibit reduced neutrophil accumulation in the skin and diminished skin blistering upon transfer of serum autoantibodies compared to wild type mice, whereas a wild type-like disease with associated neutrophil influx is restored upon mast cell reconstitution (Chen et al., 2002). Notably, administration of IL-8, a neutrophil chemoattractant, can overcome the mast cell-deficiency (Chen et al., 2001).
Evidence of mast cell involvement in EAE pathogenesis using KitW/Wv mice has been less consistent. EAE is induced by immunization of mice with myelin peptide in CFA (Mix et al., 2010). Original studies of WBB6 KitW/Wv mice and their wild type littermates in a chronic model of MS induced by myelin oligodendrocyte glycoprotein35–55 peptide (MOG)35–55 immunization revealed that the absence of mast cells does not abolish disease, but there is significantly reduced severity (Secor et al., 2000). This susceptibility is restored to wild type levels by BMMC reconstitution, a procedure that does not correct the c-kit-related anemia in these mice. Mast cells that normally reside in the meninges are essential for this fulminant disease and act to regulate BBB integrity and inflammatory cell entry into the CNS (Sayed et al., 2010). Neutrophils are among the first cells to enter the CNS in EAE and their requirement for disease pathogenesis was confirmed by studies of mice lacking the chemokine receptor, CXCR2, expressed on neutrophils. CXCR2−/− mice cannot mobilize neutrophils from the bone marrow in response to inflammatory signals and fail to develop EAE (Carlson et al., 2008). Mast cells and mast cell-derived TNF are required for neutrophil influx into the CNS (Sayed et al., 2010). Despite the convincing data in mouse models, neutrophil involvement in MS is still controversial. Neutrophils are short-lived cells and likely exert effects during the initiation of disease or in flares associated with relapsing-remitting disease. These cells are not detected in post-mortem assessment of the meninges or CNS as expected, leading many to dismiss them as participants in MS pathogenesis.
Severe PLP131–159-induced disease in SJL mice is also mast cell-dependent. This model is considered by some to be more relevant because it mimics the most commonly presenting relapsing-remitting form of human disease, one observed in at least 85% of patients. SJL KitW/Wv mice exhibit a significantly reduced disease course that corresponds to decreased infiltration of inflammatory cells to the CNS and mast cell reconstitution restores wild type disease susceptibility (Sayed et al., 2011). While there has been independent corroboration that mast cells are important in disease by other groups using KitW/Wv mice (Stelekati et al., 2009; Piconese et al., 2011), other studies report no disease differences (Bennett et al., 2009). The basis of this disparity is still not clear, but there are several potential explanations including age-related differences in susceptibility, the use of distinct disease induction protocols that result in overwhelming T cell responses and unique housing- or diet-related gut microbiota that alter the general immune responsiveness of the mice.
Studies Using KitW−sh/W−sh Mice Fail to Reveal a Mast Cell Influence in RA and EAE
C57BL/6 KitW−sh/W−sh mice were originally touted as better subjects for in vivo studies of mast cell-dependent events because they are on a pure C57BL/6 background and are fertile (Grimbaldeston et al., 2005). However, contradicting the results of studies in KitW/Wv mice, there is no decrease in EAE or RA disease severity in KitW−sh/W−sh suggesting mast cells are not important (Zhou et al., 2007; Piconese et al., 2011). These results may be explained by the fact that unlike neutropenic KitW/Wv mice, KitW−sh/W−sh mice have a baseline pro-inflammatory phenotype that includes neutrophilia, splenomegaly, and increased mast cell precursors in the spleen. Thus, their pre-existing inflammatory state, particularly the neutrophilia, likely obviates the need for the mast cell-dependent neutrophil recruitment that affects early disease-promoting events.
Studies Using Cpa3cre+/− Mice Fail to Implicate Mast Cells in RA and EAE
A recent study by Feyerabend et al. (2011) using the newly described Cpa3cre+/− mice also fails to corroborate a role for mast cells in EAE and RA. Cpa3cre+/− mice are refractory to induction of anaphylaxis and fail to exhibit the normal protective expansion of intestinal mast cells associated with N. Brasiliensis infection supporting the validity of using these mice in models in which IgE-activated mast cells are the major effector cells. Yet Cpa3cre/+ mice are susceptible to RA induction by transfer of K/BxN serum, in contrast to KitW/Wv mice. In addition, no diminution of EAE disease severity was observed in either Cpa3cre/+ or KitW/Wv mice when compared to their wild type littermate controls. Two conclusions were made based on these results: (1) mast cells are dispensable in both models of RA and MS (EAE) and, (2) other c-kit-regulated events influence the susceptibility of mice to RA. A corollary to these conclusions is that mast cells are only relevant in IgE-dependent allergic disease (Katz and Austen, 2011).
Yet these conclusions may be premature for a number of reasons. For example, mast cells express a myriad of other activating receptors that have been documented to play important physiologic roles in many inflammatory responses outside allergy (Rao and Brown, 2008; Sayed et al., 2008). In addition, as discussed earlier mast cells are not the major effector cells in T and/or B cell-dependent autoimmune responses as they are in IgE-dependent responses, thus they are relegated to accessory roles. The strong disease induction conditions used in the EAE experiments in this study (corroborated by high morbidity in all groups Feyerabend et al., 2011) make it likely that a vigorous T cell response masks the mast cell contribution. There is abundant evidence supporting mast cell contributions to autoimmune inflammation and it is important to delineate the underlying reasons for this conflicting data.
Potential Mechanisms of Mast Cell Activation in Autoimmune Disease
There are a multitude of receptors expressed on mast cells that are candidates for involvement in autoimmune modulation (Figure 2). As discussed below, many of these receptors and their ligands have been implicated in autoimmune disease, but whether they are acting to activate mast cells in the context of disease is still in question.
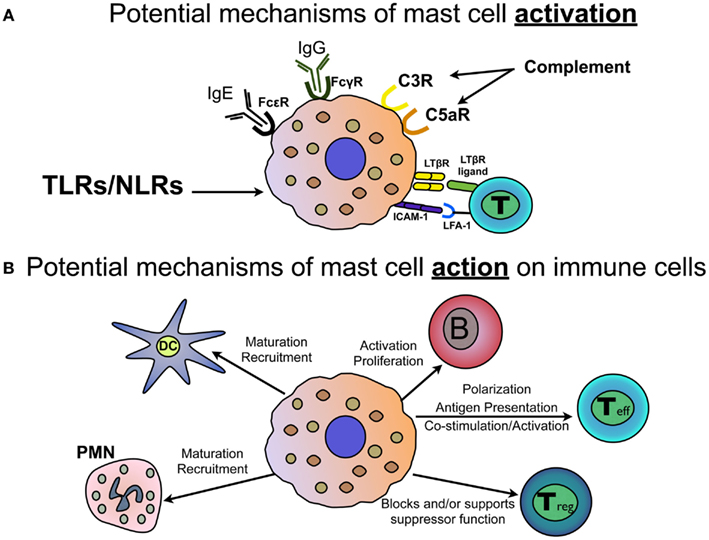
Figure 2. Mast cell activation and response in autoimmunity. (A) Mast cells express a variety of activating receptors that may play a role in modifying autoimmune disease. These include IgG receptors FcγR1, FcγRIIB, and FcγRIII; the high affinity IgE receptor, FcεR1; complement receptors C3R and C5aR; TLRs and NLRs that sense pathogen and intrinsic danger signals; as well as LTβR and ICAM-1 which can engage with LTβR-ligand and LFA-1, respectively, on T cells. (B) Mast cells, through expression of cell surface interaction molecules or mast cell-derived mediators, engage in critical cross-talk with many cells of the innate and adaptive immune system. Demonstrated effects of mast cells include elicitation of dendritic cell (DC) and neutrophil (PMN) maturation and recruitment. Mast cells interact directly with B cells to affect activation, proliferation and differentiation. Mast cells can affect the development of CD4+ T helper cells, can act as antigen presenting cells and express mediators and cell surface molecules that co-stimulate and activate T cells. Mast cells can also inhibit or support T regulatory cell function, depending on the setting.
Pathogen- and Danger-Associated Molecular Pattern Molecules in Infection-Associated Autoimmunity: Overcoming Tolerance
It is well established that the activation and expansion of self-reactive T and B cells can occur only under defined conditions: either the normal mechanisms of negative selection fail to delete particular self-reactive clones during development, there is a failure of normal peripheral mechanisms that maintain tolerance to self, the normal sequestration of antigens that prevent lymphocyte access to self antigens is breached or infection with certain pathogens elicits an adaptive immune response that cross-reacts with self antigens, a phenomenon termed molecular mimicry (Nurieva et al., 2011; Pillai et al., 2011).
Although not definitively established in humans, the development of several autoimmune diseases has been linked to exposure to infectious agents (Kivity et al., 2009). For example, prior EBV infections are postulated to increase the predisposition to Systemic lupus erythematosus (SLE), RA, MS, Sjogren’s syndrome, and polyarteritis nodosa (Barzilai et al., 2007), while autoimmune diabetes susceptibility is associated with enterovirus infections (Richardson et al., 2009). Bacterial infections including H. pylori are implicated in susceptibility to autoimmune gastritis and Sjorgrens syndrome (Annibale et al., 2001; D’Elios et al., 2004). In addition to eliciting a cross-reactive pathogen-specific adaptive immune response, infectious agents can engage a class of pathogen- and danger-associated receptors on both adaptive and innate immune cells, including mast cells. These receptors, including Toll-like receptors (TLRs) and Nacht-LRRs (NLRs), are expressed either within the plasma membrane or in endosomal compartments and recognize conserved components of bacteria, viruses, and protozoa (such as lipopolysaccharide, LPS), double stranded RNA, flagellin, and non-methylated CpG DNA, collectively termed pathogen associated molecular patterns (PAMPs; reviewed in Kawai and Akira, 2011). Activation of immune cells through TLRs and NLRs induces the expression of multiple inflammatory mediators that can ultimately result in local tissue damage causing the release of normally sequestered tissue antigen. Subsequent recognition of these antigens by T or B cells will induce activation and initiate autoimmunity. Alternatively, infection-induced activation of accessory immune cells, including mast cells, macrophages, and neutrophils, can boost inflammation and transform a relatively modest autoreactive response into one that causes detectable pathology.
Toll-like receptors/NLRs are also highly effective at sensing and responding to tissue injury, such as that induced in autoimmunity (Zhang et al., 2010; Kufer and Sansonetti, 2011). Just as pathogens are detected by virtue of “Stranger” signals, damaged cells release “Danger” signals, designated as alarmins or Damage Associated Molecular Patterns (DAMPs) that are also sensed by TLRs and NLRs (reviewed in Bianchi, 2007; Piccinini and Midwood, 2010; Marek and Kagan, 2012). In mammals, TLR3, TLR7, and TLR9 detect microbial nucleic acids, as well as those released from necrotic cells. For example, damaged cells release heat shock proteins such as Hsp60, Hsp70, and alpha-β crystalline that are recognized by TLR2 and TLR4 as well as endogenous cellular products such as uric acid crystals and aggregated peptides that activate NLRs. ATP release from damaged cells acts through purinergic receptor-regulated channels to cause cytosolic ion fluxes that are detected by NLRs. The resulting signals generated through such cell injury can activate immune cells, contributing to the persistence of the inflammatory response and promoting more tissue destruction.
There is emerging evidence that TLRs/NLRs are important receptors in autoimmune diseases (Fischer and Ehlers, 2008; Mills, 2011). Activation through TLR2, 3, 4, and 9 is associated with disease in animal models of RA (Ronaghy et al., 2002; Abdollahi-Roodsaz et al., 2008; Hoffmann et al., 2011; Goh and Midwood, 2012) and TLR4 engagement can overcome the requirement for IL-1β in the serum transfer model of this disease (Choe et al., 2003). Similarly, TLR1, 2, 3, and 7 signals mediate islet inflammation in rat and mouse models of type I diabetes (Lien and Zipris, 2009). TLR1, 2, 3, and 4 are expressed in the salivary glands of patients with Sjogren’s syndrome and when engaged can trigger the expression of a number of activation molecules, including CD40 and ICAM-1, on salivary gland epithelial cells (Spachidou et al., 2007). TLR2 ligation specifically activates Th17 cytokine production in the peripheral blood mononuclear cells of Sjogren’s syndrome patients (Kwok et al., 2012). Based on studies in MS models where TLR2, 4, 7, and 9 have been implicated in exacerbated disease (Marta et al., 2009) and TLR3 is protective (Touil et al., 2006), the targeting of these molecules for therapy has been proposed (Gambuzza et al., 2011).
With the exception of TLR5, there is evidence that most other TLRs are expressed on mast cells and are functional (Marshall et al., 2003a,b; Marshall, 2004). For example, activation of mature mast cells through TLR2 results in their production of several pro-inflammatory cytokines critical in autoimmunity including IL-17, IFNγ, TNF, and IL-1β (Mrabet-Dahbi et al., 2009). Mast cells also express the NLR, NLRP3, and activation of this intracellular receptor activates the inflammasome, a multiprotein complex that regulates the processing and secretion of the IL-1 family of cytokines including IL-1β and IL-18 through the activation of caspase 1 (Nakamura et al., 2009). Mast cells clearly are capable of responding to PAMP and DAMP signals and it will be critical to determine whether this is an important mode of activation in autoimmune disease.
FcεRI and FcγR Activation of Mast Cells: Common Mechanisms Linking Autoimmunity and Allergy
The connection between IgE- and IgG-mediated mast cell responses and autoimmunity is still a tenuous one and is just beginning to gain some traction (Valenta et al., 2009). As discussed earlier, allergy and autoimmunity are both considered hypersensitive diseases that result from inappropriate responses to innocuous antigens. However, it is notable that self-reactive IgG and IgE antibodies are commonly detected in both allergic and autoimmune diseases including atopic dermatitis, RA, systemic lupus erythematosis, Hashimoto’s thyroiditis, Graves’ disease, BP and MS, and their animal models (Permin and Wiik, 1978; Bartholomew et al., 1991; Valenta et al., 1991; Cooper et al., 1993; Ghohestani et al., 1998; Pedotti et al., 2001; Metcalfe et al., 2002; Concha et al., 2004; Smith et al., 2005; Fairley et al., 2007). In atopy, these autoreactive antibodies are primarily detected in those individuals with severe and chronic diseases such as atopic dermatitis, but not in patients with mild respiratory allergies (Valenta et al., 1991). Many of the self antigens that react with IgE are homologs of environmental antigens, including the actin binding protein, profilin, serum albumin, collagen and desmoplakin, and responses to these are thought to result from cross-reactivity (Patterson et al., 1986; Bartholomew et al., 1991; Valenta et al., 1991; Cooper et al., 1993; Spitzauer et al., 1994; Ghohestani et al., 1998). Alternatively there is speculation that inflammation associated with IgE-dependent mast cell responses to environmental allergens induces tissue damage and in a Th2 dominated immune environment these self antigens, no longer sequestered from the immune system, elicit an IgE response (Valenta et al., 2009).
Mast cells also express multiple IgG Fc receptors. Important participants in protective immune responses to pathogens, IgG receptor cross-linking elicits mast cell activation in allergy and also plays critical roles in autoimmunity. In fact, IgG autoantibodies are hallmarks of many autoimmune diseases and have been detected in BP, RA, SLE, and MS (reviewed in Sayed et al., 2008). IgG-specific Fc receptors include both the activating forms, FcγRI and FcγRIII, as well as the inhibitory FcγRIIB. FcγRIIB engagement down regulates both FcγRIII- and FcεRI-induced mast cell mediator release (Miyajima et al., 1997) and the loss of this inhibitory receptor renders mice susceptible to induced autoimmune arthritis and spontaneous SLE and leads to exacerbated EAE (Abdul-Majid et al., 2002; Lock et al., 2002; Pedotti et al., 2003; Robbie-Ryan et al., 2003). KitW/Wv mice reconstituted with either FcRγ−/− mast cells (which lack functional FcγRIII, FcεRI, and FcγRI) or with mast cells lacking only FcγRIII exhibit a reduced and delayed EAE disease course, suggesting that activation of mast cells through these receptors modulates disease (Robbie-Ryan et al., 2003). Mast cell-derived IL-1β, which is necessary for disease in the K/BxN model, is produced after stimulation through autoantibody engagement of FcγRIII (Nigrovic et al., 2007).
Clearly such findings, particularly the presence of self-reactive IgE antibodies in both allergy and autoimmunity, blur the distinction between these classes of disease and suggest that allergy and autoimmunity share similar antibody-dependent mechanisms to activate mast cells and elicit tissue damage in disease.
Complement
The complement system, named for its ability to complement the action of antibodies and phagocytes in an immune response, consists of several proteins synthesized by the liver that exist as inactive pro-proteins. Under inflammatory conditions, a proteolytic cascade is initiated that results in the cleavage and generation of active components, some of which act as direct chemoattractants or bind to specific G protein-coupled receptors on cells and stimulate their activation (reviewed in Ricklin et al., 2010). Complement activation is triggered by the direct binding of complement proteins to microbial surfaces (alternative pathway) or via complement binding to complement-fixing antibody isotypes that have been cross-linked by antigen (classical pathway). A mannose binding lectin-induced pathway also exists (lectin pathway) and these processes of complement fixation, chemoattraction, and cell activation are essential for protection from infection.
Mast cells have an intimate relationship with complement in that they can activate complement components, but they also express surface receptors that directly bind the breakdown products of this proteolytic cascade. It has been speculated that mast cell degranulation induces release of chemoattractants (e.g., LTβ4), vasoactive amines (e.g., histamine), and proteases (e.g., tryptase; (Dawicki and Marshall, 2007). C3 and C5 complement proteins are recruited through the permeable local vasculature to sites of mast cell residence and are cleaved through the action of mast cell proteases to C3a and C5a (Schwartz et al., 1983; Fukuoka et al., 2008). These byproducts can activate mast cells through the engagement of complement receptors expressed on mast cells, C3aR and C5aR, leading to further chemokine/cytokine production and release (Wyatt et al., 1979; Biesecker et al., 1981; Falk et al., 1983).
Not unexpectedly, the best evidence of complement activation in autoimmunity comes from studies of antibody-mediated diseases such as RA, BP, and SLE. Mice depleted of complement are refractory to collagen-induced arthritis (Backlund et al., 2002). Antibodies specific for double stranded DNA form immune complexes and their deposition in the glomeruli of patients with SLE activates complement, generating C3a and C5a (Wyatt et al., 1979; Biesecker et al., 1981; Falk et al., 1983). Although complement activation normally promotes immune complex clearance, the deposition of complement activation products also contributes to inflammation and local tissue injury (Rothfield and Stollar, 1967; Couser, 1985; Couser et al., 1985; Perkinson et al., 1985; Mackworth-Young et al., 1986). In BP, COL17-specific autoantibodies target human type XVII collagen (Robson et al., 2003). Access of these antibodies to the basement membrane elicits an inflammatory cascade that includes complement activation, infiltration of neutrophils and eosinophils, and degranulation of dermal mast cells (Oikarinen et al., 1983; Stahle-Backdahl et al., 1994; Chen et al., 2001; Robson et al., 2003; Liu et al., 2008). In a mouse model of BP induced by administration of anti-hCOL17 antibody, altering the capacity of the antibody to fix complement abrogates its disease-causing ability (Li et al., 2010). In SLE, complement activation deposition of complement at sites of tissue damage is often observed, however, deficiencies in the early components of the complement cascade are strongly associated with disease. In animal models of disease, the lack of C1q or C4 results in loss of apoptotic cell clearance, increasing the supply of autoantigens that are targets of the immune response and promoting a predisposition to SLE-like disease (Cook and Botto, 2006). In EAE, complement acts in concert with autoantibody to promote demyelination (Urich et al., 2006). However, while C3, but not C5, is required for disease, the deletion of both C3aR and C5aR has no effect on disease severity, perhaps illustrating another instance where immune redundancy or strong induction of disease in this experimental setting obscures the role for these molecules (Reiman et al., 2002; Boos et al., 2004; Morgan et al., 2004; Ramos et al., 2009).
There is only limited data demonstrating a direct link between complement-activated mast cells and autoimmune disease. For example, in the K/BxN model, C5aR expression on mast cells is necessary for neutrophil recruitment and subsequent disease development (Nigrovic et al., 2010). The development of BP in mice requires the expression of C5aR on mast cells, which is required for their degranulation (Heimbach et al., 2011). In EAE, the data are equivocal. Targeted deletion of receptors for C3a and C5a does not protect mice from disease, yet deletion of C3 convertase regulator complement receptor 1-related protein y (Crry) exacerbates disease (Reiman et al., 2002; Ramos et al., 2009; Ramaglia et al., 2012). There is also evidence that C5 may actually promote remyelination and prevent gliosis in the CNS (Weerth et al., 2003).
Immune cell crosstalk
The concept that cell–cell interactions result in cross-activation has now become a prevailing paradigm in immunology. There are many examples of mast cell interactions with T cells, DCs, and neutrophils, although the consequences of most of these interactions have not yet been ascribed to autoimmune disease pathogenesis. Mast cell activation is likely to be an early and initiating event that triggers cell co-localization and facilitates such interactions, given that many mast cell mediators released early in a response act to recruit cells to sites of inflammation. As alluded to above, mast cell derived TNF, CXCL2, and LTβ4, for example, recruit neutrophils to inflamed tissues (Ramos et al., 1991, 1994; Zhang et al., 1992, 1995; Mercer-Jones et al., 1999; Biedermann et al., 2000; Wang and Thorlacius, 2005; Nakamura et al., 2009). Similarly, mast dependent neutrophil recruitment to affected tissues has been demonstrated in animal models of autoimmunity including the synovium in RA, the skin in BP, and the meninges in EAE (Chen et al., 2001, 2002; Sayed et al., 2010; Pimentel et al., 2011). A subset of these same mediators, particularly TNF and LTβ4 also recruit DCs and T cells while IL-16, lymphotactin (MIP-1α), and MIP-1β are chemotactic for CD4+ and CD8+ T cells (reviewed in Mekori and Metcalfe, 1999). In a model of cutaneous contact hypersensitivity, mast cell activation is essential for DC migration to the skin and histamine and PGE2, both produced by mast cells, induce the expression of Th2-recruiting cytokines by DCs (McIlroy et al., 2006). Mast cells also affect DC function in a skin allograft model where mast cell derived GM-CSF sustains the DC population that is required for tolerance at the graft site (de Vries et al., 2011). Here we will focus on mast cell interactions with effector T cells and regulatory T cells, as these are arguably the best-described interactions and also the mostly likely to play a significant role in autoimmunity.
There is an abundance of evidence that mast cells affect the activation state and functionality of T cells. Included among the many tissue sites where mast cells reside are the secondary lymphoid organs and a number of studies have demonstrated that their numbers increase in the lymph nodes and spleen in response to some inflammatory signals (Tanzola et al., 2003; Kashiwakura et al., 2004; Sayed et al., 2010). At these sites of T cell priming as well as other tissue sites where T cells are reactivated, bystander mast cells can potentially influence the differentiation phenotype of T cells through their ability to express a myriad of T cell polarizing cytokines including IL-4 and histamine (Mekori and Metcalfe, 1999). Mast cell-derived TNF, in concert with OX40L, enhances T cell activation and proliferation and also contributes to the polarization of Th2 cells in a model of airway hyperreactivity (Nakae et al., 2006, 2007a,b). Mast cells express MHC Class I and inducible MHC Class II molecules and thus have the potential to act as antigen presenting cells (APCs) and there are several reports that substantiate mast cell APC function both in vitro and in vivo (Frandji et al., 1993, 1995; Fox et al., 1994; Malaviya et al., 1996b; Kambayashi et al., 2008, 2009). However, if mast cells serve in this capacity, it is likely that they are more relevant in recall responses and not in the priming of naïve T cells. Given the emerging appreciation of superantigens in allergic pathogenesis, particularly those derived from Staphylococcus (Barnes, 2011), it is tempting to speculate that mast cells “present” superantigens to T cells and initiate their activation. The ability of mast cells to present staphylococcal enterotoxin B to CD8+ T cells supports this idea (Frandji et al., 1996). Through the expression of co-stimulatory molecules such as CD80, CD86, CD40, CD40L, GITR, and 4-1BB as well as the inhibitory molecules PD-L1, PD-L2, and Fas, mast cells can alter T cell effector function (Nakae et al., 2005, 2007c; Lu et al., 2006; Stelekati et al., 2007, 2009; Piconese et al., 2009; Hershko and Rivera, 2010).
A newer concept is that T cells can activate and alter mast cell function. Many of the studies to support this idea have been performed in vitro using membranes isolated from activated T cells and mechanisms that underlie T cell mediated mast cell activation have not been extensively explored. However, activated T cells induce human mast cells to release MMP-9, an event that is blocked by inhibiting TNF (Baram et al., 2001). Human mast cells also produce the neutrophil chemoattract IL-8 upon interaction with activated, but not resting, T cells (Salamon et al., 2005). Oncostatin M, a multifunctional cytokine of the IL-6 family, is released by human mast cells after interaction with activated T cells, but not after cross-linking through the IgE receptor (Salamon et al., 2008). Two receptor-ligand interactions have been implicated in T cell-driven mast cell activation. Upon engagement of the lymphotoxin-β receptor (LTβR), expressed on mast cells, with its ligand, expressed on activated T cells, mast cells release pro-inflammatory cytokines including IL-4, IL-6, TNF, MIP-2, and RANTES (Stopfer et al., 2004). Interaction of LFA-1, expressed on activated T cells, and ICAM-1, expressed on mast cells, induces MC degranulation (Inamura et al., 1998).
Mast cells have a paradoxical relationship with T regulatory (Treg) cells. Most studies that have investigated mast cell-T regulatory cell crosstalk were done using a skin allograft model. In this model Treg cell-derived IL-9 recruits and activates mast cells to the site of the allograft and mast cell presence within the graft is necessary for long term allograft survival (Lu et al., 2006). Treg cells can also inhibit mast cell degranulation via OX40/OX40L interaction and thus help to maintain tolerance, as shown in both allograft transplant and allergic disease models (Gri et al., 2008; Piconese et al., 2009). In contrast, in in vitro studies, mast cell-derived histamine can block Treg cell mediated suppression through H1 receptors (Forward et al., 2009).
Future Questions
Despite the advances in our understanding of these enigmatic cells, there is still much to be learned about how mast cells impact autoimmunity. Mast cell actions in autoimmune responses are often strikingly similar to their roles in infection and allergic disease suggesting that we may gain more mechanistic insight by concentrating on these commonalities. However, in each individual disease there is likely to be variability in the response of mast cells based on the tissue site(s) they are derived from, their mode of activation and the interacting cells that are in play. In addition, it is probable that as in mice, human mast cells exhibit unique and genetically determined responses to activation that may contribute to the severity of or susceptibility to a particular autoimmune disease. Thus, the challenge in ultimately targeting these cells in therapy will be to understand how they contribute to initiating disease and to promoting chronicity and what dictates the quality and magnitude of a particular mast cell response.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdollahi-Roodsaz, S., Joosten, L. A., Koenders, M. I., Devesa, I., Roelofs, M. F., Radstake, T. R., Heuvelmans-Jacobs, M., Akira, S., Nicklin, M. J., Ribeiro-Dias, F., and van den Berg, W. B. (2008). Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J. Clin. Invest. 118, 205–216.
Abdul-Majid, K. B., Stefferl, A., Bourquin, C., Lassmann, H., Linington, C., Olsson, T., Kleinau, S., and Harris, R. A. (2002). Fc receptors are critical for autoimmune inflammatory damage to the central nervous system in experimental autoimmune encephalomyelitis. Scand. J. Immunol. 55, 70–81.
Aloe, L., Probert, L., Kollias, G., Bracci-Laudiero, L., Micera, A., Mollinari, C., and Levi-Montalcini, R. (1993). Level of nerve growth factor and distribution of mast cells in the synovium of tumour necrosis factor transgenic arthritic mice. Int. J. Tissue React. 15, 139–143.
Annibale, B., Negrini, R., Caruana, P., Lahner, E., Grossi, C., Bordi, C., and Delle Fave, G. (2001). Two-thirds of atrophic body gastritis patients have evidence of Helicobacter pylori infection. Helicobacter 6, 225–233.
Baba, T., Sonozaki, H., Seki, K., Uchiyama, M., Ikesawa, Y., and Toriisu, M. (1976). An eosinophil chemotactic factor present in blister fluids of bullous pemphigoid patients. J. Immunol. 116, 112–116.
Bach, J. F., Bendelac, A., Brenner, M. B., Cantor, H., De Libero, G., Kronenberg, M., Lanier, L. L., Raulet, D. H., Shlomchik, M. J., and von Herrath, M. G. (2004). The role of innate immunity in autoimmunity. J. Exp. Med. 200, 1527–1531.
Backlund, J., Carlsen, S., Hoger, T., Holm, B., Fugger, L., Kihlberg, J., Burkhardt, H., and Holmdahl, R. (2002). Predominant selection of T cells specific for the glycosylated collagen type II epitope (263-270) in humanized transgenic mice and in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 99, 9960–9965.
Baram, D., Vaday, G. G., Salamon, P., Drucker, I., Hershkoviz, R., and Mekori, Y. A. (2001). Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNF-alpha. J. Immunol. 167, 4008–4016.
Bartholomew, J. S., Evanson, J. M., and Woolley, D. E. (1991). Serum IgE anti-cartilage collagen antibodies in rheumatoid patients. Rheumatol. Int. 11, 37–40.
Barzilai, O., Sherer, Y., Ram, M., Izhaky, D., Anaya, J. M., and Shoenfeld, Y. (2007). Epstein-Barr virus and cytomegalovirus in autoimmune diseases: are they truly notorious? A preliminary report. Ann. N. Y. Acad. Sci. 1108, 567–577.
Bennett, J. L., Blanchet, M. R., Zhao, L., Zbytnuik, L., Antignano, F., Gold, M., Kubes, P., and McNagny, K. M. (2009). Bone marrow-derived mast cells accumulate in the central nervous system during inflammation but are dispensable for experimental autoimmune encephalomyelitis pathogenesis. J. Immunol. 182, 5507–5514.
Bianchi, M. E. (2007). DAMPs, PAMPs, and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5.
Biedermann, T., Kneilling, M., Mailhammer, R., Maier, K., Sander, C. A., Kollias, G., Kunkel, S. L., Hultner, L., and Rocken, M. (2000). Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J. Exp. Med. 192, 1441–1452.
Biesecker, G., Katz, S., and Koffler, D. (1981). Renal localization of the membrane attack complex in systemic lupus erythematosus nephritis. J. Exp. Med. 154, 1779–1794.
Binstadt, B. A., Patel, P. R., Alencar, H., Nigrovic, P. A., Lee, D. M., Mahmood, U., Weissleder, R., Mathis, D., and Benoist, C. (2006). Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat. Immunol. 7, 284–292.
Bluestone, J. A., Herold, K., and Eisenbarth, G. (2010). Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 464, 1293–1300.
Boos, L., Campbell, I. L., Ames, R., Wetsel, R. A., and Barnum, S. R. (2004). Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J. Immunol. 173, 4708–4714.
Carlson, T., Kroenke, M., Rao, P., Lane, T. E., and Segal, B. (2008). The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 205, 811–823.
Chen, M., Lam, B. K., Kanaoka, Y., Nigrovic, P. A., Audoly, L. P., Austen, K. F., and Lee, D. M. (2006). Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med. 203, 837–842.
Chen, R., Fairley, J. A., Zhao, M. L., Giudice, G. J., Zillikens, D., Diaz, L. A., and Liu, Z. (2002). Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J. Immunol. 169, 3987–3992.
Chen, R., Ning, G., Zhao, M. L., Fleming, M. G., Diaz, L. A., Werb, Z., and Liu, Z. (2001). Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J. Clin. Invest. 108, 1151–1158.
Chervonsky, A. (2009). Innate receptors and microbes in induction of autoimmunity. Curr. Opin. Immunol. 21, 641–647.
Choe, J. Y., Crain, B., Wu, S. R., and Corr, M. (2003). Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J. Exp. Med. 197, 537–542.
Concha, L. B., Chang, C. C., Szema, A. M., Dattwyler, R. J., and Carlson, H. E. (2004). IgE antithyroid antibodies in patients with Hashimoto’s disease and chronic urticaria. Allergy Asthma Proc. 25, 293–296.
Cook, H. T., and Botto, M. (2006). Mechanisms of disease: the complement system and the pathogenesis of systemic lupus erythematosus. Nat. Clin. Pract. Rheumatol. 2, 330–337.
Cooper, A. L., Snowden, N., and Woolley, D. E. (1993). IgE antibodies specific for cartilage collagens type II, IX and XI in rheumatic diseases. Scand. J. Rheumatol. 22, 207–214.
Corr, M., and Crain, B. (2002). The role of FcgammaR signaling in the K/B x N serum transfer model of arthritis. J. Immunol. 169, 6604–6609.
Couser, W. G. (1985). Mechanisms of glomerular injury in immune-complex disease. Kidney Int. 28, 569–583.
Couser, W. G., Baker, P. J., and Adler, S. (1985). Complement and the direct mediation of immune glomerular injury: a new perspective. Kidney Int. 28, 879–890.
D’Auria, L., Pietravalle, M., Cordiali-Fei, P., and Ameglio, F. (2000). Increased tryptase and myeloperoxidase levels in blister fluids of patients with bullous pemphigoid: correlations with cytokines, adhesion molecules and anti-basement membrane zone antibodies. Exp. Dermatol. 9, 131–137.
Dawicki, W., and Marshall, J. S. (2007). New and emerging roles for mast cells in host defence. Curr. Opin. Immunol. 19, 31–38.
de Vries, V. C., Pino-Lagos, K., Nowak, E. C., Bennett, K. A., Oliva, C., and Noelle, R. J. (2011). Mast cells condition dendritic cells to mediate allograft tolerance. Immunity 35, 550–561.
D’Elios, M. M., Appelmelk, B. J., Amedei, A., Bergman, M. P., and Del Prete, G. (2004). Gastric autoimmunity: the role of Helicobacter pylori and molecular mimicry. Trends. Mol. Med. 10, 316–323.
Dietsch, G. N., and Hinrichs, D. J. (1989). The role of mast cells in the elicitation of experimental allergic encephalomyelitis. J. Immunol. 142, 1476–1481.
Dimitriadou, V., Pang, X., and Theoharides, T. C. (2000). Hydroxyzine inhibits experimental allergic encephalomyelitis (EAE) and associated brain mast cell activation. Int. J. Immunopharmacol. 22, 673–684.
Dudeck, A., Dudeck, J., Scholten, J., Petzold, A., Surianarayanan, S., Kohler, A., Peschke, K., Vohringer, D., Waskow, C., Krieg, T., Muller, W., Waisman, A., Hartmann, K., Gunzer, M., and Roers, A. (2011). Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984.
Dvorak, A. M., Mihm, M. C. Jr., Osage, J. E., Kwan, T. H., Austen, K. F., and Wintroub, B. U. (1982). Bullous pemphigoid, an ultrastructural study of the inflammatory response: eosinophil, basophil and mast cell granule changes in multiple biopsies from one patient. J. Invest. Dermatol. 78, 91–101.
Fairley, J. A., Burnett, C. T., Fu, C. L., Larson, D. L., Fleming, M. G., and Giudice, G. J. (2007). A pathogenic role for IgE in autoimmunity: bullous pemphigoid IgE reproduces the early phase of lesion development in human skin grafted to nu/nu mice. J. Invest. Dermatol. 127, 2605–2611.
Falk, R. J., Dalmasso, A. P., Kim, Y., Tsai, C. H., Scheinman, J. I., Gewurz, H., and Michael, A. F. (1983). Neoantigen of the polymerized ninth component of complement. Characterization of a monoclonal antibody and immunohistochemical localization in renal disease. J. Clin. Invest. 72, 560–573.
Feyerabend, T. B., Weiser, A., Tietz, A., Stassen, M., Harris, N., Kopf, M., Radermacher, P., Moller, P., Benoist, C., Mathis, D., Fehling, H. J., and Rodewald, H. R. (2011). Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 35, 832–844.
Fischer, M., and Ehlers, M. (2008). Toll-like receptors in autoimmunity. Ann. N. Y. Acad. Sci. 1143, 21–34.
Forward, N. A., Furlong, S. J., Yang, Y., Lin, T. J., and Hoskin, D. W. (2009). Mast cells down-regulate CD4+CD25+ T regulatory cell suppressor function via histamine H1 receptor interaction. J. Immunol. 183, 3014–3022.
Fox, C. C., Jewell, S. D., and Whitacre, C. C. (1994). Rat peritoneal mast cells present antigen to a PPD-specific T cell line. Cell. Immunol. 158, 253–264.
Frandji, P., Oskeritzian, C., Cacraci, F., Lapeyre, J., Peronet, R., David, B., Guillet, J.-G., and Mecheri, S. (1993). Antigen-dependent stimulation by bone marrow-derived mast cells of MHC class II-restricted T cell hybridoma. J. Immunol. 151, 6318–6328.
Frandji, P., Tkaczk, C., Oskeritzian, C., Lapeyre, J., Peronet, R., David, B., Guillet, J.-G., and Mecheri, S. (1995). Presentation of soluble antigens by mast cells: upregulation by interleukin-4 and granulocyte/macrophage colony-stimulating factor and downregulation by interferon-γ. Cell. Immunol. 163, 37–46.
Frandji, P., Tkaczyk, C., Oskeritzian, C., David, B., Desaymard, C., and Mecheri, S. (1996). Exogenous and endogenous antigens are differentially presented by mast cells to CD4+ T lymphocytes. Eur. J. Immunol. 26, 2517–2528.
Fukuoka, Y., Xia, H. Z., Sanchez-Munoz, L. B., Dellinger, A. L., Escribano, L., and Schwartz, L. B. (2008). Generation of anaphylatoxins by human beta-tryptase from C3, C4, and C5. J. Immunol. 180, 6307–6316.
Galli, S. J., Kalesnikoff, J., Grimbaldeston, M. A., Piliponsky, A. M., Williams, C. M., and Tsai, M. (2005). Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23, 749–786.
Galli, S. J., and Kitamura, Y. (1987). Genetically mast-cell-deficient W/Wv and Sl/Sld mice. Their value for the analysis of the roles of mast cells in biologic responses in vivo. Am. J. Pathol. 127, 191–198.
Gambuzza, M., Licata, N., Palella, E., Celi, D., Foti Cuzzola, V., Italiano, D., Marino, S., and Bramanti, P. (2011). Targeting Toll-like receptors: emerging therapeutics for multiple sclerosis management. J. Neuroimmunol. 239, 1–12.
Geoffrey, R., Jia, S., Kwitek, A. E., Woodliff, J., Ghosh, S., Lernmark, A., Wang, X., and Hessner, M. J. (2006). Evidence of a functional role for mast cells in the development of type 1 diabetes mellitus in the BioBreeding rat. J. Immunol. 177, 7275–7286.
Ghohestani, R. F., Cozzani, E., Delaporte, E., Nicolas, J. F., Parodi, A., and Claudy, A. (1998). IgE antibodies in sera from patients with bullous pemphigoid are autoantibodies preferentially directed against the 230-kDa epidermal antigen (BP230). J. Clin. Immunol. 18, 202–209.
Goh, F. G., and Midwood, K. S. (2012). Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis. Rheumatology (Oxford) 51, 7–23.
Gregory, G. D., and Brown, M. A. (2006). Mast cells in allergy and autoimmunity: implications for adaptive immunity. Methods Mol. Biol. 315, 35–50.
Gri, G., Piconese, S., Frossi, B., Manfroi, V., Merluzzi, S., Tripodo, C., Viola, A., Odom, S., Rivera, J., Colombo, M. P., and Pucillo, C. E. (2008). CD4+CD25+ regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity 29, 771–781.
Grimbaldeston, M. A., Chen, C. C., Piliponsky, A. M., Tsai, M., Tam, S. Y., and Galli, S. J. (2005). Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 167, 835–848.
Hauser, S. L., and Oksenberg, J. R. (2006). The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron 52, 61–76.
Heimbach, L., Li, Z., Berkowitz, P., Zhao, M., Li, N., Rubenstein, D. S., Diaz, L. A., and Liu, Z. (2011). The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J. Biol. Chem. 286, 15003–15009.
Hershko, A. Y., and Rivera, J. (2010). Mast cell and T cell communication; amplification and control of adaptive immunity. Immunol. Lett. 128, 98–104.
Hoffmann, M. H., Skriner, K., Herman, S., Baumann, C., Steiner, C. W., Ospelt, C., Meyer, B., Gleiss, A., Pfatschbacher, J., Niederreiter, B., Tuncel, J., Zanoni, G., and Steiner, G. (2011). Nucleic acid-stimulated antigen-presenting cells trigger T cells to induce disease in a rat transfer model of inflammatory arthritis. J. Autoimmun. 36, 288–300.
Hubner, M. P., Larson, D., Torrero, M. N., Mueller, E., Shi, Y., Killoran, K. E., and Mitre, E. (2011). Anti-FcepsilonR1 antibody injections activate basophils and mast cells and delay type 1 diabetes onset in NOD mice. Clin. Immunol. 141, 205–217.
Hueber, A. J., Asquith, D. L., Miller, A. M., Reilly, J., Kerr, S., Leipe, J., Melendez, A. J., and McInnes, I. B. (2010). Mast cells express IL-17A in rheumatoid arthritis synovium. J. Immunol. 184, 3336–3340.
Inamura, N., Mekori, Y. A., Bhattacharyya, S. P., Bianchine, P. J., and Metcalfe, D. D. (1998). Induction and enhancement of Fc(epsilon)RI-dependent mast cell degranulation following coculture with activated T cells: dependency on ICAM-1- and leukocyte function-associated antigen (LFA)-1-mediated heterotypic aggregation. J. Immunol. 160, 4026–4033.
Kakizoe, E., Li, S. H., Kobayashi, Y., Nishikori, Y., Dekio, S., and Okunishi, H. (1999). Increases in mast cells and chymase in fibroproliferative paws of collagen-induced arthritic mice. Inflamm. Res. 48, 318–324.
Kambayashi, T., Allenspach, E. J., Chang, J. T., Zou, T., Shoag, J. E., Reiner, S. L., Caton, A. J., and Koretzky, G. A. (2009). Inducible MHC class II expression by mast cells supports effector and regulatory T cell activation. J. Immunol. 182, 4686–4695.
Kambayashi, T., Baranski, J. D., Baker, R. G., Zou, T., Allenspach, E. J., Shoag, J. E., Jones, P. L., and Koretzky, G. A. (2008). Indirect involvement of allergen-captured mast cells in antigen presentation. Blood 111, 1489–1496.
Kaplan, M. J. (2011). Neutrophils in the pathogenesis and manifestations of SLE. Nat. Rev. Rheumatol. 7, 691–699.
Kashiwakura, J., Yokoi, H., Saito, H., and Okayama, Y. (2004). T cell proliferation by direct cross-talk between OX40 ligand on human mast cells and OX40 on human T cells: comparison of gene expression profiles between human tonsillar and lung-cultured mast cells. J. Immunol. 173, 5247–5257.
Kasperkiewicz, M., and Zillikens, D. (2007). The pathophysiology of bullous pemphigoid. Clin. Rev. Allergy Immunol. 33, 67–77.
Katayama, I., Doi, T., and Nishioka, K. (1984). High histamine level in the blister fluid of bullous pemphigoid. Arch. Dermatol. Res. 276, 126–127.
Katz, H. R., and Austen, K. F. (2011). Mast cell deficiency, a game of kit and mouse. Immunity 35, 668–670.
Kawai, T., and Akira, S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650.
Kivity, S., Agmon-Levin, N., Blank, M., and Shoenfeld, Y. (2009). Infections and autoimmunity – friends or foes? Trends Immunol. 30, 409–414.
Kufer, T. A., and Sansonetti, P. J. (2011). NLR functions beyond pathogen recognition. Nat. Immunol. 12, 121–128.
Kumar, V., and Sharma, A. (2010). Neutrophils: cinderella of innate immune system. Int. Immunopharmacol. 10, 1325–1334.
Kwok, S. K., Cho, M. L., Her, Y. M., Oh, H. J., Park, M. K., Lee, S. Y., Woo, Y. J., Ju, J. H., Park, K. S., Kim, H. Y., and Park, S. H. (2012). TLR2 ligation induces the production of IL-23/IL-17 via IL-6, STAT3 and NF-kB pathway in patients with primary Sjogren’s syndrome. Arthritis Res. Ther. 14, R64.
Lee, D. M., Friend, D. S., Gurish, M. F., Benoist, C., Mathis, D., and Brenner, M. B. (2002). Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297, 1689–1692.
Li, Q., Ujiie, H., Shibaki, A., Wang, G., Moriuchi, R., Qiao, H. J., Morioka, H., Shinkuma, S., Natsuga, K., Long, H. A., Nishie, W., and Shimizu, H. (2010). Human IgG1 monoclonal antibody against human collagen 17 noncollagenous 16A domain induces blisters via complement activation in experimental bullous pemphigoid model. J. Immunol. 185, 7746–7755.
Lien, E., and Zipris, D. (2009). The role of Toll-like receptor pathways in the mechanism of type 1 diabetes. Curr. Mol. Med. 9, 52–68.
Lilla, J. N., Chen, C. C., Mukai, K., BenBarak, M. J., Franco, C. B., Kalesnikoff, J., Yu, M., Tsai, M., Piliponsky, A. M., and Galli, S. J. (2011). Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 118, 6930–6938.
Liu, Z., Sui, W., Zhao, M., Li, Z., Li, N., Thresher, R., Giudice, G. J., Fairley, J. A., Sitaru, C., Zillikens, D., Ning, G., Marinkovich, M. P., and Diaz, L. A. (2008). Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J. Autoimmun. 31, 331–338.
Lock, C., Hermans, G., Pedotti, R., Brendolan, A., Schadt, E., Garren, H., Langer-Gould, A., Strober, S., Cannella, B., Allard, J., Klonowski, P., Austin, A., Lad, N., Kaminski, N., Galli, S. J., Oksenberg, J. R., Raine, C. S., Heller, R., and Steinman, L. (2002). Gene microarray analysis of multiple sclerosis lesions yield new targets validated in autoimmune encephalomyelitis. Nat. Med. 8, 500–508.
Lohr, J., Knoechel, B., Nagabhushanam, V., and Abbas, A. K. (2005). T-cell tolerance and autoimmunity to systemic and tissue-restricted self-antigens. Immunol. Rev. 204, 116–127.
Louvet, C., Szot, G. L., Lang, J., Lee, M. R., Martinier, N., Bollag, G., Zhu, S., Weiss, A., and Bluestone, J. A. (2008). Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 105, 18895–18900.
Lu, L. F., Lind, E. F., Gondek, D. C., Bennett, K. A., Gleeson, M. W., Pino-Lagos, K., Scott, Z. A., Coyle, A. J., Reed, J. L., Van Snick, J., Strom, T. B., Zheng, X. X., and Noelle, R. J. (2006). Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 442, 997–1002.
Maciejewska Rodrigues, H., Jungel, A., Gay, R. E., and Gay, S. (2009). Innate immunity, epigenetics and autoimmunity in rheumatoid arthritis. Mol. Immunol. 47, 12–18.
Mackay, I. R., and Anderson, W. H. (2010). What’s in a name? Experimental encephalomyelitis: “allergic” or “autoimmune.” J. Neuroimmunol. 223, 1–4.
Mackworth-Young, C. G., Chan, J. K., Bunn, C. C., Hughes, G. R., and Gharavi, A. E. (1986). Complement fixation by anti-dsDNA antibodies in SLE: measurement by radioimmunoassay and relationship with disease activity. Ann. Rheum. Dis. 45, 314–318.
Malaviya, R., Ikeda, T., Ross, E., and Abraham, S. (1996a). Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature 381, 77–80.
Malaviya, R., Twesten, N. J., Ross, E. A., Abraham, S. N., and Pfeifer, J. D. (1996b). Mast cells process bacterial Ags through a phagocytic route for class I MHC presentation to T cells. J. Immunol. 156, 1490–1496.
Mantovani, A., Cassatella, M. A., Costantini, C., and Jaillon, S. (2011). Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11, 519–531.
Marek, L. R., and Kagan, J. C. (2012). Deciphering the function of nucleic acid sensing TLRs one regulatory step at a time. Front. Biosci. 17, 2060–2068.
Marshak-Rothstein, A., and Ohashi, P. S. (2007). Intricate connections between innate and adaptive autoimmunity. Curr. Opin. Immunol. 19, 603–605.
Marshall, J. S., McCurdy, J. D., and Olynych, T. (2003a). Toll-like receptor-mediated activation of mast cells: implications for allergic disease? Int. Arch. Allergy Immunol. 132, 87–97.
Marshall, J. S., King, C. A., and McCurdy, J. D. (2003b). Mast cell cytokine and chemokine responses to bacterial and viral infection. Curr. Pharm. Des. 9, 11–24.
Marta, M., Meier, U. C., and Lobell, A. (2009). Regulation of autoimmune encephalomyelitis by toll-like receptors. Autoimmun. Rev. 8, 506–509.
McIlroy, A., Caron, G., Blanchard, S., Fremaux, I., Duluc, D., Delneste, Y., Chevailler, A., and Jeannin, P. (2006). Histamine and prostaglandin E up-regulate the production of Th2-attracting chemokines (CCL17 and CCL22) and down-regulate IFN-gamma-induced CXCL10 production by immature human dendritic cells. Immunology 117, 507–516.
Mekori, Y. A., and Metcalfe, D. D. (1999). Mast cell-T cell interactions. J. Allergy Clin. Immunol. 104, 517–523.
Mercer-Jones, M. A., Shrotri, M. S., Heinzelmann, M., Peyton, J. C., and Cheadle, W. G. (1999). Regulation of early peritoneal neutrophil migration by macrophage inflammatory protein-2 and mast cells in experimental peritonitis. J. Leukoc. Biol. 65, 249–255.
Metcalfe, R., Jordan, N., Watson, P., Gullu, S., Wiltshire, M., Crisp, M., Evans, C., Weetman, A., and Ludgate, M. (2002). Demonstration of immunoglobulin G, A, and E autoantibodies to the human thyrotropin receptor using flow cytometry. J. Clin. Endocrinol. Metab. 87, 1754–1761.
Metz, M., Grimbaldeston, M. A., Nakae, S., Piliponsky, A. M., Tsai, M., and Galli, S. J. (2007). Mast cells in the promotion and limitation of chronic inflammation. Immunol. Rev. 217, 304–328.
Mills, K. H. (2011). TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 11, 807–822.
Mix, E., Meyer-Rienecker, H., Hartung, H. P., and Zettl, U. K. (2010). Animal models of multiple sclerosis – potentials and limitations. Prog. Neurobiol. 92, 386–404.
Miyajima, I., Dombrowicz, D., Martin, T. R., Ravetch, J. V., Kinet, J. P., and Galli, S. J. (1997). Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and Fc gammaRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J. Clin. Invest. 99, 901–914.
Monach, P. A., Nigrovic, P. A., Chen, M., Hock, H., Lee, D. M., Benoist, C., and Mathis, D. (2010). Neutrophils in a mouse model of autoantibody-mediated arthritis: critical producers of Fc receptor gamma, the receptor for C5a, and lymphocyte function-associated antigen 1. Arthritis Rheum. 62, 753–764.
Morgan, B. P., Griffiths, M., Khanom, H., Taylor, S. M., and Neal, J. W. (2004). Blockade of the C5a receptor fails to protect against experimental autoimmune encephalomyelitis in rats. Clin. Exp. Immunol. 138, 430–438.
Mrabet-Dahbi, S., Metz, M., Dudeck, A., Zuberbier, T., and Maurer, M. (2009). Murine mast cells secrete a unique profile of cytokines and prostaglandins in response to distinct TLR2 ligands. Exp. Dermatol. 18, 437–444.
Nakae, S., Lunderius, C., Ho, L. H., Schafer, B., Tsai, M., and Galli, S. J. (2007a). TNF can contribute to multiple features of ovalbumin-induced allergic inflammation of the airways in mice. J. Allergy Clin. Immunol. 119, 680–686.
Nakae, S., Suto, H., Berry, G. J., and Galli, S. J. (2007b). Mast cell-derived TNF can promote Th17 cell-dependent neutrophil recruitment in ovalbumin-challenged OTII mice. Blood 109, 3640–3648.
Nakae, S., Ho, L. H., Yu, M., Monteforte, R., Iikura, M., Suto, H., and Galli, S. J. (2007c). Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J. Allergy Clin. Immunol. 120, 48–55.
Nakae, S., Suto, H., Iikura, M., Kakurai, M., Sedgwick, J. D., Tsai, M., and Galli, S. J. (2006). Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J. Immunol. 176, 2238–2248.
Nakae, S., Suto, H., Kakurai, M., Sedgwick, J. D., Tsai, M., and Galli, S. J. (2005). Mast cells enhance T cell activation: importance of mast cell-derived TNF. Proc. Natl. Acad. Sci. U.S.A. 102, 6467–6472.
Nakamura, Y., Kambe, N., Saito, M., Nishikomori, R., Kim, Y. G., Murakami, M., Nunez, G., and Matsue, H. (2009). Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J. Exp. Med. 206, 1037–1046.
Navi, D., Saegusa, J., and Liu, F. T. (2007). Mast cells and immunological skin diseases. Clin. Rev. Allergy Immunol. 33, 144–155.
Nigrovic, P. A., Binstadt, B. A., Monach, P. A., Johnsen, A., Gurish, M., Iwakura, Y., Benoist, C., Mathis, D., and Lee, D. M. (2007). Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc. Natl. Acad. Sci. U.S.A. 104, 2325–2330.
Nigrovic, P. A., Gray, D. H., Jones, T., Hallgren, J., Kuo, F. C., Chaletzky, B., Gurish, M., Mathis, D., Benoist, C., and Lee, D. M. (2008). Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 173, 1693–1701.
Nigrovic, P. A., and Lee, D. M. (2007). Synovial mast cells: role in acute and chronic arthritis. Immunol. Rev. 217, 19–37.
Nigrovic, P. A., Malbec, O., Lu, B., Markiewski, M. M., Kepley, C., Gerard, N., Gerard, C., Daeron, M., and Lee, D. M. (2010). C5a receptor enables participation of mast cells in immune complex arthritis independently of Fcgamma receptor modulation. Arthritis Rheum. 62, 3322–3333.
Nurieva, R. I., Liu, X., and Dong, C. (2011). Molecular mechanisms of T-cell tolerance. Immunol. Rev. 241, 133–144.
Oikarinen, A. I., Zone, J. J., Ahmed, A. R., Kiistala, U., and Uitto, J. (1983). Demonstration of collagenase and elastase activities in the blister fluids from bullous skin diseases. Comparison between dermatitis herpetiformis and bullous pemphigoid. J. Invest. Dermatol. 81, 261–266.
Otsuka, A., Kubo, M., Honda, T., Egawa, G., Nakajima, S., Tanizaki, H., Kim, B., Matsuoka, S., Watanabe, T., Nakae, S., Miyachi, Y., and Kabashima, K. (2011). Requirement of interaction between mast cells and skin dendritic cells to establish contact hypersensitivity. PLoS ONE 6, e25538. doi:10.1371/journal.pone.0025538
Palmer, H. S., Kelso, E. B., Lockhart, J. C., Sommerhoff, C. P., Plevin, R., Goh, F. G., and Ferrell, W. R. (2007). Protease-activated receptor 2 mediates the proinflammatory effects of synovial mast cells. Arthritis Rheum. 56, 3532–3540.
Patterson, R., Pateras, V., Grammer, L. C., and Harris, K. E. (1986). Human antibodies against formaldehyde-human serum albumin conjugates or human serum albumin in individuals exposed to formaldehyde. Int. Arch. Allergy Appl. Immunol. 79, 53–59.
Pedotti, R., DeVoss, J. J., Youssef, S., Mitchell, D., Wedemeyer, J., Madanat, R., Garren, H., Fontoura, P., Tsai, M., Galli, S. J., Sobel, R. A., and Steinman, L. (2003). Multiple elements of the allergic arm of the immune response modulate autoimmune demyelination. Proc. Natl. Acad. Sci. U.S.A. 100, 1867–1872.
Pedotti, R., Mitchell, D., Wedemeyer, J., Karpuj, M., Chabas, D., Hattab, E. M., Tsai, M., Galli, S. J., and Steinman, L. (2001). An unexpected version of horror autotoxicus: anaphylactic shock to a self-peptide. Nat. Immunol. 2, 216–222.
Pennell, L. M., Galligan, C. L., and Fish, E. N. (2012). Sex affects immunity. J. Autoimmun. 38, 282–291.
Perkinson, D. T., Baker, P. J., Couser, W. G., Johnson, R. J., and Adler, S. (1985). Membrane attack complex deposition in experimental glomerular injury. Am. J. Pathol. 120, 121–128.
Permin, H., and Wiik, A. (1978). The prevalence of IgE antinuclear antibodies in rheumatoid arthritis and systemic lupus erythematosus. Acta Pathol. Microbiol. Scand. C 86C, 245–249.
Piccinini, A. M., and Midwood, K. S. (2010). DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010. doi: 10.1155/2010/672395
Piconese, S., Costanza, M., Musio, S., Tripodo, C., Poliani, P. L., Gri, G., Burocchi, A., Pittoni, P., Gorzanelli, A., Colombo, M. P., and Pedotti, R. (2011). Exacerbated experimental autoimmune encephalomyelitis in mast-cell-deficient Kit W-sh/W-sh mice. Lab. Invest. 91, 627–641.
Piconese, S., Gri, G., Tripodo, C., Musio, S., Gorzanelli, A., Frossi, B., Pedotti, R., Pucillo, C. E., and Colombo, M. P. (2009). Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood 114, 2639–2648.
Pillai, S., Mattoo, H., and Cariappa, A. (2011). B cells and autoimmunity. Curr. Opin. Immunol. 23, 721–731.
Pimentel, T. A., Sampaio, A. L., D’Acquisto, F., Perretti, M., and Oliani, S. M. (2011). An essential role for mast cells as modulators of neutrophils influx in collagen-induced arthritis in the mouse. Lab. Invest. 91, 33–42.
Pisetsky, D. S. (2008). The role of innate immunity in the induction of autoimmunity. Autoimmun. Rev. 8, 69–72.
Pitman, N., Asquith, D. L., Murphy, G., Liew, F. Y., and McInnes, I. B. (2011). Collagen-induced arthritis is not impaired in mast cell-deficient mice. Ann. Rheum. Dis. 70, 1170–1171.
Rai, E., and Wakeland, E. K. (2011). Genetic predisposition to autoimmunity – what have we learned? Semin. Immunol. 23, 67–83.
Ramaglia, V., Hughes, T. R., Donev, R. M., Ruseva, M. M., Wu, X., Huitinga, I., Baas, F., Neal, J. W., and Morgan, B. P. (2012). C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 109, 965–970.
Ramos, B. F., Zhang, Y., and Jakschik, B. A. (1994). Neutrophil elicitation in the reverse passive Arthus reaction. Complement-dependent and -independent mast cell involvement. J. Immunol. 152, 1380–1384.
Ramos, B. F., Zhang, Y., Qureshi, R., and Jakschik, B. A. (1991). Mast cells are critical for the production of leukotrienes responsible for neutrophil recruitment in immune complex-induced peritonitis in mice. J. Immunol. 147, 1636–1641.
Ramos, T. N., Wohler, J. E., and Barnum, S. R. (2009). Deletion of both the C3a and C5a receptors fails to protect against experimental autoimmune encephalomyelitis. Neurosci. Lett. 467, 234–236.
Rao, K. N., and Brown, M. A. (2008). Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann. N. Y. Acad. Sci. 1143, 83–104.
Reiman, R., Gerard, C., Campbell, I. L., and Barnum, S. R. (2002). Disruption of the C5a receptor gene fails to protect against experimental allergic encephalomyelitis. Eur. J. Immunol. 32, 1157–1163.
Richardson, S. J., Willcox, A., Bone, A. J., Foulis, A. K., and Morgan, N. G. (2009). The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 52, 1143–1151.
Ricklin, D., Hajishengallis, G., Yang, K., and Lambris, J. D. (2010). Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797.
Robbie-Ryan, M., Tanzola, M. B., Secor, V. H., and Brown, M. A. (2003). Cutting edge: both activating and inhibitory Fc receptors expressed on mast cells regulate experimental allergic encephalomyelitis disease severity. J. Immunol. 170, 1630–1634.
Robson, M. G., Cook, H. T., Pusey, C. D., Walport, M. J., and Davies, K. A. (2003). Antibody-mediated glomerulonephritis in mice: the role of endotoxin, complement and genetic background. Clin. Exp. Immunol. 133, 326–333.
Ronaghy, A., Prakken, B. J., Takabayashi, K., Firestein, G. S., Boyle, D., Zvailfler, N. J., Roord, S. T., Albani, S., Carson, D. A., and Raz, E. (2002). Immunostimulatory DNA sequences influence the course of adjuvant arthritis. J. Immunol. 168, 51–56.
Rothfield, N. F., and Stollar, B. D. (1967). The relation of immunoglobulin class, pattern of anti-nuclear antibody, and complement-fixing antibodies to DNA in sera from patients with systemic lupus erythematosus. J. Clin. Invest. 46, 1785–1794.
Rozniecki, J. J., Hauser, S. L., Stein, M., Lincoln, R., and Theoharides, T. C. (1995). Elevated mast cell tryptase in cerebrospinal fluid of multiple sclerosis patients. Ann. Neurol. 37, 63–66.
Rubtsov, A. V., Rubtsova, K., Kappler, J. W., and Marrack, P. (2010). Genetic and hormonal factors in female-biased autoimmunity. Autoimmun. Rev. 9, 494–498.
Salamon, P., Shoham, N. G., Gavrieli, R., Wolach, B., and Mekori, Y. A. (2005). Human mast cells release Interleukin-8 and induce neutrophil chemotaxis on contact with activated T cells. Allergy 60, 1316–1319.
Salamon, P., Shoham, N. G., Puxeddu, I., Paitan, Y., Levi-Schaffer, F., and Mekori, Y. A. (2008). Human mast cells release oncostatin M on contact with activated T cells: possible biologic relevance. J. Allergy Clin. Immunol. 121, 448–455 e5.
Sawamukai, N., Yukawa, S., Saito, K., Nakayamada, S., Kambayashi, T., and Tanaka, Y. (2010). Mast cell-derived tryptase inhibits apoptosis of human rheumatoid synovial fibroblasts via rho-mediated signaling. Arthritis Rheum. 62, 952–959.
Sayed, B. A., Christy, A., Quirion, M. R., and Brown, M. A. (2008). The master switch: the role of mast cells in autoimmunity and tolerance. Annu. Rev. Immunol. 26, 705–739.
Sayed, B. A., Christy, A. L., Walker, M. E., and Brown, M. A. (2010). Meningeal mast cells affect early T cell central nervous system infiltration and blood-brain barrier integrity through TNF: a role for neutrophil recruitment? J. Immunol. 184, 6891–6900.
Sayed, B. A., Walker, M. E., and Brown, M. A. (2011). Cutting edge: mast cells regulate disease severity in a relapsing-remitting model of multiple sclerosis. J. Immunol. 186, 3294–3298.
Schwartz, L. B., Kawahara, M. S., Hugli, T. E., Vik, D., Fearon, D. T., and Austen, K. F. (1983). Generation of C3a anaphylatoxin from human C3 by human mast cell tryptase. J. Immunol. 130, 1891–1895.
Secor, V. H., Secor, W. E., Gutekunst, C. A., and Brown, M. A. (2000). Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J. Exp. Med. 191, 813–822.
Shin, K., Gurish, M. F., Friend, D. S., Pemberton, A. D., Thornton, E. M., Miller, H. R., and Lee, D. M. (2006). Lymphocyte-independent connective tissue mast cells populate murine synovium. Arthritis Rheum. 54, 2863–2871.
Shin, K., Nigrovic, P. A., Crish, J., Boilard, E., McNeil, H. P., Larabee, K. S., Adachi, R., Gurish, M. F., Gobezie, R., Stevens, R. L., and Lee, D. M. (2009). Mast cells contribute to autoimmune inflammatory arthritis via their tryptase/heparin complexes. J. Immunol. 182, 647–656.
Smith, C. E., Eagar, T. N., Strominger, J. L., and Miller, S. D. (2005). Differential induction of IgE-mediated anaphylaxis after soluble vs. cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 102, 9595–9600.
Sospedra, M., and Martin, R. (2005). Immunology of multiple sclerosis. Annu. Rev. Immunol. 23, 683–747.
Spachidou, M. P., Bourazopoulou, E., Maratheftis, C. I., Kapsogeorgou, E. K., Moutsopoulos, H. M., Tzioufas, A. G., and Manoussakis, M. N. (2007). Expression of functional Toll-like receptors by salivary gland epithelial cells: increased mRNA expression in cells derived from patients with primary Sjogren’s syndrome. Clin. Exp. Immunol. 147, 497–503.
Spitzauer, S., Schweiger, C., Sperr, W. R., Pandjaitan, B., Valent, P., Muhl, S., Ebner, C., Scheiner, O., Kraft, D., Rumpold, H., and Valenta, R. (1994). Molecular characterization of dog albumin as a cross-reactive allergen. J. Allergy Clin. Immunol. 93, 614–627.
Stahle-Backdahl, M., Inoue, M., Guidice, G. J., and Parks, W. C. (1994). 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J. Clin. Invest. 93, 2022–2030.
Stelekati, E., Bahri, R., D’Orlando, O., Orinska, Z., Mittrucker, H. W., Langenhaun, R., Glatzel, M., Bollinger, A., Paus, R., and Bulfone-Paus, S. (2009). Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity 31, 665–676.
Stelekati, E., Orinska, Z., and Bulfone-Paus, S. (2007). Mast cells in allergy: innate instructors of adaptive responses. Immunobiology 212, 505–519.
Stopfer, P., Mannel, D. N., and Hehlgans, T. (2004). Lymphotoxin-beta receptor activation by activated T cells induces cytokine release from mouse bone marrow-derived mast cells. J. Immunol. 172, 7459–7465.
Tanzola, M. B., Robbie-Ryan, M., Gutekunst, C. A., and Brown, M. A. (2003). Mast cells exert effects outside the central nervous system to influence experimental allergic encephalomyelitis disease course. J. Immunol. 171, 4385–4391.
Touil, T., Fitzgerald, D., Zhang, G. X., Rostami, A., and Gran, B. (2006). Cutting edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J. Immunol. 177, 7505–7509.
Tuomisto, L., Kilpelainen, H., and Riekkinen, P. (1983). Histamine and histamine-N-methyltransferase in the CSF of patients with multiple sclerosis. Agents Actions 13, 255–257.
Urich, E., Gutcher, I., Prinz, M., and Becher, B. (2006). Autoantibody-mediated demyelination depends on complement activation but not activatory Fc-receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 18697–18702.
Valenta, R., Duchene, M., Pettenburger, K., Sillaber, C., Valent, P., Bettelheim, P., Breitenbach, M., Rumpold, H., Kraft, D., and Scheiner, O. (1991). Identification of profilin as a novel pollen allergen; IgE autoreactivity in sensitized individuals. Science 253, 557–560.
Valenta, R., Mittermann, I., Werfel, T., Garn, H., and Renz, H. (2009). Linking allergy to autoimmune disease. Trends Immunol. 30, 109–116.
van den Broek, M. F., van den Berg, W. B., and van de Putte, L. B. (1988). The role of mast cells in antigen induced arthritis in mice. J. Rheumatol. 15, 544–551.
Verri, W. A. Jr., Souto, F. O., Vieira, S. M., Almeida, S. C., Fukada, S. Y., Xu, D., Alves-Filho, J. C., Cunha, T. M., Guerrero, A. T., Mattos-Guimaraes, R. B., Oliveira, F. R., Teixeira, M. M., Silva, J. S., McInnes, I. B., Ferreira, S. H., Louzada-Junior, P., Liew, F. Y., and Cunha, F. Q. (2010). IL-33 induces neutrophil migration in rheumatoid arthritis and is a target of anti-TNF therapy. Ann. Rheum. Dis. 69, 1697–1703.
Walker, M. E., Hatfield, J. K., and Brown, M. A. (2011). New insights into the role of mast cells in autoimmunity: evidence for a common mechanism of action? Biochim. Biophys. Acta. 1822, 57–65.
Wang, Y., and Thorlacius, H. (2005). Mast cell-derived tumor necrosis factor-alpha mediates macrophage inflammatory protein-2-induced recruitment of neutrophils in mice. Br. J. Pharmacol. 145, 1062–1068.
Warde, N. (2011). Autoimmunity: the role of neutrophils in SLE: untangling the NET. Nat. Rev. Rheumatol. 7, 252.
Weerth, S. H., Rus, H., Shin, M. L., and Raine, C. S. (2003). Complement C5 in experimental autoimmune encephalomyelitis (EAE) facilitates remyelination and prevents gliosis. Am. J. Pathol. 163, 1069–1080.
Wipke, B. T., Wang, Z., Kim, J., McCarthy, T. J., and Allen, P. M. (2002). Dynamic visualization of a joint-specific autoimmune response through positron emission tomography. Nat. Immunol. 3, 366–372.
Wipke, B. T., Wang, Z., Nagengast, W., Reichert, D. E., and Allen, P. M. (2004). Staging the initiation of autoantibody-induced arthritis: a critical role for immune complexes. J. Immunol. 172, 7694–7702.
Wyatt, R. J., McAdams, A. J., Forristal, J., Snyder, J., and West, C. D. (1979). Glomerular deposition of complement-control proteins in acute and chronic glomerulonephritis. Kidney Int. 16, 505–512.
Xu, D., Jiang, H. R., Kewin, P., Li, Y., Mu, R., Fraser, A. R., Pitman, N., Kurowska-Stolarska, M., McKenzie, A. N., McInnes, I. B., and Liew, F. Y. (2008). IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. U.S.A. 105, 10913–10918.
Xu, D., Jiang, H. R., Li, Y., Pushparaj, P. N., Kurowska-Stolarska, M., Leung, B. P., Mu, R., Tay, H. K., McKenzie, A. N., McInnes, I. B., Melendez, A. J., and Liew, F. Y. (2010). IL-33 exacerbates autoantibody-induced arthritis. J. Immunol. 184, 2620–2626.
Zhang, Q., Itagaki, K., and Hauser, C. J. (2010). Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34, 55–59.
Zhang, Y., Ramos, B. F., Jakschik, B., Baganoff, M. P., Deppeler, C. L., Meyer, D. M., Widomski, D. L., Fretland, D. J., and Bolanowski, M. A. (1995). Interleukin 8 and mast cell-generated tumor necrosis factor-alpha in neutrophil recruitment. Inflammation 19, 119–132.
Zhang, Y., Ramos, B. F., and Jakschik, B. A. (1992). Neutrophil recruitment by tumor necrosis factor from mast cells in immune complex peritonitis. Science 258, 1957–1959.
Keywords: mast cells, autoimmunity, neutrophils, MC-T cell crosstalk, mast cell deficient mice
Citation: Brown MA and Hatfield JK (2012) Mast cells are important modifiers of autoimmune disease: with so much evidence, why is there still controversy? Front. Immun. 3:147. doi: 10.3389/fimmu.2012.00147
Received: 20 March 2012; Accepted: 19 May 2012;
Published online: 07 June 2012.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Miriam Wittmann, University of Leeds, UKJohn Ryan, Virginia Commonwealth University, USA
Copyright: © 2012 Brown and Hatfield. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Melissa A. Brown, Department of Microbiology and Immunology, Northwestern University Feinberg School of Medicine, 303 E. Chicago Avenue, Tarry 6-701, Chicago, IL 60611, USA. e-mail: m-brown12@northwestern.edu