 Sabine Mellor-Heineke1 Joyce Villanueva1 Michael B. Jordan1,2 Rebecca Marsh1,2 Kejian Zhang2,3 Jack J. Bleesing1,2 Alexandra H. Filipovich1,2 Kimberly A. Risma2,4*
Sabine Mellor-Heineke1 Joyce Villanueva1 Michael B. Jordan1,2 Rebecca Marsh1,2 Kejian Zhang2,3 Jack J. Bleesing1,2 Alexandra H. Filipovich1,2 Kimberly A. Risma2,4*- 1Immunodeficiency and Histiocytosis Program, Division of Bone Marrow Transplantation, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
- 2Department of Pediatrics, College of Medicine, University of Cincinnati, Cincinnati, OH, USA
- 3Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
- 4Division of Allergy and Immunology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA
Patients with hemophagocytic lymphohistiocytosis (HLH) exhibit immune hyper-activation as a consequence of genetic defects in secretory granule proteins of cytotoxic T lymphocytes (CTL) and natural killer (NK) cells. Murine models of HLH demonstrate significant activation of CTL as central to the disease pathogenesis, but evaluation of CTL and NK activation in children with HLH or inflammatory conditions is not well described. CD8 T cells only express granzyme B (GrB) following stimulation and differentiation into CTL; therefore, we reasoned that GrB expression may serve as a signature of CTL activation. It is unknown whether human NK cells are similarly activated in vivo, as marked by increased granule proteins. Perforin and GrB levels are measured in both CTL and NK cells by flow cytometry to diagnose perforin deficiency. We retrospectively compared GrB expression in blood samples from 130 children with clinically suspected and/or genetically defined HLH to age-matched controls. As predicted, CD8 expressing GrB cells were increased in HLH, regardless of genetic etiology. Remarkably, the GrB protein content also increased in NK cells in patients with HLH and decreased following immunosuppressive therapy. This suggests that in vivo activation of NK cells occurs in hyper-inflammatory conditions. We conclude that increased detection of GrB in CTL and NK are an immune signature for lymphocyte activation in HLH, irrespective of genetic subtype and may also be a useful measure of immune activation in other related conditions.
Introduction
Cytotoxic lymphocytes utilize the perforin/granzyme pathway for target-cell killing in both the innate and adaptive immune response (Russell and Ley, 2002; Voskoboinik et al., 2006; Lieberman, 2010). Cytotoxic T lymphocytes (CTL) and natural killer (NK) cells contain specialized secretory lysosomes, cytotoxic granules, which degranulate upon contact with a target-cell. These granules contain perforin (Prf), a membrane-disrupting protein that facilitates the delivery of granzymes, granule proteases that initiate apoptotic death in target-cells.
In humans, genetic defects in the pathway for Prf-mediated lymphocyte cytotoxicity is associated with a primary immunodeficiency presenting with profound systemic inflammation in infancy, familial hemophagocytic lymphohistiocytosis (HLH) (Filipovich, 2009; Risma and Jordan, 2012). Mutations in seven genes have been associated with familial HLH phenotypes (PRF1, UNC13D, STX11, RAB27A, STXBP2, SH2D1A, and BIRC4). Five of these genes express proteins (Prf, Munc 13-4, Rab 27a, Syntaxin binding protein 2, and Syntaxin 11) involved in the function of cytotoxic secretory granules, and biallelic genetic mutations lead to impaired Prf-dependent cytotoxicity by impairing Prf function or degranulation of cytotoxic granules (de Saint Basile et al., 2011). The latter two genes (SH2D1A and BIRC4) encode slam associated protein (SAP) and X-linked inhibitor of apoptosis protein (XIAP) and defects are identified in boys with X-linked lymphoproliferative disease (XLP types 1 and 2), a primary immunodeficiency that may present with HLH, especially upon infection with Epstein Barr Virus (EBV) (Marsh and Filipovich, 2011). SAP deficiency impairs NK and CTL mediated killing of EBV-infected B cells (Cannons et al., 2011). Although clearly associated with HLH (Marsh et al., 2010; Pachlopnik Schmid et al., 2011), the impact of XIAP deficiency on Prf-mediated lymphocyte cytotoxicity remains unclear.
The clinical symptoms of HLH (irrespective of genotype) include fever, lymphadenopathy, jaundice, and mental status changes, and the pathologic signs include pancytopenia, hypercytokinemia, and multisystem organ failure. Hemophagocytosis is frequently seen on bone marrow or liver biopsy, but is variably present on a single biopsy. An immune trigger, such as a viral infection, may or may not be identified. Genetically defined, or primary, HLH is generally fatal unless treated with immune suppression and ultimately bone marrow transplant (Jordan and Filipovich, 2008; Trottestam et al., 2011).
The pathophysiology of HLH has been modeled in Prf1-deficient and Unc13d deficient mice (Jordan et al., 2004; Crozat et al., 2007). Severe immune dysregulation occurs in response to lymphocytic choriomeningitis virus (LCMV) infection of these genetically defined mice. Jordan and colleagues have shown that the development of HLH phenotype in Prf1-deficient mice is dependent upon and defined by dramatic hyper-activation of CTL (Lykens et al., 2011), suggesting that CTL activation may be a reproducible immune signature for HLH. It is unknown whether human CTL and NK are similarly hyper-activated during the clinical course of HLH. Indeed, studies of immune activation in HLH and other systemic inflammatory disorders have focused primarily on non-specific markers of lymphocyte and monocyte activation such as soluble IL-2 receptor alpha (sIL2R or sCD25) and soluble CD163 (Komp et al., 1989; Imashuku et al., 1995; Bleesing et al., 2007; Johnson et al., 2011). Elevated sIL2R levels are utilized as a gold standard, measure of immune activation for HLH, but may be elevated in other lymphoproliferative disorders as well (Bien and Balcerska, 2008). Therefore, we considered whether detection of granule proteins induced in cytotoxic lymphocytes central to the pathogenesis of HLH may herald the immune hyper-activation that is characteristic of HLH.
Granzyme B (GrB) protein levels in human CTL and NK increase following activation in vitro (Salcedo et al., 1993; Grossman et al., 2004; Sedelies et al., 2004; Buzza and Bird, 2006; Skak et al., 2008; Kim et al., 2011; Wang et al., 2012). Recent studies have focused on the utility of soluble GrB in the blood as a biomarker of immune activation in humans with a variety of disorders, including autoimmune disease, graft rejection, and viral infection (Spaeny-Dekking et al., 1998; Tak et al., 1999; Goldbach-Mansky et al., 2005; Buzza and Bird, 2006; Altimari et al., 2008; Bem et al., 2008; Truong et al., 2008; Boivin et al., 2009). It is assumed that the elevated blood granzyme levels are due to increased GrB expression by CTL activation; however the contribution from NK cells is unknown. We hypothesized that GrB protein levels in both CD8 T cells (activated CTL) and NK cells would increase secondary to immune activation in patients with HLH. Compared to age-matched controls, we observed that GrB protein expression was increased in both CTL and NK cells of patients with HLH, irrespective of genetic etiology. GrB levels in NK cells also correlated with disease activity. Therefore, human NK cells, like CTL, acquire enhanced GrB protein expression in pro-inflammatory conditions, suggesting a mechanism for up-regulation of cytotoxic function.
Materials and Methods
Patients
With approval from the institutional review board (IRB), we retrospectively reviewed clinical data listed on Diagnostic Immunology Laboratory (DIL) test requisition forms, immune testing results, and genetic testing results from patients referred for testing to our center between 2001 and 2011. We defined sub-groups of patients with genetically defined HLH accordingly (see Table 1): (1) FHL2- biallelic PRF1 mutations, (2) FHL3- biallelic UNC13D mutations, (3) FHL5-biallelic STXPB2 mutations, and (4) HLHxlp associated with XLP (mutations in SH2D1A or BIRC4). Data from five patients with XLP but no evidence of HLH were also compared to boys with HLHxlp.
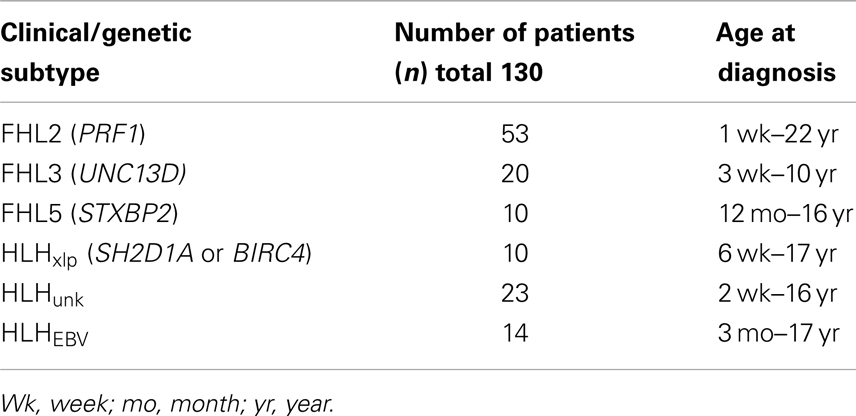
Table 1. Group cohort with HLH.
For comparison, we evaluated a group of patients whose genetic diagnoses were not established HLHunk, by reviewing data from over 800 patient blood samples (ages 0–22 years) submitted to this institution’s DIL between 06/2001 and 07/2007 for Prf detection by flow cytometry as part of an evaluation for suspected HLH. Our goal was to utilize stringent criteria for inclusion of patients so as to eliminate samples where the diagnosis of HLH was not confirmed. Therefore, in the HLHunk group we included only patients with a diagnosis of HLH documented by the ordering physician on the sample requisition (or documented following evaluation at our hospital) and an elevated sIL2R. The requirement for elevated sIL2R was not utilized as a criterion for inclusion of patients with documented gene mutations to include the maximum number of children with genetically defined HLH. For the majority of cases, it was not known if a patient had already begun treatment at the time of sample submission. A subset of patients with a clinical history of HLH due to EBV infection documented on the test requisition (HLHEBV) and unknown genotype was considered separately (n = 14).
In a follow-up analysis, 10 children with HLH were evaluated following and/or during clinical treatment for HLH at this institution. These patients were included if they had repeat testing with both sIL2R and Prf/GrB staining on the same day at a discrete time following treatment. A description of their clinical features is included in Table 2.
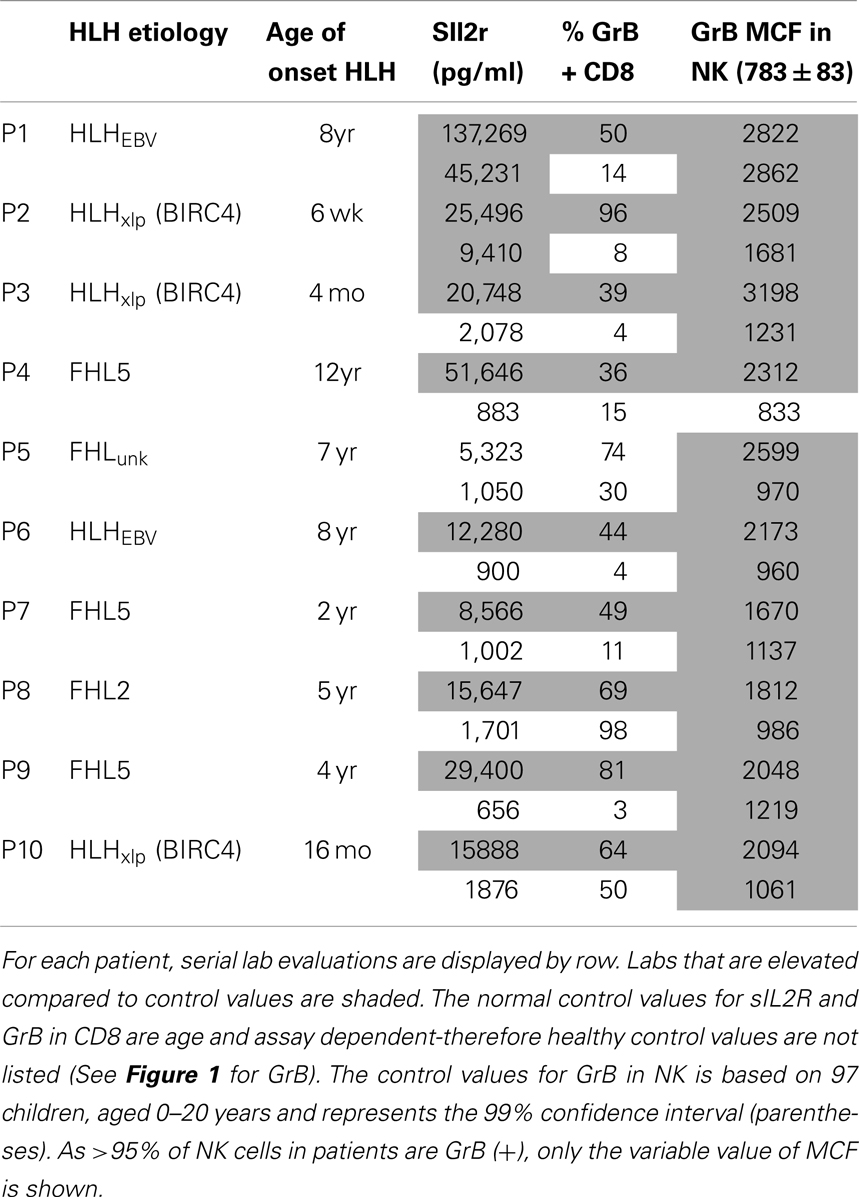
Table 2. Granzyme B detection in NK and CTL before and after treatment for HLH, prior to BMT.
Control Group
Samples from healthy children (age 0–20) and adults (>20 years) were obtained and analyzed for GrB expression in NK cells and cytotoxic lymphocytes to establish normal ranges in our laboratory as we previously described for Prf (Kogawa et al., 2002). Residual peripheral blood samples obtained in ethylenediaminetetraacetic acid for complete blood counts (CBC) were retrieved from an outpatient clinic at our institution under an agreement approved by the IRB. The samples included the age and gender of the patient and the result of the CBC. Those samples with CBC or differential parameters outside of the normal range for age were excluded from the study and discarded. All samples were held at room temperature and processed within 24 h of collection.
Flow Cytometric Analysis (Perforin/Granzyme B Staining)
Perforin and GrB staining was performed by using standard procedures as described in previous publications (Kogawa et al., 2002; Molleran Lee et al., 2004). The antibodies used were either PE-conjugated anti-Prf (deltaG9, BD Pharmingen) or PE-conjugated anti-GrB (GB12, Caltag Laboratories, Burlingame, CA, USA). Detection of Prf and GrB expression by flow cytometry in patients and healthy controls was performed using anti-TCRαβ FITC, anti-CD8PerCP, and anti-CD56 APC antibodies (BD) for surface staining and isotype controls for intracellular staining. CD56+ T cells were excluded from the analysis of NK cells. Samples were analyzed using a FACSCalibur or FACSCanto flow cytometer (Becton Dickinson, San Jose, CA, USA). The normal range for the percentage and mean channel fluorescence (MCF) of Prf and GrB in NK cells is age-independent (see Results) and was constant for all daily controls from 2001 to 2009 using FACSCalibur machines. Daily standardization takes place on the flow cytometers using commercial beads with established amounts of fluorescence. With use of the FACSCanto introduced in 2009, a new normal range for the MCF of Prf and GrB was established. Therefore, in the group analysis (Figures 1–4) only samples prior to the change in cytometer were included for NK data. The data from the FACSCanto was included only for the pre/post therapy group (Figure 5). As expected the normal range for the percentages of CD8 cells expressing Prf and GrB did not change from 2001 to 2011.
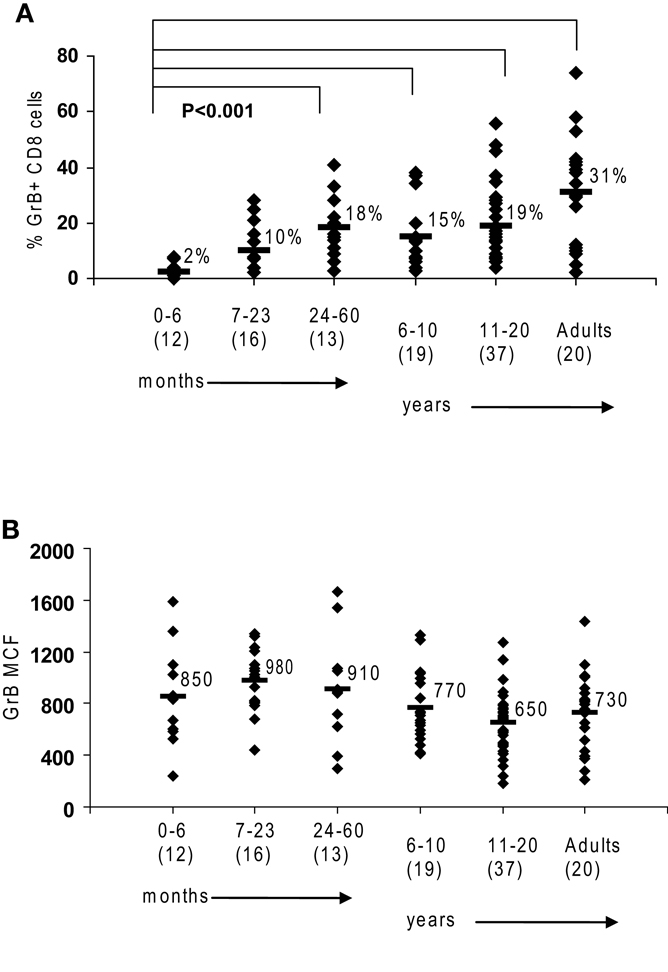
Figure 1. Granzyme B in human cytotoxic lymphocytes. Flow cytometry was used to measure protein expression of granzyme B in healthy controls. (A) There is an age-dependent increase in CD8 cells expressing granzyme B. (B) NK expression of granzyme B as measured by mean channel fluorescence (MCF) does not increase with age. The mean values are listed for each group, with standard deviation. p Values were calculated assuming non-parametric analysis. The number of individuals sampled is shown in parentheses under the age range.
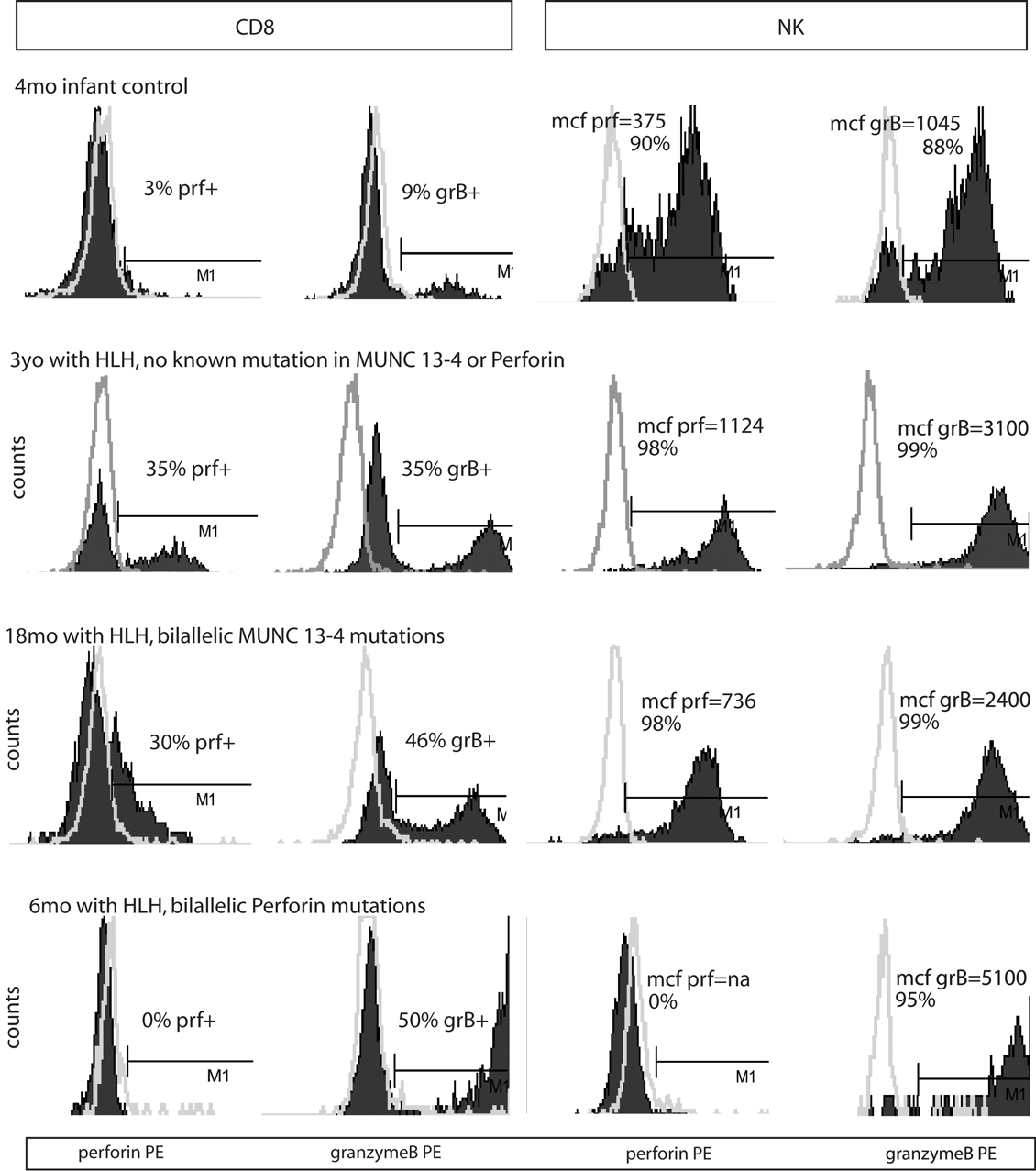
Figure 2. Representative histograms of flow cytometry for Prf and GrB in patients with HLH. Dark shading indicates the test antibody. Light gray line is the isotype control.
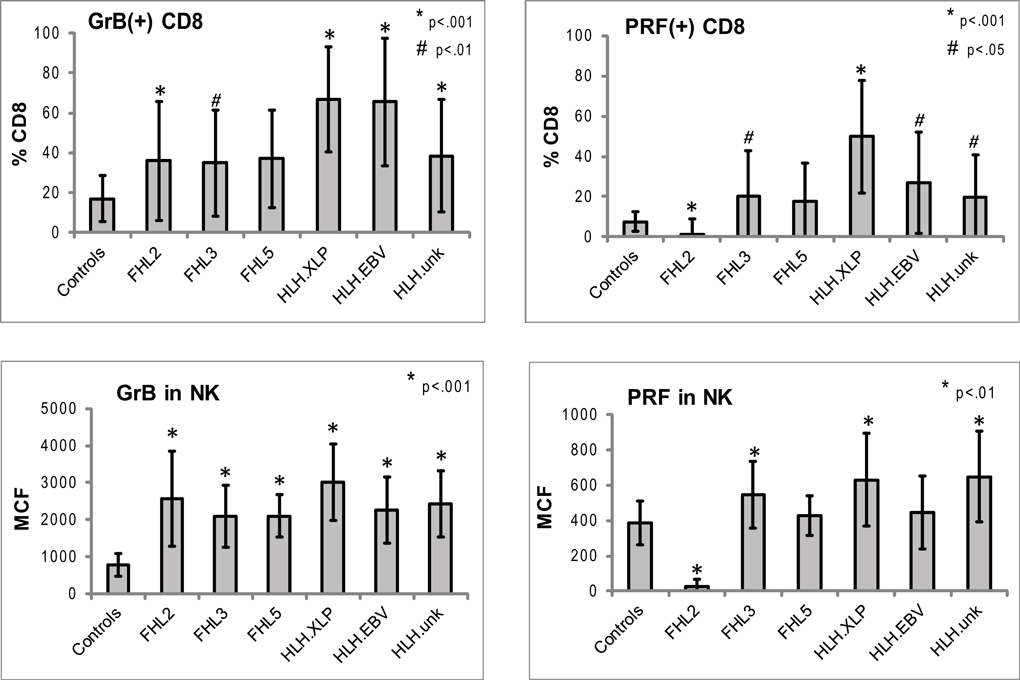
Figure 3. Expression of Prf and GrB in NK and CD8+ lymphocytes in various subtypes of HLH. CD8+ cells: shown are the percentages of Prf and GrB positive CD8 cells. GrB and Prf expression was statistically different than controls in all groups except FHL5. For CD8 data, the p Values are compared to healthy pediatric controls aged 1–20 years (n = 49 for Prf, n = 76 for GrB). NK cells: to evaluate the protein content we analyzed the intensity of the fluorescent signal per cell, i.e., MCF. The MCF of GrB was increased in patients with all subtypes of HLH. MCF of Prf was elevated only in patients in HLHxlp, HLHunk and FHL3 groups. For the NK data, pediatric and adult controls were used as there is no age dependence (n = 116 for Prf, n = 97 for GrB). The error bars represent the standard deviation from the mean. All p values assume two tails, non-parametric data.
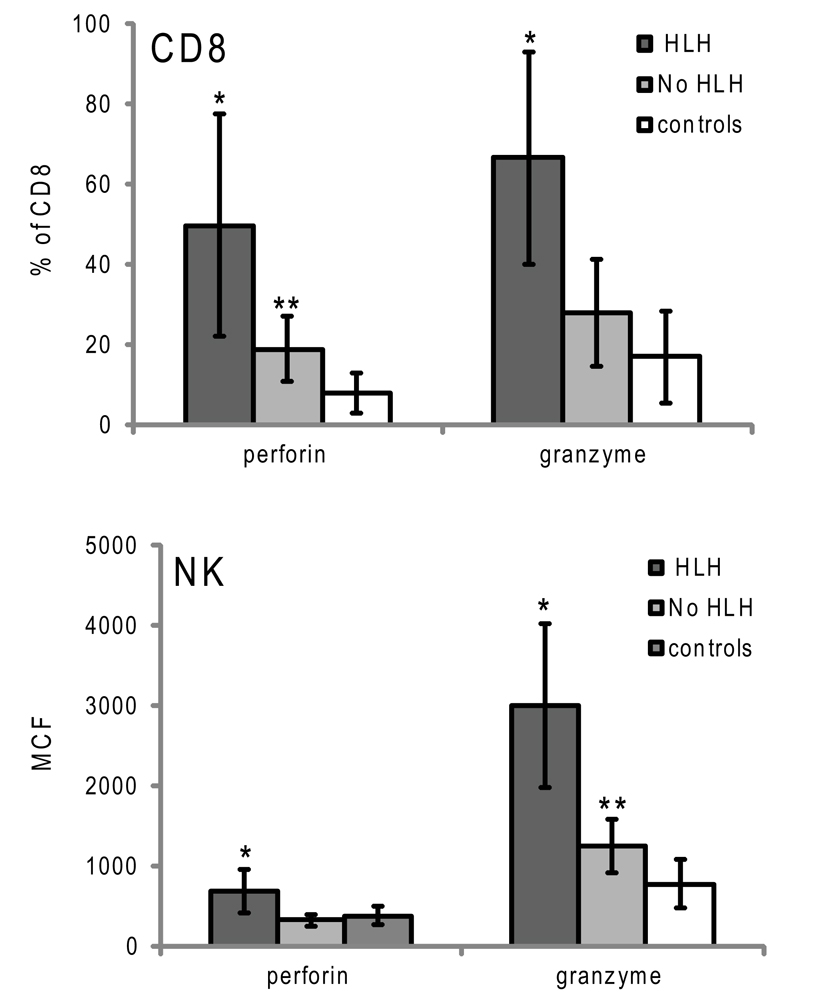
Figure 4. Granzyme B and perforin expression in cytotoxic lymphocytes from patients with XLP presenting with or without HLH. Error bars represent standard deviation from the mean. *p < 0.001 compared to no HLH and controls, **p < 0.05 compared to controls.
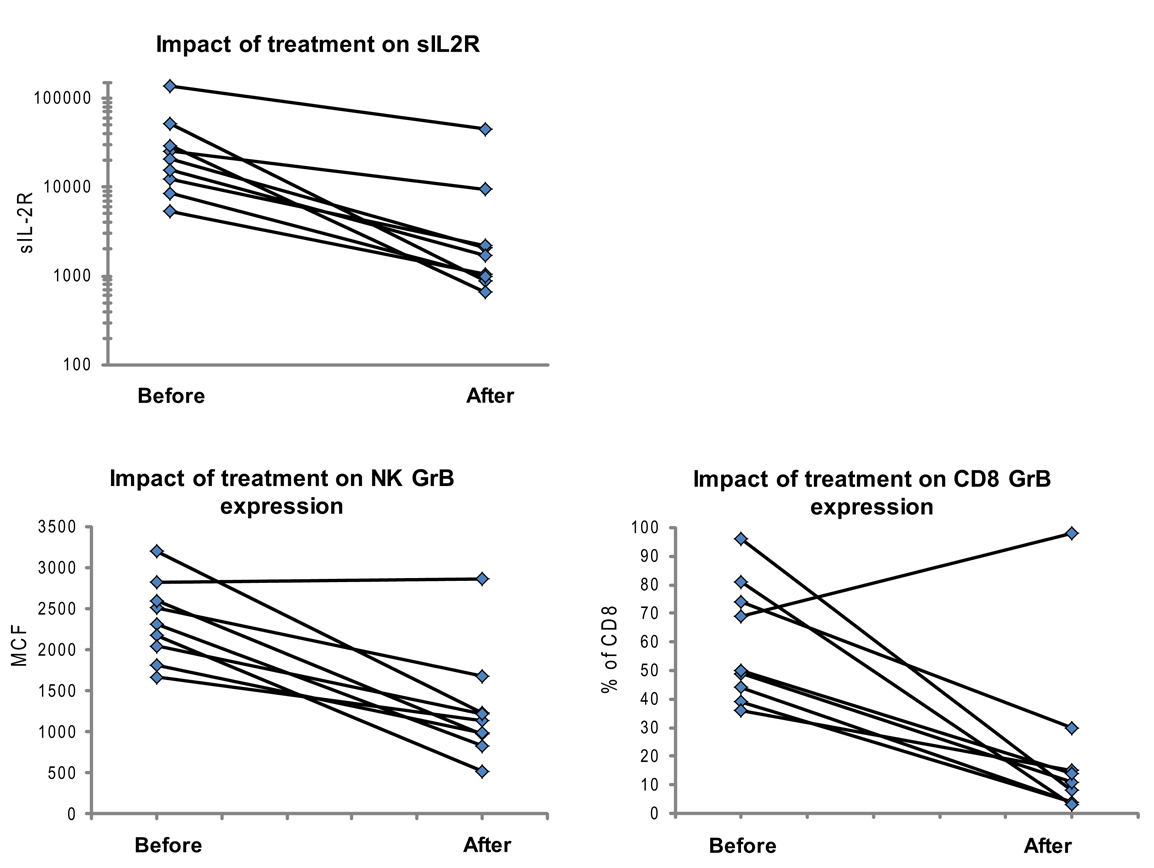
Figure 5. Granzyme B protein content in cytotoxic lymphocytes is a marker of HLH disease activity. Ten patients had a follow-up test performed after initiating treatment for HLH (for description of patients see Table 2). Shown are the first and second data points for each patient. Two patients had persistent immune activation, as measured by the gold standard, sIL2R. These two patients also had persistent GrB elevation in the NK compartment (Table 2).
Soluble IL 2 Receptor
The plasma concentration of the sIL2R was determined using the Human sIL2R (sCD25) ELISA Set (BD OptEIA from BD Biosciences #559104) from 2001 to 2004. From 2004 to 2007, this was changed to a Human sIL2R ELISA Set from R&D. Since February, 2007, the soluble IL2 receptor has been determined using the Immulite 1000 platform, a solid-phase, two-site chemiluminescent immunometric assay system. Soluble sIL2R values were normalized to account for the method utilized. To establish an age-dependent normal range for each method, ethylenediaminetetraacetic acid peripheral blood was obtained from control children of all ages as described above.
Statistical Analysis
Statistical comparisons between two groups were performed using the student t-test, assuming two tails and unequal variance as reported in figure legends.
Results
Granzyme B Expression in Cytotoxic Lymphocytes from Healthy Children
We first established the normal ranges for GrB protein expression by flow cytometry using residual blood samples collected from control children of all age groups as previously described. Similar to the observations by Kogawa et al. (2002) that the percentage of CD8 cells expressing Prf increases with age in healthy children, we observed that the percentage of CD 8 cells expressing GrB is very low in young infants and increases with age to an average 31% in adults (p < 0.001) (Figure 1A). This is due to the fact that CD8 T cells in a newborn are naive and therefore have not differentiated yet into CTL or memory T cells. With antigen exposure in the first 24 months, there is rapid acquisition of CTL and memory cells, to the same level as adults.
Just as the proportion of Prf-containing NK cells has been reported to be constant from birth to adulthood in humans (Kogawa et al., 2002) we found that GrB expression is detectable in >85% of NK cells at all age groups (not shown). Additionally, the content of GrB in NK cells is constant, with a MCF of 850 in children 0–6-month-old, 730 in adults, and 783 (99% confidence interval is 783 + 83) with all ages combined (obtained in our laboratory between 2001 and 2009) (Figure 1B).
Granzyme B Expression in Patients with HLH
We analyzed GrB expression in 93 patients who were identified with known genetic causes of HLH (FHL2, FHL3, FHL5, and HLHxlp) (Table 1). In order to determine if patients without clearly established, genetic diagnoses (HLHunk) also exhibited the same immune signature, we included 37 additional patients who reported by their physicians as having HLH, of which 14 had EBV-associated HLH (see Table 1). All patients were referred with a clinical diagnosis of HLH by their primary physician and had flow cytometric evaluation of Prf and GrB performed by the DIL.
Typical histograms of Prf and GrB expression in cytotoxic cells are shown in Figure 2 and the results from all patients with HLH summarized in Figure 3. Consistent with our hypothesis, we noted an increased percentage of GrB positive CD8 cells in patients with HLH, in all genetic subtypes. This reached statistical significance for all groups except FHL5, as compared to age-matched healthy normal controls. The lack of statistical significance in FHL5 is due to the lower number of children in this group (n = 10), as the mean level was elevated to the same degree as other groups with HLH that did reach a p value < 0.01. Prf expression was low (as expected in FHL2 due to biallelic PRF1 mutations), and statistically elevated in all other groups except FHL5. We compared all groups with HLH to controls aged 1–20 years, as the patients with FHL5 were 1 year and older. For the other groups with younger children, we performed an independent analysis including children aged 0–20 years, but this did not change the statistical significance.
We analyzed the MCF of prf and grB in NK cells as a measure of protein content per cell to determine if NK cells similarly upregulated granule proteins. Indeed, GrB content was elevated in NK cells in patients with HLH regardless of genetic subtype. The mean GrB signal (MCF) in NK cells was threefold elevated in all subtypes of HLH, including boys with HLH due to XLP. Therefore, the immunologic phenotype was not dependent upon a granule protein pathway defect per se, but instead reflected the common final pathway for immune activation of CTL and NK. Interestingly, the Prf was minimally elevated in some, but not all patients with HLH. It was low, as expected in patients with FHL2 (PRF1 mutations). As the GrB expression was more consistently (and dramatically) elevated than Prf, we focused on this granule protein as a marker for immune activation.
Granzyme B Expression Correlates with Immune Activation in HLH
Although the data demonstrate that elevated GrB content in NK and an increased percentage of GrB expressing CTLs are present in children with HLH, the correlation of increased GrB detection for immune activation was unclear. To address this, we reviewed data from boys diagnosed with XLP but no evidence of HLH at the time of evaluation. These patients were diagnosed with XLP after a sibling’s diagnosis or after multiple malignancies. As shown in Figure 4, patients with XLP without HLH exhibited subtle increases in GrB detection; whilst the group with HLH had signals that were three times higher than the control group. This data suggests that not only is GrB a signature for immune activation in HLH, but it also increases subtly in individuals with XLP whose baseline immune activation has not yet reached a critical threshold for development of HLH.
We also evaluated whether GrB content decreased with immunosuppression as a second confirmation that protein levels correlated with immune activation. Therefore, we analyzed data from 10 patients who had been treated for HLH at our institution and had at least two samples sent for GrB and Prf testing, along with concomitant sIL2R measurements. All 10 patients had Prf/GrB testing at diagnosis and at a second time point, i.e., during treatment for HLH therapy and prior to bone marrow transplantation. As shown in Table 2, all patients exhibited a decrease in the gold standard for immune activation, sIL2R, during therapy, as expected. The GrB expression in CD8+ cells decreased to normal in 8/10 patients; however, in P8 and P10, GrB expression in CTL remained markedly elevated despite normalization of other markers. Likewise, expression of GrB in NK cells decreased in all but one patient (P1), whose sIL2R remained persistently elevated. Thus, elevations of GrB may serve as a biomarker of immune activation in patients with HLH. Interestingly, although the NK GrB levels dropped, they did not typically normalize, despite a normalized sIL2R. This is reminiscent of the subtle increases in GrB seen in boys with XLP but no evidence of HLH. This suggests that NK expression of GrB may be a more sensitive marker of persistent, dysregulated cytotoxic lymphocyte activation.
Discussion
Our data in patients with HLH shows for the first time that increased detection of GrB in both CTL and NK are a signature of HLH-associated immune activation, irrespective of genetic etiology. Although expressed constitutively in all human NK cells, GrB protein levels (as measured by flow cytometry) in human NK cells were elevated in patients with HLH and decreased with treatment. This suggests that elevated GrB levels in NK cells may also be a biomarker of disease activity. As a result of this finding, our physicians now consider elevated NK and/or CTL GrB expression as a marker of disease activity, providing additional evidence for evidence of immune activation in the cytotoxic compartment. This is feasible as the diagnostic lab calibrates flow cytometers daily to a fluorescent standard (beads) and defines age-matched ranges prior to testing clinical samples. We also noted increased Prf expressing CD8. In FHL2, due to PRF1 mutations, the Prf levels are generally absent or severely reduced (Figure 3), but in other forms of HLH, PRF levels were also increased in NK cells (FHL3, HLHxlp, and HLHunk).
Occasional samples submitted from patients with HLH exhibited normal levels of GrB expression in cytotoxic lymphocytes. As blood is sent from outside physicians for testing without information regarding treatment, it is likely that these patients had already started immunosuppressive therapy, but this could not be confirmed. Additionally, newborns are occasionally tested prior to the onset of HLH symptomatology if an older sibling has died from HLH. The inclusion of patients with clinically silent disease in our analysis of 130 patients may have led to an underestimation of the maximum GrB expression possible in all subtypes of HLH.
Based on the current understanding of the pathophysiology of HLH, we would predict to find increased Prf/GrB expression in CD8 cells in patients with HLH. The murine model has clearly shown activation of this T cell compartment and CD8 cells acquire both granzymes and Prf during activation and differentiation. As CTL differentiation is dependent upon T cell receptor stimulation, our data implies that antigen presentation via MHC class I is a primary determinant of the immune activation. Viral triggers are presumed in HLH, although not always identified in young infants presenting with HLH. In contrast to CTL activation, elevated NK levels of Prf and GrB have not previously been described under pathologic conditions. Our pre/post evaluation of patients with HLH and the data for individuals with XLP suggests the elevation in these protein markers are a result of immune activation and/or dysregulation.
The finding of NK activation in patients with HLH due to BIRC4 mutations is interesting in that no clear secretory granule defect has been identified in these patients. It is consistent with the fact that HLH is often a presenting symptom in these patients. (Marsh et al., 2010) Therefore, elevated GrB levels may not simply reflect a failure of secretion or degranulation of this granule associated protein. If this were true, we would expect the highest values to be found in cytotoxic lymphocytes with an inability to degranulate (Munc 13-4 and STXBP2 deficiency). There was no clear difference between sub-groups of HLH. We have not yet addressed the mechanism for increased Prf/GrB detection in NK, but possibilities include increased transcription, post-translational regulation of mRNA stability, increased secretory granule production, and/or increased half-life of the proteins.
Regulation of GrB in NK has been described at the level of mRNA transcription and post-translational regulation through microRNAs, largely through in vitro studies of cytokine activation (Kim et al., 2011; Trotta et al., 2011; Wang et al., 2012). As elevated pro-inflammatory cytokines have been described in HLH, it is possible that blood and tissue-derived cytokines drive NK and CTL activation of GrB. It is also likely that ongoing antigenic stimulation (due to a failure to eliminate antigen presenting or virally infected cells) provides an ongoing activating stimulus to NK and CTL. Further studies in murine models of HLH will help clarify these remaining questions.
Although ours is the first study to report GrB protein expression in CTL and NK cells in patients with immune activation due to HLH, GrB transcripts in the blood and GrB ELISPOT measurements of PBMC have been proposed as a biomarker of immune activation to screen for CTL activation leading to graft rejection following solid organ transplants (Altimari et al., 2008; Truong et al., 2008). For example, in patients who received intestinal transplantation, blood monitoring of GrB and Prf RNA were diagnostic markers of acute graft rejection, increasing prior to histological diagnosis of a rejection, and then returning back to baseline with initiation of therapy (Altimari et al., 2008). In addition to investigations following solid organ transplants, studies have evaluated whether GrB levels are elevated in patients with active viral infections such as children with Respiratory Syncytial Virus infection and patients infected with EBV or HIV (Spaeny-Dekking et al., 1998; Bem et al., 2008). Finally, GrB levels in the blood have been associated with disease activity in patients with rheumatoid arthritis (Goldbach-Mansky et al., 2005). No direct comparison have been made of flow cytometry measurements to ELISA or RNA measurements of GrB, making comparisons with these studies challenging to interpret. Evaluation of NK/CTL GrB detection in patients with macrophage activation syndrome (MAS) in patients with juvenile idiopathic arthritis at our center revealed variable results, with increased detection in some, but not all patients with active disease (Grom et al., 2003) and data not shown), highlighting the subtle differences between HLH and MAS.
In summary, we have shown that elevated GrB expression in NK and CTL is a signature of immune activation in patients with HLH regardless of the genetic background. GrB staining of cytotoxic lymphocytes is included routinely as an internal control in the diagnostic immunology lab at our center in the assessment of Prf protein. Our data suggest that it may serve as a useful biomarker of disease activity. As a retrospective study limited to a large cohort of patients with HLH, we have not addressed the specificity of our findings to HLH. Future studies are needed to determine the utility of our findings in other inflammatory conditions such as critically ill patients with systemic inflammatory response syndrome, graft versus host disease, or autoimmune diseases. We would predict that any disorder with profound CTL/NK immune activation secondary to systemic viral activation may share a similar immune profile. Characterization of cytotoxic lymphocyte activation by assessment of GrB detection may thus be a useful tool clinically and in research settings.
Authors Contribution
Sabine Mellor-Heineke conducted the retrospective chart review, performed data analysis, and wrote the manuscript. Kejian Zhang developed the genetic testing and analyzed the genetic data. Alexandra H. Filipovich, Michael B. Jordan, Jack J. Bleesing, and Rebecca Marsh analyzed the immune data and evaluated the patients treated at this institution. Each of these physicians also contributed to the writing of the manuscript. Joyce Villanueva developed the assays in the DIL and analyzed the immunologic data on patients and controls. Kimberly A. Risma designed the study, interpreted all data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Kimberly A. Risma was funded by the Doris Duke Charitable Foundation Clinical Scientist Development Award. R.A.M was supported by a grant from the Clinical Immunology Society.
Abbreviations
CMV, cytomegalovirus; CTL, cytotoxic T lymphocytes; EBV, Epstein Barr virus; GrB, granzyme B protein; HLH, hemophagocytic lymphohistiocytosis; MCF, mean channel fluorescence; NK, natural killer; Prf, perforin protein; sIL2R, soluble IL-2 receptor alpha (sCD25).
References
Altimari, A., Gruppioni, E., Capizzi, E., Bagni, A., Corti, B., Fiorentino, M., et al. (2008). Blood monitoring of granzyme B and perforin expression after intestinal transplantation: considerations on clinical relevance. Transplantation 85, 1778–1783.
Bem, R. A., Bos, A. P., Bots, M., Wolbink, A. M., van Ham, S. M., Medema, J. P., et al. (2008). Activation of the granzyme pathway in children with severe respiratory syncytial virus infection. Pediatr. Res. 63, 650–655.
Bien, E., and Balcerska, A. (2008). Serum soluble interleukin 2 receptor alpha in human cancer of adults and children: a review. Biomarkers 13, 1–26.
Bleesing, J., Prada, A., Siegel, D. M., Villanueva, J., Olson, J., Ilowite, N. T., et al. (2007). The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 56, 965–971.
Boivin, W. A., Cooper, D. M., Hiebert, P. R., and Granville, D. J. (2009). Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab. Invest. 89, 1195–1220.
Buzza, M. S., and Bird, P. I. (2006). Extracellular granzymes: current perspectives. Biol. Chem. 387, 827–837.
Cannons, J. L., Tangye, S. G., and Schwartzberg, P. L. (2011). SLAM family receptors and SAP adaptors in immunity. Annu. Rev. Immunol. 29, 665–705.
Crozat, K., Hoebe, K., Ugolini, S., Hong, N. A., Janssen, E., Rutschmann, S., et al. (2007). Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J. Exp. Med. 204, 853–863.
de Saint Basile, G., Menasche, G., and Latour, S. (2011) Inherited defects causing hemophagocytic lymphohistiocytic syndrome. Ann. N. Y. Acad. Sci. 1246, 64–76.
Filipovich, A. H. (2009). Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am. Soc. Hematol. Educ. Program 2009, 127–131.
Goldbach-Mansky, R., Suson, S., Wesley, R., Hack, C. E., El-Gabalawy, H. S., and Tak, P. P. (2005). Raised granzyme B levels are associated with erosions in patients with early rheumatoid factor positive rheumatoid arthritis. Ann. Rheum. Dis. 64, 715–721.
Grom, A. A., Villanueva, J., Lee, S., Goldmuntz, E. A., Passo, M. H., and Filipovich, A. (2003). Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J. Pediatr. 142, 292–296.
Grossman, W. J., Verbsky, J. W., Tollefsen, B. L., Kemper, C., Atkinson, J. P., and Ley, T. J. (2004). Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood 104, 2840–2848.
Imashuku, S., Hibi, S., Sako, M., Ishida, Y., Mugishima, H., Chen, J., et al. (1995). Soluble interleukin-2 receptor: a useful prognostic factor for patients with hemophagocytic lymphohistiocytosis. Blood 86, 4706–4707.
Johnson, T. S., Villanueva, J., Filipovich, A. H., Marsh, R. A., and Bleesing, J. J. (2011). Contemporary diagnostic methods for hemophagocytic lymphohistiocytic disorders. J. Immunol. Methods 364, 1–13.
Jordan, M. B., and Filipovich, A. H. (2008). Hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis: a journey of a thousand miles begins with a single (big) step. Bone Marrow Transplant. 42, 433–437.
Jordan, M. B., Hildeman, D., Kappler, J., and Marrack, P. (2004). An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood 104, 735–743.
Kim, T. D., Lee, S. U., Yun, S., Sun, H. N., Lee, S. H., Kim, J. W., et al. (2011). Human microRNA-27a* targets Prf1 and GzmB expression to regulate NK-cell cytotoxicity. Blood 118, 5476–5486.
Kogawa, K., Lee, S. M., Villanueva, J., Marmer, D., Sumegi, J., and Filipovich, A. H. (2002). Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood 99, 61–66.
Komp, D. M., McNamara, J., and Buckley, P. (1989). Elevated soluble interleukin-2 receptor in childhood hemophagocytic histiocytic syndromes. Blood 73, 2128–2132.
Lieberman, J. (2010). Anatomy of a murder: how cytotoxic T cells and NK cells are activated, develop, and eliminate their targets. Immunol. Rev. 235, 5–9.
Lykens, J. E., Terrell, C. E., Zoller, E. E., Risma, K., and Jordan, M. B. (2011). Perforin is a critical physiologic regulator of T-cell activation. Blood 118, 618–626.
Marsh, R. A., and Filipovich, A. H. (2011). Familial hemophagocytic lymphohistiocytosis and X-linked lymphoproliferative disease. Ann. N. Y. Acad. Sci. 1238, 106–121.
Marsh, R. A., Madden, L., Kitchen, B. J., Mody, R., McClimon, B., Jordan, M. B., et al. (2010). XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood 116, 1079–1082.
Molleran Lee, S., Villanueva, J., Sumegi, J., Zhang, K., Kogawa, K., Davis, J., et al. (2004). Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J. Med. Genet. 41, 137–144.
Pachlopnik Schmid, J., Canioni, D., Moshous, D., Touzot, F., Mahlaoui, N., Hauck, F., et al. (2011). Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood 117, 1522–1529.
Risma, K., and Jordan, M. B. (2012). Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr. Opin. Pediatr. 24, 9–15.
Russell, J. H., and Ley, T. J. (2002). Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 20, 323–370.
Salcedo, T. W., Azzoni, L., Wolf, S. F., and Perussia, B. (1993). Modulation of perforin and granzyme messenger RNA expression in human natural killer cells. J. Immunol. 151, 2511–2520.
Sedelies, K. A., Sayers, T. J., Edwards, K. M., Chen, W., Pellicci, D. G., Godfrey, D. I., et al. (2004). Discordant regulation of granzyme H and granzyme B expression in human lymphocytes. J. Biol. Chem. 279, 26581–26587.
Skak, K., Frederiksen, K. S., and Lundsgaard, D. (2008). Interleukin-21 activates human natural killer cells and modulates their surface receptor expression. Immunology 123, 575–583.
Spaeny-Dekking, E. H., Hanna, W. L., Wolbink, A. M., Wever, P. C., Kummer, J. A., Swaak, A. J., et al. (1998). Extracellular granzymes A and B in humans: detection of native species during CTL responses in vitro and in vivo. J. Immunol. 160, 3610–3616.
Tak, P. P., Spaeny-Dekking, L., Kraan, M. C., Breedveld, F. C., Froelich, C. J., and Hack, C. E. (1999). The levels of soluble granzyme A and B are elevated in plasma and synovial fluid of patients with rheumatoid arthritis (RA). Clin. Exp. Immunol. 116, 366–370.
Trotta, R., Ciarlariello, D., Dal Col, J., Mao, H., Chen, L., Briercheck, E., et al. (2011). The PP2A inhibitor SET regulates granzyme B expression in human natural killer cells. Blood 117, 2378–2384.
Trottestam, H., Horne, A., Arico, M., Egeler, R. M., Filipovich, A. H., Gadner, H., et al. (2011). Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood 118, 4577–4584.
Truong, D. Q., Cornet, A., Wieers, G., Robert, A., Reding, R., and Latinne, D. (2008). Pre- and post-transplant monitoring of granzyme B enzyme-linked immunosorbent spot assay in pediatric liver recipients. Transpl. Immunol. 19, 215–219.
Voskoboinik, I., Smyth, M. J., and Trapani, J. A. (2006). Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 6, 940–952.
Keywords: granzyme B, hemophagocytic lymphohistiocytosis, natural killer cells, clinical immunology
Citation: Mellor-Heineke S, Villanueva J, Jordan MB, Marsh R, Zhang K, Bleesing JJ, Filipovich AH and Risma KA (2013) Elevated granzyme B in cytotoxic lymphocytes is a signature of immune activation in hemophagocytic lymphohistiocytosis. Front. Immunol. 4:72. doi: 10.3389/fimmu.2013.00072
Received: 13 December 2012; Accepted: 05 March 2013;
Published online: 22 March 2013.
Edited by:
Eric Vivier, Centre d’Immunologie de Marseille-Luminy, FranceReviewed by:
Nadia Guerra, Imperial College London, UKDebby Burshtyn, University of Alberta, Canada
Gilles Kaplanski, Assiatance Publique Hôpitaux de Marseille, France
Copyright: © 2013 Mellor-Heineke, Villanueva, Jordan, Marsh, Zhang, Bleesing, Filipovich and Risma. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Kimberly A. Risma, Division of Allergy and Immunology, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229-2000, USA. e-mail: kimberly.risma@cchmc.org