 Christopher J. Holland1
Christopher J. Holland1- 1Institute of Infection and Immunity, Cardiff University School of Medicine, Cardiff, UK
- 2Department of Integrated Medicine, University Hospital of Wales, Cardiff, UK
Recombinant αβ T cell receptors, expressed on T cell membranes, recognize short peptides presented at the cell surface in complex with MHC molecules. There are two main subsets of αβ T cells: CD8+ T cells that recognize mainly cytosol-derived peptides in the context of MHC class I (pMHC-I), and CD4+ T cells that recognize peptides usually derived from exogenous proteins presented by MHC class II (pMHC-II). Unlike the more uniform peptide lengths (usually 8–13mers) bound in the MHC-I closed groove, MHC-II presented peptides are of a highly variable length. The bound peptides consist of a core bound 9mer (reflecting the binding motif for the particular MHC-II type) but with variable peptide flanking residues (PFRs) that can extend from both the N- and C-terminus of the MHC-II binding groove. Although pMHC-I and pMHC-II play a virtually identical role during T cell responses (T cell antigen presentation) and are very similar in overall conformation, there exist a number of subtle but important differences that may govern the functional dichotomy observed between CD8+ and CD4+ T cells. Here, we provide an overview of the impact of structural differences between pMHC-I and pMHC-II and the molecular interactions with the T cell receptor including the functional importance of MHC-II PFRs. We consider how factors such as anatomical location, inflammatory milieu, and particular types of antigen presenting cell might, in theory, contribute to the quantitative (i.e., pMHC ligand frequency) as well as qualitative (i.e., variable PFR) nature of peptide epitopes, and hence offer a means of control and influence of a CD4+ T cell response. Lastly, we review our recent findings showing how modifications to MHC-II PFRs can modify CD4+ T cell antigen recognition. These findings may have novel applications for the development of CD4+ T cell peptide vaccines and diagnostics.
Introduction
T cell immunity is mediated primarily by the membrane bound T cell receptor (TCR) that interacts with peptide epitopes presented by major histocompatibility molecules (pMHC) (1). This interaction governs T cell specificity and leads to downstream T cell activation. Classical MHC exists in two forms: MHC class I (MHC-I) and MHC class II (MHC-II), which differ in both their subunit composition and functional expression pattern. MHC-I presents peptides derived mainly from endogenous cytosolic proteins and is expressed upon the cell surface of most nucleated cells allowing cognate CD8+ T cells to scan cells for intracellular infections or abnormal proteins in cancerous cells (2, 3). In contrast, MHC-II is expressed mainly upon antigen presenting cells (APCs) e.g., dendritic cells and macrophages, that patrol the extracellular space, actively endocytosing potentially immunogenic proteins that are proteolysed and complexed with MHC-II (pMHC-II). Activated APCs enter the lymphatic system and travel to secondary lymphoid nodes allowing naive CD4+ T cells to interrogate cell surface expressed pMHC-II enabling CD4+ T cell activation and initiation of immune responses (3– 5).
Peptides Presented by MHC-I and MHC-II have Distinct Structural Characteristics
In spite of the differing subunit compositions of the two MHC classes, they are structurally very similar (Figures 1A,B). The peptide binding groove, in both cases, is comprised of two anti-parallel α-helices that form a channel in which the peptide can bind in an extended conformation, and eight anti-parallel β-sheets that provide specific peptide binding pockets in the base of the groove (Figures 1C,D) (3, 6). Peptides are selected according to their ability to bind to these MHC allele specific pockets within the floor of the peptide binding groove using peptide anchor residues. All of the currently available structural data suggest the TCRs bind to both pMHC-I and II with a fixed polarity (TCRα chain over the N-terminus of the peptide and the TCRβ chain over the C-terminus) and make similar interactions with the bound peptide and MHC surface (Figures 1E,F). Thus, the overall mechanism by which TCRs interact with MHC-I and II to initiate T cell activation is closely matched.
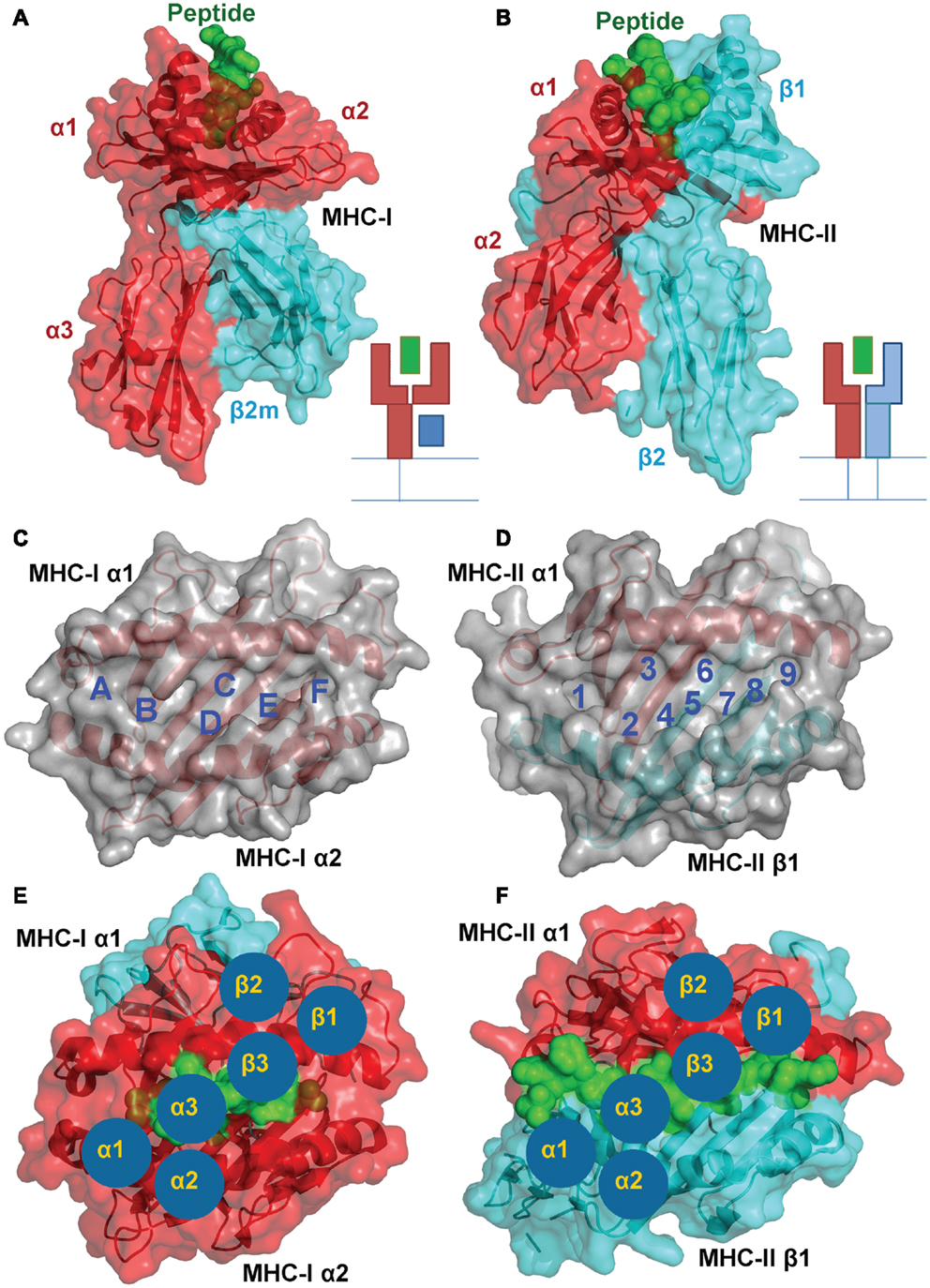
Figure 1. A structural comparison of pMHC-I and pMHC-II. Although the subunit compositions of MHC-I (PDB: 1ZHL) (A) and MHC-II (PDB: 1KG0) (B) are different, the structural conformation they assume is very similar, illustrating their shared role in presenting antigenic peptides (green) to T cells. (A) MHC-I is comprised of three α-chain domains (1, 2, and 3 in red) and β2m (cyan), whereas (B) MHC-II is comprised of a two domain α-chain (red) and a two domain β-chain (cyan). A top down view of the MHC-I (C) and MHC-II (D) demonstrates the two molecules form similar peptide binding grooves comprised of two anti-parallel α-helices that form a channel in which the peptide can bind in an extended conformation, and eight anti-parallel β-sheets that provide specific peptide binding pockets in the base of the groove. These pockets are lined with polymorphic residues that define the size and chemical characteristics of each pocket, and therefore the specific peptide binding motif and register that can be accommodated by different MHC alleles. TCR binding to pMHC-I (E) and pMHC-II (F) is also conserved. The three complementarity determining loops (CDRs) of the TCR (blue circles) bind in a very similar overall orientation with the TCR α-chain over the N-terminus of the peptide and the TCR β-chain over the C-terminus
Despite these similarities, MHC-I and II present peptides in a distinct manner that is governed by the composition of the MHC peptide binding groove. The closed conformation of the MHC-I α1α2 binding grove (Figure 2A) restricts peptide length to ∼8–13 amino acids (most commonly 9 or 10mers) (3, 7). In contrast, the MHC-II α1β1 binding groove comprises an open-ended conformation (Figure 2B) that allows variable length peptides to bind. The core binding 9mer contains the motif for binding to the particular MHC-II heterodimer, but eluted and sequenced peptides often reveal families of processed peptides ∼12–20 amino acids (referred to as nested sets) sharing the core binding region (3, 8, 9). MHC-I-restricted peptides usually bind to the MHC surface using anchor residues located at, or near, the N- and C-termini of the peptide. Depending on the length of the peptide, this binding mode squeezes the central peptide residues up so that they extend out the groove (central bulge), exposing peptide side chains for direct interaction with the TCR (3, 10). Longer MHC-I peptides can only be accommodated by forming a larger central bulge, which presumably constrains the length of the peptide beyond a certain threshold (Figure 2C; Table 1).
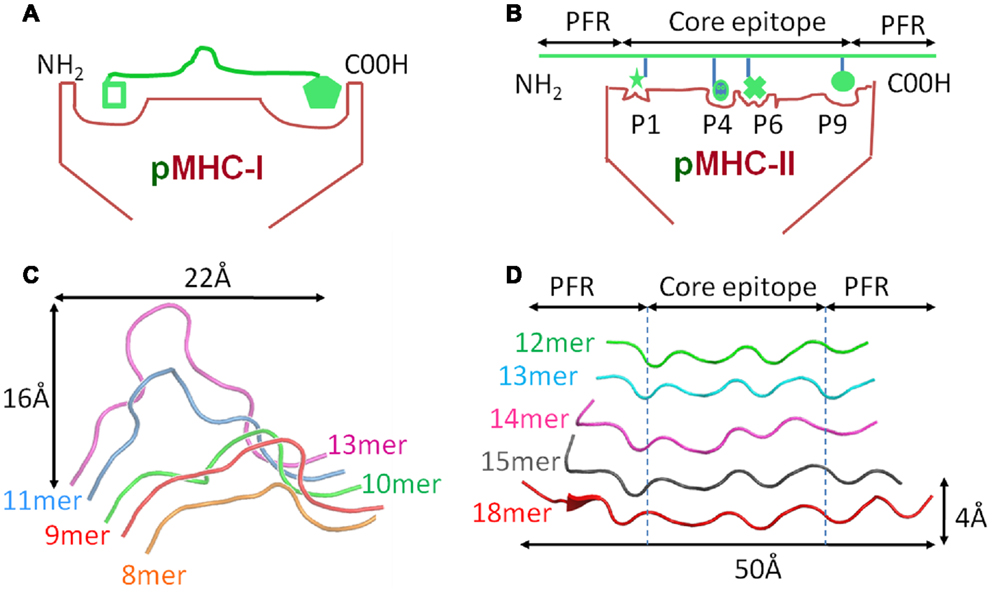
Figure 2. Comparison of peptide conformations presented by MHC-I and MHC-II. Cartoon cross sections of the pMHC-I (A) and pMHC-II (B) binding grooves, show the key anchor sites in the floor of each groove determine which peptide can associate and the conformation it can assume. (C) The structural database of pMHC-I complexes shows that peptides presented by a MHC-I molecules (represented as ribbon cartoons) generally assume a central bulged conformation. As peptide length increases, the “closed” nature of the pMHC-I binding groove forces the central residues of the peptide up out of the groove to accommodate the extra residues. (D) In contrast, the pMHC-II binding groove is “open” enabling longer peptide to extend out of the groove at form peptide flanking regions. Thus, peptides presented by MHC-II molecules (represented as ribbon cartoons) generally assume a much flatter conformation in the MHC-II binding groove, irrespective of the length of the peptide presented.
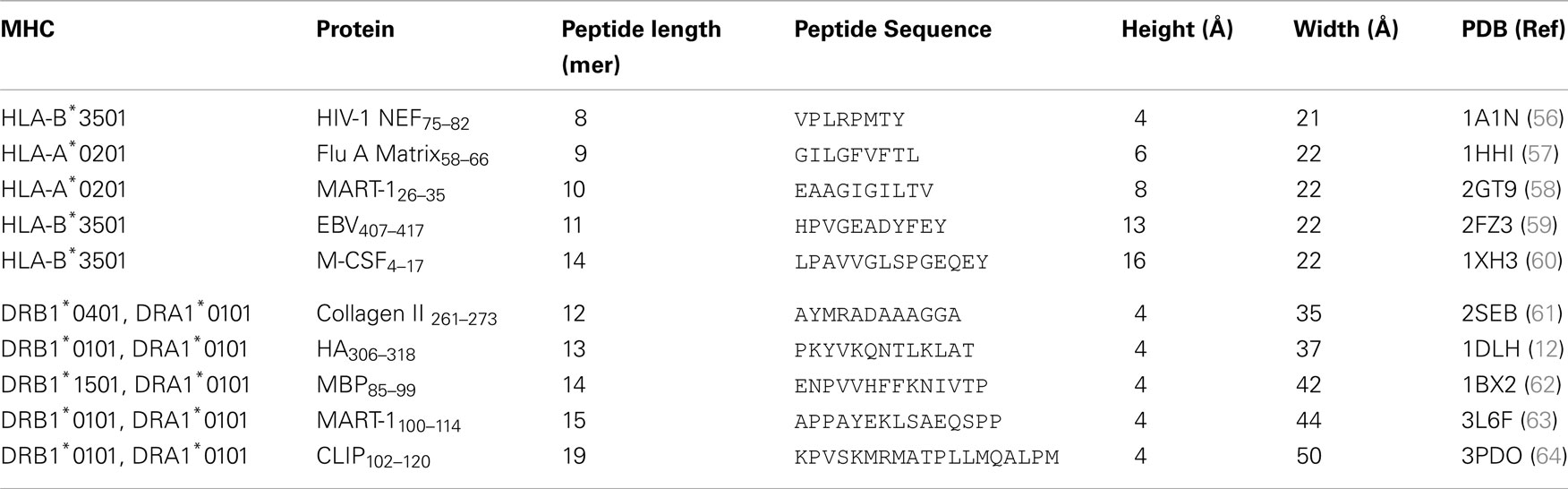
Table 1. Comparison of peptide conformations presented by MHC-I and MHC-II.
MHC-II restricted peptides contain a central binding motif of nine “core” amino acids that bind to the MHC-II groove via an extensive hydrogen bond network between the MHC-II groove and the peptide backbone (Figure 2B). Peptide side chains also form contacts with allelic specific pockets of the MHC-II binding groove. These pockets, usually P1, P4, P6, and P9, are lined with polymorphic residues that define the size and chemical characteristics of each pocket, and therefore the specific peptide binding motif and register that can be accommodated by different MHC-II alleles (Figure 1D) (11, 12). Amino acids that are outside of the “core” peptide region can extend out of the open MHC-II binding groove forming so called “peptide flanking regions” (PFRs) at both the N- and C-terminus (Figure 2D; Table 1). Thus, although pMHC-I and pMHC-II are similar in their overall structure and function, the nature of peptide presentation is generally distinct (e.g., bulged versus flat peptides). These differences present different challenges for TCR binding at the atomic level. For example, the flat binding surface and lack of a central peptide bulge may enable MHC-II restricted TCRs to adopt a more flexible binding mode compared to MHC-I restricted TCRs. In support of this notion, the structures of a number of TCR-MHC-II complexes have shown that, although the binding mode can be very similar to the classical diagonal TCR-MHC binding mode (13– 17), some MHC-II restricted TCR bind with highly unorthodox conformations (18, 19).
MHC-II Restricted TCRs have Weaker Binding Affinity Compared to pMHC-I Restricted TCRs
Biophysical studies have shown that TCR/pMHC affinity is relatively weak (KD = 100 nM–270 μM), with fast kinetics, compared to antibody binding (usually nM–pM affinity) (20, 21). We have recently shown that TCR/pMHC-I binding affinities are, on average, fives times stronger compared to equivalent TCR/pMHC-II interactions (i.e., viral pMHC-I restricted TCRs versus viral pMHC-II restricted TCRs) (Figures 3A,B), which has limited the usefulness of pMHC-II multimers for the identification, isolation and detection of antigen specific CD4+ T cells. This distinction in affinity was mainly due to a significantly faster on-rate for TCR/pMHC-I binding compared to that of TCR/pMHC-II, while the off-rate or half-lives of all of the TCR/pMHC interactions were relatively conserved, possibly indicating a more important role for off-rate in determining T cell activation. The consistent difference in binding affinity between TCRs restricted to either pMHC-I or pMHC-II is extraordinary when considering that the same pools of genes, on chromosome 9, encode the human TCR for both types of αβ T cell (22). The TCR itself is expressed before positive selection, at which point immature T cells express both the CD4 and CD8 co-receptors (double positive). Once positively selected, immature thymocytes become single positive for either CD4 or CD8 (22). Until this point, the thymocyte, which has already developed antigen specificity through its TCR, can theoretically have either cell fate (23). Considering these shared genetic and developmental processes, it is possible that the differences in MHC restricted TCR binding is conferred by the variations in the “antigenic landscape” of pMHC-I versus pMHC-II.
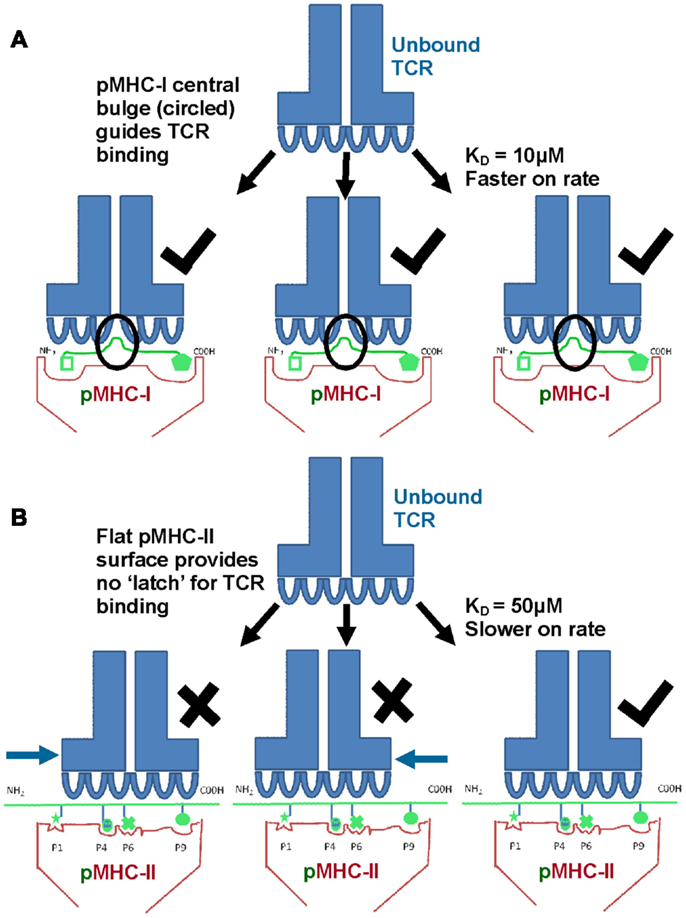
Figure 3. TCRs bind with stronger affinity to pMHC-I compared to pMHC-II. Biophysical studies have shown that TCR/pMHC-I binding affinities are, on average, five times stronger compared to equivalent TCR/pMHC-II interactions (i.e., viral pMHC-I restricted TCRs versus viral pMHC-II restricted TCRs) because of faster on-rate for TCR/pMHC-I binding compared to that of TCR/pMHC-II. These differences could be due to the structural differences in peptide presentation between MHC-I and MHC-II. (A) Cartoon of TCR binding to pMHC-I. The presence of a solvent exposed central bulge for MHC-I peptide presentation may represent a structurally advantageous feature for TCR binding, providing an anchor point that can guide the TCR into the correct binding orientation to engage its cognate ligand. (B) Cartoon of TCR binding to the flatter surface of pMHC-II. This relatively featureless surface provides no dominant structural feature for the TCR to “latch” onto, and may reduce the chance of a productive TCR/pMHC-II interaction occurring (explaining the slower on-rate and weaker affinity compared to TCR/pMHC-I interactions).
As discussed above, peptides presented by MHC-I molecules generally assume a central bulged conformation, often requiring conformational adjustments in the binding regions of the TCR during ligand engagement (Figure 2C) (24– 26). An extreme example of this observation is a 13mer Epstein-Barr virus derived peptide presented by HLA-B∗3508 which forms a “superbulge” extending nearly 20Å out of the MHC-I binding groove (26). In contrast, MHC-II presented peptides generally assume a much flatter conformation in the MHC-II binding groove (13, 14) (Figure 2D). The presence of a solvent exposed central bulge for MHC-I peptide presentation may represent a structurally advantageous feature for TCR binding, providing an anchor point that can guide the TCR into the correct binding orientation to engage its cognate ligand (Figure 3A). Conversely, the flat, and relatively featureless surface of pMHC-II confers no dominant structural feature for the TCR to “latch” onto, and may reduce the chance of a productive TCR/pMHC-II interaction occurring (explaining the slower on-rate and weaker affinity compared to TCR/pMHC-I interactions) (Figure 3B). This notion is consistent with our recent observation that a MHC-II restricted TCR underwent minimal conformational adjustments during binding compared to most MHC-I restricted TCRs (27). The immunological significance of these topological and biophysical distinctions between MHC-I and MHC-II is still unclear. However, the difference in binding affinity between MHC restricted TCRs may represent a biophysical characteristic that relates to cellular function.
MHC-II Antigen Processing Generates Variable Length Peptides
The “flat” surface of pMHC-II may contribute to the reduced affinity of MHC-II restricted TCRs. However, a striking difference in the peptides bound to MHC-II is the presence of non-bound PFRs, which may be available to interact with adjacent membrane proteins on the same or different cells. These PFRs can vary in length generating nested sets of peptides that are presented on the surface of APCs (28, 29). One consequence of having a longer PFR is an increased binding affinity of peptides to the MHC-II (30– 33), and therefore an increased probability of a meaningful interaction with a cognate T cell.
These variable PFRs are generated by proteolytic processes during the exogenous antigen processing pathway that has been reviewed in detail elsewhere (34, 35). Briefly, extracellular protein antigens are endocytosed by tissue resident APCs (Figure 4A). The pH of the endosome containing potential antigens progressively decreases, activating proteases which cleave captured proteins (Figure 4B). Newly synthesized MHC-II molecules reside in the endoplasmic reticulum (ER) in complex with a stabilizing chaperon, calnexin. To prevent premature peptide association with the MHC-II binding groove by ER derived proteins, the groove is “plugged” with a protein known as the MHC-II associated invariant chain (Ii) (36) (Figure 4C). Exocytic vesicles containing precursor Ii:MHC-II complexes then combine with endosomes containing exogenous peptide fragments forming the MHC-II compartment (Figure 4D). The acidic pH of the MHC-II compartment and presence of the chaperon, HLA-DM (37), allows peptide exchange between the class II-associated invariant-chain peptide (CLIP) and high affinity complementary peptides proteolysed in the endosomal compartment. Peptide selection, that presumably plays a strong role in determining the characteristics of PFRs, is also facilitated by HLA-DM in a process termed “peptide-editing” which ensures that only stable MHC-II peptide complexes are expressed and transported to the cell surface for potential TCR interactions (38, 39) (Figure 4E). Structural modeling of HLA-DM association with pMHC-II indicated that peptide editing was achieved through conformational changes around pocket 1 (P1) of the binding groove, a pocket crucial for the stability of the peptide-MHC-II complex (40). Such conformational changes induced by HLA-DM were thought to weaken the hydrogen bond network between the bound peptide and MHC-II molecule and facilitate peptide release (41). A recent co-complex structure of HLA-DM with HLA-DRα∗0101; β∗0101 (HLA-DR1) has confirmed this experimentally, revealing that HLA-DM binding induced a conformational change in the α-helix of the DRα chain in the peptide binding groove (42). This change enabled two HLA-DR1 residues, DRα phenylalanine 51 and DRβ phenylalanine 89, to bind to, and stabilize, the P1 binding pocket of the MHC-II binding groove, presumably blocking the association of weakly binding peptides. Thus, only the association of high affinity peptides with MHC-II results in displacement of these residues, enabling a revision in the conformation of MHC-II and the dissociation of HLA-DM (40, 42).
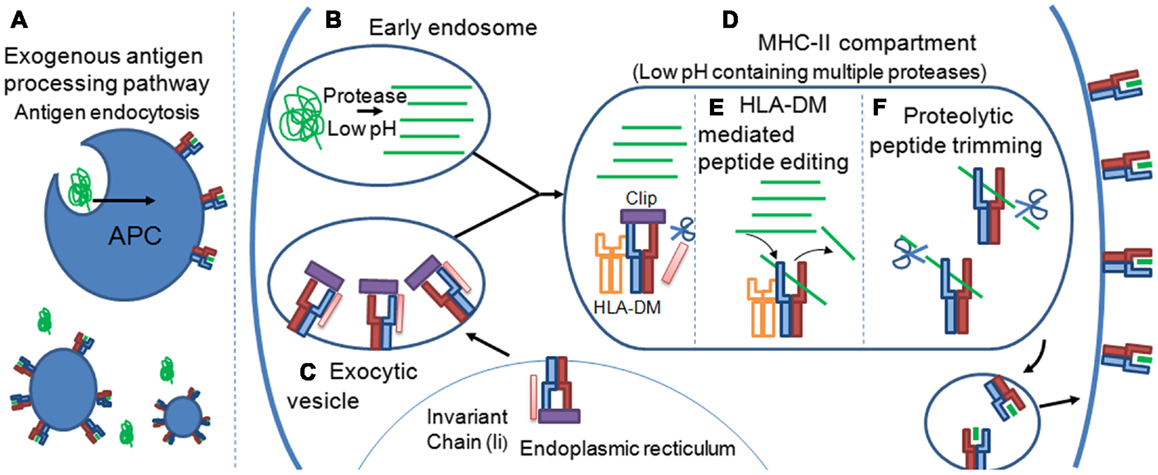
Figure 4. Peptide flanking regions are determined during the MHC-II antigen processing pathway. (A) Extracellular protein antigens are endocytosed by tissue resident APCs. (B) The pH of the endosome containing potential antigens progressively decreases, activating proteases which cleave captured proteins. (C) Newly synthesized MHC-II molecules reside in the endoplasmic reticulum (ER) in complex with the MHC-II associated invariant chain (Ii), which “plugs” the MHC-II binding groove, preventing ER derived peptides from premature peptide association. (D) Exocytic vesicles containing precursor Ii:MHC-II complexes then combine with endosomes containing exogenous peptide fragments forming the MHC-II compartment. Formation of the MHC-II compartment results in proteolytic cleavage of the Ii chain leaving a 24 amino acid remnant called the class II-associated invariant-chain peptide (CLIP) within the binding groove of the MHC-II molecule. The acidic pH of the MHC-II compartment and presence of the chaperon, HLA-DM, allows peptide exchange between CLIP and high affinity complementary peptides proteolysed in the endosomal compartment. (E) Peptide selection, that presumably plays a strong role in determining the characteristics of PFRs, is also facilitated by HLA-DM in a process termed “peptide-editing” which ensures that only stable MHC-II peptide complexes are expressed and transported to the cell surface for potential TCR interactions. (F) The final pMHC-II, loaded with exogenous peptide, can also be modified further in a process termed peptide trimming that may play a role in governing PFR length. pMHC-II molecules are then transported to the cell surface for interrogation by CD4+ T cells.
It is possible that the proteolytic events that occur before peptide-MHC-II loading govern the final pool of peptides available for selection during MHC-II peptide loading. However, it has also been suggested that the final pMHC-II, loaded with exogenous peptide, can be modified further in a process termed peptide trimming, whereby the length of the PFRs can be edited (Figure 4F) (43, 44). These processes demonstrate a remarkable degree of complexity and control during MHC-II peptide selection that is still not fully understood. The antigen processing by cellular proteases and the generation of pMHC-II may also be influenced by cell extrinsic factors such as inflammatory cytokines, e.g., IFNs (45), as well as cell intrinsic factors reflecting the type/subtype of APC (46, 47). The cellular machinery involved in antigen processing and presentation is different between cell types (48), hence the determinants resulting from protein digestion may vary depending on cell type or subtype (e.g., B cell versus macrophage; CD8+ versus CD8- dendritic cells etc); and context (e.g., inflamed versus non-inflamed tissues, anatomical location). It is conceivable that a range of determinants presented in a lymph node may differ from those presented at the primary site of infection in both a quantitative fashion, i.e., the number of pMHC-II complexes per cell, and qualitative fashion i.e., length and type of PFRs and hence offer a local control of CD4+ T cell responses accordingly.
Modulating CD4+ T Cell Responses via Altered Peptide Flanking Residues
There is convincing evidence that PFRs can modulate T cell function (49). A study of a HIV-I p24 (GAG) epitope, presented by HLA-DR1, revealed that antigen specific T cell activation was enhanced with longer flanking residues. Structural analyzes showed that the C-terminal flank could form a hairpin turn, raising the possibility that MHC-II PFRs may form more complex conformations that could directly impinge on TCR binding (50). Thus, the open ended nature of the MHC-II binding groove, that allows long peptides to extend beyond the binding region at both the N- and C-terminus (Figures 2 and 3), may play a direct role during T cell antigen recognition. In support of this notion, it has been demonstrated that removal of C-terminus PFRs from the immunodominant epitope in hen egg lysosome52–61 (HEL) significantly altered the immunogenicity of the epitope, reducing T cell sensitivity (9).
Our previous work, using sequence analysis of eluted peptide ligands from a range of allelic variants of MHC-II molecules, has identified allele-transcending enrichments in PFRs at the peptide C- and N-terminus (51, 52). These data show that a range of different modifications to PFRs could modulate specific CD4+ T cell responses including amino acids with biochemically distinct side chains (52, 53). The identification of these PFR amino acid enrichment patterns suggests that they play a role during CD4+ T cell activation and can modulate antigen recognition. Further studies using antigen specific CD4+ T cell clones demonstrated that PFR modifications could enhance CD4+ T cell activation (53). Although a wide range of different amino acid substitutions in PFRs could generate stronger CD4+ T cell responses, we observed that basic residues at the peptide C-terminus, or acidic residues in the N-terminus, were most commonly enriched and generated enhanced CD4+ T cell responses across different MHC-II alleles and different peptides. Studies focusing on the C-terminal PFRs, in which the basic amino acid, arginine was substituted into the C-terminal flank (at position 10 or 11) of known T cell epitopes from haemaggluttinin (HA) and myelin basic protein (MBP), demonstrated that these alterations led to a significant increase in CD4+ T cell responses (52, 53). Screening T cells which recognized this same MBP-derived epitope with a combinatorial library also revealed a preference for C-terminal basic residues (54).
However, in all of these examples, the mechanism for the effect of PFRs on T cell responsiveness had remained elusive. Two possibilities can be considered. Firstly, PFR modifications may alter the stability of pMHC-II molecules (a notion that has been experimentally observed (30– 33), altering their expression levels at the surface of APCs. However, we have demonstrated that, although the substitution of basic residues in the C-terminus increased T cell activation, they actually reduced peptide/MHC binding (52). Secondly, if the TCR can directly contact the PFRs, then modifications in the PFR could alter TCR binding affinity and subsequent T cell activation. In order to investigate the second possibility, we conducted biophysical experiments by surface plasmon resonance using cloned TCRs specific for an influenza epitope (HA305–320) presented by HLA-DR1 (53). The substitution of arginine into either position 10 (HA10R) or 11 (HA11R) of HA306–318 generated approximately twofold increase in TCR binding affinity (Figures 5A,B). Intriguingly, analysis of the TCR clonotypic repertoire of peptide-expanded influenza-specific CD4+ T cells from HLA-DR1+ donors in response to HA305-320 or arginine altered variants (HA10R and HA11R) demonstrated a marked alteration in TCR usage, with a striking focusing of the response when using the peptides which are known to increase TCR binding i.e., number of clonotypes for HA > HA10R > HA11R. The structure of HLA-DR1-HA306–318 in complex with the MHC-II restricted TCR, HA1.7, has been solved by X-ray crystallography (55). This structure demonstrated that the TCR could not directly contact the short side chains of either Alanine at P10, or Threonine at P11 in the universal HA306–318 epitope (Figure 5C). The closest proximity between the TCR and either P10 or P11 of the HA306–318 peptide was over 8Å, which was beyond the limits for atomic contacts. However, structural modeling of the substitution of arginine, which has a long acidic side chain, at either P10 or P11 indicated this gap could be closed allowing additional interactions to form between the peptide and the TCR (Figure 5D) (53). These potential new contacts could offer an explanation for the stronger binding affinity between the HA1.7 TCR and the P10 or P11 substituted DR1-HA306–318 epitope.
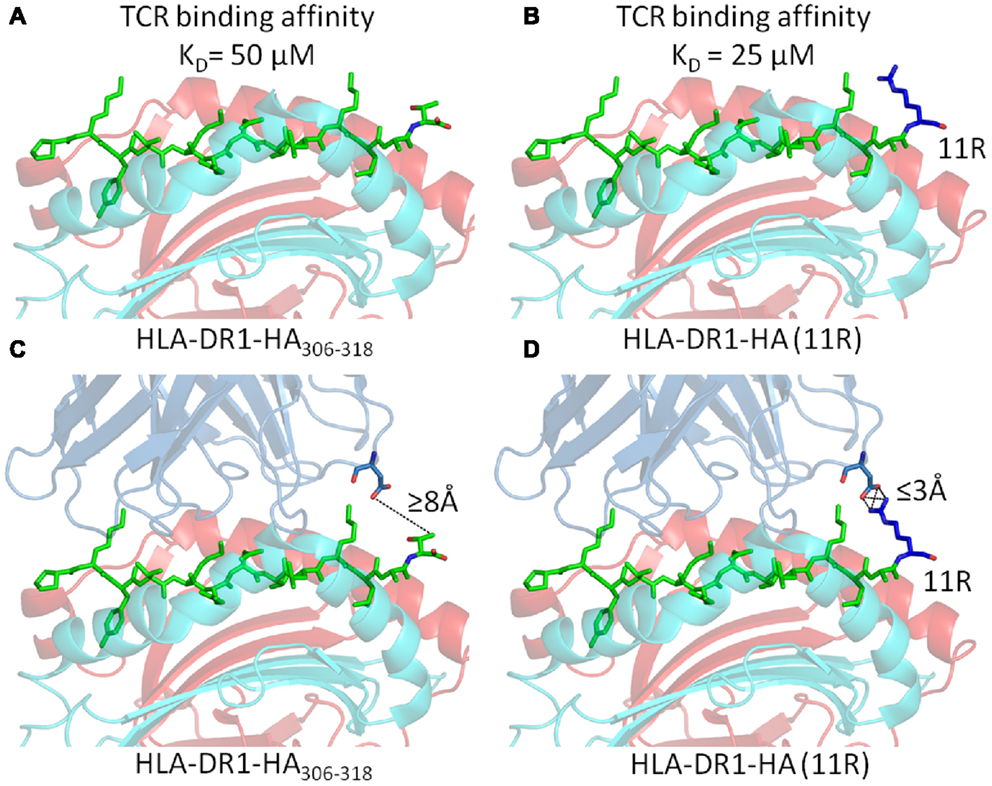
Figure 5. Substitution of Arginine substitutions in the C-terminal flanking region of the native Flu1 peptide increases binding affinity. (A,B) Substitution of arginine at position 11 (blue) in the HA305–320 epitope generates around a twofold increase in TCR binding affinity. (C,D) Cartoon representation of the interaction between the TCR and C-terminal PFR (modeled from PDB: 1FYT). (C) The TCR β-chain is beyond the limits for atomic contacts with HA305–320 P11 (dotted line). (D) Modeling shows that a new interaction, possibly a salt bridge, could be formed between the TCR β-chain and arginine (blue) substituted at position 11 of the HA305–320 peptide. This new interaction could explain the increase in affinity observed for cognate TCR binding to the HA305–320 peptide and HA11R.
Conclusion
The function of classical MHC molecules is presentation of peptide epitopes to the cell mediated arm of adaptive immune response. However, the subtle differences that exist between the two classical forms of MHC, with respect to antigen processing and structural architecture, significantly alters the nature of the peptide each class of MHC can present upon the cell surface. In particular, the closed binding groove of MHC-I forces bound peptides to bulge in the center, compared to the open binding groove of MHC-II that allow PFRs to form. In vitro experiments using a variety of antigens in mice and human systems, including HA, GAG, MBP, and HEL, have demonstrated that the PFRs of an epitope can profoundly affect CD4+ T cell function. Generation and selection of different PFRs might be governed according to the anatomical location, inflammatory milieu and particular types of APC involved during antigen processing. Thus, a key question that remains is whether particular changes in PFRs occur through a random, stochastic process, or whether changes are purposefully intentioned to control or alter the nature of a specific immune response. Irrespective of the answer to this question, our recent data revealed that experimental PFR modifications could enhance TCR/pMHC-II affinities closer to the range typically observed for TCR/pMHC-I interactions. This exciting observation suggests that augmentation of pMHC-II antigens through C-terminal PFR modifications might be a useful strategy to enhance MHC-II restricted TCR binding affinity and CD4+ T cell responsiveness, with attendant implications for vaccination and other immune system interventions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
David K. Cole is a Wellcome Trust Research Career Development Fellow (WT095767).
References
1. Katz DR. Antigen presentation, antigen-presenting cells and antigen processing. Curr Opin Immunol (1988) 1:213–9. doi:10.1016/0952-7915(88)90004-0
2. Kumar V, Mcnerney ME. A new self: MHC-class-I-independent natural-killer-cell self-tolerance. Nat Rev Immunol (2005) 5:363–74. doi:10.1038/nri1603
3. Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol (2006) 24:419–66. doi:10.1146/annurev.immunol.23.021704.115658
4. Garcia KC, Degano M, Speir JA, Wilson IA. Emerging principles for T cell receptor recognition of antigen in cellular immunity. Rev Immunogenet (1999) 1:75–90.
5. Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol (2006) 24:519–40. doi:10.1146/annurev.immunol.23.021704.115825
6. Van Der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol (2003) 21:659–84. doi:10.1146/annurev.immunol.21.120601.141036
7. Rammensee HG. Chemistry of peptides associated with MHC class I and class II molecules. Curr Opin Immunol (1995) 7:85–96. doi:10.1016/0952-7915(95)80033-6
8. Altman JD, Moss PA, Goulder PJ, Barouch DH, Mcheyzer-Williams MG, Bell JI, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science (1996) 274:94–6. doi:10.1126/science.274.5284.94
9. Carson RT, Vignali KM, Woodland DL, Vignali DA. T cell receptor recognition of MHC class II-bound peptide flanking residues enhances immunogenicity and results in altered TCR V region usage. Immunity (1997) 7:387–99. doi:10.1016/S1074-7613(00)80360-X
10. Rudolph MG, Luz JG, Wilson IA. Structural and thermodynamic correlates of T cell signaling. Annu Rev Biophys Biomol Struct (2002) 31:121–49. doi:10.1146/annurev.biophys.31.082901.134423
11. Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature (1993) 364:33–9. doi:10.1038/364033a0
12. Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature (1994) 368:215–21. doi:10.1038/368215a0
13. Hennecke J, Carfi A, Wiley DC. Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J (2000) 19:5611–24. doi:10.1093/emboj/19.21.5611
14. Hennecke J, Wiley DC. Structure of a complex of the human alpha/beta T cell receptor (TCR) HA1.7, influenza hemagglutinin peptide, and major histocompatibility complex class II molecule, HLA-DR4 (DRA*0101 and DRB1*0401): insight into TCR cross-restriction and alloreactivity. J Exp Med (2002) 195:571–81. doi:10.1084/jem.20011194
15. Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J (2005) 24:2968–79. doi:10.1038/sj.emboj.7600771
16. Deng L, Langley RJ, Brown PH, Xu G, Teng L, Wang Q, et al. Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor. Nat Immunol (2007) 8:398–408. doi:10.1038/ni1447
17. Yin Y, Li Y, Kerzic MC, Martin R, Mariuzza RA. Structure of a TCR with high affinity for self-antigen reveals basis for escape from negative selection. EMBO J (2011) 30:1137–48. doi:10.1038/emboj.2011.21
18. Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol (2005) 6:490–6. doi:10.1038/ni1187
19. Sethi DK, Schubert DA, Anders AK, Heroux A, Bonsor DA, Thomas CP, et al. A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med (2011) 208:91–102. doi:10.1084/jem.20100725
20. Cole DK, Pumphrey NJ, Boulter JM, Sami M, Bell JI, Gostick E, et al. Human TCR-binding affinity is governed by MHC class restriction. J Immunol (2007) 178:5727–34.
21. Bridgeman JS, Sewell AK, Miles JJ, Price DA, Cole DK. Structural and biophysical determinants of alphabeta T-cell antigen recognition. Immunology (2012) 135:9–18. doi:10.1111/j.1365-2567.2011.03515.x
22. Sebzda E, Mariathasan S, Ohteki T, Jones R, Bachmann MF, Ohashi PS. Selection of the T cell repertoire. Annu Rev Immunol (1999) 17:829–74. doi:10.1146/annurev.immunol.17.1.829
23. Jameson SC, Hogquist KA, Bevan MJ. Positive selection of thymocytes. Annu Rev Immunol (1995) 13:93–126. doi:10.1146/annurev.iy.13.040195.000521
24. Miles JJ, El-Hassen D, Borg NA, Silins SL, Tynan FE, Burrows JM, et al. CTL recognition of a bulged viral peptide involves biased TCR selection. J Immunol (2005) 175:3826–34.
25. Tynan FE, Borg NA, Miles JJ, Beddoe T, El-Hassen D, Silins SL, et al. High resolution structures of highly bulged viral epitopes bound to major histocompatibility complex class I. Implications for T-cell receptor engagement and T-cell immunodominance. J Biol Chem (2005) 280:23900–9. doi:10.1074/jbc.M503060200
26. Tynan FE, Reid HH, Kjer-Nielsen L, Miles JJ, Wilce MC, Kostenko L, et al. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat Immunol (2007) 8:268–76. doi:10.1038/ni1432
27. Holland CJ, Rizkallah PJ, Vollers S, Calvo-Calle JM, Madura F, Fuller A, et al. Minimal conformational plasticity enables TCR cross-reactivity to different MHC class II heterodimers. Sci Rep (2012) 2:629. doi:10.1038/srep00629
28. Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med (1993) 178:27–47. doi:10.1084/jem.178.1.27
29. Vignali DA, Urban RG, Chicz RM, Strominger JL. Minute quantities of a single immunodominant foreign epitope are presented as large nested sets by major histocompatibility complex class II molecules. Eur J Immunol (1993) 23:1602–7. doi:10.1002/eji.1830230731
30. Nelson CA, Petzold SJ, Unanue ER. Identification of two distinct properties of class II major histocompatibility complex-associated peptides. Proc Natl Acad Sci U S A (1993) 90:1227–31. doi:10.1073/pnas.90.4.1227
31. Nelson CA, Petzold SJ, Unanue ER. Peptides determine the lifespan of MHC class II molecules in the antigen-presenting cell. Nature (1994) 371:250–2. doi:10.1038/371250a0
32. Sant’angelo DB, Robinson E, Janeway CA Jr., Denzin LK. Recognition of core and flanking amino acids of MHC class II-bound peptides by the T cell receptor. Eur J Immunol (2002) 32:2510–20. doi:10.1002/1521-4141(200209)32:9<2510::AID-IMMU2510>3.0.CO;2-Q
33. O’brien C, Flower DR, Feighery C. Peptide length significantly influences in vitro affinity for MHC class II molecules. Immunome Res (2008) 4:6. doi:10.1186/1745-7580-4-6
34. Mohan JF, Unanue ER. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat Rev Immunol (2012) 12:721–8. doi:10.1038/nri3294
35. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol (2013) 31:443–73. doi:10.1146/annurev-immunol-032712-095910
36. Romagnoli P, Layet C, Yewdell J, Bakke O, Germain RN. Relationship between invariant chain expression and major histocompatibility complex class II transport into early and late endocytic compartments. J Exp Med (1993) 177:583–96. doi:10.1084/jem.177.3.583
37. Weber DA, Evavold BD, Jensen PE. Enhanced dissociation of HLA-DR-bound peptides in the presence of HLA-DM. Science (1996) 274:618–20. doi:10.1126/science.274.5287.618
38. Busch R, Doebele RC, Patil NS, Pashine A, Mellins ED. Accessory molecules for MHC class II peptide loading. Curr Opin Immunol (2000) 12:99–106. doi:10.1016/S0952-7915(99)00057-6
39. Anders AK, Call MJ, Schulze MS, Fowler KD, Schubert DA, Seth NP, et al. HLA-DM captures partially empty HLA-DR molecules for catalyzed removal of peptide. Nat Immunol (2011) 12:54–61. doi:10.1038/ni.1967
40. Painter CA, Negroni MP, Kellersberger KA, Zavala-Ruiz Z, Evans JE, Stern LJ. Conformational lability in the class II MHC 310 helix and adjacent extended strand dictate HLA-DM susceptibility and peptide exchange. Proc Natl Acad Sci U S A (2011) 108:19329–34. doi:10.1073/pnas.1108074108
41. Narayan K, Chou CL, Kim A, Hartman IZ, Dalai S, Khoruzhenko S, et al. HLA-DM targets the hydrogen bond between the histidine at position beta81 and peptide to dissociate HLA-DR-peptide complexes. Nat Immunol (2007) 8:92–100. doi:10.1038/ni1414
42. Pos W, Sethi DK, Call MJ, Schulze MS, Anders AK, Pyrdol J, et al. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell (2012) 151:1557–68. doi:10.1016/j.cell.2012.11.025
43. Manoury B, Hewitt EW, Morrice N, Dando PM, Barrett AJ, Watts C. An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation. Nature (1998) 396:695–9. doi:10.1038/25379
44. Lippolis JD, White FM, Marto JA, Luckey CJ, Bullock TN, Shabanowitz J, et al. Analysis of MHC class II antigen processing by quantitation of peptides that constitute nested sets. J Immunol (2002) 169:5089–97.
45. O’donnell PW, Haque A, Klemsz MJ, Kaplan MH, Blum JS. Cutting edge: induction of the antigen-processing enzyme IFN-gamma-inducible lysosomal thiol reductase in melanoma cells Is STAT1-dependent but CIITA-independent. J Immunol (2004) 173:731–5.
46. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (2007) 315:107–11. doi:10.1126/science.1136080
47. Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang DE, Trilling M, et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol (2012) 13:51–7. doi:10.1038/ni.2169
48. Sercarz EE, Lehmann PV, Ametani A, Benichou G, Miller A, Moudgil K. Dominance and crypticity of T cell antigenic determinants. Annu Rev Immunol (1993) 11:729–66. doi:10.1146/annurev.iy.11.040193.003501
49. Larsen SL, Pedersen LO, Buus S, Stryhn A. T cell responses affected by aminopeptidase N (CD13)-mediated trimming of major histocompatibility complex class II-bound peptides. J Exp Med (1996) 184:183–9. doi:10.1084/jem.184.1.183
50. Zavala-Ruiz Z, Strug I, Walker BD, Norris PJ, Stern LJ. A hairpin turn in a class II MHC-bound peptide orients residues outside the binding groove for T cell recognition. Proc Natl Acad Sci U S A (2004) 101:13279–84. doi:10.1073/pnas.0403371101
51. Godkin AJ, Davenport MP, Willis A, Jewell DP, Hill AV. Use of complete eluted peptide sequence data from HLA-DR and -DQ molecules to predict T cell epitopes, and the influence of the nonbinding terminal regions of ligands in epitope selection. J Immunol (1998) 161:850–8.
52. Godkin AJ, Smith KJ, Willis A, Tejada-Simon MV, Zhang J, Elliott T, et al. Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide-MHC interactions. J Immunol (2001) 166:6720–7.
53. Cole DK, Gallagher K, Lemercier B, Holland CJ, Junaid S, Hindley JP, et al. Modification of the carboxy-terminal flanking region of a universal influenza epitope alters CD4(+) T-cell repertoire selection. Nat Commun (2012) 3:665. doi:10.1038/ncomms1665
54. Hemmer B, Fleckenstein BT, Vergelli M, Jung G, Mcfarland H, Martin R, et al. Identification of high potency microbial and self ligands for a human autoreactive class II-restricted T cell clone. J Exp Med (1997) 185:1651–9. doi:10.1084/jem.185.9.1651
55. Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science (1999) 286:1913–21. doi:10.1126/science.286.5446.1913
56. Smith KJ, Reid SW, Stuart DI, Mcmichael AJ, Jones EY, Bell JI. An altered position of the alpha 2 helix of MHC class I is revealed by the crystal structure of HLA-B*3501. Immunity (1996) 4:203–13. doi:10.1016/S1074-7613(00)80429-X
57. Madden DR, Garboczi DN, Wiley DC. The antigenic identity of peptide-MHC complexes: a comparison of the conformations of five viral peptides presented by HLA-A2. Cell (1993) 75:693–708. doi:10.1016/0092-8674(93)90490-H
58. Borbulevych OY, Insaidoo FK, Baxter TK, Powell DJ Jr., Johnson LA, Restifo NP, et al. Structures of MART-126/27-35 Peptide/HLA-A2 complexes reveal a remarkable disconnect between antigen structural homology and T cell recognition. J Mol Biol (2007) 372:1123–36. doi:10.1016/j.jmb.2007.07.025
59. Miles JJ, Borg NA, Brennan RM, Tynan FE, Kjer-Nielsen L, Silins SL, et al. TCR alpha genes direct MHC restriction in the potent human T cell response to a class I-bound viral epitope. J Immunol (2006) 177:6804–14.
60. Probst-Kepper M, Hecht HJ, Herrmann H, Janke V, Ocklenburg F, Klempnauer J, et al. Conformational restraints and flexibility of 14-meric peptides in complex with HLA-B*3501. J Immunol (2004) 173:5610–6.
61. Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC. X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity (1997) 7:473–81. doi:10.1016/S1074-7613(00)80369-6
62. Smith KJ, Pyrdol J, Gauthier L, Wiley DC, Wucherpfennig KW. Crystal structure of HLA-DR2 (DRA*0101, DRB1*1501) complexed with a peptide from human myelin basic protein. J Exp Med (1998) 188:1511–20. doi:10.1084/jem.188.8.1511
63. Li Y, Depontieu FR, Sidney J, Salay TM, Engelhard VH, Hunt DF, et al. Structural basis for the presentation of tumor-associated MHC class II-restricted phosphopeptides to CD4+ T cells. J Mol Biol (2010) 399:596–603. doi:10.1016/j.jmb
Keywords: modified peptide, peptide flanking residue, peptide-major histocompatibility complex class II, T cell receptor, T cell repertoire, vaccine, crystal structure, MHC processing
Citation: Holland CJ, Cole DK and Godkin A (2013) Re-directing CD4+ T cell responses with the flanking residues of MHC class II-bound peptides: the core is not enough. Front. Immunol. 4:172. doi: 10.3389/fimmu.2013.00172
Received: 24 April 2013; Accepted: 14 June 2013;
Published online: 01 July 2013.
Edited by:
Bruno Laugel, Cardiff University, UKReviewed by:
Balbino Alarcon, Consejo Superior de Investigaciones Cientificas, SpainDaniel M Altmann, Imperial College, UK
Copyright: © 2013 Holland, Cole and Godkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: David K. Cole and Andrew Godkin, Cardiff University School of Medicine, The Henry Wellcome Building, Cardiff, CF14 4XN, UK e-mail: coledk@cf.ac.uk, godkinaj@cardiff.ac.uk
†David K. Cole and Andrew Godkin have contributed equally to this work.