
- 1Molecular Immunology Unit, Department of Experimental Medicine, Fondazione IRCCS “Istituto Nazionale Tumori,”, Milan, Italy
- 2Cellular and Molecular Immunology Unit, Dipartimento di Medicina Interna e Specialità Mediche, “Sapienza” Università di Roma, Rome, Italy
The expansion of regulatory T cells (Treg) is a common event characterizing the vast majority of human and experimental tumors and it is now well established that Treg represent a crucial hurdle for a successful immunotherapy. Treg are currently classified, according to their origin, into thymus-derived Treg (tTreg) or peripherally induced Treg (pTreg) cells. Controversy exists over the prevalent mechanism accounting for Treg expansion in tumors, since both tTreg proliferation and de novo pTreg differentiation may occur. Since tTreg and pTreg are believed as preferentially self-specific or broadly directed to non-self and tumor-specific antigens, respectively, the balance between tTreg and pTreg accumulation may impact on the repertoire of antigen specificities recognized by Treg in tumors. The prevalence of tTreg or pTreg may also affect the outcome of immunotherapies based on tumor-antigen vaccination or Treg depletion. The mechanisms dictating pTreg induction or tTreg expansion/stability are a matter of intense investigation and the most recent results depict a complex landscape. Indeed, selected Treg subsets may display peculiar characteristics in terms of stability, suppressive function, and cytokine production, depending on microenvironmental signals. These features may be differentially distributed between pTreg and tTreg and may significantly affect the possibility of manipulating Treg in cancer therapy. We propose here that innovative immunotherapeutic strategies may be directed at diverting unstable/uncommitted Treg, mostly enriched in the pTreg pool, into tumor-specific effectors, while preserving systemic immune tolerance ensured by self-specific tTreg.
Treg Suppress Pro-Tumoral Inflammation or Anti-Tumor Response
Introduction
Tumor onset is a very complex process, in which cells of both innate and adaptive immune system play crucial roles in inhibiting or fostering tumor development. The awareness that the immune system could act as an extrinsic tumor suppressor or as a tumor-sculpting player resulted in the cancer immunoediting theory, which described the interaction between tumor and host as consisting of three different phases: elimination, equilibrium, and escape (1). During the last phase of this process, transformed cells acquire the ability to subvert the control exerted by immune cells thus originating the clinically evident pathology. The escape is due to different mechanisms, including reduced immunogenicity (low expression level of MHC class I and loss of antigen expression), acquired resistance to the cytotoxic functions of immune cells, and accumulation in the tumor microenvironment of immunosuppressive mediators, like regulatory T cells (Treg) (1). The first marker to be identified as distinguishing Treg from the other CD4+ T lymphocytes was CD25 (2) and depletion of CD25-positive cells unveiled anti-tumor immunity in experimental models (3). Few years later, the transcription factor Forkhead Box P3 (Foxp3) was indicated as the master regulator of Treg (4, 5). In support of the crucial roles played by Foxp3 in Treg fate determination and immune homeostasis, Foxp3 mutations have been recognized as responsible for human Immune Dysfunction, Polyendocrinopathy, Enteropathy, X-linked (IPEX) syndrome (6, 7), and for the phenotype of scurfy mutant mice (8), both characterized by fatal autoimmune lymphoproliferation linked to severe defects in Treg development/functions. However, very recent data have demonstrated that the complete development of the Treg lineage requires the concomitant, Foxp3-independent, establishment of a Treg-specific pattern of DNA hypomethylation (9).
According to recently proposed recommendations (10), Treg are classified into two principal subsets based on their developmental origin: thymus-derived Treg (tTreg) develop in the thymus, while peripherally induced Treg (pTreg) develop in vivo in the periphery from conventional T cells (Tconv), through a process called “conversion” (11). Treg can also be induced in vitro (and are called iTreg) under TGF-β and/or retinoic acid exposure, but their complete commitment into fully differentiated Treg is still under debate (12). In physiological conditions, the pool of Treg, encompassing both tTreg and pTreg, which represents about the 5–10% of the circulating CD4+ T lymphocytes, assures peripheral self-tolerance and prevents exacerbated immune responses (7, 8). A huge amount of data now demonstrates that tumor onset and progression perturb Treg homeostasis and lead to increased Treg/Tconv and Treg/CD8 ratios both in peripheral blood and in the tumor microenvironment (13). The accumulation of Treg at tumor sites may be due to the concomitant or the preferential occurrence of distinct events, such as the recruitment of Treg from periphery, the proliferation of pre-existing Treg in the tumor microenvironment, and the de novo conversion of tumor-infiltrating CD4+ lymphocytes (TIL) into pTreg (14, 15). Despite controversy existing over the dominant suppression mechanism, and despite the incomplete understanding of the biological meaning of Treg accumulation in cancer, it is well accepted that Treg are crucial players in tumor biology and that the modulation of their function is an indispensable requisite for the development of successful anti-tumor immune-therapies.
Mechanisms of Treg Suppression in Tumors
It was recently demonstrated that Treg infiltrating different tissues have a specific gene signature (16), thus Treg may use peculiar suppression mechanisms in response to microenvironmental stimuli. This specialization may represent the basis for designing immune interventions targeting specific Treg functions in a given tissue while sparing systemic immune homeostasis. Even though a tumor Treg-specific gene signature has not been delineated yet, some mechanisms have been described to contribute to Treg suppression in tumors, which can be clustered in three main types: surface molecules, enzymatic activities, and cytokines (Figure 1).
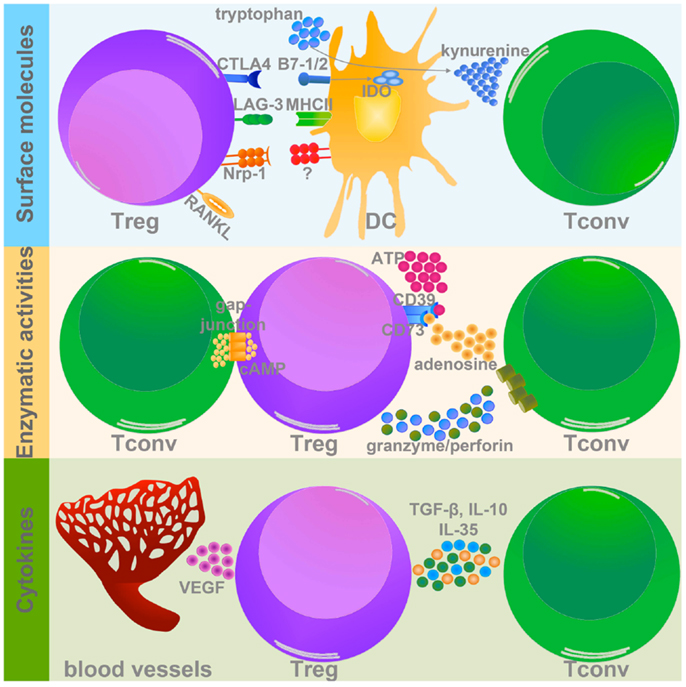
Figure 1. Treg suppressive mechanisms in tumor microenvironment. Treg use different strategies to inhibit target cells within the tumor mass. Three types of Treg-related molecules can mediate these suppressive mechanisms: (1) surface molecules (upper panel); (2) enzymes (middle panel), and (3) cytokines (lower panel). (1) Among the surface molecules expressed by Treg, CTLA-4, LAG-3, Nrp-1, and RANKL have a well-demonstrated role in promoting tumor progression, mainly modulating DC activation and function. In particular CTLA-4 and LAG-3, binding to CD80/CD86 (B7-1/2), and MHCII respectively, significantly impair DC capacity to activate Tconv. In addition CTLA-4 promotes IDO expression and the production of the pro-apoptotic metabolite kynurenine. Nrp-1 instead stabilizes Treg-DC contact, allowing Treg to adequately suppress DC. Although the course of action of RANKL is not yet well defined, its expression is associated to tumor metastatization. (2) The two ecto-enzymes CD39 and CD73 generate from ATP pericellular adenosine, which is endowed with strong tolerogenic effects. Also cAMP, similarly to adenosine, interferes with Tconv activation and survival. Granzyme and perforin induce the apoptosis of target cells by cytolysis. (3) Treg secrete several immune-modulatory cytokines, which could directly modulate Tconv functions (TGF-β, IL-10, and IL-35), or indirectly promote the establishment of pro-tumorigenic microenvironment (VEGF).
Both human and mouse Treg constitutively express on their surface cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), a co-inhibitory member of the CD28/B7 family, endowed with strong immune-regulatory properties (17). The relevance of CTLA-4 in regulating Treg function was demonstrated in several settings, including autoimmune diseases and different tumor types (18). A comparative gene expression profile between Treg and Tconv revealed that Treg specifically up-modulate lymphocyte activation gene 3 (LAG-3) (19), a homolog of CD4, that binds to MHC class II on antigen-presenting cells (APCs). LAG-3 is upregulated in tumor-infiltrating Treg and experiments with anti-LAG-3 mAb demonstrated that functional LAG-3 is required for maximal Treg suppressive function (20, 21). Treg-DC interaction is also mediated by the transmembrane protein neuropilin-1 (Nrp-1), expressed on Treg membrane, which ensures the stability of Treg-DC interaction and allows Treg to efficaciously suppress DC (22). A study conducted on patients with early-stage cervical tumor showed that Treg infiltrating the tumor-draining lymph node of patients with metastasis have a higher expression of several immune-modulatory molecules, including Nrp-1 (23). The receptor activator of nuclear factor-kB ligand (RANKL), member of the tumor necrosis factor family, was found to be highly expressed on Treg isolated from tumor-bearing hosts, and substantial evidence indicates that RANKL expressed by Treg is involved in the onset of metastasis in both mammary (24) and prostate tumors (25).
Regulatory T cell suppression may be mediated by enzymatic activities, such as CD39/CD73 (26, 27), granzyme B, and perforin (28). CD39 and CD73 are two ecto-enzymes that dephosphorylate extracellular ATP and generate pericellular adenosine, which in turn exerts a strong pro-tumorigenic role modulating the function of numerous tumor-infiltrating immune cells. CD73-deficient mice develop a stronger anti-tumor immune response compared to CD73-sufficient mice (29). Treg are also able to control the proliferation and function of different immune cells via the Perforin pathway (30). In mouse models of melanoma, lymphoma, and acute myeloid leukemia it has been demonstrated that tumor-infiltrating Treg, but not naïve Treg, secrete high amounts of both perforin and granzyme B, which in turn induce NK and CD8+ T cell death (28).
Immunosuppressive cytokines, like TGF-β and IL-10, are critical players in Treg biology, being involved in both their differentiation and suppressive potential, especially in tumors. Treg-derived TGF-β was found relevant in suppression of anti-tumor T cell response in both mouse (31) and human (32, 33) tumors. IL-10 is a well-known immunosuppressive mediator, and several pieces of evidence highlight the relevance of Treg-derived IL-10 in controlling inflammation at the mucosal interfaces such as gut and lung (34, 35). Despite these data, little information is available about the roles of Treg-derived IL-10 in tumor microenvironments. We have recently demonstrated that tumor-associated Treg secrete high amounts of IL-10, which in turn impairs DC migration to the draining lymph nodes and the mounting of a specific anti-tumor immune response. This phenotype could be reverted by stimulating the receptor OX40 on the surface of intratumoral Treg. Indeed, OX40-triggered Treg showed reduced secretion of IL-10 as a consequence of the down-modulation of the interferon regulatory factor 1 (IRF1), a transcription factor active in the IL-10 promoter (36). Another cytokine recently described as critical for the full Treg suppressive function is IL-35, formed by Epstein–Barr-virus-induced gene 3 (Ebi3) and IL-12α (p35) (37). In two different mouse transplantable tumor models (melanoma and colon carcinoma), it was observed that Treg secrete abundant IL-35, thus promoting the differentiation of induced IL-35-secreting Treg (37). It is well known that tumor growth is associated with a consistent process of new angiogenesis in response to hypoxia. A circuit involving tumor hypoxia, Treg recruitment, and angiogenesis has been recently discovered (38). In the hypoxic tumor microenvironment, the chemokine axis CCL28–CCR10 plays a determinant role in the recruitment of Treg, which secrete huge amounts of vascular endothelial growth factor (VEGF), further stimulating the new angiogenesis process and the establishment of a tolerogenic microenvironment (38).
Double Effects of Treg on Prognosis
Since their discovery, Treg were considered one of the main obstacles for tumor clearance, according to their tolerogenic properties and their accumulation along tumor progression. In this view, several anti-tumor immune-therapies focus on Treg depletion or inhibition, in order to “contrasuppress” Treg inhibitory functions and to block the conversion of non-regulatory cells (non-Treg) into regulatory cells (15). Reduced Tconv/Treg ratio was observed in patients with pancreatic tumor (39, 40), breast cancer (39), ovarian cancer (41), Hodgkin lymphoma (42), and melanoma (43). Increased Treg frequency is generally associated to advanced tumor stage and poor prognosis, as recently demonstrated in a study on ovarian cancer (44). In the ascites of patients with advanced tumor, the percentage of Treg was increased compared to the ascites of patients with early-stage tumor. Same results were obtained with the mouse WF-3 transplantable ovarian tumor model, showing augmented percentages of Treg in both spleen and tumor-associated cells of mice with advanced tumors, compared to naïve or mice with early lesions. In addition, the treatment of tumor-bearing mice with the Treg-depleting mAb PC61 (αCD25) reduced tumor growth and prolonged mice survival (44). Similarly, it has been demonstrated that Treg number inversely correlated with the therapy outcome in melanoma patients treated with non-myeloablative chemotherapy, in combination or not with total body irradiation, followed by adoptive T cell transfer (45). Responder patients had a lower frequency of Treg in peripheral blood compared to non-responder patients (45). A study conducted on patients affected by invasive ductal carcinoma showed a positive correlation among Treg, Th17, and tumor aggressiveness. These data imply that Treg and Th17 cells may concomitantly expand during tumor progression, with Treg mainly suppressing protective effector T cell proliferation while sparing Th17 proangiogenic activities, fostering cooperatively tumor progression, and the metastatic process (46).
Nevertheless, recent data, in particular tumor systems, point out that Treg may exert a protective role for the host (13, 47, 48). The connection between tumor and inflammation is a well-assessed process (49), but now it is clearly emerging that the type of tumor-associated inflammation imprints the behavior of Treg, becoming detrimental or beneficial for the host. Type-1 inflammation, characterized by high concentration of IFN-γ and IL-12 and fully active cytotoxic cells, represents efficient anti-tumor immunity (49). In this setting, the inhibitory properties of Treg may promote tumor escape and aggressiveness (47). On the contrary, immune responses dominated by cytokines like TNF-α, IL-1β, IL-6, IL-23, and IL-17 act as pro-tumoral mediators (47). In this environment Treg may suppress a pro-tumoral inflammatory status, thus playing a protective role for the host (47).
These unexpected anti-tumoral Treg properties were observed in patients with colorectal cancer (CRC) (50–52). In these patients, with different tumor stages, a better prognosis and an increased overall survival were associated with higher infiltration of FOXP3+ T cells compared to patients with a poor tumor outcome. These data suggested the hypothesis that FOXP3+ T cells could be considered as an independent prognostic factor for CRC. Following a strong activation, both conventional CD4+ (53) and CD8+ (54) lymphocytes up-regulate Foxp3 expression in colonic tissue. These observations indicated that the CRC prognosis positively correlated with non-regulatory FOXP3+ cells rather than to Treg. However in vitro suppression assays demonstrated that FOXP3+ cells, isolated from CRC tissues, were endowed with suppressive functions, confirming their nature as regulatory cells (55). In a recent study conducted on 65 patients with different stages of CRC, FOXP3 expression was systematically evaluated in both tumor-infiltrating lymphocytes and neoplastic cells, and was correlated to tumor progression and clinical-pathological features (56). From this study the notion emerged that high FOXP3 expression in tumor cells correlated with poor tumor outcome, compared to tumors poorly expressing FOXP3; on the contrary, no correlation was observed between CRC prognosis and FOXP3 expression by T cells (56).
A protective role of Treg was also found in head and neck squamous cell carcinoma (HNSCC) (57). Univariate and multivariate analysis demonstrated that the locoregional control of the tumor was positively associated with CD4+FOXP3+ regulatory cell infiltration (57). However, also for this type of neoplasia, there are some discordant data regarding the role of Treg in tumor progression. Indeed, another study showed that Treg frequency and suppressive function were higher in the peripheral blood of tumor-bearing patients than in healthy volunteers (58).
The discrepancies observed in these studies may be due to the number of patients included, different strategies of analysis and non-homogeneity of tumor samples (stage, metastasis, etiology). Certainly, to properly define the role of Treg in tumor outcome, the new studies should take into account the tumor stage and the related inflammatory features, depending on the anatomical localization. In general, those tumors arising from chronic inflammation, almost at their initial stage, can benefit from the suppressive properties of Treg. In fact, during the inflammatory process, Treg highly accumulate in the site of inflammation such to prevent exacerbated immune responses and tissue damage, which are the prelude to neoplastic transformation. On the contrary, in the presence of an established tumor, Treg may reduce anti-tumor immunity thus favoring tumor escape. A more specific definition of Treg contribution in tumor development and progression is desirable for the design of new and more effective immunotherapies, allowing the discrimination among tumors that will benefit or not from Treg depletion/inhibition.
Evidence for pTreg or tTreg Accumulation in Tumors
Distinguishing Features of pTreg and tTreg
Many efforts have been recently addressed to the identification of phenotypic, molecular, and functional features distinguishing tTreg and pTreg, besides their site of origin (11). Some markers have been proposed to distinguish pTreg and tTreg, even though with some limitations: the initial enthusiasm for the suggestion of Helios as able to identify tTreg (59) has been soon moldered by the observation of Helios expression in pTreg (60); the recent finding of the Nrp-1 as a marker of tTreg (61, 62) has application limited to murine cells, being not expressed on human Treg (63). Several attempts have been conducted to identify genetic (64–66) and/or epigenetic signatures distinguishing pTreg and tTreg. The Treg-specific demethylated region (TSDR) is involved in the stable commitment of the Treg lineage, and controversy still remains on whether iTreg or pTreg can efficiently demethylate this region and become fully committed Treg (66–69). Despite this growing amount of information, distinguishing the relative contribution of pTreg and tTreg to immune suppression in physiological and pathological conditions remains hard. However, some pieces of evidence have accumulated in the last years that speak in favor of tTreg or pTreg prevalence or concomitance in tumors.
Evidence for tTreg Accumulation in Cancer
One of the first attempts to distinguish between pTreg conversion and tTreg expansion in cancer was pursued by Bui and colleagues who adoptively transferred CD4+CD25+ cells, mixed at 1:10 ratio with CD25-depleted Thy1.1-congenic splenocytes, into immunodeficient mice bearing a progressive sarcoma (70). The analysis performed 10 days after tumor injection showed that the vast majority (around 80%) of tumor-infiltrating CD4+CD25+ cells derived from expansion/recruitment of the transferred Treg, rather than from conversion of non-Treg. This and other reports, appeared in the “pre-Foxp3” era, were biased by the idea that CD25 was the most stringent Treg marker and that CD25-depleted cells represented a suitable precursor population to efficiently detect de novo generation of pTreg. However, subsequent studies have demonstrated that the CD25+ subset of Foxp3− Tconv is enriched in pTreg precursors (69, 71), thus the extent of pTreg differentiation from CD25-depleted cells represents probably an underestimation of the actual contribution of pTreg induction in the tumor context. Other authors have shown that tTreg may dominate pTreg not only quantitatively but also qualitatively, in terms of suppressive function: indeed, IDO+ plasmacytoid dendritic cells, derived from mouse tumor-draining lymph nodes, were capable to induce Foxp3+ pTreg at very high levels but were unable to activate the suppressive function of these cells to an extent comparable to tTreg (72). Many studies have clearly shown Treg proliferation (in terms of de novo DNA synthesis and/or cell division) in tumor-bearing mice or cancer patients, thus indirectly supporting the idea that expansion of pre-existing tTreg might prevail over pTreg differentiation in building the tumor-associated Treg pool. For instance, Treg have been shown to incorporate high levels of BrdU in tumor-draining lymph nodes and at cancer sites in several experimental models (73, 74). A study conducted in patients with multiple myeloma showed that the TREC content in naive cells was significantly lower in Treg (identified as CD4+CD25high cells) than CD4+CD25− or CD25low cells, suggesting that the Treg pool mainly derived from peripheral expansion rather than recent thymic emigration (75). However, the observation of high Treg proliferation at tumor sites cannot be considered as an unequivocal proof of tTreg prevalence over pTreg, since both subsets could be endowed with the same proliferative potential in vivo. Indeed, several pieces of evidence indicate that conversion and proliferation may represent uncoupled and independent events (see pTreg Development and tTreg Expansion as Independent Processes).
Evidence for pTreg Induction in Cancer
Some studies support the idea that pTreg conversion actually occurs in tumor-bearing hosts at higher efficiency than in physiological conditions, even if controversy still exists on whether pTreg may prevail numerically over tTreg at the tumor site. We have in the past demonstrated that thymectomized and CD25-depleted mice, subsequently transplanted with carcinoma cells, showed a significantly higher Treg recovery in tumor-draining than in contralateral lymph nodes, suggesting that in tumor-bearing mice the Treg pool might be replenished mostly by newly derived pTreg than by proliferation of residual Treg. To prove this possibility, CD25-depleted CD4-purified T cells were transferred into immunocompetent, Thy1.1-congenic, CT26 tumor-bearing mice. In this setting, we could show that the transferred cells acquired CD25 and Foxp3 at significantly higher levels in draining lymph nodes, compared to contralateral lymph nodes of tumor-bearing mice, or to the lymph nodes of tumor-free mice (76). This result clearly showed that tumor progression actively promoted the conversion of non-regulatory precursors into pTreg. Some tumor-derived molecular signals were found to be involved in tumor-associated conversion. For instance, in different mouse models, tumor cells have been shown to induce in vitro Treg conversion through TGF-β, and TGF-β neutralization abrogated Treg accumulation at the tumor site (77). Human leukemic cells converted in vitro non-Treg into Treg through the tumor cell-restricted IDO activity, and IDO blockade prevented pTreg induction in vivo in a leukemia mouse model (78). A confirmation of extensive pTreg infiltration in murine tumors has been recently obtained thanks to the recent discovery of Nrp-1 as a tTreg-restricted marker (61, 62). The analysis of Nrp-1 expression has indeed revealed that Nrp-1-negative bona fide pTreg cells were significantly enriched at tumor site compared to spleen, ranging around 40–90% of total tumor-infiltrating Treg depending on the tumor type (61). These Nrp-1-negative cells also presented a gene signature (Helioslow, SWAP-70low, and Dapl1high) compatible with the pTreg identity (61). Unfortunately, human Treg do not express Nrp-1 (63), thus this marker cannot be used to estimate the relative contribution of tTreg or pTreg in human cancers.
Developmental and Functional Relations between pTreg and tTreg in Cancer
pTreg Development and tTreg Expansion as Independent Processes
Many attempts have been made to understand whether tTreg accumulation and pTreg development are mutually exclusive or rather cooperative in establishing immune suppression. The evidence that tTreg may “educate” Tconv to convert into Treg through the secretion of cytokines, such as TGF-β and IL-10 (79), may support the latter possibility. This event would generate a cascade of suppressive function transmitted from Treg to bystander cells, establishing a loop of immunosuppression, reminiscent of a phenomenon called as “infectious tolerance” (80). Zhou and coworkers have addressed this issue in the tumor setting, and have demonstrated that tumor-antigen-specific pTreg could indeed arise from Treg-depleted cells (adoptively transferred in mice carrying the cognate antigen-expressing tumor), but that the extent of pTreg induction was not affected by the concomitant presence of tTreg, either exogenous (adoptively co-transferred) or endogenous (pre-existing in the host) (81, 82). This result indicated that tTreg and pTreg accumulate in tumors in a reciprocally independent fashion and that “infectious tolerance” may play minor roles in shaping the tumor-associated Treg pool.
A comprehensive scenario of Treg accumulation in tumors should take into account, beside de novo conversion, the active proliferation of not only tTreg but also pTreg. Proliferation plays opposite roles in the differentiation of Tconv into pTreg versus the expansion of already differentiated pTreg. Regarding the former aspect, we have demonstrated that Tconv proliferation was not required for their conversion into pTreg, since CD25+Foxp3+ cells could develop in tumor-bearing mice from CD25-depleted cells treated with an anti-proliferative agent (76). A seminal study by Kretschmer and colleagues showed that Tconv proliferation was not only dispensable but also detrimental to conversion: indeed, low levels of T cell proliferation, in conditions of suboptimal antigen presentation, lack of co-stimulation, and IL-2 paucity, favored TGF-β-mediated pTreg induction, thus suggesting that an inverse relationship might exist between Tconv proliferation and conversion into pTreg (83). However, once developed, pTreg promptly proliferated in response to experimental antigens (83) and, more importantly, in response to tumor antigens (81, 82). Experiments performed in CNS1-mutated mice, which are genetically unable to generate pTreg, have shown that pTreg and tTreg may occupy distinct “niches”: indeed, the efficiency of pTreg differentiation from Tconv was higher when those Tconv were co-transferred, in lymphopenic recipients, with a CNS1-deficient (non-containing pTreg) compared to a CNS1-sufficient (containing pTreg) counterpart, suggesting that not only the tTreg pool, but also the pTreg niche, may be controlled by autonomous homeostatic mechanisms (84).
Division of Labor between tTreg and pTreg in Cancer
Both tTreg and pTreg have been generally recognized as immune suppressive cells in a variety of in vivo and in vitro experimental settings (12). However, whether the two subsets are endowed with peculiar activities remains unclear and is a matter of intense investigation.
Gene expression profiling revealed that the pTreg and tTreg signatures were mostly overlapping but also presented some differentially expressed genes, encoding for proteins involved in Treg suppressive function, suggesting that pTreg may preferentially exploit different molecules and related mechanisms to exert suppression (64–66). The Nrp-1 itself is not only a marker discriminating murine tTreg from pTreg, but also plays a role in Treg suppression: since this molecule prolongs Treg interactions with immature dendritic cells, tTreg may benefit from this pathway in preferentially modulating dendritic cell and cognate T cell activation (22). Many data suggest that pTreg may be specialized suppressors of immune responses at interfaces with external environments, such as airways, gut, and maternal-fetal interface (64, 84–87). Of note, a peculiar Treg suppressive molecule, IL-10, plays crucial roles at environmental interfaces, therefore pTreg may perform their specialized activity through IL-10 secretion (34, 88). IL-10 is critically involved in the establishment of tumor-associated immune suppression, and we have clearly demonstrated IL-10 production by around 40% of tumor-infiltrating Treg in murine transplanted tumors (36). It would be interesting to understand whether the fraction of IL-10-producing Treg is enriched in pTreg, rather than tTreg, in tumors. One study has directly addressed the issue of induced Treg functional specialization in tumors, by generating in vitro tumor-specific iTreg and co-culturing these cells, or tTreg as control, with NK cells: these authors found that iTreg and tTreg equally suppressed IL-2-induced NK activation, but only iTreg were endowed with the surprising ability not to suppress, but to enhance, NK cytotoxicity induced by tumor target cell contact (89). This observation may speak in favor of differential roles played by tTreg and pTreg in cancer, with the former more involved in preventing target cell-independent, and possibly self-directed, unwanted immune responses, and the latter concurrently enhancing tumor-specific immunity.
This division of labor may result in the progressive shaping of the immune response toward an effective anti-tumor immunity with minimal side effects. Such dichotomy is also reminiscent of the double role played by Treg in different cancers, according to the hypothesis that high Treg frequency is associated to poor or good prognosis in non-inflammatory or inflammatory cancer onset, respectively (13, 47). In the former case, i.e., non-inflammatory cancers in which protective type-1 responses are suppressed by high Treg infiltration, Treg may mainly recognize tumor-associated self-antigens, and mostly include tTreg; conversely, in the case of inflammatory cancers, related to chronic low-dose type-17 cytokines, which are typical of mucosal tissues, high numbers of pTreg may suppress pro-tumoral inflammation through IL-10, relevantly produced by pTreg at those sites. We are tempting to speculate that tTreg may dominate in suppressing anti-tumor type-1 responses, while pTreg may prevail in shaping pro-tumor type-2 and type-17 inflammatory responses. Notably, the prototypical example of an inflammation-related tumor in which Treg accumulation associates to good prognosis is CRC (50), developing in the gastrointestinal mucosa, in which immune tolerance is under the control of pTreg (84).
Antigen Specificity of tTreg Versus pTreg in Cancer
Antigen recognition may play a crucial role in dictating whether tTreg or pTreg will prevail in the tumor context. Controversy still exists on the antigen specificity of these two populations. On the one side, tTreg are generally believed to recognize self-antigens encountered during thymic selection (90). On the other side, pTreg, deriving from Tconv, are thought to display the same TCR repertoire of Tconv and thus to mainly recognize foreign antigens. Indeed, only a small overlap exists between TCR repertoires of pTreg and tTreg (66), and pTreg are believed to recognize non-self-antigens such as commensal microbiota, allergens, and fetal alloantigens (84, 87).
Tumor cells can express a variety of antigens that can be broadly classified into: (i) self-antigens physiologically expressed as in the tissue of origin, (ii) self-antigens aberrantly expressed, in terms of expression level, developmental stage, or histotype (called tumor-associated antigens or TAAs), and (iii) neo-antigens uniquely expressed by tumor cells, mostly derived from oncogenic mutations (named tumor-specific antigens or TSAs). Based on the above considerations, self-antigens and TAA should be recognized by tTreg, while TSA would induce and activate pTreg. However, a complex picture arises from studies analyzing the TCR specificity of tumor-associated Treg.
Treg can recognize TAA and TSA in tumors
In different tumor models, TCR-transgenic Treg have been shown to promptly proliferate in response to the cognate antigen specifically expressed by tumor cells, suggesting that Treg can undergo tumor-antigen-driven activation and expansion (74, 81, 82, 91). Antigen presentation in the tumor context may favor Treg expansion: in a mouse model of spontaneous prostate cancer, an efficient Treg induction/expansion occurred only when TCR-transgenic, antigen-specific CD4 T cells encountered the cognate antigen expressed in the context of prostate cancer cells, rather than non-transformed cells or viral vector-infected cells (91). In this model, TAA-specific Treg were recognized as pTreg induced in vivo in a TGF-β-independent fashion.
This evidence of TAA-responding Treg has been confirmed in human tumors. CD4 clones derived from cancer patients resulted to be regulatory cells and to recognize peptides derived from TAAs, such as LAGE1 (92), ARTC1 (93), TRAG-3, NY-ESO-1 (94–96), Melan-A (97), survivin, TRP1, and gp100 (94) in melanoma patients, and WT-1 in leukemia patients (98). By using MHCII/peptide tetramer technology, other authors failed to detect Treg specific for NY-ESO-1 in the peripheral blood of ovarian cancer patients (99). Bonertz et al. developed an in vitro system to screen the suppressive function of Treg in response to single peptides and, with this approach, could detect Treg specific for several TAA in the peripheral blood of colon carcinoma patients but not in healthy donors; notably, Treg depletion in vitro unveiled TAA-specific Tconv responses (100).
The possibility that tumor-associated Treg may recognize not only TAA but also TSA is demonstrated by the observation that Treg specific for exogenous viral antigens, acting as TSA, may arise in virus-related cancers. Treg clones specific for human papilloma virus (HPV), and suppressing the cognate antigen-directed T cell response, have been obtained from tumor-draining lymph nodes and tumor biopsies of cervical cancer patients (101). Treg clones specific for antigens of the Epstein–Barr virus (EBV), associated to several hematological and solid malignancies, can be generated from the peripheral blood of healthy donors (102).
Treg can recognize self-antigens in tumors
Several data in mouse models confirm that Treg responding to self-antigens can play a role in suppressing the anti-tumor responses. Immunization with serologically defined auto-antigens was found to enhance tumor growth in different mouse models, and this event was dependent on the expansion of Treg responding to those self-antigens (103). This study confirmed that self-antigens-specific Treg could suppress anti-tumor immunity, but did not explore the Treg response to self-antigens expressed by tumor cells themselves during tumor progression. This issue has been instead addressed in an experimental model in which a foreign antigen, artificially expressed in transgenic mice under tissue-specific promoter, was seen (peripherally and/or thymically) by the immune system as a self-antigen and elicited the generation of a pool of memory Treg specific for that antigen (74). If those mice were injected with the cognate antigen-bearing tumor, the memory Treg pool specific for that self-antigen was hugely expanded in tumors and tumor-draining lymph nodes, confirming that self-specific Treg can respond to self-antigens expressed by tumor cells (74). A seminal paper has recently reported the immunoscope analysis of Treg infiltrating spontaneous prostate tumors in a mouse transgenic model, and described the clonal enrichment of a single Treg specificity that was directed not to a unique TSA but to a self-antigen expressed also by normal prostate cells (104). The development of Treg specific for peripheral tissue-restricted self-antigens occurred in the thymus under the control of the Aire molecule, which allows the expression of peripheral antigens in thymic epithelial cells (104). These findings clearly demonstrate that Treg can recognize self-antigens in cancer and suggest that maintaining self-antigen expression may help transformed cells to overcome the immune surveillance through self-specific Treg expansion.
Repertoire analysis as an estimation of pTreg/tTreg balance
The direct comparison between the repertoires of tumor-associated Treg and Tconv may help understanding the processes underlying Treg enrichment in cancer. Some authors have reported that the analysis of TCR diversity (performed with the immunoscope technology) showed that Treg infiltrating murine transplanted tumors displayed a biased TCR repertoire toward “public” CDR3 sequences (i.e., shared by different mice), suggesting Treg intra-tumor clonal expansion driven by the recognition of dominant antigens (105). Also tumor-infiltrating activated Tconv showed a biased TCR repertoire, but it was distinct from the Treg spectrum, and the minimal overlap between the two subsets was mainly confined to “private” specificities (105). By using a similar approach, others have reported distinct and not overlapping TCR repertoires of Treg and Tconv infiltrating prostate tumors in a genetically engineered mouse model of spontaneous prostate carcinogenesis (104). Lack of overlap between Tconv and Treg repertoires was also found in tumors and tumor-draining lymph nodes in a mouse model of chemical carcinogenesis (106). Overall, the lack of overlap between Treg and Tconv has been interpreted in many cases as the result of negligible pTreg conversion at the tumor site; however, pTreg and tTreg may share more specificities than expected, since tTreg-associated antigens may preferentially drive Tconv fate decision toward the conversion into pTreg rather than toward the conventional activation; moreover, already established pTreg may then undergo intra-tumor clonal expansion together with tTreg in response to the same antigens. Therefore, the overall overlap between Tconv and Treg specificities may not accurately estimate the extent of pTreg induction in tumors. Indeed, in one study performed in advanced melanoma patients, TAA-specific TCRs, expressed by tumor Treg clones, could be detected in both Treg and Tconv populations, demonstrating that TAA-specific Treg may be comprised of pTreg derived from the conversion of Tconv (95).
Indirect data support the notion that TAA-specific Treg may contain pTreg. TAA-specific Treg clones, obtained from patients with advanced melanoma, suppressed in vitro the cognate antigen-specific T cell response, but produced high levels of Th1 and/or Th2 cytokines (95), and showed low FOXP3 expression and TSDR demethylation, indicating that these cells may represent an incompletely uncommitted Treg population, which more likely belongs to the pTreg than to the tTreg pool (95).
A recent study has directly evaluated the consequences of pTreg and tTreg antigen specificities in tumor-bearing hosts. Indeed, Schreiber et al. have shown that, if purified polyclonal tTreg and Tconv, differentially labeled with green or red fluorescence, were co-transferred in CD4-null mice, the tTreg progeny exceeded the newly Tconv-derived pTreg population in tumor-draining lymph nodes as well as in the spleen; conversely, when transgenic, tumor-antigen-specific, tTreg and Tconv were injected, tTreg and pTreg reached comparable frequencies in tumor-draining lymph nodes (107). These results suggest that tTreg and pTreg are mostly specific for self- or tumor-antigens respectively, and that the balance between pTreg and tTreg may be fine-tuned by the relative prevalence of TSAs versus self-antigens expressed by tumor cells.
Heterogeneity and Plasticity of tTreg and pTreg
Treg Heterogeneity in Cancer: Relations with the pTreg/tTreg Dichotomy
During the latest years, it has become increasingly clear that Treg, meant as Foxp3-positive cells, are not a homogeneous lineage, but rather represent a mixture of subpopulations. Indeed, beside the tTreg/pTreg distinction based on their developmental origin, diverse Treg subsets can be identified endowed with peculiar features in terms of suppression, proliferation, and stability, even though not properly classifiable as developmentally distinct lineages (Figure 2). Tumor microenvironmental signals may differentially affect these subsets, thus shaping Treg heterogeneity to the advantage of tumor progression.
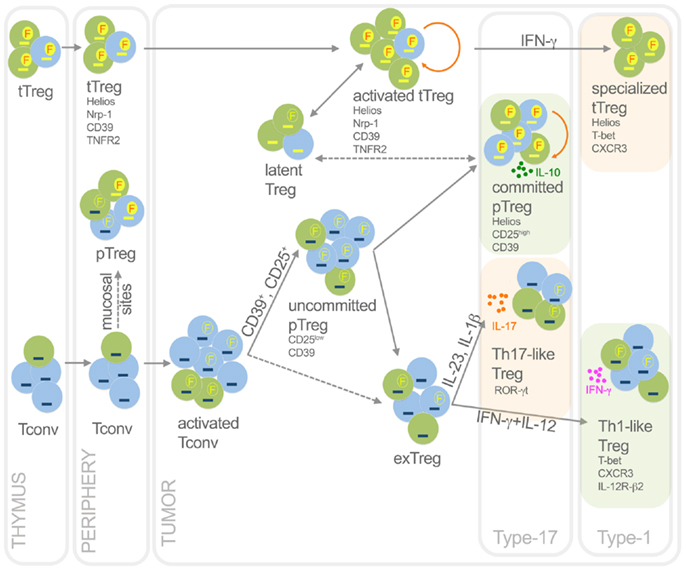
Figure 2. Functional dynamics of tTreg and pTreg in cancer. This picture summarizes development, heterogeneity, plasticity, antigen specificity, and function of pTreg and tTreg in cancer. Activated Treg, which are epigenetically committed and mostly self- and TAA-specific, can transiently lose Foxp3 without methylating TSDR thus becoming latent Treg; in some conditions, they can acquire T-bet expression thus becoming specialized suppressors, detrimental to the anti-tumor type-1 response. Activated Tconv, mostly foreign (TSA) antigen-specific, can promiscuously express Foxp3 without demethylating TSDR. However, a fraction (CD25+, or CD39+) of activated Tconv can convert into pTreg, progressively moving from an uncommitted to a committed stage. Through IL-10, committed pTreg can suppress pro-tumoral inflammatory and type-17 responses, thus exerting beneficial roles for the host in some cancer types. In some contexts, uncommitted pTreg (and possibly activated Tconv) can move back to exTreg stage, acquiring the ability to produce inflammatory cytokines. Therefore, in some tumors such as colon cancer, Th17-like Treg may foster type-17 inflammation thus supporting tumor growth; in other tumor contexts, Th1-like Treg can favor type-1 responses that rather block tumor growth. Green, cells specific for self-antigens and TAA; light blue, cells specific for foreign antigens including TSA. Yellow dash, demethylated TSDR; blue dash, methylated TSDR. Red “F” in yellow circles, stable Foxp3; yellow “F” in empty circles, unstable Foxp3. Dashed arrows, unclear events. Orange rounded arrows, proliferation in the tTreg or the pTreg homeostatic niche. Light green frames, conditions in which Treg are beneficial to the host; light orange frames, conditions in which Treg are detrimental to the host.
Treg stability and epigenetic commitment in cancer
Foxp3 inherent stability, rather than Foxp3 expression in a given moment and tissue, is intimately linked to an actual commitment to the Treg lineage and therefore to the maintenance of immune suppression. Pioneer studies have demonstrated that Foxp3 stability is strictly related to an epigenetic imprinting of CpG demethylation in the TSDR region of the Foxp3 locus (67, 86, 108). TSDR demethylation was then recognized as the mechanism featuring the distinction between committed (demethylated) and uncommitted (methylated) Treg, irrespective of Foxp3 expression (9). Controversy exists on whether pTreg show complete or partial TSDR demethylation and can then be considered as committed Treg. Many studies show that iTreg have a partially or completely methylated TSDR (9, 67–69), while pTreg have been described as TSDR-demethylated (68), TSDR-methylated (66), or as a mixed population of stable and unstable cells, characterized by CD25 high or low expression respectively (69).
Few data exist on the extent of TSDR demethylation in tumor-associated Treg. The frequency of TSDR-demethylated cells is higher in peripheral and intratumoral leukocytes of lung, colon, prostate, or breast cancer patients, in relation to a higher Treg frequency as determined by flow cytometry or immunohistochemistry (109). Of note, the extent of TSDR demethylation in CRC patients was only slightly higher than in healthy volunteers, in contrast to the significantly increased Treg frequency in these samples shown by previous studies (110, 111). This discrepancy may be explained with the peculiar nature of this inflammation-related and mucosal tissue-located cancer, in which the inflammatory response may specifically involve pTreg, possibly containing more uncommitted (TSDR-methylated) cells than tTreg.
The evaluation of TSDR demethylation has been used as a reliable analytical tool for the estimation of committed Treg in some tumor conditions and therapies. An increased frequency of epigenetically committed (TSDR-demethylated) Treg has been determined in tumor-infiltrating cells of ovarian, colorectal, and bronchial cancers compared to non-tumoral tissue counterparts (112). TSDR demethylation was decreased in the peripheral blood of metastatic renal carcinoma patients receiving tumor vaccination (113), and increased in patients treated with dendritic cell vaccination and cytokine therapy (114).
Treg functional dynamics in cancer
The idea of Foxp3 as the master transcription factor of Treg lineage has been challenged by the observation that some Treg features are Foxp3-independent, and that Foxp3 plays Treg-unrelated functions (115). This is especially true for human FOXP3-positive cells, since human activated Tconv can transiently express this transcription factor that acts as an intrinsic T cell regulator (116). The concomitant analysis of CD45RA and FOXP3 in human Treg in both physiological and pathological contexts allowed delineating a classification into three subsets: CD45RA+FOXP3low resting Treg (rTreg), CD45RA−FOXP3low non-Treg, and CD45RA−FOXP3high (CD45RO+) activated Treg (aTreg), endowed with different potentials of proliferation, suppression, stability, and plasticity (117). Whether each subset mainly contains tTreg or pTreg is unclear. While rTreg were recognized as CD31+ recent thymic emigrants, thus belonging to the tTreg pool, aTreg can be considered as a mixed population of activated tTreg (derived from rTreg) and pTreg (derived from non-Treg or Tconv). The CD45RA−FOXP3low non-Treg subset may represent a mixture of activated Tconv (promiscuously and unstably expressing FOXP3), latent Treg (transiently downregulating FOXP3), and recently converted pTreg (117).
The three human Treg subsets can be differentially expanded in distinct pathologies. In conditions characterized by exacerbated immune responses, such as autoimmune diseases, rTreg and non-Treg are expanded; conversely, in diseases associated to immune unresponsiveness, such as sarcoidosis, the aTreg subset is instead enriched in the peripheral blood (117). The tumor context, which conceivably belongs to the latter category, should be characterized by aTreg expansion. In line with this hypothesis, CD45RO+FOXP3high aTreg were found significantly expanded in the peripheral blood, and much more at the tumor site, in patients with malignant melanoma (118). Also the non-Treg and the rTreg fractions were increased, but only in the peripheral blood, in cancer patients compared to healthy controls, and both subsets positively correlated with tumor progression (118). The non-Treg pool produced some IFN-γ and its frequency returned to normal levels after tumor removal, thus probably representing aberrantly activated Tconv, or Treg with attenuated FOXP3 activity (118). A much deeper knowledge on Treg dynamics in cancer is needed to better understand the role played by each specialized component in suppressing anti-tumor type-1, or pro-tumor inflammatory, responses.
Treg subsets specified by functional/developmental markers
Several surface or intracellular markers have been suggested to identify Treg subsets endowed with peculiar abilities other than suppressive functions. A portion of human Treg with an effector/memory-like phenotype (26, 119, 120) expresses CD39, which has been proposed as a target to enrich human suppressive Treg (119). CD39 was found overrepresented in peripheral and tumor-infiltrating Treg from HNSCC, and was further increased in patients with advanced-stage disease or after radiochemotherapy (120, 121). CD39 is also expressed by a Tconv subset, which produces lower levels of pro-inflammatory cytokines than the bulk Tconv population, and is more prone to convert, at least in vitro, into Treg (120). Both CD39+ Treg and CD39+ Tconv were enriched in peripheral blood, and further increased at tumor site, in HNSCC patients, and a positive correlation existed between frequencies of these two populations (120). Therefore, these data suggest that tumor-associated CD39+ Tconv may represent a reservoir of CD39+ Treg precursors. As a consequence, it could be suggested that CD39+ Treg may include both tTreg and pTreg, and that both Treg subsets can exploit the CD39-mediated suppressive mechanisms of ATP degradation and adenosine generation.
Not only the functional arms of suppression, but also the activation requirements may differ in tTreg and pTreg: for instance, TNFR2 expression is needed to activate tTreg but not iTreg suppressive ability in experimental colitis (122). Of note, TNFR2-positive Treg have been found enriched in murine tumors, in association with a higher suppressive ability, ex vivo, in the standard suppression assay (123). In a mouse model of metastatic melanoma, TNF-α caused enhanced tumor progression through the TNFR2-mediated Treg expansion at the site of metastasis (124). These data suggest that TNFR2 expression may label tumor-infiltrating Treg of thymic origin, and that TNF-α at the tumors site may preferentially expand and activate tTreg. Supporting the idea that tTreg may represent more stable cells, TNFR2 was found to be involved in the maintenance of Foxp3 stability in mouse models of inflammation (125). Also in human peripheral blood, CD25 and TNFR2 co-expression identifies cells highly expressing FOXP3, showing an effector/memory phenotype and strong suppression, ex vivo (126). The TNF-α/TNFR2 pathway may amplify Treg activation also through the induction of a NF-kB-driven transcriptional program enriched for other members of TNF superfamily, such as 4-1BB, FAS, and OX40 (127).
The early idea that Helios could differentiate tTreg from pTreg (59, 128) prompted the use of this marker in delineating tTreg accumulation in cancer. The vast majority of tumor-infiltrating Treg were found to express Helios in a mouse model of glioblastoma (129), in glioblastoma multiforme patients (129), and renal cell carcinoma patients (130). However, the value of Helios as univocal marker of tTreg has been questioned by other studies that showed Helios also expressed in pTreg (60, 131), and in association to Treg suppression (128, 131) and commitment. Indeed, Helios−FOXP3+ cells freshly isolated from healthy donors or autoimmune disease patients showed decreased TSDR demethylation compared to Helios+FOXP3+ (132, 133), and also displayed a higher plasticity in terms of cytokine production (133). In a murine transplanted tumor model, tumor-infiltrating Treg were enriched in Helios+ cells, representing the subset with the highest proliferative potential (as shown by Ki67 staining) (131). In summary, the well-recognized enrichment of Helios+ Treg in several human and mouse tumors may be attributed, rather than to preferential attraction and expansion of tTreg, to the tumor-driven local activation and/or commitment of both tTreg and pTreg.
Specialization and Plasticity of tTreg/pTreg in Cancer
It is now well established that Treg (or better, their specific subsets) adapt their molecular programs to optimize their in vivo suppressive function in distinct inflammatory milieus, which may be alternatively dominated by Th1, Th2, Th17, or TFH responses. Strikingly, these Treg specialized programs are orchestrated by the same transcription factors that drive the polarization of the targeted T-helper subset: therefore, T-bet, IRF4, Stat3, and Bcl6 expression are respectively and selectively required for the Treg specialized suppression of Th1 (134, 135), Th2 (136), Th17 (137), and TFH (138, 139) responses. Indeed, by acquiring master T-helper genes, Treg may gain the expression of chemokine receptors driving the recruitment of specialized Treg into inflamed tissues. However, in some contexts, Treg (or, again, some Treg subsets) can express not only T-helper-related transcription factors and migratory molecules, but also cytokines such as IFN-γ or IL-17, thus turning from specialized suppressors into so-called Th1-like or Th17-like Treg that may rather contribute to inflammation (140). Some data, mostly from mouse experimental models, suggest that such Treg plasticity is not a lineage reprograming of committed Treg, which appear instead quite stable; rather, Th1-like or Th17-like Treg may derive from uncommitted cells expanded in inflammatory conditions (69, 141). However, other studies have shown that in both mouse and human pathologies Treg can produce relevant amounts of type-1 and type-17 cytokines even though preserving Foxp3 expression (142–146).
Th17-like Treg in cancer
Regulatory T cells may shift to a Th17-like phenotype in inflamed microenvironments dominated by type-17 cytokines, thus favoring, rather than suppressing, pro-tumoral mechanisms of chronic inflammation. According to this idea, human Treg have been found to spontaneously secrete IL-17 in the intestine of patients carrying inflammatory bowel disease (145, 147) and colon carcinoma (147). In epithelial ovarian cancer, a malignancy associated to chronic inflammation, Tconv were found to secrete high levels of IL-17 (and other cytokines) when cultured ex vivo with IL-2; under similar conditions, tumor-infiltrating Treg were prone to FOXP3 downregulation, attenuation of suppressive function, and prompt IL-17 production (148). In human lung adenocarcinoma, FOXP3 message amounts correlated with Th17-related transcripts enriched at the tumor site, where IL-17 antagonized the development of the anti-tumor, T-bet-dependent, Th1 response (149). Myeloid antigen-presenting cells and cytokines such as IL-2, TGF-β, IL-1, IL-23, and IL-6 may initiate Treg polarization into Th17-like cells in these tumor contexts (147–149). In a mouse model of hereditary colon polyposis, as well as in human colon cancer, the Th17-like Treg co-expressed the Th17-related transcription factor ROR-γt, and fostered tumor progression, also through the promotion of mast cell local expansion (150, 151). This study clearly demonstrated that microenvironmental signals could direct Treg plasticity toward pro-inflammatory and pro-tumoral activities.
One group has demonstrated that Th17-like Treg can also arise in experimental tumors as an outcome of vaccination strategies (152). In this study, vaccination with antigen plus TLR-9 ligand induced Treg reprograming into polyfunctional T-helper-like cells, producing a wide array of cytokines including IL-2, TNF-α, and IL-17, and expressing cell-surface CD40L, thus providing efficient T cell help for tumor-antigen cross-presentation and development of anti-tumor response (152). The IDO immunosuppressive enzymatic activity was responsible for preventing this anti-tumor Treg polarization, which was instead enhanced using an IDO blocker (152).
Little data exist on the precursors of Th17-like Treg in cancer. In the peripheral blood of healthy subjects, the CD45RA−FOXP3low non-Treg subset was found enriched in Th17-related transcripts and in cells actively secreting IL-17, even at higher levels than naïve or memory Tconv, a data suggesting that this population contains Th17 or Th17-like precursors (117). It would be interesting to understand whether the Th17 potential resides, within the non-Treg gate, in activated Tconv, in latent Treg, and/or in recently induced pTreg, possibly co-expressing FOXP3 and RORγt and thus pre-committed to the Th17 lineage.
Th1-like Treg in cancer
Pioneer studies from Koch and colleagues demonstrated that, following exposure to IFN-γ in Th1-dominated microenvironments, a subset of Treg can up-regulate the Th1-related transcription factor T-bet, which drives Treg expansion, migration (CXCR3-mediated), and function specifically during type-1 inflammation (134). Further experiments have shed light on the developmental requirements and the alternative fates of murine T-bet+ Treg: following IFN-γ stimulation, Treg could gain T-bet expression but failed to fully polarize into IFN-γ-producing Th1-like Treg, due to an impaired Treg susceptibility to IL-12. Indeed, IL-12 receptor β2, which is inducible in Tconv in an IFN-γ- and T-bet-dependent fashion, is epigenetically inaccessible in Treg (135). Only long-lasting IFN-γ preconditioning could unlock IL-12 responsiveness, thus allowing the complete Treg polarization into Th1-like cells (135). Presumably, in contexts characterized by chronic IFN-γ and IL-12 abundance, such as autoimmune, inflammatory, and viral diseases, Treg will be oriented to a full reprograming into Th1-like cells. Supporting this idea, IFN-γ-producing Treg have been reported in mouse models of graft-versus-host disease (153), viral (154) or parasite (142) infection, in human multiple sclerosis (144), and diabetes (146, 155). In one of these systems, IFN-γ-producing Treg were recognized to be specific for a foreign (viral) antigen (154). Whether such Th1-like Treg can be yet considered as classical regulatory cells is still debated. One study has shown that in vitro polarized Th1-like Treg were less suppressive than conventional Treg in the standard in vitro suppression assay, and that suppression could be partially rescued with concomitant IL-10 and IFN-γ neutralization (144). Another study has proven that IFN-γ-producing human iTreg were equally functional as natural Treg in suppressing both proliferation and cytokine production of responder T cells (156). In a mouse model of graft-versus-host disease, IFN-γ produced by stable (TSDR-demethylated) Treg was shown to be even required for Treg protective effect (153), suggesting that IFN-γ-releasing Treg can display in vivo unexpected functions depending on the context.
Conversely, it could be envisaged that, in the tumor context, the low levels of IFN-γ derived from Tconv, NK, and CD8 cells, and the paucity of IL-12 production by tumor-associated APCs, may concur to induce a pool of Treg expressing T-bet but not secreting IFN-γ, thus specialized in suppressing anti-tumor type-1 immunity. In line with this possibility, TAA-specific Tconv, but not TAA-specific Treg, produced IFN-γ in patients with epithelial ovarian cancer (99). In both healthy subjects (117) and malignant melanoma patients (118), IFN-γ-producing cells were enriched within the circulating CD45RA−FOXP3low (CD45RO+) non-Treg subset, mostly including activated Tconv and/or uncommitted Treg. Conversely, in ovarian cancer, tumor-infiltrating Treg were enriched in CXCR3+ cells, highly expressing T-bet but poorly producing IFN-γ, and strongly suppressing Th1 response ex vivo (157). Tumor-associated CXCR3+ Treg were mostly Helios-positive, and T-bet+ Treg could be generated in vitro by culturing CD45RA+CCR7+ rTreg (mostly containing tTreg) under Th1-polarizing conditions (157), suggesting their derivation from committed tTreg. This finding was in accordance to Koch et al. who showed that T-bet+ Treg derived from T-bet− Treg, rather than from activated Tconv (134). These data support the idea that tTreg, rather than pTreg, may contain the precursors for Th1-specialized suppressors, thus playing critical roles in suppressing protective responses in tumors whose high Treg frequency correlates with poor prognosis (13).
Some therapeutic interventions can force tumor-associated Treg toward a fully differentiated Th1-like phenotype. For instance, circulating Treg from melanoma patients showed significantly higher IFN-γ secretion following a protocol of tumor peptide vaccination plus IL-2 and cyclophosphamide, in line with enhanced serum IL-12 (158). On the whole, these data suggest that, especially in the human system, the transition from T-bet+ Treg, specialized Th1 suppressors, into T-bet+ IFN-γ+ Treg, Th1-like plastic cells, may not only depend on the availability and the responsiveness to exogenous stimuli, but may differentially occur in distinct Treg precursors: on the one side, tTreg, enriched in committed and self-specific cells, may be forced to arrest to the specialization (T-bet+) endpoint; on the other side, pTreg, containing less committed and foreign antigens-specific cells, may be more prone to the complete reprograming into pro-inflammatory (T-bet+ IFN-γ+) cells. Future studies will elucidate the mechanisms by which different growing tumors may favor the expansion of pro-tumoral specialized Th1 suppressors or the induction of Th1-like plastic Treg.
Implications for Cancer Immunotherapy
The initial enthusiasm on the use of therapeutic cancer vaccines has been soon disappointed by the observation of a low response rate in many trials (159). After the discovery of Treg as potent immune suppressive cells hampering the establishment of anti-tumor immunity, it soon became clear that anti-tumor vaccination might fail to elicit an effective immune response and to achieve successful tumor eradication, because of the immune suppressive barrier created by Treg. In addition, since Treg may recognize TAA and TSA at higher frequency than Tconv, tumor-antigen-based vaccines may expand/induce Treg rather than effector cells, thus inhibiting rather than boosting the anti-tumor response. Indeed, Zhou et al. first demonstrated that TCR-transgenic CD4 T cells specific for a TAA, adoptively transferred into mice bearing TAA-expressing tumor cells, proliferated extensively after administration of a therapeutic tumor vaccine (in the form of a recombinant vaccinia virus encoding the antigen), but tumor-antigen-experienced cells were mostly regulatory cells, ex vivo suppressive, and anergic to subsequent stimulation (81).
In cancer patients receiving tumor-antigen vaccination, the expansion of antigen-specific Treg has been documented. Circulating NY-ESO-1-specific Treg spontaneously develop in late-stage melanoma patients and are expanded following immunization with NY-ESO-1 protein supplemented with adjuvants (96). Therapeutic vaccination with an HPV synthetic long peptide vaccine, administered to patients with HPV-positive cervical carcinoma, induced both CD8 and CD4 T cell immunity, but also enhanced the HPV-specific Treg pool (160). The pool of vaccine-specific Treg may derive not only from the expansion of pre-existing tumor-antigen-specific clones, but also from de novo generation of vaccine-specific pTreg. This is suggested by results obtained vaccinating melanoma patients with an HLA-DP4-restricted MAGE-A3 peptide: in this setting, a subset of vaccine-specific Treg becomes detectable only after vaccination (161). Vaccine-elicited Treg showed some degree of heterogeneity: out of five CD25+ regulatory clones isolated from vaccinated patients, four expressed high FOXP3 mRNA levels, produced TGF-β, and showed demethylated TSDR; one clone expressed less FOXP3, had methylated TSDR and produced some Th2 cytokines (161). These data suggest that antigen-specific Treg, induced in the periphery following antigen exposure and thus recognizable as pTreg, can contain both committed and uncommitted cells.
The concomitant and detrimental Treg expansion in anti-tumor vaccination can be avoided by using CD8 T cell-targeted approaches. A melanoma vaccination protocol based on an MHCI-restricted Melan-A peptide significantly decreased the frequency of Melan-A-specific Treg, in association with an improved and more diverse Th1 response (97).
Some attempts have also been made to combine active immunotherapy with Treg depletion or functional blockade. Several studies showed that depletion of CD25+ cells in vivo in cancer patients could enhance the tumor-specific T cell responses induced by cancer vaccines (15). However, CD25-directed strategies may fail to achieve sustained results, since activated effector cells may be concomitantly eliminated and pTreg may replenish the Treg pool after depletion (15). Interestingly, a recent study has demonstrated that different Treg-depleting agents, either CD25-targeted (IL-2/diphtheria toxin fusion protein, or anti-CD25 antibody) or not (low-dose cyclophosphamide), failed to consistently eliminate more than 50% of committed Treg, as identified by TSDR demethylation (162).
Therefore, alternative strategies are needed to counteract the “hard core” of immune suppression that is represented by epigenetically committed Treg. We have proposed in the past that Treg functional inactivation, rather than depletion, may represent a successful strategy to prevent massive pTreg induction and concomitantly block Treg suppression (15). This idea may be corroborated by the observation that markers associated to Treg suppressive functions, and therapeutically targetable, may show enriched or restricted expression in epigenetically committed Treg. For instance, GITR stimulation has been shown to attenuate Treg suppression and favor the rejection of experimental tumors (163). A recent study has demonstrated that GITR engagement in vivo led to the downregulation of Foxp3 expression in intratumoral Treg (164). Of note, GITR+ Treg were found enriched in Helios+ cells, representing highly committed Treg (131), thus GITR targeting may preferentially block the strongest suppressors among the Treg pool. A similar possibility could be envisaged for therapeutic strategies aimed at TNF-α/TNFR2 blockade, since this axis may be mainly involved in the activation of more committed and stable cells (122–126). Committed Treg may also be targeted by virtue of their high proliferative potential: indeed, high proliferation rates, in terms of Ki67 positivity, were detected in healthy subjects within the aTreg subset, enriched in stable and committed Treg (117), and also in murine tumor-infiltrating Helios+ Treg (131). Therefore, treatments based on the depletion of proliferating cells, such as low-dose cyclophosphamide, may efficiently target committed Treg.
An innovative way to improve immunotherapy would be to reprogram tumor-associated Treg into fully armed effector cells, which would then become “exTreg.” Different from other vaccine-based approaches, Treg reprograming is expected to trigger anti-tumor response very rapidly, since Treg are already located at the tumor site and already tumor-antigen-experienced, thus not requiring a de novo T cell priming. Therefore, exTreg may function in an “innate-like” manner, promptly providing co-stimulatory and pro-inflammatory signals when adequately modulated, before a novel adaptive anti-tumor response develops (140). An example of this approach has been proposed by Sharma et al. who demonstrated that reprograming of mature pre-existing tumor-associated Treg into CD40L-expressing helper effector cells was needed to achieve tumor regression in a model of immunotherapy combining antigen vaccination, TLR-9 stimulation, and IDO blockade (152).
The above-discussed data overall indicate that tTreg and pTreg may not be equally susceptible to functional reprograming, but this dichotomy may turn into a benefit for the efficacy and safety of the evoked response. Indeed, on the one hand, tTreg, predominantly self-specific, highly committed, and hard to be reprogrammed into T-helper-like cells, would be preserved, thus ensuring immune tolerance to self-antigens and maintaining systemic immune homeostasis. On the other hand, pTreg, mainly representing tumor-specific and uncommitted cells, may be more easily converted into exTreg, thus mounting an immediate helper and/or effector response in a mostly tumor-antigen-specific fashion.
Reprograming into exTreg may be achieved by immunotherapies aimed at subverting the immune suppression mechanisms established by innate cells in tumor microenvironments. For instance, in the above-reported model of tumor vaccination, CD40L upregulation by Treg following TLR-9 stimulation was strictly dependent on host-derived MyD88 and IL-6 signals (152). In melanoma patients, tumor peptide antigen vaccination combined with low-dose cyclophosphamide and low-dose IL-2 evoked Th1-like Treg accumulation, in line with a less tolerogenic microenvironment and with enhanced IL-12 availability (158). Of note, in this system, depletion of proliferating (conceivably committed and thymus-derived) Treg by means of cyclophosphamide allowed the functional reshuffling of innate cells that in turn unveiled the emergence of Th1-like exTreg.
However, it is arguable that microenvironmental rearrangements would better accomplish full Treg reprograming with the concomitant direct modulation of Treg activities, aimed especially at enhancing Treg susceptibility to external signals. For instance, expression of IL-12 receptor, which is epigenetically regulated in Treg (135), could be artificially boosted by pharmacological approaches. Also, targeting with monoclonal antibodies some receptors expressed on Treg surface and correlated with Treg stability (such as TNFR2 and GITR) could result in enhancing Treg propensity to reprograming. In line with this idea, treatment of murine melanomas with a GITR agonistic antibody resulted in the accumulation of exTreg at the tumor site (164). Suppressor of cytokine signaling (SOCS) 1 and 2, which maintain Foxp3 stability and prevent Treg polarization into effector cells (165, 166), may be pharmacologically inhibited to unlock Treg responsiveness to pro-inflammatory microenvironmental cytokines.
Conclusion
Even though discrimination between pTreg and tTreg by simple surface phenotyping is not yet possible many pieces of evidence indicate that both subsets contribute to the Treg pool conditioning the tumor microenvironment. Nevertheless, the development/expansion of pTreg and tTreg are independent processes, possibly resulting from disparate antigens and signals, and their activities seem characterized by very peculiar features in terms of specificity, stability, and specialization. On the one side, tTreg may expand at tumor site in response to self-antigens expressed by tumor cells, mostly include committed (TSDR-demethylated) Helios- and TNFR2-expressing cells, and contain the precursors of specialized T-bet+ Th1-suppressing cells, thus representing not only the guardians of systemic immune homeostasis but also the “hard core” of tumor immune escape. On the other side, pTreg may mostly develop following local encounter with TAA or TSA antigens, possibly represent a mixed population of committed (TSDR-demethylated) and uncommitted (TSDR-methylated) cells, and are more prone to be reprogramed into Th1-like or Th17-like effector cells. We envisage that future successful immunotherapies may not only target committed Treg but also favor “recycling” uncommitted Treg into prompt anti-tumor effectors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from Associazione Italiana Ricerca sul Cancro (MFAG 8726 to Silvia Piconese), from Ministero dell’Istruzione, dell’Università e della Ricerca (FIRB-Futuro in ricerca RBFR12I3UB_002 to Silvia Piconese), and Italian Ministry of Health. Alessia Burocchi is recipient of a FIRC fellowship.
References
1. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi:10.1146/annurev.immunol.22.012703.104803
2. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
3. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol (1999) 163:5211–8.
4. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
5. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
6. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi:10.1038/83713
7. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27:18–20. doi:10.1038/83707
8. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27:68–73. doi:10.1038/83784
9. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37:785–99. doi:10.1016/j.immuni.2012.09.010
10. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14:307–8. doi:10.1038/ni.2554
11. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol (2012) 30:733–58. doi:10.1146/annurev-immunol-020711-075043
12. Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity (2009) 30:626–35. doi:10.1016/j.immuni
13. deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res (2012) 18:3022–9. doi:10.1158/1078-0432.CCR-11-3216
14. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6:295–307. doi:10.1038/nri1806
15. Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer (2007) 7:880–7. doi:10.1038/nrc2250
16. Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat Immunol (2009) 10:689–95. doi:10.1038/ni.1760
17. Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res (2007) 13:5238–42. doi:10.1158/1078-0432.CCR-07-0813
18. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi:10.1126/science.1160062
19. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21:503–13. doi:10.1016/j.immuni.2004.08.010
20. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol (2010) 184:6545–51. doi:10.4049/jimmunol.0903879
21. Park HJ, Kusnadi A, Lee EJ, Kim WW, Cho BC, Lee IJ, et al. Tumor-infiltrating regulatory T cells delineated by upregulation of PD-1 and inhibitory receptors. Cell Immunol (2012) 278:76–83. doi:10.1016/j.cellimm.2012.07.001
22. Sarris M, Andersen KG, Randow F, Mayr L, Betz AG. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity (2008) 28:402–13. doi:10.1016/j.immuni.2008.01.012
23. Battaglia A, Buzzonetti A, Baranello C, Ferrandina G, Martinelli E, Fanfani F, et al. Metastatic tumour cells favour the generation of a tolerogenic milieu in tumour draining lymph node in patients with early cervical cancer. Cancer Immunol Immunother (2009) 58:1363–73. doi:10.1007/s00262-008-0646-7
24. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature (2011) 470:548–53. doi:10.1038/nature09707
25. Zhao E, Wang L, Dai J, Kryczek I, Wei S, Vatan L, et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology (2012) 1:152–61. doi:10.4161/onci.1.2.18480
26. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110:1225–32. doi:10.1182/blood-2006-12-064527
27. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
28. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27:635–46. doi:10.1016/j.immuni.2007.08.014
29. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res (2011) 71:2892–900. doi:10.1158/0008-5472.CAN-10-4246
30. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity (2004) 21:589–601. doi:10.1016/j.immuni.2004.09.002
31. Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A (2005) 102:419–24. doi:10.1073/pnas.0408197102
32. Hilchey SP, De A, Rimsza LM, Bankert RB, Bernstein SH. Follicular lymphoma intratumoral CD4+CD25+GITR+ regulatory T cells potently suppress CD3/CD28-costimulated autologous and allogeneic CD8+CD25− and CD4+CD25− T cells. J Immunol (2007) 178:4051–61.
33. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res (2007) 13:4345–54. doi:10.1158/1078-0432.CCR-07-0472
34. Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity (2008) 28:546–58. doi:10.1016/j.immuni.2008.02.017
35. Chaudhry A, Samstein RM, Treuting P, Liang Y, Pils MC, Heinrich JM, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity (2011) 34:566–78. doi:10.1016/j.immuni.2011.03.018
36. Burocchi A, Pittoni P, Gorzanelli A, Colombo MP, Piconese S. Intratumor OX40 stimulation inhibits IRF1 expression and IL-10 production by Treg cells while enhancing CD40L expression by effector memory T cells. Eur J Immunol (2011) 41:3615–26. doi:10.1002/eji.201141700
37. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450:566–9. doi:10.1038/nature06306
38. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475:226–30. doi:10.1038/nature10169
39. Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol (2002) 169:2756–61.
40. Liyanage UK, Goedegebuure PS, Moore TT, Viehl CT, Moo-Young TA, Larson JW, et al. Increased prevalence of regulatory T cells (Treg) is induced by pancreas adenocarcinoma. J Immunother (2006) 29:416–24. doi:10.1097/01.cji.0000205644.43735.4e
41. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res (2001) 61:4766–72.
42. Marshall NA, Christie LE, Munro LR, Culligan DJ, Johnston PW, Barker RN, et al. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood (2004) 103:1755–62. doi:10.1182/blood-2003-07-2594
43. Correll A, Tuettenberg A, Becker C, Jonuleit H. Increased regulatory T-cell frequencies in patients with advanced melanoma correlate with a generally impaired T-cell responsiveness and are restored after dendritic cell-based vaccination. Exp Dermatol (2010) 19:e213–21. doi:10.1111/j.1600-0625.2009.01055.x
44. Chen YL, Chang MC, Chen CA, Lin HW, Cheng WF, Chien CL. Depletion of regulatory T lymphocytes reverses the imbalance between pro- and anti-tumor immunities via enhancing antigen-specific T cell immune responses. PLoS ONE (2012) 7:e47190. doi:10.1371/journal.pone.0047190
45. Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, et al. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood (2012) 119:5688–96. doi:10.1182/blood-2011-10-386482
46. Benevides L, Cardoso CR, Tiezzi DG, Marana HR, Andrade JM, Silva JS. Enrichment of regulatory T cells in invasive breast tumor correlates with the upregulation of IL-17A expression and invasiveness of the tumor. Eur J Immunol (2013) 43(6): 1518–28. doi:10.1002/eji.201242951
47. Gallimore AM, Simon AK. Positive and negative influences of regulatory T cells on tumour immunity. Oncogene (2008) 27:5886–93. doi:10.1038/onc.2008.269
48. Whiteside TL. What are regulatory T cells (Treg) regulating in cancer and why? Semin Cancer Biol (2012) 22:327–34. doi:10.1016/j.semcancer.2012.03.004
49. Chow MT, Moller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol (2012) 22:23–32. doi:10.1016/j.semcancer.2011.12.004
50. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol (2009) 27:186–92. doi:10.1200/JCO.2008.18.7229
51. Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer (2010) 126:2635–43.
52. Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother (2011) 60:909–18. doi:10.1007/s00262-011-1046-y
53. Sinicrope FA, Rego RL, Ansell SM, Knutson KL, Foster NR, Sargent DJ. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology (2009) 137:1270–9. doi:10.1053/j.gastro.2009.06.053
54. Chaput N, Louafi S, Bardier A, Charlotte F, Vaillant JC, Menegaux F, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut (2009) 58:520–9. doi:10.1136/gut.2008.158824
55. Kryczek I, Liu R, Wang G, Wu K, Shu X, Szeliga W, et al. FOXP3 defines regulatory T cells in human tumor and autoimmune disease. Cancer Res (2009) 69:3995–4000. doi:10.1158/0008-5472.CAN-08-3804
56. Kim M, Grimmig T, Grimm M, Lazariotou M, Meier E, Rosenwald A, et al. Expression of Foxp3 in colorectal cancer but not in Treg cells correlates with disease progression in patients with colorectal cancer. PLoS ONE (2013) 8:e53630. doi:10.1371/journal.pone.0053630
57. Badoual C, Hans S, Rodriguez J, Peyrard S, Klein C, Agueznay Nel H, et al. Prognostic value of tumor-infiltrating CD4+ T-cell subpopulations in head and neck cancers. Clin Cancer Res (2006) 12:465–72. doi:10.1158/1078-0432.CCR-05-1886
58. Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res (2007) 13:6301–11. doi:10.1158/1078-0432.CCR-07-1403
59. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184:3433–41. doi:10.4049/jimmunol.0904028
60. Gottschalk RA, Corse E, Allison JP. Expression of Helios in peripherally induced Foxp3+ regulatory T cells. J Immunol (2012) 188:976–80. doi:10.4049/jimmunol.1102964
61. Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med (2012) 209:1723–42, S1. doi:10.1084/jem.20120914
62. Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med (2012) 209:1713–22, S1–19. doi:10.1084/jem.20120822
63. Milpied P, Renand A, Bruneau J, Mendes-Da-Cruz DA, Jacquelin S, Asnafi V, et al. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol (2009) 39:1466–71. doi:10.1002/eji.200839040
64. Haribhai D, Lin W, Edwards B, Ziegelbauer J, Salzman NH, Carlson MR, et al. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J Immunol (2009) 182:3461–8. doi:10.4049/jimmunol.0802535
65. Feuerer M, Hill JA, Kretschmer K, von Boehmer H, Mathis D, Benoist C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc Natl Acad Sci U S A (2010) 107:5919–24. doi:10.1073/pnas.1002006107
66. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity (2011) 35:109–22. doi:10.1016/j.immuni.2011.03.029
67. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol (2007) 5:e38. doi:10.1371/journal.pbio.0050038
68. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38:1654–63. doi:10.1002/eji.200838105
69. Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, et al. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity (2012) 36:262–75. doi:10.1016/j.immuni.2011.12.012
70. Bui JD, Uppaluri R, Hsieh CS, Schreiber RD. Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res (2006) 66:7301–9. doi:10.1158/0008-5472.CAN-06-0556
71. Schallenberg S, Tsai PY, Riewaldt J, Kretschmer K. Identification of an immediate Foxp3(-) precursor to Foxp3(+) regulatory T cells in peripheral lymphoid organs of nonmanipulated mice. J Exp Med (2010) 207:1393–407. doi:10.1084/jem.20100045
72. Sharma MD, Baban B, Chandler P, Hou DY, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest (2007) 117:2570–82. doi:10.1172/JCI31911
73. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med (2005) 202:919–29. doi:10.1084/jem.20050463
74. Darrasse-Jeze G, Bergot AS, Durgeau A, Billiard F, Salomon BL, Cohen JL, et al. Tumor emergence is sensed by self-specific CD44hi memory Tregs that create a dominant tolerogenic environment for tumors in mice. J Clin Invest (2009) 119:2648–62. doi:10.1172/JCI36628
75. Beyer M, Kochanek M, Giese T, Endl E, Weihrauch MR, Knolle PA, et al. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood (2006) 107:3940–9. doi:10.1182/blood-2005-09-3671
76. Valzasina B, Piconese S, Guiducci C, Colombo MP. Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25− lymphocytes is thymus and proliferation independent. Cancer Res (2006) 66:4488–95. doi:10.1158/0008-5472.CAN-05-4217
77. Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor evasion of the immune system by converting CD4+CD25− T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol (2007) 178:2883–92.
78. Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25− into CD25+ T regulatory cells. Blood (2007) 109:2871–7.
79. Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA. Natural and induced CD4+CD25+ cells educate CD4+CD25− cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J Immunol (2004) 172:5213–21.
80. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. “Infectious” transplantation tolerance. Science (1993) 259:974–7. doi:10.1126/science.8094901
81. Zhou G, Drake CG, Levitsky HI. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood (2006) 107:628–36. doi:10.1182/blood-2005-07-2737
82. Zhou G, Levitsky HI. Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J Immunol (2007) 178:2155–62.
83. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol (2005) 6:1219–27. doi:10.1038/ni1265
84. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature (2012) 482:395–9. doi:10.1038/nature10772
85. Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity (2008) 29:114–26. doi:10.1016/j.immuni.2008.05.010
86. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463:808–12. doi:10.1038/nature08750
87. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell (2012) 150:29–38. doi:10.1016/j.cell.2012.05.031
88. Schmitt EG, Haribhai D, Williams JB, Aggarwal P, Jia S, Charbonnier LM, et al. IL-10 produced by induced regulatory T cells (iTregs) controls colitis and pathogenic ex-iTregs during immunotherapy. J Immunol (2012) 189:5638–48. doi:10.4049/jimmunol.1200936
89. Bergmann C, Wild CA, Narwan M, Lotfi R, Lang S, Brandau S. Human tumor-induced and naturally occurring Treg cells differentially affect NK cells activated by either IL-2 or target cells. Eur J Immunol (2011) 41:3564–73. doi:10.1002/eji.201141532
90. Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol (2012) 12:157–67.
91. Getnet D, Maris CH, Hipkiss EL, Grosso JF, Harris TJ, Yen HR, et al. Tumor recognition and self-recognition induce distinct transcriptional profiles in antigen-specific CD4 T cells. J Immunol (2009) 182:4675–85. doi:10.4049/jimmunol.0803400
92. Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, et al. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity (2004) 20:107–18. doi:10.1016/S1074-7613(03)00359-5
93. Wang HY, Peng G, Guo Z, Shevach EM, Wang RF. Recognition of a new ARTC1 peptide ligand uniquely expressed in tumor cells by antigen-specific CD4+ regulatory T cells. J Immunol (2005) 174:2661–70.
94. Vence L, Palucka AK, Fay JW, Ito T, Liu YJ, Banchereau J, et al. Circulating tumor antigen-specific regulatory T cells in patients with metastatic melanoma. Proc Natl Acad Sci U S A (2007) 104:20884–9. doi:10.1073/pnas.0710557105
95. Fourcade J, Sun Z, Kudela P, Janjic B, Kirkwood JM, El-Hafnawy T, et al. Human tumor antigen-specific helper and regulatory T cells share common epitope specificity but exhibit distinct T cell repertoire. J Immunol (2010) 184:6709–18. doi:10.4049/jimmunol.0903612
96. Ebert LM, Macraild SE, Zanker D, Davis ID, Cebon J, Chen W. A cancer vaccine induces expansion of NY-ESO-1-specific regulatory T cells in patients with advanced melanoma. PLoS ONE (2012) 7:e48424. doi:10.1371/journal.pone.0048424
97. Jandus C, Bioley G, Dojcinovic D, Derre L, Baitsch L, Wieckowski S, et al. Tumor antigen-specific FOXP3+ CD4 T cells identified in human metastatic melanoma: peptide vaccination results in selective expansion of Th1-like counterparts. Cancer Res (2009) 69:8085–93. doi:10.1158/0008-5472.CAN-09-2226
98. Lehe C, Ghebeh H, Al-Sulaiman A, Al Qudaihi G, Al-Hussein K, Almohareb F, et al. The Wilms’ tumor antigen is a novel target for human CD4+ regulatory T cells: implications for immunotherapy. Cancer Res (2008) 68:6350–9. doi:10.1158/0008-5472.CAN-08-0050
99. Redjimi N, Duperrier-Amouriaux K, Raimbaud I, Luescher I, Dojcinovic D, Classe JM, et al. NY-ESO-1-specific circulating CD4+ T cells in ovarian cancer patients are prevalently T(H)1 type cells undetectable in the CD25+ FOXP3+ Treg compartment. PLoS ONE (2011) 6:e22845. doi:10.1371/journal.pone.0022845
100. Bonertz A, Weitz J, Pietsch DH, Rahbari NN, Schlude C, Ge Y, et al. Antigen-specific Tregs control T cell responses against a limited repertoire of tumor antigens in patients with colorectal carcinoma. J Clin Invest (2009) 119:3311–21.
101. van der Burg SH, Piersma SJ, de Jong A, van der Hulst JM, Kwappenberg KM, van den Hende M, et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc Natl Acad Sci U S A (2007) 104:12087–92. doi:10.1073/pnas.0704672104
102. Voo KS, Peng G, Guo Z, Fu T, Li Y, Frappier L, et al. Functional characterization of EBV-encoded nuclear antigen 1-specific CD4+ helper and regulatory T cells elicited by in vitro peptide stimulation. Cancer Res (2005) 65:1577–86. doi:10.1158/0008-5472.CAN-04-2552
103. Nishikawa H, Kato T, Tanida K, Hiasa A, Tawara I, Ikeda H, et al. CD4+ CD25+ T cells responding to serologically defined autoantigens suppress antitumor immune responses. Proc Natl Acad Sci U S A (2003) 100:10902–6. doi:10.1073/pnas.1834479100
104. Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science (2013) 339:1219–24.
105. Sainz-Perez A, Lim A, Lemercier B, Leclerc C. The T-cell receptor repertoire of tumor-infiltrating regulatory T lymphocytes is skewed toward public sequences. Cancer Res (2012) 72:3557–69. doi:10.1158/0008-5472.CAN-12-0277
106. Hindley JP, Ferreira C, Jones E, Lauder SN, Ladell K, Wynn KK, et al. Analysis of the T-cell receptor repertoires of tumor-infiltrating conventional and regulatory T cells reveals no evidence for conversion in carcinogen-induced tumors. Cancer Res (2011) 71:736–46. doi:10.1158/0008-5472.CAN-10-1797
107. Schreiber TH, Wolf D, Bodero M, Podack E. Tumor antigen specific iTreg accumulate in the tumor microenvironment and suppress therapeutic vaccination. Oncoimmunology (2012) 1:642–8. doi:10.4161/onci.20298
108. Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med (2007) 204:1543–51.
109. Wieczorek G, Asemissen A, Model F, Turbachova I, Floess S, Liebenberg V, et al. Quantitative DNA methylation analysis of FOXP3 as a new method for counting regulatory T cells in peripheral blood and solid tissue. Cancer Res (2009) 69:599–608. doi:10.1158/0008-5472.CAN-08-2361
110. Clarke SL, Betts GJ, Plant A, Wright KL, El-Shanawany TM, Harrop R, et al. CD4+CD25+FOXP3+ regulatory T cells suppress anti-tumor immune responses in patients with colorectal cancer. PLoS ONE (2006) 1:e129. doi:10.1371/journal.pone.0000129
111. Ling KL, Pratap SE, Bates GJ, Singh B, Mortensen NJ, George BD, et al. Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun (2007) 7:7.
112. Sehouli J, Loddenkemper C, Cornu T, Schwachula T, Hoffmuller U, Grutzkau A, et al. Epigenetic quantification of tumor-infiltrating T-lymphocytes. Epigenetics (2011) 6:236–46. doi:10.4161/epi.6.2.13755
113. Pohla H, Buchner A, Stadlbauer B, Frankenberger B, Stevanovic S, Walter S, et al. High immune response rates and decreased frequencies of regulatory T cells in metastatic renal cell carcinoma patients after tumor cell vaccination. Mol Med (2012) 18:1499–508. doi:10.2119/molmed.2012.00221
114. Schwarzer A, Wolf B, Fisher JL, Schwaab T, Olek S, Baron U, et al. Regulatory T-cells and associated pathways in metastatic renal cell carcinoma (mRCC) patients undergoing DC-vaccination and cytokine-therapy. PLoS ONE (2012) 7:e46600. doi:10.1371/journal.pone.0046600
115. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
116. McMurchy AN, Gillies J, Gizzi MC, Riba M, Garcia-Manteiga JM, Cittaro D, et al. A novel function for FOXP3 in humans: intrinsic regulation of conventional T cells. Blood (2013) 121:1265–75. doi:10.1182/blood-2012-05-431023
117. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30:899–911. doi:10.1016/j.immuni.2009.03.019
118. Fujii H, Arakawa A, Kitoh A, Miyara M, Kato M, Kore-Eda S, et al. Perturbations of both nonregulatory and regulatory FOXP3+ T cells in patients with malignant melanoma. Br J Dermatol (2011) 164:1052–60. doi:10.1111/j.1365-2133.2010.10199.x
119. Schuler PJ, Harasymczuk M, Schilling B, Lang S, Whiteside TL. Separation of human CD4+CD39+ T cells by magnetic beads reveals two phenotypically and functionally different subsets. J Immunol Methods (2011) 369:59–68. doi:10.1016/j.jim.2011.04.004
120. Schuler PJ, Schilling B, Harasymczuk M, Hoffmann TK, Johnson J, Lang S, et al. Phenotypic and functional characteristics of CD4+ CD39+ FOXP3+ and CD4+ CD39+ FOXP3neg T-cell subsets in cancer patients. Eur J Immunol (2012) 42:1876–85. doi:10.1002/eji.201142347
121. Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Lenzner DE, Jackson EK, et al. Increased ectonucleotidase expression and activity in regulatory T cells of patients with head and neck cancer. Clin Cancer Res (2009) 15:6348–57. doi:10.1158/1078-0432.CCR-09-1143
122. Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, et al. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol (2011) 186:6779–87. doi:10.4049/jimmunol.1003868
123. Chen X, Subleski JJ, Kopf H, Howard OM, Mannel DN, Oppenheim JJ. Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J Immunol (2008) 180:6467–71.
124. Chopra M, Riedel SS, Biehl M, Krieger S, von Krosigk V, Bauerlein CA, et al. Tumor necrosis factor receptor 2-dependent homeostasis of regulatory T cells as a player in TNF-induced experimental metastasis. Carcinogenesis (2013) 34(6): 1296–303. doi:10.1093/carcin/bgt038
125. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T cell phenotype in the inflammatory environment. J Immunol (2013) 190:1076–84. doi:10.4049/jimmunol.1202659
126. Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur J Immunol (2010) 40:1099–106. doi:10.1002/eji.200940022
127. Nagar M, Jacob-Hirsch J, Vernitsky H, Berkun Y, Ben-Horin S, Amariglio N, et al. TNF activates a NF-kappaB-regulated cellular program in human CD45RA− regulatory T cells that modulates their suppressive function. J Immunol (2010) 184:3570–81. doi:10.4049/jimmunol.0902070
128. Getnet D, Grosso JF, Goldberg MV, Harris TJ, Yen HR, Bruno TC, et al. A role for the transcription factor Helios in human CD4(+)CD25(+) regulatory T cells. Mol Immunol (2010) 47:1595–600. doi:10.1016/j.molimm
129. Wainwright DA, Sengupta S, Han Y, Lesniak MS. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro-oncology (2011) 13:1308–23. doi:10.1093/neuonc/nor134
130. Elkord E, Sharma S, Burt DJ, Hawkins RE. Expanded subpopulation of FoxP3+ T regulatory cells in renal cell carcinoma co-express Helios, indicating they could be derived from natural but not induced Tregs. Clin Immunol (2011) 140:218–22. doi:10.1016/j.clim.2011.04.014
131. Zabransky DJ, Nirschl CJ, Durham NM, Park BV, Ceccato CM, Bruno TC, et al. Phenotypic and functional properties of Helios+ regulatory T cells. PLoS ONE (2012) 7:e34547. doi:10.1371/journal.pone.0034547
132. Alexander T, Sattler A, Templin L, Kohler S, Gross C, Meisel A, et al. Foxp3+ Helios+ regulatory T cells are expanded in active systemic lupus erythematosus. Ann Rheum Dis (2013) 72:1549–58. doi:10.1136/annrheumdis-2012-202216
133. Kim YC, Bhairavabhotla R, Yoon J, Golding A, Thornton AM, Tran DQ, et al. Oligodeoxynucleotides stabilize Helios-expressing Foxp3+ human T regulatory cells during in vitro expansion. Blood (2012) 119:2810–8. doi:10.1182/blood-2011-09-377895
134. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol (2009) 10:595–602. doi:10.1038/ni.1731
135. Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity (2012) 37:501–10. doi:10.1016/j.immuni.2012.05.031
136. Zheng Y, Chaudhry A, Kas A, Deroos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature (2009) 458:351–6. doi:10.1038/nature07674
137. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science (2009) 326:986–91. doi:10.1126/science.1172702
138. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med (2011) 17:983–8. doi:10.1038/nm.2426
139. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17:975–82. doi:10.1038/nm.2425
140. Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol (2009) 21:281–5. doi:10.1016/j.coi.2009.05.007
141. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science (2010) 329:1667–71. doi:10.1126/science.1191996
142. Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity (2009) 31:772–86. doi:10.1016/j.immuni.2009.10.001
143. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol (2009) 10:1000–7. doi:10.1038/ni.1774
144. Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med (2011) 17:673–5. doi:10.1038/nm.2389
145. Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology (2011) 140:957–65. doi:10.1053/j.gastro.2010.12.002
146. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol (2011) 186:3918–26. doi:10.4049/jimmunol.1003099
147. Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol (2011) 186:4388–95. doi:10.4049/jimmunol.1003251
148. Leveque L, Deknuydt F, Bioley G, Old LJ, Matsuzaki J, Odunsi K, et al. Interleukin 2-mediated conversion of ovarian cancer-associated CD4+ regulatory T cells into proinflammatory interleukin 17-producing helper T cells. J Immunother (2009) 32:101–8. doi:10.1097/CJI.0b013e318195b59e
149. Reppert S, Boross I, Koslowski M, Tureci O, Koch S, Lehr HA, et al. A role for T-bet-mediated tumour immune surveillance in anti-IL-17A treatment of lung cancer. Nat Commun (2011) 2:600. doi:10.1038/ncomms1609
150. Blatner NR, Bonertz A, Beckhove P, Cheon EC, Krantz SB, Strouch M, et al. In colorectal cancer mast cells contribute to systemic regulatory T-cell dysfunction. Proc Natl Acad Sci U S A (2010) 107:6430–5. doi:10.1073/pnas.0913683107
151. Blatner NR, Mulcahy MF, Dennis KL, Scholtens D, Bentrem DJ, Phillips JD, et al. Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Sci Transl Med (2012) 4:164ra159. doi:10.1126/scitranslmed.3004566
152. Sharma MD, Hou DY, Baban B, Koni PA, He Y, Chandler PR, et al. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity (2010) 33:942–54. doi:10.1016/j.immuni.2010.11.022
153. Koenecke C, Lee CW, Thamm K, Fohse L, Schafferus M, Mittrucker HW, et al. IFN-gamma production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol (2012) 189:2890–6. doi:10.4049/jimmunol.1200413
154. Zhao J, Fett C, Trandem K, Fleming E, Perlman S. IFN-gamma- and IL-10-expressing virus epitope-specific Foxp3(+) T reg cells in the central nervous system during encephalomyelitis. J Exp Med (2011) 208:1571–7. doi:10.1084/jem.20110236
155. Du W, Shen YW, Lee WH, Wang D, Paz S, Kandeel F, et al. Foxp3+ Treg expanded from patients with established diabetes reduce Helios expression while retaining normal function compared to healthy individuals. PLoS ONE (2013) 8:e56209. doi:10.1371/journal.pone.0056209
156. Chowdary Venigalla RK, Guttikonda PJ, Eckstein V, Ho AD, Sertel S, Lorenz HM, et al. Identification of a human Th1-like IFNgamma-secreting Treg subtype deriving from effector T cells. J Autoimmun (2012) 39:377–87. doi:10.1016/j.jaut.2012.06.004
157. Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunsi K, et al. CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res (2012) 72:4351–60. doi:10.1158/0008-5472.CAN-12-0579
158. Camisaschi C, Filipazzi P, Tazzari M, Casati C, Beretta V, Pilla L, et al. Effects of cyclophosphamide and IL-2 on regulatory CD4 T cell frequency and function in melanoma patients vaccinated with HLA-class I peptides: impact on the antigen-specific T cell response. Cancer Immunol Immunother (2013) 62(5): 897–908. doi:10.1007/s00262-013-1397-7
159. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med (2004) 10:909–15. doi:10.1038/nm1100
160. Welters MJ, Kenter GG, Piersma SJ, Vloon AP, Lowik MJ, Berends-van Der Meer DM, et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res (2008) 14:178–87. doi:10.1158/1078-0432.CCR-07-1880
161. Francois V, Ottaviani S, Renkvist N, Stockis J, Schuler G, Thielemans K, et al. The CD4(+) T-cell response of melanoma patients to a MAGE-A3 peptide vaccine involves potential regulatory T cells. Cancer Res (2009) 69:4335–45. doi:10.1158/0008-5472.CAN-08-3726
162. de Vries IJ, Castelli C, Huygens C, Jacobs JF, Stockis J, Schuler-Thurner B, et al. Frequency of circulating Tregs with demethylated FOXP3 intron 1 in melanoma patients receiving tumor vaccines and potentially Treg-depleting agents. Clin Cancer Res (2011) 17:841–8. doi:10.1158/1078-0432.CCR-10-2227
163. Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med (2005) 202:885–91. doi:10.1084/jem.20050940
164. Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS ONE (2010) 5:e10436. doi:10.1371/journal.pone.0010436
165. Takahashi R, Nishimoto S, Muto G, Sekiya T, Tamiya T, Kimura A, et al. SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-{gamma} and IL-17A production. J Exp Med (2011) 208:2055–67. doi:10.1084/jem.20110428
Keywords: Treg development, heterogeneity, specialization, plasticity, epigenetic commitment, tumor antigens
Citation: Burocchi A, Colombo MP and Piconese S (2013) Convergences and divergences of thymus- and peripherally derived regulatory T cells in cancer. Front. Immunol. 4:247. doi: 10.3389/fimmu.2013.00247
Received: 23 May 2013; Accepted: 08 August 2013;
Published online: 27 August 2013.
Edited by:
Eyad Elkord, United Arab Emirates University, UAE; University of Salford, UK; University of Manchester, UKReviewed by:
Masahide Tone, Cedars-Sinai Medical Center, USAMuriel Moser, Université Libre de Bruxelles, Belgium
Bin Li, Chinese Academy of Sciences, China
Eyad Elkord, United Arab Emirates University, UAE; University of Salford, UK; University of Manchester, UK
Copyright: © 2013 Burocchi, Colombo and Piconese. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario P. Colombo, Molecular Immunology Unit, Department of Experimental Medicine, Fondazione IRCCS “Istituto Nazionale Tumori,” Via Amadeo 42, I-20133 Milan, Italy e-mail: mario.colombo@istitutotumori.mi.it