
- 1Arthritis and Clinical Immunology Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK, USA
- 2Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA
Sjögren’s syndrome (SS) is a common, progressive autoimmune exocrinopathy distinguished by dry eyes and mouth and affects ∼0.7% of the European population. Overexpression of transcripts induced by interferons (IFN), termed as an “IFN signature,” has been found in SS patients. Four microarray studies have been published in SS that identified dysregulated genes within type I IFN signaling in either salivary glands or peripheral blood of SS patients. The mechanism of this type I IFN activation is still obscure, but several possible explanations have been proposed, including virus infection-initiated and immune complex-initiated type I IFN production by plasmacytoid dendritic cells. Genetic predisposition to increased type I IFN signaling is supported by candidate gene studies showing evidence for association of variants within IFN-related genes. Once activated, IFN signaling may contribute to numerous aspects of SS pathophysiology, including lymphocyte infiltration into exocrine glands, autoantibody production, and glandular cell apoptosis. Thus, dysregulation of IFN pathways is an important feature that can be potentially used as a serum biomarker for diagnosis and targeting of new treatments in this complex autoimmune disease.
Introduction
Sjögren’s syndrome (SS) is a chronic autoimmune disease that primarily affects middle-aged women with an estimated prevalence of ∼0.7% in European populations (1, 2). SS is characterized by infiltration of lymphocytes into glandular tissues, typically the salivary and lacrimal glands, leading to xerostomia (dry mouth) and keratoconjunctivitis sicca (dry eye). The resulting pathology can be debilitating, and target organ damage may be so severe that moisture production is virtually non-existent. However, manifestations of SS are not limited to exocrinopathy; other common extraglandular features include fatigue, arthritis, Raynaud’s phenomenon, and an increased incidence of non-Hodgkin B cell lymphoma (3). Two autoantibodies targeting ribonucleoproteins, anti-Ro/SSA, and anti-La/SSB, are detected in 60–70% of SS patients and are important to disease diagnosis (4, 5).
The etiology and pathogenesis of SS are still unclear, partially due to the complexity and heterogeneity of disease mechanisms. Recently, the dysregulation of interferon (IFN) signaling pathways, especially upregulation of type I IFN-inducible genes, has been observed in salivary glands and peripheral blood in a subset of SS patients (6– 10). Type I IFNs, including IFNα and IFNβ, are key immune mediators involved in viral defense and activation of immune responses (11). Viral infection has long been suspected to trigger SS (12), and abnormal elevations in type I IFN signaling may reflect an important role for viral infection in disease pathogenesis. Additionally, genetic association studies indicate the importance of multiple genetic loci within IFN pathways. Here, we review the identification of the type I IFN “signature” through high-throughput techniques, and discuss potential mechanisms and functions of dysregulated IFN signaling in SS.
Identification of the “IFN Signature” in SS
Many powerful, high-throughput techniques have emerged in the last few decades and have revealed important insights into mechanisms of complex human diseases. Gene expression profiling (GEP) studies using microarrays represent one of the most widely used approaches to determine global transcriptome differences between patients and healthy controls. “Signatures” of disease have been defined that represent clusters of co-expressed genes, often within a biological network, that may serve as biomarkers for disease diagnosis, classification, and drug response prediction. The “IFN signature,” first described in a GEP study of systemic lupus erythematosus (SLE), has been defined by the overexpression of type I IFN-inducible genes (13). Subsequent studies have demonstrated similar signatures in other autoimmune diseases, such as rheumatoid arthritis (RA), systemic sclerosis (SSc), myositis, and SS (14).
Four microarray studies have been published in SS to date that describe the overexpression of type I IFN-inducible transcripts in minor salivary glands or peripheral blood from SS patients (7–10). Hjelmervik et al. (7) published the first GEP in SS using a relatively low-density 16K microarray to identify a whole transcriptome signature in biopsy samples of minor salivary glands from 10 SS patients and 10 controls who experienced subjective oral dryness. They successfully clustered 19 out of the 20 subjects into the correct group by using the top 200 differentially expressed transcripts, in which numerous type I IFN-regulated genes were represented, including IFI27, ISG12, GBP2, IFITM1, and IRF8. Subsequently, Gottenberg et al. (8) also identified distinct gene expression patterns involving IFN pathways (both type I and type II) in salivary glands of SS patients by comparing seven cases and seven controls. Specifically, 23 genes were IFN-inducible, including genes in the antiviral IFN-induced transmembrane protein (IFITM) family (IFITM1, IFITM2, and IFITM3) and genes in the Toll-like receptor (TLR) family (TLR8 and TLR9) that play a fundamental role in pathogen recognition and activation of innate immunity (15). All showed significantly increased expression in SS patients. Interestingly, the only two known IFN-inducible genes that showed decreased expression in SS salivary glands, CCL18 and SOCS3, are involved in the inhibition of inflammatory processes. The overexpression of IFN-inducible genes was supported by the detection of plasmacytoid dendritic cells (pDC), the most potent producer of type I IFNs (16), in salivary glands of patients with SS, but none in the glands of controls. These results suggest that pDC activation may play a role in SS pathogenesis, which is discussed later in this review.
Upregulation of 11 IFN-inducible genes has been identified by Pérez et al. (10) through a microarray study of epithelial cells from salivary glands of nine SS patients and six controls. Notably, three of these genes belong to the IFN regulatory factor (IRF) family (IRF7, IRF8 or ICSBP1, and IRF9). IRFs are pivotal transcriptional regulators of type I IFN and IFN-inducible genes, and are important in cellular differentiation of hematopoietic cells (17). This GEP study also identified dysregulation of apoptotic pathways in SS epithelial cells, which are now thought to be involved in local auto-antigen production and tissue damage in the salivary glands of SS patients (18, 19). Additionally, they identified six genetic loci associated with SS using microsatellite markers, with five of the association signals falling within regions where differentially expressed genes were found, such as IL6, CD44, and IRF9. These results support a genetic contribution to the dysregulated IFN pathways observed in SS.
The IFN signature has also been observed in peripheral blood of SS patients. Emamian et al. (9) detected upregulation of IFN-inducible genes in peripheral blood of a subset of SS patients by comparing 21 cases and 23 controls followed by replication in an independent dataset of 17 SS cases and 22 controls. IFI35, MX1, OAS1, IRF7, and OAS2 were among the top differentially expressed genes and are known to be induced by IFNs. The authors also showed that the expression levels of most IFN-inducible genes were positively correlated with anti-Ro/SSA and anti-La/SSB titers. Although the relationship between IFN pathway activation and autoantibody production is unclear, these results provide a link for both innate and adaptive immune responses to the pathogenesis of disease. These results also suggest that the IFN signature can be potentially used as a disease biomarker for a subgroup of SS patients with certain clinical features that includes the production of anti-Ro/SSA and anti-La/SSB.
All of these microarray studies have consistently identified differentially expressed genes in IFN-mediated signaling pathways. As shown in Table 1, there are several differentially expressed genes found to be common across multiple studies and multiple tissue types, such as IFITM1, IFI44, MX1, IRF7, and IRF8, suggesting both local and systemic dysregulation of IFN signaling pathways in SS patients (Table 1). Each study also revealed unique dysregulated genes, partially due to the different types of arrays, relatively limited sample sizes, different quality control processes, and sample heterogeneity between studies.
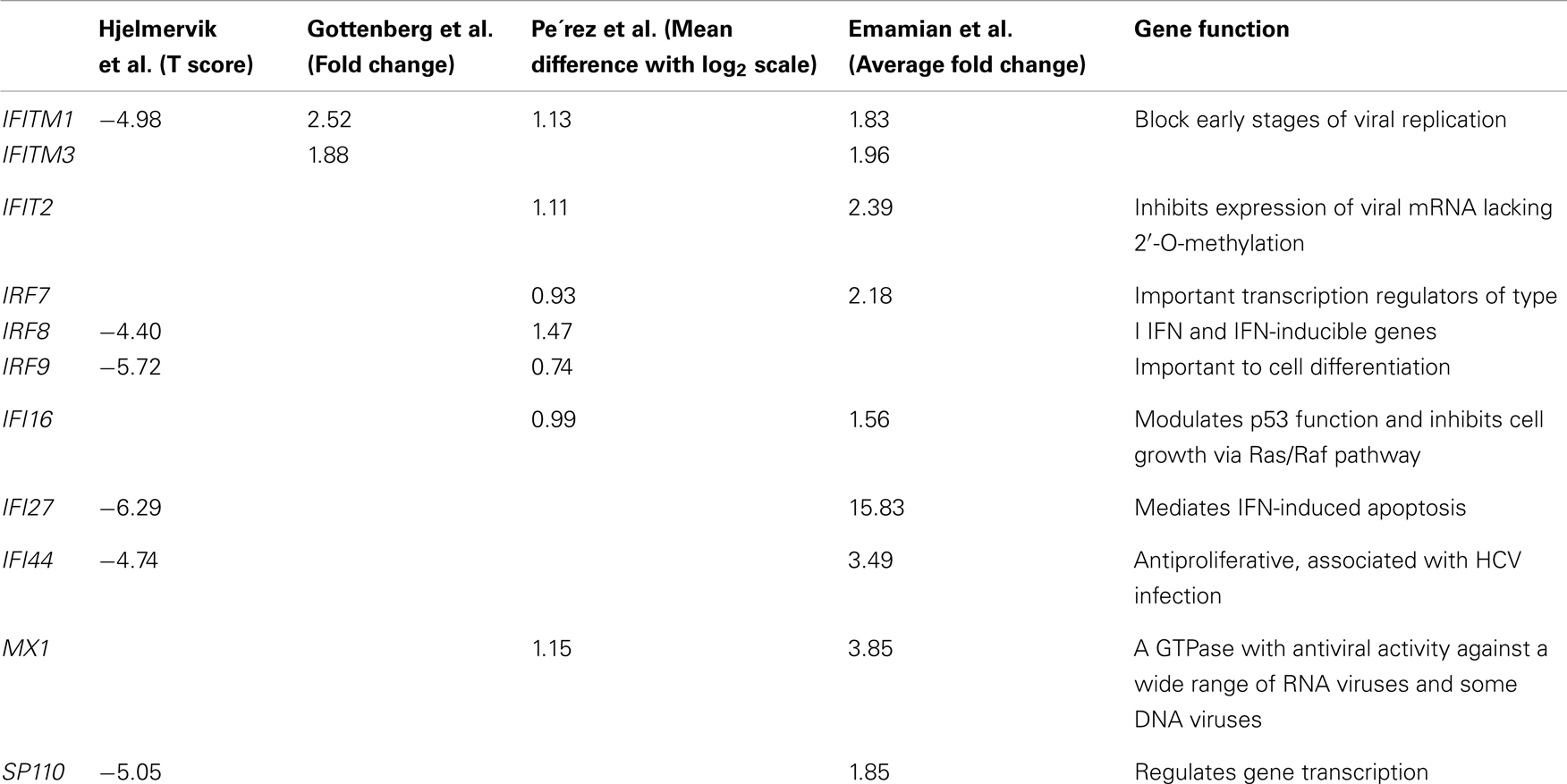
Table 1. Differentially expressed IFN-inducible genes found in common from the gene expression profiling studies in SS.
Additional studies have identified increased activation of type I IFN-mediated genes in SS patients by candidate gene approaches (20–24). Increased IFN-inducible gene expression profiles have been detected in saliva and tears as well as particular cell types from SS patients (25, 26). However, results for the expression levels of IFNα itself in SS patients are controversial (6, 27). This may be due to the different techniques used in separate studies, a mixture of cell types in each experiment, and the heterogeneity of SS patients involved.
Mechanisms of Elevated IFN-Mediated Signaling in SS
Although virtually all cells can produce type I IFNs in response to viral and bacterial infection, pDCs are the most potent IFN-producing cells, making up to 1000-fold more type I IFNs than other cell types (28). The detection of activated pDCs in salivary glands of SS patients but not in controls makes pDCs prominent candidates for the local production of type I IFNs that may promote the formation of inflammatory foci (8). Activated pDCs have also been found in the target organs of other autoimmune diseases (29, 30). Interestingly, Wildenberg et al. (26) found that, although the number of pDCs is decreased in the blood of SS patients, supposedly due to the migration of pDCs to peripheral sites, the cell surface activation marker CD40 is significantly overexpressed on pDCs from SS patients. Possible explanations of sustained activation of pDCs in SS include chronic exogenous stimulation and constitutive expression of pro-inflammatory transcription factors, such as IRF5 and IRF7, in SS patients (31).
Type I IFNs are induced transiently by viral infection and elicit antiviral effects. The pDCs can be rapidly induced to produce IFNα upon stimulation by RNA and DNA through TLR7 and TLR9, respectively (32). Thus, an initial viral infection is suspected to trigger the production of type I IFN by pDCs. The contribution of viral infection to elevated IFN signaling in SS is unknown; however, a number of viruses have been thought to contribute to SS pathogenesis, including Epstein–Barr virus (EBV), cytomegalovirus (CMV), hepatitis B virus (HBV), and hepatitis C virus (HCV) (12). Several mechanisms have been hypothesized regarding possible infectious triggers of SS, such as antigenic molecular mimicry. For example, infection with EBV results in the production of EBV nuclear antigen-1 (EBNA-1). Immune response against EBNA-1 can generate antibodies that cross-react with SS-associated autoantigens, such as anti-Ro/SSA (33). These antibodies may undergo epitope spreading and may ultimately become pathogenic in SS.
Autoantibodies and autoantigen-specific B cells have been detected in the salivary glands of SS patients (34, 35) and may involve in the production the type I IFNs through the formation of immune complexes. Båve et al. (6) have found that the combination of autoantibodies to RNA-binding proteins and material released by apoptotic cells can induce IFNα production by pDCs. This event is probably triggered by the interaction of RNA-containing immune complexes with Fcγ receptor IIa (FCGRIIA) on the surface of pDCs. Lövgren et al. (36) have described the production of IFNα by pDCs stimulated using U1 snRNA combined with IgG from patients with SLE. This response can be inhibited by FCGRIIA antagonists or RNase, suggesting a role for the RNA component as well as FCGRIIA in the immune complex-induced IFNα production by pDCs.
Another role of autoantibodies, especially anti-Ro52, in promoting IFN signaling is based on the function of their target autoantigens. Ro52, also known as tripartite motif-containing protein 21 (TRIM21), is an IFN-inducible E3 ubiquitin-protein ligase that promotes ubiquitination and proteasomal degradation of IRF3 and IRF7 (37, 38). After induction by IFNs following TLR signaling, Ro52 exerts a negative role on IFN signaling and prevents further inflammatory damage. Therefore, autoantibodies against Ro52 may interrupt the negative feedback of type I IFN signaling. Indeed, anti-Ro52 from SS patients is able to inhibit the E3 ligase activity of Ro52 by blocking the E3/E2 interface (39). Additionally, tissue inflammation and systemic autoimmunity in Ro52 knockout mice is thought to be induced by overproduction of pro-inflammatory cytokines (40). These results may well explain the correlation between the IFN signature and autoantibody positivity in SS patients (9). However, anti-Ro alone does not seem sufficient to induce high IFNα activity, given the fact that patients with disease are more likely to have high serum IFN activity than asymptomatic individuals with autoantibodies (41). Therefore, the contribution of autoantibodies to the elevated IFN signaling warrants further study.
Genetics Risk Factors in IFN Pathways
A possible model for SS development is that an initial viral infection induces the production of type I IFNs and genetic susceptibility factors in certain individuals promote prolonged activation of the IFN system. Genetic predisposition to SS is supported by family aggregation of disease as well as a few twin studies (42–45). Genetic risk variants within or near IFN-regulated genes could possibly predispose patients to increased IFN signaling by (1) the constitutive expression of IFN-inducible genes or (2) the induction of loss-of-function inhibitors within IFN pathways. Genetic studies in SS have relied primarily on candidate gene approaches, focusing on those genes with biological plausibility for a role in SS etiology or evidence of association in other autoimmune diseases (46). The most convincing associations outside the HLA in SS found by candidate gene studies are within the regions of IRF5 and STAT4 (47–49), both of which are involved in IFN signaling. IRF5 is a transcription factor mediating type I IFN responses in various immune-related cells (17). Upon viral infection, IRF5 induces the transcription of IFNα and other pro-inflammatory cytokines, including IL12 p40 subunit, IL6, and TNFα (50). Genetic association within the IRF5 region has been established in other autoimmune diseases, including SLE, RA, ulcerative colitis, primary biliary cirrhosis (PBC), and SSc (51–55). STAT4 is also a critical transcription factor involved in signaling initiated by type I and type II IFNs. It is required for the development of Th1 cells from naive CD4+ T cells and IFNγ production in response to IL12 (56, 57). The association of variants in STAT4 has also been well established in other inflammatory diseases (58–60). Ongoing genome-wide association studies in SS have firmly established these loci as well as other genes that promote susceptibility to disease and may contribute to the dysregulation of IFN-inducible genes (61).
Effects of the Overexpression of Type I IFN-Inducible Genes in SS
Type I IFNs are key regulators of human immune systems and exert a broad effect on immune responses and autoimmunity (11). Overexpression of type I IFN-inducible genes in the salivary glands and peripheral blood of SS patients may influence many aspects of SS pathophysiology. Epithelial cells from the salivary glands of SS patients play an active role in promoting immune responses, including increased expression of MHC molecules and co-stimulatory molecules, such as B7 and CD40 (62–65). Many T cell-attracting and germinal center-forming chemokines, such as CXCL10, IL-8, and CXCL13, have also been found to be expressed in epithelial cells from the salivary glands of SS patients (22, 66, 67). Thus, these cells acquire antigen-presenting characteristics, mediating the recruitment, activation, and differentiation of the infiltrating inflammatory cells (68). Many of these molecules are induced by IFNα and IFNβ.
Another cytokine induced by type I IFNs in both salivary gland epithelial cells and peripheral blood monocytes is B cell activating factor (BAFF) (69, 70). BAFF is important in B cell activation, proliferation, and differentiation and has been found to be overexpressed in SS patients (71). Increased expression of BAFF has been observed in salivary gland epithelial cells from SS patients compared with those from healthy controls upon stimulation by IFNα, but not IFNγ or TNFα, suggesting a specific role of type I IFNs in B cell dysfunction in SS (72).
As mentioned above, autoantigen Ro52, or TRIM21 can be induced by IFNα in cultured human B cells and peripheral blood mononuclear cells (73). After upregulation, Ro52 translocates from the cytoplasm to the nucleus and initiates IFNα-induced apoptosis through intrinsic caspase-3. IFNα can also induce the expression of pro-apoptotic molecules, including Fas and FasL (74), and the increased expression of Fas and FasL has been identified in salivary glands from SS patients (18, 75). But this effect is not limited to type I IFNs: IFNγ and TNFα are also potent inducers of these pro-apoptotic molecules in salivary glands (18, 76). Concordantly, elevated levels of epithelial apoptotic cell death have been detected in the minor salivary gland tissues of SS patients (77). In addition, Ro52, Ro60, and La48 redistribute to apoptotic blebs and are exposed to the surface of cells undergoing apoptosis (78). Thus, apoptotic cells are also a main source of the autoantigens that form immune complexes in the salivary gland tissue of SS patients that further boost local IFNα production.
Conclusion
It is widely acknowledged that SS patients have elevated type I IFN signaling that may contribute to the pathogenesis of this complex disease. Hundreds of genes can be induced by IFNs and are mediated by IFN-related pathways in different cell types, which makes it difficult to attribute disease pathology to the malfunction of a single gene or certain gene family. An efficient approach for unraveling mechanisms of complex diseases is to use multiple tools integrated into systems approaches to answer a specific biological question. High-throughput techniques, such as microarray and genome-wide genotyping arrays, provide us with exceptional opportunities to comprehensively analyze dysregulated IFN networks.
It is worth noting that the IFN signature is only observed in a subset of SS patients who tend to be autoantibody positive (9). This heterogeneity is obviously important to consider and may explain the controversial results of recent IFN-targeting therapies and drug trials for SS (79–83). Eventually the IFN signature in SS may be used as a tool to identify high-risk individuals for preventative strategies, to help diagnose SS through its use as a serum biomarker in lieu of cumbersome and costly routine diagnostic processes currently used in research and clinical practice, and to predict the efficacy of treatment by IFN-related therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Alamanos Y, Tsifetaki N, Voulgari PV, Venetsanopoulou AI, Siozos C, Drosos AA. Epidemiology of primary Sjogren’s syndrome in north-west Greece, 1982–2003. Rheumatology (Oxford) (2006) 45(2):187–91. doi:10.1093/rheumatology/kei107
2. Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum (2008) 58(1):15–25. doi:10.1002/art.23177
3. Jonsson R, Vogelsang P, Volchenkov R, Espinosa A, Wahren-Herlenius M, Appel S. The complexity of Sjogren’s syndrome: novel aspects on pathogenesis. Immunol Lett (2011) 141(1):1–9. doi:10.1016/j.imlet.2011.06.007
4. Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, et al. Classification criteria for Sjogren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis (2002) 61(6):554–8. doi:10.1136/ard.61.6.554
5. Hernandez-Molina G, Leal-Alegre G, Michel-Peregrina M. The meaning of anti-Ro and anti-La antibodies in primary Sjogren’s syndrome. Autoimmun Rev (2011) 10(3):123–5. doi:10.1016/j.autrev.2010.09.001
6. Båve U, Nordmark G, Lovgren T, Ronnelid J, Cajander S, Eloranta ML, et al. Activation of the type I interferon system in primary Sjogren’s syndrome: a possible etiopathogenic mechanism. Arthritis Rheum (2005) 52(4):1185–95. doi:10.1002/art.20998
7. Hjelmervik TO, Petersen K, Jonassen I, Jonsson R, Bolstad AI. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren’s syndrome patients from healthy control subjects. Arthritis Rheum (2005) 52(5):1534–44. doi:10.1002/art.21006
8. Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren’s syndrome. Proc Natl Acad Sci USA (2006) 103(8):2770–5. doi:10.1073/pnas.0510837103
9. Emamian ES, Leon JM, Lessard CJ, Grandits M, Baechler EC, Gaffney PM, et al. Peripheral blood gene expression profiling in Sjogren’s syndrome. Genes Immun (2009) 10(4):285–96. doi:10.1038/gene.2009.20
10. Pérez P, Anaya JM, Aguilera S, Urzua U, Munroe D, Molina C, et al. Gene expression and chromosomal location for susceptibility to Sjogren’s syndrome. J Autoimmun (2009) 33(2):99–108. doi:10.1016/j.jaut.2009.05.001
11. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12(2):125–35. doi:10.1038/nri3133
12. Igoe A, Scofield RH. Autoimmunity and infection in Sjogren’s syndrome. Curr Opin Rheumatol (2013) 25(4):480–7. doi:10.1097/BOR.0b013e32836200d2
13. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA (2003) 100(5):2610–5. doi:10.1073/pnas.0337679100
14. Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis (2011) 70(11):2029–36. doi:10.1136/ard.2011.150326
15. O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors – redefining innate immunity. Nat Rev Immunol (2013) 13(6):453–60. doi:10.1038/nri3446
16. Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol (2005) 23:275–306. doi:10.1146/annurev.immunol.23.021704.115633
17. Savitsky D, Tamura T, Yanai H, Taniguchi T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol Immunother (2010) 59(4):489–510. doi:10.1007/s00262-009-0804-6
18. Abu-Helu RF, Dimitriou ID, Kapsogeorgou EK, Moutsopoulos HM, Manoussakis MN. Induction of salivary gland epithelial cell injury in Sjogren’s syndrome: in vitro assessment of T cell-derived cytokines and Fas protein expression. J Autoimmun (2001) 17(2):141–53. doi:10.1006/jaut.2001.0524
19. Nordmark G, Alm GV, Ronnblom L. Mechanisms of disease: primary Sjogren’s syndrome and the type I interferon system. Nat Clin Pract Rheumatol (2006) 2(5):262–9. doi:10.1038/ncprheum0173
20. Ohyama Y, Nakamura S, Matsuzaki G, Shinohara M, Hiroki A, Fujimura T, et al. Cytokine messenger RNA expression in the labial salivary glands of patients with Sjogren’s syndrome. Arthritis Rheum (1996) 39(8):1376–84. doi:10.1002/art.1780390816
21. Mitsias DI, Tzioufas AG, Veiopoulou C, Zintzaras E, Tassios IK, Kogopoulou O, et al. The Th1/Th2 cytokine balance changes with the progress of the immunopathological lesion of Sjogren’s syndrome. Clin Exp Immunol (2002) 128(3):562–8. doi:10.1046/j.1365-2249.2002.01869.x
22. Wakamatsu E, Matsumoto I, Yasukochi T, Naito Y, Goto D, Mamura M, et al. Overexpression of phosphorylated STAT-1alpha in the labial salivary glands of patients with Sjogren’s syndrome. Arthritis Rheum (2006) 54(11):3476–84. doi:10.1002/art.22176
23. Giron-Gonzalez JA, Baturone R, Soto MJ, Marquez M, Macias I, Montes de Oca M, et al. Implications of immunomodulatory interleukins for the hyperimmunoglobulinemia of Sjogren’s syndrome. Cell Immunol (2009) 259(1):56–60. doi:10.1016/j.cellimm.2009.05.013
24. Szodoray P, Papp G, Horvath IF, Barath S, Sipka S, Nakken B, et al. Cells with regulatory function of the innate and adaptive immune system in primary Sjogren’s syndrome. Clin Exp Immunol (2009) 157(3):343–9. doi:10.1111/j.1365-2249.2009.03966.x
25. Hu S, Wang J, Meijer J, Ieong S, Xie Y, Yu T, et al. Salivary proteomic and genomic biomarkers for primary Sjogren’s syndrome. Arthritis Rheum (2007) 56(11):3588–600. doi:10.1002/art.22954
26. Wildenberg ME, van Helden-Meeuwsen CG, van de Merwe JP, Drexhage HA, Versnel MA. Systemic increase in type I interferon activity in Sjogren’s syndrome: a putative role for plasmacytoid dendritic cells. Eur J Immunol (2008) 38(7):2024–33. doi:10.1002/eji.200738008
27. Zheng L, Zhang Z, Yu C, Tu L, Zhong L, Yang C. Association between IFN-alpha and primary Sjogren’s syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod (2009) 107(1):e12–8. doi:10.1016/j.tripleo.2008.09.015
28. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol (2005) 23:307–36. doi:10.1146/annurev.immunol.23.021704.115843
29. Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol (2001) 159(1):237–43. doi:10.1016/S0002-9440(10)61689-6
30. Van Krinks CH, Matyszak MK, Gaston JS. Characterization of plasmacytoid dendritic cells in inflammatory arthritis synovial fluid. Rheumatology (Oxford) (2004) 43(4):453–60. doi:10.1093/rheumatology/keh115
31. Nordmark G, Eloranta ML, Ronnblom L. Primary Sjogren’s syndrome and the type I interferon system. Curr Pharm Biotechnol (2012) 13(10):2054–62. doi:10.2174/138920112802273290
32. Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev (2008) 19(1):3–19. doi:10.1016/j.cytogfr.2007.10.006
33. Poole BD, Scofield RH, Harley JB, James JA. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity (2006) 39(1):63–70. doi:10.1080/08916930500484849
34. Salomonsson S, Wahren-Herlenius M. Local production of Ro/SSA and La/SSB autoantibodies in the target organ coincides with high levels of circulating antibodies in sera of patients with Sjogren’s syndrome. Scand J Rheumatol (2003) 32(2):79–82. doi:10.1080/03009740310000076
35. Wahren-Herlenius M, Salomonsson S. Detection of antigen specific B-cells in tissues. Methods Mol Med (2007) 136:19–24. doi:10.1007/978-1-59745-402-5_2
36. Lövgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, Ronnblom L. Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjogren’s syndrome autoantigen-associated RNA. Arthritis Rheum (2006) 54(6):1917–27. doi:10.1002/art.21893
37. Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol (2008) 181(3):1780–6.
38. Higgs R, Lazzari E, Wynne C, Ni Gabhann J, Espinosa A, Wahren-Herlenius M, et al. Self protection from anti-viral responses – Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral Toll-Like receptors. PLoS ONE (2010) 5(7):e11776. doi:10.1371/journal.pone.0011776
39. Espinosa A, Hennig J, Ambrosi A, Anandapadmanaban M, Abelius MS, Sheng Y, et al. Anti-Ro52 autoantibodies from patients with Sjogren’s syndrome inhibit the Ro52 E3 ligase activity by blocking the E3/E2 interface. J Biol Chem (2011) 286(42):36478–91. doi:10.1074/jbc.M111.241786
40. Espinosa A, Dardalhon V, Brauner S, Ambrosi A, Higgs R, Quintana FJ, et al. Loss of the lupus autoantigen Ro52/Trim21 induces tissue inflammation and systemic autoimmunity by disregulating the IL-23-Th17 pathway. J Exp Med (2009) 206(8):1661–71. doi:10.1084/jem.20090585
41. Niewold TB, Rivera TL, Buyon JP, Crow MK. Serum type I interferon activity is dependent on maternal diagnosis in anti-SSA/Ro-positive mothers of children with neonatal lupus. Arthritis Rheum (2008) 58(2):541–6. doi:10.1002/art.23191
42. Scofield RH, Kurien BT, Reichlin M. Immunologically restricted and inhibitory anti-Ro/SSA in monozygotic twins. Lupus (1997) 6(4):395–8. doi:10.1177/096120339700600409
43. Sestak AL, Shaver TS, Moser KL, Neas BR, Harley JB. Familial aggregation of lupus and autoimmunity in an unusual multiplex pedigree. J Rheumatol (1999) 26(7):1495–9.
44. Sabio JM, Milla E, Jimenez-Alonso J. A multicase family with primary Sjogren’s syndrome. J Rheumatol (2001) 28(8):1932–4.
45. Houghton KM, Cabral DA, Petty RE, Tucker LB. Primary Sjogren’s syndrome in dizygotic adolescent twins: one case with lymphocytic interstitial pneumonia. J Rheumatol (2005) 32(8):1603–6.
46. Ice JA, Li H, Adrianto I, Lin PC, Kelly JA, Montgomery CG, et al. Genetics of Sjogren’s syndrome in the genome-wide association era. J Autoimmun (2012) 39(1–2):57–63. doi:10.1016/j.jaut.2012.01.008
47. Korman BD, Alba MI, Le JM, Alevizos I, Smith JA, Nikolov NP, et al. Variant form of STAT4 is associated with primary Sjogren’s syndrome. Genes Immun (2008) 9(3):267–70. doi:10.1038/gene.2008.1
48. Nordmark G, Kristjansdottir G, Theander E, Eriksson P, Brun JG, Wang C, et al. Additive effects of the major risk alleles of IRF5 and STAT4 in primary Sjogren’s syndrome. Genes Immun (2009) 10(1):68–76. doi:10.1038/gene.2008.94
49. Nordmark G, Kristjansdottir G, Theander E, Appel S, Eriksson P, Vasaitis L, et al. Association of EBF1, FAM167A(C8orf13)-BLK and TNFSF4 gene variants with primary Sjogren’s syndrome. Genes Immun (2011) 12(2):100–9. doi:10.1038/gene.2010.44
50. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature (2005) 434(7030):243–9. doi:10.1038/nature03308
51. Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet (2005) 76(3):528–37. doi:10.1086/428480
52. Dideberg V, Kristjansdottir G, Milani L, Libioulle C, Sigurdsson S, Louis E, et al. An insertion-deletion polymorphism in the interferon regulatory Factor 5 (IRF5) gene confers risk of inflammatory bowel diseases. Hum Mol Genet (2007) 16(24):3008–16. doi:10.1093/hmg/ddm259
53. Sigurdsson S, Padyukov L, Kurreeman FA, Liljedahl U, Wiman AC, Alfredsson L, et al. Association of a haplotype in the promoter region of the interferon regulatory factor 5 gene with rheumatoid arthritis. Arthritis Rheum (2007) 56(7):2202–10. doi:10.1002/art.22704
54. Dieude P, Guedj M, Wipff J, Avouac J, Fajardy I, Diot E, et al. Association between the IRF5 rs2004640 functional polymorphism and systemic sclerosis: a new perspective for pulmonary fibrosis. Arthritis Rheum (2009) 60(1):225–33. doi:10.1002/art.24183
55. Liu X, Invernizzi P, Lu Y, Kosoy R, Bianchi I, Podda M, et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet (2010) 42(8):658–60. doi:10.1038/ng.627
56. Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O’Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev (2004) 202:139–56. doi:10.1111/j.0105-2896.2004.00211.x
57. Kaplan MH. STAT4: a critical regulator of inflammation in vivo. Immunol Res (2005) 31(3):231–42. doi:10.1385/IR:31:3:231
58. Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med (2007) 357(10):977–86. doi:10.1056/NEJMoa073003
59. Rueda B, Broen J, Simeon C, Hesselstrand R, Diaz B, Suarez H, et al. The STAT4 gene influences the genetic predisposition to systemic sclerosis phenotype. Hum Mol Genet (2009) 18(11):2071–7. doi:10.1093/hmg/ddp119
60. Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet (2011) 43(4):329–32. doi:10.1038/ng.789
61. Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary Sjogren’s syndrome. Nat Rev Rheumatol (2013) 9(9):544–56. doi:10.1038/nrrheum.2013.110
62. Rowe D, Griffiths M, Stewart J, Novick D, Beverley PC, Isenberg DA. HLA class I and II, interferon, interleukin 2, and the interleukin 2 receptor expression on labial biopsy specimens from patients with Sjogren’s syndrome. Ann Rheum Dis (1987) 46(8):580–6. doi:10.1136/ard.46.8.580
63. Thrane PS, Halstensen TS, Haanaes HR, Brandtzaeg P. Increased epithelial expression of HLA-DQ and HLA-DP molecules in salivary glands from patients with Sjogren’s syndrome compared with obstructive sialadenitis. Clin Exp Immunol (1993) 92(2):256–62. doi:10.1111/j.1365-2249.1993.tb03389.x
64. Manoussakis MN, Dimitriou ID, Kapsogeorgou EK, Xanthou G, Paikos S, Polihronis M, et al. Expression of B7 costimulatory molecules by salivary gland epithelial cells in patients with Sjogren’s syndrome. Arthritis Rheum (1999) 42(2):229–39. doi:10.1002/1529-0131(199902)42:2<229::AID-ANR4>3.0.CO;2-X
65. Dimitriou ID, Kapsogeorgou EK, Moutsopoulos HM, Manoussakis MN. CD40 on salivary gland epithelial cells: high constitutive expression by cultured cells from Sjogren’s syndrome patients indicating their intrinsic activation. Clin Exp Immunol (2002) 127(2):386–92. doi:10.1046/j.1365-2249.2002.01752.x
66. Amft N, Bowman SJ. Chemokines and cell trafficking in Sjogren’s syndrome. Scand J Immunol (2001) 54(1–2):62–9. doi:10.1046/j.1365-3083.2001.00970.x
67. Barone F, Bombardieri M, Rosado MM, Morgan PR, Challacombe SJ, De Vita S, et al. CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with Sjogren’s syndrome and MALT lymphoma: association with reactive and malignant areas of lymphoid organization. J Immunol (2008) 180(7):5130–40.
68. Tsunawaki S, Nakamura S, Ohyama Y, Sasaki M, Ikebe-Hiroki A, Hiraki A, et al. Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjogren’s syndrome. J Rheumatol (2002) 29(9):1884–96.
69. Ittah M, Miceli-Richard C, Gottenberg JE, Sellam J, Eid P, Lebon P, et al. Viruses induce high expression of BAFF by salivary gland epithelial cells through TLR- and type-I IFN-dependent and -independent pathways. Eur J Immunol (2008) 38(4):1058–64. doi:10.1002/eji.200738013
70. Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. B-cell activating factor of the tumour necrosis factor family expression in blood monocytes and T cells from patients with primary Sjogren’s syndrome. Scand J Immunol (2008) 67(2):185–92. doi:10.1111/j.1365-3083.2007.02049.x
71. Lavie F, Miceli-Richard C, Quillard J, Roux S, Leclerc P, Mariette X. Expression of BAFF (BLyS) in T cells infiltrating labial salivary glands from patients with Sjogren’s syndrome. J Pathol (2004) 202(4):496–502. doi:10.1002/path.1533
72. Ittah M, Miceli-Richard C, Eric Gottenberg J, Lavie F, Lazure T, Ba N, et al. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjogren’s syndrome. Arthritis Res Ther (2006) 8(2):R51. doi:10.1186/ar1912
73. Strandberg L, Ambrosi A, Espinosa A, Ottosson L, Eloranta ML, Zhou W, et al. Interferon-alpha induces up-regulation and nuclear translocation of the Ro52 autoantigen as detected by a panel of novel Ro52-specific monoclonal antibodies. J Clin Immunol (2008) 28(3):220–31. doi:10.1007/s10875-007-9157-0
74. Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis (2003) 8(3):237–49. doi:10.1023/A:1023668705040
75. Gannot G, Bermudez D, Lillibridge D, Fox PC. Fas and Fas-mediated effects on a human salivary cell line in vitro: a model for immune-mediated exocrine damage in Sjogren’s syndrome. Cell Death Differ (1998) 5(9):743–50. doi:10.1038/sj.cdd.4400414
76. Matsumura R, Umemiya K, Goto T, Nakazawa T, Ochiai K, Kagami M, et al. Interferon gamma and tumor necrosis factor alpha induce Fas expression and anti-Fas mediated apoptosis in a salivary ductal cell line. Clin Exp Rheumatol (2000) 18(3):311–8.
77. Polihronis M, Tapinos NI, Theocharis SE, Economou A, Kittas C, Moutsopoulos HM. Modes of epithelial cell death and repair in Sjogren’s syndrome (SS). Clin Exp Immunol (1998) 114(3):485–90. doi:10.1046/j.1365-2249.1998.00705.x
78. Ohlsson M, Jonsson R, Brokstad KA. Subcellular redistribution and surface exposure of the Ro52, Ro60 and La48 autoantigens during apoptosis in human ductal epithelial cells: a possible mechanism in the pathogenesis of Sjogren’s syndrome. Scand J Immunol (2002) 56(5):456–69. doi:10.1046/j.1365-3083.2002.01072_79.x
79. Unoki H, Moriyama A, Tabaru A, Masumoto A, Otsuki M. Development of Sjogren’s syndrome during treatment with recombinant human interferon-alpha-2b for chronic hepatitis C. J Gastroenterol (1996) 31(5):723–7. doi:10.1007/BF02347624
80. Cummins MJ, Papas A, Kammer GM, Fox PC. Treatment of primary Sjogren’s syndrome with low-dose human interferon alfa administered by the oromucosal route: combined phase III results. Arthritis Rheum (2003) 49(4):585–93. doi:10.1002/art.11199
81. Khurshudian AV. A pilot study to test the efficacy of oral administration of interferon-alpha lozenges to patients with Sjogren’s syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod (2003) 95(1):38–44. doi:10.1067/moe.2003.30
82. De Santi L, Costantini MC, Annunziata P. Long time interval between multiple sclerosis onset and occurrence of primary Sjogren’s syndrome in a woman treated with interferon-beta. Acta Neurol Scand (2005) 112(3):194–6. doi:10.1111/j.1600-0404.2005.00455.x
Keywords: interferon signature, Sjögren’s syndrome, gene expression profiling, microarrays, type I interferon, genetic association, mechanisms, biomarker
Citation: Li H, Ice JA, Lessard CJ and Sivils KL (2013) Interferons in Sjögren’s syndrome: genes, mechanisms, and effects. Front. Immunol. 4:290. doi: 10.3389/fimmu.2013.00290
Received: 08 July 2013; Paper pending published: 26 July 2013;
Accepted: 04 September 2013; Published online: 20 September 2013.
Edited by:
Timothy B. Niewold, Mayo Clinic, USACopyright: © 2013 Li, Ice, Lessard and Sivils. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathy L. Sivils, Arthritis and Clinical Immunology Research Program, Oklahoma Medical Research Foundation, 825 N.E. 13th Street, MS57, Oklahoma City, OK 73104, USA e-mail: sivilsk@omrf.org