 Priyanka Nair-Gupta
Priyanka Nair-Gupta J. Magarian Blander
J. Magarian Blander- 1Department of Medicine, Immunology Institute, Graduate School of Biological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA
- 2Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Cross-presentation involves the presentation of peptides derived from internalized cargo on major histocompatibility complex class I molecules by dendritic cells, a process critical for tolerance and immunity. Detailed studies of the pathways mediating cross-presentation have revealed that this process takes place in a specialized subcellular compartment with a unique set of proteins. In this review, we focus on the recently appreciated role for intracellular vesicular traffic, which serves to equip compartments such as endosomes and phagosomes with the necessary apparatus for conducting the various steps of cross-presentation. We also consider how these pathways may integrate with inflammatory signals particularly from pattern recognition receptors that detect the presence of microbial components during infection. We discuss the consequences of such signals on initiating cross-presentation to stimulate adaptive CD8 T cell responses.
Introduction
Classically, endogenous antigens such as proteins synthesized by virally infected cells or tumor cells, are presented on major histocompatibility class I (MHC I) molecules for detection by CD8 T cells. However, a seminal study by Bevan (1) showed that in animals immunized with fully allogeneic cells, cytotoxic CD8 T cell responses were seen specific for minor antigens from the graft that were presented on MHC I molecules of the host (1). This finding indicated that antigens from the transplanted cells could be internalized by antigen presenting cells (APC) of the host and presented on the host MHC I molecules. Bevan termed the activation of CD8 T cells to this process of antigen transfer as “cross-priming,” and later the actual process of antigen transfer was called “cross-presentation” (2). A once poorly defined phenomenon, cross-presentation is now considered to be a critical mechanism to mediate immune responses against infectious agents and tumors as well as to induce peripheral tolerance (3).
The importance of cross-presentation becomes apparent given the existence of several viruses that exhibit strict tissue tropisms such as papilloma virus where infection is mainly confined to epithelial cells in the skin barrier (4). Other examples for viruses that do not infect APC include encephalomyocarditis virus (EMCV) and semliki forest virus (SFV) (5). Additionally, some viruses such as herpes simplex virus (HSV), measles, retrovirus, canarypox virus, vaccinia virus, and lymphocytic choriomeningitis virus infect APC, but impair direct presentation of antigen (6–13). Additionally, cross-presentation has been demonstrated to play a critical role in mediating CD8 T cell immune responses against parasitic infections such as Toxoplasma gondii (14). Cross-priming has also been studied in the context of bacterial infections such as Listeria monocytogenes and Mycobacterium tuberculosis. In these infections, host defense is primarily mediated by dendritic cells (DC) that phagocytose infected apoptotic cells and mediate cross-priming, thus allowing for effective cytotoxic T lymphocytes (CTL) responses against the pathogens (15–17). Hence, cross-presentation allows for a mechanism through which the antigen can be presented by the APC without the need for direct infection.
Although other phagocytes have been reported to cross-present antigen, DC are considered to be the primary cross-presenting cell. The superior ability of DC to cross-present is largely attributed to their antigen processing capacity. Endocytic pathways in DC preserve and retain antigen epitopes via low lysosomal proteolysis and expression of protease inhibitors (18). This aspect makes sense when one considers that DC pick up antigen in the peripheral tissue and migrate for several hours-days (12–24 h for dermal DC and 3 days for Langerhans cells) reaching the lymph nodes (19). Thus, instead of being processed and degraded prematurely, retention of antigen would allow optimal cross-presentation for subsequent recognition by lymph node resident naïve CD8 T cells. However, DC subsets are heterogeneous in their ability to cross-present antigens. Subsets such as conventional splenic and lymph node resident CD8α+ DC, migratory DC populations such as lung and dermal CD103+ DC as well as monocyte-derived inflammatory DC excel at cross-presentation (20–24). It is still unclear why these DC subsets are specialized for cross-presentation. Interestingly, conventional CD8α+ DC as well migratory CD103+ DC populations appear to rely exclusively on Batf3 and IRF8 transcription factors for their development (25–28). Therefore, it is curious to ask if the ability to cross-present is developmentally controlled by the Batf3/IRF8 transcription programs that enable these subsets to have unique and specialized compartments geared toward cross-presentation. On the other hand, in vitro studies argue that the ability of splenic CD8α+ DC to cross-present antigen is induced as a subsequent step in maturation aided by cytokines such as granulocyte-macrophage colony-stimulating factor (GMCSF) or exposure to microbial products (29).
Recent studies in human DC have proposed the lymphoid and non-lymphoid resident BDCA3+ (CD141+) DC to be the human counterparts of the cross-presenting murine lymphoid CD8α+ DC and non-lymphoid CD103+ DC (30–34). The BDCA3+ DC subset is indeed an attractive candidate for the human homolog as it shares with the murine CD8α+ and CD103+ DC several cell surface markers, including DNGR1 and XCR1, transcription factors such as Batf3 and IRF8, along with excelling at in vitro cross-presentation assays. However, evidence also exists contradicting the superiority of the BDCA3+ DC subset at cross-presentation (35–37). Furthermore, patients harboring an autosomal dominant mutation in IRF8 selectively lose BDCA-1+ DCs but not BDCA3+ DCs in the peripheral blood, indicating that BDCA3+ DC is at least not developmentally regulated in the same manner as murine cross-presenting subsets (38). Additionally although Batf3 deficiency impairs development of BDCA3+ DC in vitro, humanized mice reconstituted with Batf3 deficient progenitors still display sufficient and comparable numbers of BDCA3+ DCs (39). Thus, even though the human BDCA3+ DC subset appears to be functionally related to the murine CD8α+ and CD103+ DC, further studies are warranted to determine their developmental program and subsequent specialization for cross-presentation.
Several groups have also focused their efforts on revealing the intracellular pathways and molecular mechanisms mediating cross-presentation at steady state. Three major mechanisms have been determined. The phagosome-to-cytosol pathway involves escape of the antigen into the cytosol, possibly mediated by Sec61, followed by degradation by the proteasome and transport of peptides into the endoplasmic reticulum (ER) by the transporter associated with antigen presentation (TAP) to be loaded onto MHC I molecules (40, 41). The ER-phagosome fusion pathway involves fusion of the ER-Golgi intermediate compartment (ERGIC) with the phagosome, leading to the formation of a so called “ERgosome,” where ERGIC components such as TAP and components of the peptide loading complex are recruited directly to the phagosome. This recruitment is mediated through pairing of the ER SNARE Sec22b with the plasma membrane SNARE syntaxin-4, which is also found on phagosomes (42). In this model, the antigen still requires escape into the cytosol for proteasomal degradation and is then imported back to the “ERgosome” by TAP to be loaded onto MHC I molecules (43, 44). In contrast to the cytosolic pathways, the vacuolar pathway involves direct processing of the antigen within the phagosome by endocytic proteases such as cathepsins and subsequent loading of peptides onto MHC I molecules (45).
Some of the above pathways suggest that cross-presentation takes place in a specialized intracellular compartment. This compartment could be endosomes or phagosomes depending on the size of the exogenous antigen and mode of internalization. For instance, in the ERgosome model, cross-presentation takes place in a subcellular compartment bearing characteristics of both the ERGIC and the endosome/phagosome. In this review, we elaborate on the vesicular pathways that serve to bring various components of the cross-presentation machinery to such specialized intracellular compartments. We discuss the unique combination of proteins in these compartments that make it attractive for cross-presentation at steady state and how this compartment might be modified and further optimized for efficient cross-presentation in the scenario of infection. We also review evidence for regulation of cross-presentation during microbial stimulation and discuss if this process can still take place at steady state.
Nature of the Cross-Presenting Compartment at Steady State
In recent years, several groups have elucidated the architecture of the “cross-presenting” compartment at steady state. This compartment contains several identified proteins brought in by distinct vesicular pathways. These proteins may all be present in the compartment. Alternatively, these vesicular pathways may be mutually exclusive of one another, culminating in the presence of only some of these identified proteins. Regardless, these proteins serve as important players in executing different steps of the cross-presentation response (Figure 1).
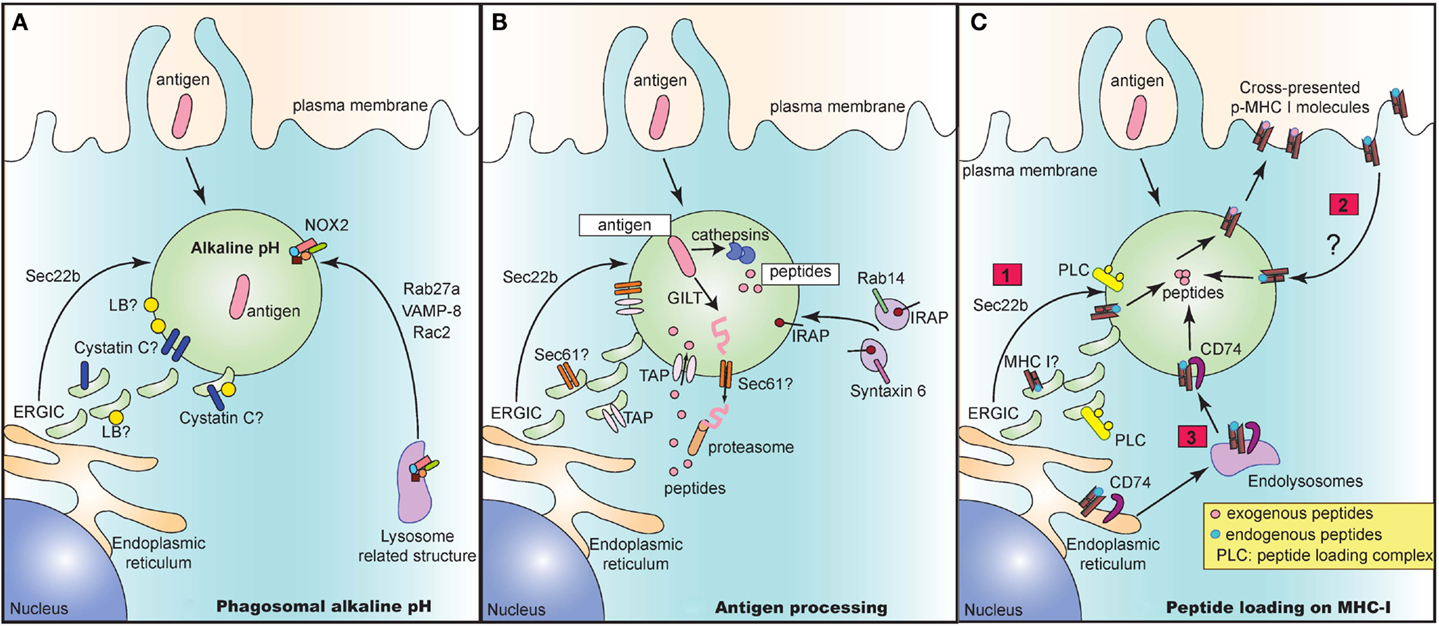
Figure 1. The cross-presentation compartment at steady state. Several vesicular pathways have been proposed to mediate delivery of key proteins that play an integral role in the crucial steps of cross-presentation including maintenance of alkaline pH (A), processing of antigen to peptides (B) and subsequent loading of peptides onto MHC I molecules (C). (A) Delivery of NOX2 via Rab27a, VAMP-8, and Rac2 is critical for maintaining an alkaline pH in intracellular cross-presenting compartments. Alternatively, recruitment of ERGIC via Sec22b also regulates pH and proteolytic activity in phagosomes. The ERGIC may contain protease inhibitors such as Cystatin C and lipid bodies (LB) that could directly alter phagosomal pH and enzymatic activity of proteases. (B) In the vacuolar pathway, antigen is directly processed into peptides by phagosome resident proteases such as cathepsins. In the cytosolic pathway, the antigen may need to be first unfolded by GILT prior to exit into the cytosol via a channel. This mystery translocon may be Sec61 and could be present in ERGIC, thereby recruited to the cross-presenting compartment via Sec22b. Once in the cytosol, antigen is then degraded into peptides and shuttled back into the phagosome via phagosomal TAP. TAP is dependent on Sec22b for its recruitment from ERGIC to endocytic compartments. Finally, IRAP, which is present on Rab14+ and Syntaxin 6+ endosomes, is recruited to phagosomes and mediates trimming of imported peptides that are further optimized for cross-presentation. (C) Lastly, processed and trimmed peptides have to now be loaded onto MHC I molecules. Source of MHC I in such compartments is unclear and could either be recruited from (1) ERGIC via Sec22b or (2) recycling from plasma membrane or (3) recruitment from endolysosomal compartments via CD74. In all cases, the peptide loading complex (PLC) is recruited from ERGIC via Sec22b to the compartment and can chaperone loading of exogenous foreign peptides to create “cross-presentable” peptide-MHC complexes.
Maintenance of Optimal Alkaline pH
As antigens are internalized by endocytosis or phagocytosis, they undergo gradual proteolytic degradation along their journey from early endosomes and phagosomes to lysosomes (Figure 1A). Once in lysosomes, antigens are degraded by lysosomal proteases, which could destroy potential peptide epitopes crucial for T cell activation. DC circumvent this problem by expressing low levels of lysosomal proteases (18). Additionally, since most of these proteases function optimally at acidic pH (46), maintenance of a strongly alkaline pH in the cross-presentation compartment would inhibit protease activity, thus preventing overt and premature degradation of antigens. To this end, DC were reported to have high phagosomal pH, reaching values of 7.5–8 in contrast to macrophages which rapidly acidified their phagosomes, reaching values of 4.5–5 following phagocytosis of inert latex beads (47). Alkalinization of phagosomes in DC was attributed to selective recruitment, assembly and functioning of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase NOX2. Finally, NOX2 activity was shown to be crucial for cross-presentation as genetic deletion of NOX2 subunit gp91phox led to abrogation of cross-presentation.
How does NOX2 reach the cross-presenting compartment? Recruitment of NOX2 is facilitated by Rab27a (48), a small guanosine tri-phosphatase (GTPase), which was initially characterized to mediate regulatory exocytosis of secretory vesicles in hematopoietic and non-hematopoietic cells (49). In a separate study, the Rac2 GTPase was demonstrated to also control the recruitment and assembly of phagosomal NOX2 in splenic CD8α+ DC as well as in in vitro derived bone-marrow derived DC (50). Additionally, VAMP-8, a SNARE protein which interacts with plasma membrane and phagosomal SNAREs syntaxin-4 and SNAP-23 (51), has also been recently reported to play a role in NOX2 recruitment and in mediating cross-presentation of phagocytic antigen (52). Interestingly, the protozoan Leishmania specifically cleaves VAMP-8 in phagocytes to prevent NOX2 assembly, thereby acidifying the phagosomes, in order to evade the cross-presentation response. However, it is still unclear if these GTPases and SNARE proteins act in concert or independently of one another to mediate recruitment of NOX2 and in turn to control cross-presentation. Given that VAMP-8 also participates in trafficking of secretory vesicles (53, 54), it is tempting to speculate that VAMP-8 and Rab27a might be present in similar secretory granules and are routed to the cross-presenting compartment upon entry of antigen.
In a second pathway, Sec22b mediated recruitment of ERGIC components has additionally been implicated in the maintenance of an alkaline pH (42). Sec22b silenced DC phagosomes have higher levels of mature cathepsin D, increased proteolytic activity, leading to accelerated degradation of antigen. These results therefore suggest that the ERGIC contains protease inhibitors. Which protease inhibitors could be involved? The cystatin family of protease inhibitors has been implicated to play a role in antigen presentation. Cystatin C was demonstrated to inhibit degradation of CD74, leading to enhanced accumulation of MHC II in endolysosomal compartments (31). Interestingly, cystatin C is abundantly expressed by CD8α+ DC compared to CD8α− DC from the spleen (32), and only partially colocalizes with endolysosomal compartments (31, 32). Given that the cellular localization of cystatin C as well as its role in cross-presentation is still unclear, a feasible possibility is that cystatin C could perhaps colocalize with ERGIC and play a role in cross-presentation.
Another explanation for why recruitment of ERGIC would delay phagosome maturation is that the ERGIC may contain lipid bodies (LB) that have been implicated in regulating phagosomal alkalinization and antigen cross-presentation (55). These LB accumulate in the cytosol and on DC phagosomes in an interferon (IFN)-inducible ER-resident GTPase (Igtp) dependent manner. Specifically, Igtp was shown to interact with LB resident adipose differentiation related protein (ADFP) to mediate formation of LB, which were crucial for cross-presentation (55).
Antigen Processing
The cytosolic pathway model of cross-presentation stipulates that once antigen is internalized, it has to make its way out of the endosome/phagosome and into the cytosol for proteasomal degradation (Figure 1B). It is generally thought that prior to export into the cytosol, antigens may need to be unfolded. For certain antigens, this is a challenge owing to specific di-sulfide bonds holding the structure of the antigen together. In this case, gamma-interferon-inducible lysosomal thiolreductase (GILT) has been shown to be critical for cross-presentation of di-sulfide bonds containing antigen derived from HSV infected cells (56). Once unfolded, antigen is then routed to the cytosol through a channel, the identity of which still remains enigmatic and controversial. Sec61, a translocon involved in the ER associated degradation pathway (ERAD) was regarded as a top candidate given that blocking Sec61 activity by using Pseudomonas aeruginosa bacteria exotoxin A resulted in loss of cross-presentation of soluble OVA antigen (41). However, the evidence for exotoxin A directly and solely blocking Sec61 channel activity is still lacking. Interestingly, DC lacking Sec22b SNARE protein via short hairpin ribonucleic acid (shRNA) targeted deletion, showed impaired antigen export from endocytic compartments, thus arguing for the recruitment of an ERGIC resident translocon channel (42). Further studies analyzing phagosomal proteomics of these Sec22b sufficient and deficient DC would be integral to revealing the identity of the enigmatic translocon.
Once in the cytosol, it is well accepted that the antigen undergoes proteasomal degradation resulting in the generation of peptides. These peptides are then reimported by TAP into the lumen of the cross-presenting compartment (57). TAP is an ER and ERGIC resident protein that has been demonstrated to be recruited to phagosomes in a Sec22b dependent manner (42). Several groups have confirmed the dependence of cross-presentation on TAP, although in the case of certain bacterial antigens, cross-presentation can take place even in the absence of TAP via the vacuolar pathway where antigens are processed within endosomes and phagosomes by resident proteases (3).
Upon internalization of exogenous antigens, newly generated peptides can be trimmed by endosomal insulin-regulated aminopeptidase (IRAP) to be further optimized for cross-presentation (58). IRAP−/− DC are impaired in their ability to cross-present soluble and particulate antigen, thus implicating the importance of IRAP in cross-presentation. IRAP colocalizes with Rab14+ and syntaxin 6+ endosomes at steady state, and is recruited to phagosomes after antigen uptake (58, 59). Whether IRAP depends on Rab14 GTPase and syntaxin 6 SNARE proteins for its delivery to the phagosomes remains to be studied.
Peptide Loading on MHC I
Processed and trimmed peptides are now faced with the possibility of being loaded on MHC I molecules that are present within endosomes and phagosomes (Figure 1C). A question that remains is where these MHC I molecules are recruited from. An attractive possibility is that MHC I molecules are present in the ERGIC and that perhaps Sec22b can deliver MHC I along with its chaperone proteins such as calreticulin and tapasin to the cross-presenting compartment. However, analysis of MHC I molecules in cell lines has revealed that MHC I molecules are transiently trafficked through the ERGIC at steady state. In fact, MHC I accumulated in ERGIC only in conditions where these molecules are not bound to high affinity peptides which could occur in the absence of TAP or calreticulin or when traffic out of the ERGIC is blocked (60–64). Whether MHC I trafficking in APC occurs similarly is still unclear.
An alternative possibility is that MHC I may be derived from the plasma membrane. Indeed, endocytosis and subsequent recycling of MHC I has been extensively documented in several cell lines where internalized MHC I are delivered to endosomal recycling compartments (ERC) in a step prior to being re-routed to the plasma membrane (63). Trafficking patterns of MHC I in APC are less clear. Some studies in APC confirm the reliance on plasma membrane derived MHC I, where internalization of surface MHC I molecules was shown to be dependent on a conserved tyrosine within the cytosolic domain of the MHC I, and to a lesser extent on a conserved serine phosphorylation site (65, 66). Mutations in these conserved sites resulted in inhibition of cross-presentation both in vitro and in vivo. However, given the strong evidence in cell lines for accumulation of MHC I in ERC, further studies are warranted to determine the contribution of MHC I molecules recycling through the ERC to cross-presentation.
Finally, it was also recently shown that CD74, which was originally characterized to route MHC II molecules from the ER to lysosomal compartments (67), was also important in mediating cross-presentation of viral and cell-associated antigen (68). CD74 was found in complex with immature endoglycosidase sensitive MHC I, indicating that it associates with newly synthesized MHC I in the ER (69). In CD74−/− DC, MHC I molecules were present to a lesser extent in LAMP-1+ compartments, implying that CD74 delivers MHC I from the ER to endolysosomal compartments.
Nature of the Cross-Presenting Compartment during Infection
In spite of these studies detailing the molecular and cellular makeup of cross-presenting compartments at steady state, the mechanisms underlying regulation and remodeling of this compartment during infection remain largely undefined. Upon uptake of microbial or infected cellular cargo, the phagosomal or endosomal compartment can be substantially modified by the acquisition of pattern recognition receptors (PRRs) (16). PRRs are evolutionarily conserved receptors that recognize and respond to conserved pathogen-associated molecular patterns (PAMPs) which are unique to microbes (70). Upon PRR engagement, intracellular signal transduction pathways are initiated such as those mediated by nuclear factor-κB (NF-κB), mitogen-activated protein kinases (MAPKs), and IFN-regulatory factors (IRFs) (71). These pathways are critical for providing immunity against several pathogen infections.
Several families of mammalian PRRs have been identified, namely Toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like (NOD-like) receptors (NLRs), RIG-I like receptors (RLRs), and C-type lectin receptors (CLRs) (72). Indeed, TLRs were the first family of PRRs to be studied in detail (73–75). We focus here on recent developments in the biology of TLR that enable the recruitment of these receptors and assembly of their signaling machinery. This recruitment aids in the transduction of responses from the compartment itself, allowing localized phagosomal or endosomal specific control of responses, including presentation of exogenous antigen on MHC I and MHC II molecules. To signal and regulate presentation of exogenous antigen from endosomal compartments, TLRs first need to be recruited to the relevant compartment.
TLR4
TLR4 is a plasma membrane resident “surface” TLR that can be endocytosed and also signal from intracellular compartments upon interaction with its ligand lipopolysaccharide (LPS). TLR4 initially engages toll-interleukin 1 receptor (TIR) domain-containing adaptor protein (TIRAP) and myeloid differentiation primary-response protein (MyD88) to initiate signal transduction from the plasma membrane (76). Subsequently, TLR4 is internalized into endocytic compartments and engages TRIF-related adaptor molecule (TRAM) and TIR-domain-containing adaptor inducing IFN-β (TRIF) (76). In fact, blocking TLR4 endocytosis using dynamin inhibitors selectively inhibits TRIF-TRAM mediated IRF3 dependent type I IFN responses without affecting TIRAP-MyD88 dependent signaling (77).
Recently, CD14 was shown to regulate the endocytosis of TLR4 from the plasma membrane during stimulation with LPS (78). TLR4 acts as a cargo for CD14, which transports the receptor and LPS to endosomes in a Syk and PLCγ2 dependent process where TRIF signaling leads to IFN-β production. While CD14 is critical for TLR4 endocytosis and IFN-β production in response to soluble LPS, TLR4 endocytosis in DC can proceed in the absence of CD14 during phagocytosis of E. coli or LPS-coated beads, although a lower percent of TLR4 is internalized compared to soluble LPS. Notably, despite lower percent TLR4 internalization in response to these particles, IFN-β production, which relies on TLR4 signaling from endocytic compartments, was unaffected in the absence of CD14. This result suggests that in the case of phagocytosed cargo, TLR4 can accumulate on phagosomes from another source independently of the plasma membrane.
Indeed, the small GTPase Rab11a was shown to play a crucial role in trafficking TLR4 from ERC to phagosomes containing gram-negative and not gram-positive bacteria, leading to IRF3 activation and IFN-β transcription (79). Additionally, adaptor protein 3 (AP-3) also plays a role in recruiting TLR4 and MyD88 from intracellular stores to phagosomes containing TLR4 ligands (80). Interestingly, AP-3 dependent recruitment of TLR4 and MyD88 was crucial for mediating production of pro-inflammatory cytokines selectively in response to phagocytic cargo and not to soluble LPS. Hence, the mode of uptake can dictate the pathway of delivery of TLR4.
It is curious to ask if the recruitment of TLR4 via these regulatory transport proteins contributes toward antigen presentation and adaptive immunity. For MHC II presentation, phagosome autonomous TLR4 signaling led to accelerated phagosome maturation and subsequent degradation of CD74 specifically in phagosomes bearing TLR ligands and not other phagosomes in the same DC (81, 82). Additionally, impaired recruitment of TLR4 to phagosomes in AP-3 deficient mice also led to decreased MHC II presentation (80). However, whether any of these proteins regulate cross-presentation of antigen internalized via endocytosis or phagocytosis remains to be investigated.
TLR9
TLR9 is an endosomal receptor and begins its journey in the ER, where it associates with the chaperone protein Unc-93 homolog B (UNC93B), which mediates its transport to endosomes (83). Recently, recruitment of TLR9 and UNC93B was demonstrated selectively to phagosomes that contained DNA and anti-DNA immunoglobulin (Ig) complexes (84). Importantly, phagosomal TLR9 recruitment did not depend on its ability to sense the presence of TLR9 ligand DNA but instead relied on Fc receptor γ (FcRγ) mediated engagement by Ig complexes. These data suggest that Ig mediated FcRγ signaling leads to recruitment of TLR9 to phagosomes. When these phagosomes also contain the TLR9 ligand DNA, TLR9 signaling is engaged, resulting in IFN-α secretion. Given that engagement of FcRγ prepares phagosomes for optimal TLR9 signaling, it is tempting to speculate that this synergy between FcRγ and TLR9 may also impact subsequent cross-presentation responses of antigen complexed with DNA-Ig aggregates.
TLR2
TLR2 is a cell surface TLR that synergizes with other surface TLRs such as TLR1 and TLR6 to mediate MyD88 dependent signal transduction responses. Similar to TLR4, there are reports showing TLR2 localization to endosomal compartments specifically early endosomes, lysosomes, and Rab11a+ vesicles in monocytes (85). Some studies also indicate TLR2 dependent induction of type I IFN signaling from endocytic compartments (86, 87). How TLR2 is directed to such compartments is still unclear.
PRR Regulation of Cross-Presentation
Studies looking at the role of PRRs in cross-presentation have been largely limited to TLRs and CLRs. These receptors are well suited to regulate cross-presentation as they are present at the plasma membrane as well as along the endocytic pathway, where they encounter microbial antigen and initiate signaling to regulate adaptive immune responses such as cross-presentation. Here, we present evidence supporting regulation by these receptors.
Toll-Like Receptors
There are several studies that show that TLR signaling enhances cross-priming of CD8 T cells (88). The in vivo contribution of TLR3 to cross-priming became clear after an elegant study demonstrated that signaling via TLR3 leads to maturation of DC and therefore promotes virus-specific CD8 T cell responses (5). Another study showed that injecting mice with apoptotic vesicles derived from M. tuberculosis infected macrophages, activates DC via TLR2 in a MyD88 dependent manner and can cross-prime CD8 T cells, thereby protecting mice from developing tuberculosis infection (17). Moreover, by employing biodegradable microspheres for the delivery of phagocytic cargo to DC, Schlosser et al. were able to demonstrate that the presence of both TLR ligand and antigen within the same phagosome yielded efficient CTL responses as compared to when the ligand and antigen are located in separate phagosomes (89).
One caveat of the studies above is the inability to distinguish the effects of TLR signaling on cross-presentation versus cross-priming. For example, a couple of studies revealed that TLR3 and TLR9 ligands could both induce cross-presentation of OVA by DC (90, 91). This induction of cross-presentation was found to be dependent on TLR signaling as DC from Tlr9−/− and Myd88−/− mice were unable to cross-present antigen after exposure to TLR ligands. However, the authors of these studies relied exclusively on T cell activation as a measure of displayed peptide-MHC I complexes. Given that TLR signaling also controls DC maturation and expression of co-stimulatory molecules that are pivotal for T cell activation, it is difficult to rule out the confounding factor of impaired co-stimulation and decreased DC maturation seen with TLR deficient DC. In fact, the inability of Myd88−/−DC to activate CD8 T cells after phagocytosis of virally infected cells was fully restored by treatment with a CD40 cross-linking antibody suggesting that defective cross-priming was due to impaired TLR induced co-stimulation rather than cross-presentation per se in this particular case (92).
The most direct way to assess cross-presentation is to use a conformation dependent antibody directly against preformed peptide-MHC I complexes on the surface of the APC. However, these antibodies are quite insensitive and work successfully only when DC were pulsed with large amounts of antigen. Nevertheless, Christian Kurts’ group successfully used 25D1.16 antibody to detect SIINFEKL-MHC I complexes and thus demonstrated increased cross-presentation with TLR signaling after uptake of soluble OVA in the presence of LPS (93). This increase in cross-presentation was mediated by TLR4, MyD88 and not TRIF signaling. However, this study was focused on the cross-presentation of soluble antigen and hence whether TLR signaling enhances cross-presentation of phagocytosed particulate antigen remains to be determined.
C-Type Lectin Receptors
C-type lectin receptors contain at least one carbohydrate recognition domain via which they bind to sugar moieties on self or microbial derived antigens. CLRs can regulate cross-presentation, although most of them do so by regulating antigen uptake. For example, CD209 and mannose receptor have been reported to increase internalization of cargo and target antigens to early endosomes for cross-presentation (94, 95). DNGR1 was also reported to enhance cross-presentation of cellular antigens derived from necrotic dying cells (96). Interestingly, despite intact maturation phenotypes and maintenance of signals to relay co-stimulation in DNGR1 deficient DC, these DC were impaired in cross-presentation, suggesting specific regulation of cross-presentation rather than cross-priming (97). DNGR1 is also expressed at high levels in subsets of DC specialized for cross-presentation including murine CD8α+ DCs and tissue-resident CD103+ CD11b− DCs as well as in human counterparts BDCA3+ DC (39).
Can Cross-Presentation Still Take Place at Steady State?
In the absence of inflammation or infection, cross-presentation of self-antigens at steady state can take place, leading to tolerance to host antigens and deletion of potentially auto-reactive CD8 T cells. Indeed, generation and activity of CTL must be tightly controlled to avoid auto-reactivity to self, given the potency of CTL in killing infected target host cells (98, 99). Here, we review existing evidence for the role of cross-presentation in both central and peripheral tolerance mechanisms.
Peripheral Tolerance
Peripheral tolerance constitutes mechanisms of tolerance that take place after mature lymphocytes enter into the periphery. There are several studies that argue for the constitutive TLR or PRR-independent nature of cross-presentation for the induction of peripheral cross-tolerance to non-inflammatory self-antigens, leading to deletion of self-reactive CTL. Many of these models employed the expression of neo-self-antigens under the control of tissue-specific promoters like the rat insulin promoter (RIP). These models ensure that the antigens are expressed outside of the thymus, allowing researchers to specifically study peripheral tolerance.
Cross-tolerance was first demonstrated when OVA specific OT-I CD8 T cells were efficiently deleted after being adoptively transferred into a mouse expressing OVA under control of the RIP (RIP-mOVA) (100). Cross-tolerance was also shown to be important for the control of endogenous auto-reactive CD8 T cells specific for naturally expressed self-antigen (101). In this study, the authors bred the RIP-mOVA mice with mice lacking GTPase Rac1 in CD11c+ cells to generate Rac1-RIP mice. Conveniently, deficiency in Rac1 GTPase selectively affected the ability of CD8α+ DC to internalize antigen (33), resulting in impaired cross-presentation while leaving the classical MHC II and MHC I pathways of antigen presentation unaffected (101). Consequently, they were able to demonstrate that DC in Rac1-RIP mice failed to cross-present transgenic self-antigen and hence failed to delete transferred OT-I T cells. Moreover these mice developed symptoms of diabetes. Interestingly, mice that just had Rac1 deleted in CD11c+ cells also had higher numbers of endogenous CD8 T cells, although the mice seemed healthy and did not develop autoimmunity. However, when CD25 depleted T cells from these mice were transferred into lymphopenic hosts, the hosts developed several signs of autoimmunity as a result of homeostatic T cell proliferation. Hence, the above studies clearly demonstrate the role of cross-presentation under steady state to induce peripheral tolerance.
An interesting study by Christian Kurts’ group looked to see if tolerogenic DC could be converted into autoimmunogenic DC after exposure to stimulating conditions such as TLR ligands (102). TLR ligands were able to induce CTL mediated autoimmunity only in cases where antigen specific CD4 T cell help was provided concomitantly. These results demonstrated that the mere presence of TLR ligands, such as those present in commensal bacteria or those derived from the use of vaccine adjuvants, is not sufficient to break cross-tolerance mechanisms.
Central Tolerance
In addition to peripheral tolerance, cross-presentation was also implicated in the induction of central tolerance (103). Central tolerance is induced in the thymus where developing thymocytes that recognize peptide-MHC complexes are positively selected to express either CD4 or CD8 molecules. Subsequently, thymocytes that are able to recognize self-peptide-MHC complexes with high affinity are efficiently deleted via negative selection. Medullary thymic epithelial cells (mTECs) and DC play a critical role in mediating negative selection. mTECs exclusively express a broad range of tissue-specific antigens (TSA) (104, 105). In spite of expressing both MHC II and MHC I molecules, mTECs are poor APC. Bevan’s group was the first to show that bone-marrow derived cells in the thymus were capable of cross-presenting antigen captured from mTECs (103). However, this study also showed that mTECs, by themselves, were capable of direct presentation on MHC I molecules and were sufficient to induce CD8 T cell deletion, thus diminishing the importance of cross-presentation in central tolerance. A recent report does point to the relevance of cross-presentation by human thymic DC. Upon analyzing peptides eluted from both MHC I and MHC II molecules of human thymic DC, the authors observed that around 22% of the MHC I ligands were derived from proteins present in the vesicular/extracellular compartment the presentation of which would typically be associated with the classical MHC II pathway (30).
Conclusion
The studies we reviewed here certainly point to the constitutive nature of cross-presentation, however, an increasingly large body of work now provides strong evidence for the capacity to enhance cross-presentation by signals from inflammatory PRRs. Having mechanisms of regulation in place allows for the generation of robust CTL responses during an infection while maintaining induction of tolerance at steady state. Further insight into these regulatory mechanisms may potentially help in tailoring better therapeutic strategies to combat infectious agents as well as tumors, while preventing autoimmunity. Hence, elucidating the mechanistic differences in vesicular trafficking between steady state and inflammatory cross-presentation would be important for developing new rationales in the design of safe and effective vaccines for anti-viral, anti-bacterial as well as anti-tumor immunity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bevan MJ. Minor H antigens introduced on H-2 different stimulating cells cross-react at the cytotoxic T cell level during in vivo priming. J Immunol (1976) 117:2233–8.
2. Carbone FR, Kurts C, Bennett SR, Miller JF, Heath WR. Cross-presentation: a general mechanism for CTL immunity and tolerance. Immunol Today (1998) 19:368–73. doi:10.1016/S0167-5699(98)01301-2
3. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol (2012) 12:557–69. doi:10.1038/nri3254
4. Tindle RW, Frazer IH. Immune response to human papillomaviruses and the prospects for human papillomavirus-specific immunisation. Curr Top Microbiol Immunol (1994) 186:217–53. doi:10.1007/978-3-642-78487-3_12
5. Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature (2005) 433:887–92. doi:10.1038/nature03326
6. Gabrilovich DI, Patterson S, Harvey JJ, Woods GM, Elsley W, Knight SC. Murine retrovirus induces defects in the function of dendritic cells at early stages of infection. Cell Immunol (1994) 158:167–81. doi:10.1006/cimm.1994.1265
7. Fugier-Vivier I, Servet-Delprat C, Rivailler P, Rissoan MC, Liu YJ, Rabourdin-Combe C. Measles virus suppresses cell-mediated immunity by interfering with the survival and functions of dendritic and T cells. J Exp Med (1997) 186:813–23. doi:10.1084/jem.186.6.813
8. Engelmayer J, Larsson M, Subklewe M, Chahroudi A, Cox WI, Steinman RM, et al. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol (1999) 163:6762–8.
9. Salio M, Cella M, Suter M, Lanzavecchia A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol (1999) 29:3245–53. doi:10.1002/(SICI)1521-4141(199910)29:10<3245::AID-IMMU3245>3.3.CO;2-O
10. Sigal LJ, Crotty S, Andino R, Rock KL. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature (1999) 398:77–80. doi:10.1038/18038
11. Ignatius R, Marovich M, Mehlhop E, Villamide L, Mahnke K, Cox WI, et al. Canarypox virus-induced maturation of dendritic cells is mediated by apoptotic cell death and tumor necrosis factor alpha secretion. J Virol (2000) 74:11329–38. doi:10.1128/JVI.74.23.11329-11338.2000
12. Servet-Delprat C, Vidalain PO, Bausinger H, Manie S, Le Deist F, Azocar O, et al. Measles virus induces abnormal differentiation of CD40 ligand-activated human dendritic cells. J Immunol (2000) 164:1753–60.
13. Sevilla N, Kunz S, Holz A, Lewicki H, Homann D, Yamada H, et al. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J Exp Med (2000) 192:1249–60. doi:10.1084/jem.192.9.1249
14. John B, Harris TH, Tait ED, Wilson EH, Gregg B, Ng LG, et al. Dynamic Imaging of CD8(+) T cells and dendritic cells during infection with Toxoplasma gondii. PLoS Pathog (2009) 5:e1000505. doi:10.1371/journal.ppat.1000505
15. Jung S, Unutmaz D, Wong P, Sano G, De Los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity (2002) 17:211–20. doi:10.1016/S1074-7613(02)00365-5
16. Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med (2003) 9:1039–46. doi:10.1038/nm906
17. Winau F, Weber S, Sad S, De Diego J, Hoops SL, Breiden B, et al. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity (2006) 24:105–17. doi:10.1016/j.immuni.2005.12.001
18. Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science (2005) 307:1630–4. doi:10.1126/science.1108003
19. Kamath AT, Henri S, Battye F, Tough DF, Shortman K. Developmental kinetics and lifespan of dendritic cells in mouse lymphoid organs. Blood (2002) 100:1734–41.
20. Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol (2009) 10:488–95. doi:10.1038/ni.1724
21. Segura E, Villadangos JA. Antigen presentation by dendritic cells in vivo. Curr Opin Immunol (2009) 21:105–10. doi:10.1016/j.coi.2009.03.011
22. Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell (2010) 143:416–29. doi:10.1016/j.cell.2010.09.039
23. Henri S, Poulin LF, Tamoutounour S, Ardouin L, Guilliams M, De Bovis B, et al. CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J Exp Med (2010) 207:189–206. doi:10.1084/jem.20091964
24. Shortman K, Heath WR. The CD8+ dendritic cell subset. Immunol Rev (2010) 234:18–31. doi:10.1111/j.0105-2896.2009.00870.x
25. Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reis E, Sousa C, et al. Essential role for ICSBP in the in vivo development of murine CD8alpha + dendritic cells. Blood (2003) 101:305–10. doi:10.1182/blood-2002-04-1088
26. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi:10.1126/science.1164206
27. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med (2009) 206:3115–30. doi:10.1084/jem.20091756
28. Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med (2010) 207:823–36. doi:10.1084/jem.20091627
29. Sathe P, Pooley J, Vremec D, Mintern J, Jin JO, Wu L, et al. The acquisition of antigen cross-presentation function by newly formed dendritic cells. J Immunol (2011) 186:5184–92. doi:10.4049/jimmunol.1002683
30. Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med (2010) 207:1273–81. doi:10.1084/jem.20100348
31. Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207:1283–92. doi:10.1084/jem.20100223
32. Jongbloed SL, Kassianos AJ, Mcdonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med (2010) 207:1247–60. doi:10.1084/jem.20092140
33. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207:1261–71. doi:10.1084/jem.20092618
34. Haniffa M, Shin A, Bigley V, Mcgovern N, Teo P, See P, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity (2012) 37:60–73. doi:10.1016/j.immuni.2012.04.012
35. Mittag D, Proietto AI, Loudovaris T, Mannering SI, Vremec D, Shortman K, et al. Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. J Immunol (2011) 186:6207–17. doi:10.4049/jimmunol.1002632
36. Cohn L, Chatterjee B, Esselborn F, Smed-Sorensen A, Nakamura N, Chalouni C, et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J Exp Med (2013) 210:1049–63. doi:10.1084/jem.20121251
37. Segura E, Durand M, Amigorena S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med (2013) 210:1035–47. doi:10.1084/jem.20121103
38. Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med (2011) 365:127–38. doi:10.1056/NEJMoa1100066
39. Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, Murphy KM, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood (2012) 119:6052–62. doi:10.1182/blood-2012-01-406967
40. Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science (1995) 267:243–6. doi:10.1126/science.7809629
41. Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity (2006) 25:607–17. doi:10.1016/j.immuni.2006.08.017
42. Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell (2011) 147:1355–68. doi:10.1016/j.cell.2011.11.021
43. Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature (2003) 425:397–402. doi:10.1038/nature01911
44. Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, et al. Phagosomes are competent organelles for antigen cross-presentation. Nature (2003) 425:402–6. doi:10.1038/nature01912
45. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity (2004) 21:155–65. doi:10.1016/j.immuni.2004.07.004
46. Claus V, Jahraus A, Tjelle T, Berg T, Kirschke H, Faulstich H, et al. Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J Biol Chem (1998) 273:9842–51. doi:10.1074/jbc.273.16.9842
47. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell (2006) 126:205–18. doi:10.1016/j.cell.2006.05.035
48. Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PM, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol (2007) 9:367–78. doi:10.1038/ncb1552
49. Tolmachova T, Anders R, Stinchcombe J, Bossi G, Griffiths GM, Huxley C, et al. A general role for Rab27a in secretory cells. Mol Biol Cell (2004) 15:332–44. doi:10.1091/mbc.E03-07-0452
50. Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, et al. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity (2009) 30:544–55. doi:10.1016/j.immuni.2009.01.013
51. Stow JL, Murray RZ. Intracellular trafficking and secretion of inflammatory cytokines. Cytokine Growth Factor Rev (2013) 24:227–39. doi:10.1016/j.cytogfr.2013.04.001
52. Matheoud D, Moradin N, Bellemare-Pelletier A, Shio MT, Hong WJ, Olivier M, et al. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe (2013) 14:15–25. doi:10.1016/j.chom.2013.06.003
53. Puri N, Roche PA. Mast cells possess distinct secretory granule subsets whose exocytosis is regulated by different SNARE isoforms. Proc Natl Acad Sci U S A (2008) 105:2580–5. doi:10.1073/pnas.0707854105
54. Tiwari N, Wang CC, Brochetta C, Ke G, Vita F, Qi Z, et al. VAMP-8 segregates mast cell-preformed mediator exocytosis from cytokine trafficking pathways. Blood (2008) 111:3665–74. doi:10.1182/blood-2007-07-103309
55. Bougneres L, Helft J, Tiwari S, Vargas P, Chang BH, Chan L, et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity (2009) 31:232–44. doi:10.1016/j.immuni.2009.06.022
56. Singh R, Cresswell P. Defective cross-presentation of viral antigens in GILT-free mice. Science (2010) 328:1394–8. doi:10.1126/science.1189176
57. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol (2013) 31:443–73. doi:10.1146/annurev-immunol-032712-095910
58. Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science (2009) 325:213–7. doi:10.1126/science.1172845
59. Weimershaus M, Maschalidi S, Sepulveda F, Manoury B, Van Endert P, Saveanu L. Conventional dendritic cells require IRAP-Rab14 endosomes for efficient cross-presentation. J Immunol (2012) 188:1840–6. doi:10.4049/jimmunol.1101504
60. Raposo G, Van Santen HM, Leijendekker R, Geuze HJ, Ploegh HL. Misfolded major histocompatibility complex class I molecules accumulate in an expanded ER-Golgi intermediate compartment. J Cell Biol (1995) 131:1403–19. doi:10.1083/jcb.131.6.1403
61. Spiliotis ET, Pentcheva T, Edidin M. Probing for membrane domains in the endoplasmic reticulum: retention and degradation of unassembled MHC class I molecules. Mol Biol Cell (2002) 13:1566–81. doi:10.1091/mbc.01-07-0322
62. Garstka M, Borchert B, Al-Balushi M, Praveen PV, Kuhl N, Majoul I, et al. Peptide-receptive major histocompatibility complex class I molecules cycle between endoplasmic reticulum and cis-Golgi in wild-type lymphocytes. J Biol Chem (2007) 282:30680–90. doi:10.1074/jbc.M701721200
63. Donaldson JG, Williams DB. Intracellular assembly and trafficking of MHC class I molecules. Traffic (2009) 10:1745–52. doi:10.1111/j.1600-0854.2009.00979.x
64. Van Hateren A, James E, Bailey A, Phillips A, Dalchau N, Elliott T. The cell biology of major histocompatibility complex class I assembly: towards a molecular understanding. Tissue Antigens (2010) 76:259–75. doi:10.1111/j.1399-0039.2010.01550.x
65. Lizee G, Basha G, Tiong J, Julien JP, Tian M, Biron KE, et al. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol (2003) 4:1065–73. doi:10.1038/ni989
66. Basha G, Lizee G, Reinicke AT, Seipp RP, Omilusik KD, Jefferies WA. MHC class I endosomal and lysosomal trafficking coincides with exogenous antigen loading in dendritic cells. PLoS One (2008) 3:e3247. doi:10.1371/journal.pone.0003247
67. Bakke O, Dobberstein B. MHC class II-associated invariant chain contains a sorting signal for endosomal compartments. Cell (1990) 63:707–16. doi:10.1016/0092-8674(90)90137-4
68. Basha G, Omilusik K, Chavez-Steenbock A, Reinicke AT, Lack N, Choi KB, et al. A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nat Immunol (2012) 13:237–45. doi:10.1038/ni.2225
69. Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem (1985) 54:631–64. doi:10.1146/annurev.bi.54.070185.003215
70. Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol (2012) 12:215–25. doi:10.1038/nri3167
71. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
72. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol (2011) 30:16–34. doi:10.3109/08830185.2010.529976
73. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell (1996) 86:973–83. doi:10.1016/S0092-8674(00)80172-5
74. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature (1997) 388:394–7. doi:10.1038/41131
75. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282:2085–8. doi:10.1126/science.282.5396.2085
76. Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med (2007) 13:552–9. doi:10.1038/nm1589
77. Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol (2008) 9:361–8. doi:10.1038/ni1569
78. Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell (2011) 147:868–80. doi:10.1016/j.cell.2011.09.051
79. Husebye H, Aune MH, Stenvik J, Samstad E, Skjeldal F, Halaas O, et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity (2010) 33:583–96. doi:10.1016/j.immuni.2010.09.010
80. Mantegazza AR, Guttentag SH, El-Benna J, Sasai M, Iwasaki A, Shen H, et al. Adaptor protein-3 in dendritic cells facilitates phagosomal toll-like receptor signaling and antigen presentation to CD4(+) T cells. Immunity (2012) 36:782–94. doi:10.1016/j.immuni.2012.02.018
81. Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science (2004) 304:1014–8. doi:10.1126/science.1096158
82. Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature (2006) 440:808–12. doi:10.1038/nature04596
83. Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature (2008) 452:234–8. doi:10.1038/nature06726
84. Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity (2012) 37:986–97. doi:10.1016/j.immuni.2012.09.014
85. Nilsen NJ, Deininger S, Nonstad U, Skjeldal F, Husebye H, Rodionov D, et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol (2008) 84:280–91. doi:10.1189/jlb.0907656
86. Ip WK, Takahashi K, Moore KJ, Stuart LM, Ezekowitz RA. Mannose-binding lectin enhances Toll-like receptors 2 and 6 signaling from the phagosome. J Exp Med (2008) 205:169–81. doi:10.1084/jem.20071164
87. Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol (2009) 10:1200–7. doi:10.1038/ni.1792
88. Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol (2010) 10:403–14. doi:10.1038/nri2780
89. Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, et al. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine (2008) 26:1626–37. doi:10.1016/j.vaccine.2008.01.030
90. Maurer T, Heit A, Hochrein H, Ampenberger F, O’Keeffe M, Bauer S, et al. CpG-DNA aided cross-presentation of soluble antigens by dendritic cells. Eur J Immunol (2002) 32:2356–64. doi:10.1002/1521-4141(200208)32:8<2356::AID-IMMU2356>3.0.CO;2-Z
91. Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, et al. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol (2003) 170:4102–10.
92. Chen M, Barnfield C, Naslund TI, Fleeton MN, Liljestrom P. MyD88 expression is required for efficient cross-presentation of viral antigens from infected cells. J Virol (2005) 79:2964–72. doi:10.1128/JVI.79.5.2964-2972.2005
93. Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol (2008) 9:558–66. doi:10.1038/ni.1601
94. Burgdorf S, Kautz A, Bohnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science (2007) 316:612–6. doi:10.1126/science.1137971
95. Tacken PJ, Ginter W, Berod L, Cruz LJ, Joosten B, Sparwasser T, et al. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood (2011) 118:4111–9. doi:10.1182/blood-2011-04-346957
96. Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature (2009) 458:899–903. doi:10.1038/nature07750
97. Zelenay S, Keller AM, Whitney PG, Schraml BU, Deddouche S, Rogers NC, et al. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J Clin Invest (2012) 122:1615–27. doi:10.1172/JCI60644
98. Bendelac A, Carnaud C, Boitard C, Bach JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J Exp Med (1987) 166:823–32. doi:10.1084/jem.166.4.823
99. Miller BJ, Appel MC, O’Neil JJ, Wicker LS. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol (1988) 140:52–8.
100. Kurts C, Kosaka H, Carbone FR, Miller JF, Heath WR. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8(+) T cells. J Exp Med (1997) 186:239–45. doi:10.1084/jem.186.2.239
101. Luckashenak N, Schroeder S, Endt K, Schmidt D, Mahnke K, Bachmann MF, et al. Constitutive crosspresentation of tissue antigens by dendritic cells controls CD8+ T cell tolerance in vivo. Immunity (2008) 28:521–32. doi:10.1016/j.immuni.2008.02.018
102. Hamilton-Williams EE, Lang A, Benke D, Davey GM, Wiesmuller KH, Kurts C. Cutting edge: TLR ligands are not sufficient to break cross-tolerance to self-antigens. J Immunol (2005) 174:1159–63.
103. Gallegos AM, Bevan MJ. Central tolerance to tissue-specific antigens mediated by direct and indirect antigen presentation. J Exp Med (2004) 200:1039–49. doi:10.1084/jem.20041457
104. Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol (2001) 2:1032–9. doi:10.1038/ni723
Keywords: cross-presentation, major histocompatibility complex class I, pattern recognition receptor, Toll-like receptor, vesicular traffic, phagosomes, endosomes, dendritic cells
Citation: Nair-Gupta P and Blander JM (2013) An updated view of the intracellular mechanisms regulating cross-presentation. Front. Immunol. 4:401. doi: 10.3389/fimmu.2013.00401
Received: 31 August 2013; Paper pending published: 25 September 2013;
Accepted: 08 November 2013; Published online: 22 November 2013.
Edited by:
Penelope Anne Morel, University of Pittsburgh, USAReviewed by:
Juan J. Garcia-Vallejo, VU University Medical Center, NetherlandsFlorent Ginhoux, Singapore Immunology Network, Singapore
Guillaume Hoeffel, Singapore Immunology Network, Singapore
Copyright: © 2013 Nair-Gupta and Blander. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Magarian Blander, Department of Medicine, Immunology Institute, Tisch Cancer Institute, and the Graduate School of Biological Sciences, Icahn School of Medicine at Mount Sinai, 1425 Madison Avenue, 12-20D, New York, NY 10029, USA e-mail: julie.blander@mssm.edu