 Marc Hilhorst
Marc Hilhorst Tsuyoshi Shirai
Tsuyoshi Shirai Gerald Berry
Gerald Berry Jörg J. Goronzy
Jörg J. Goronzy Cornelia M. Weyand
Cornelia M. Weyand- 1Division of Immunology and Rheumatology, Department of Medicine, Stanford University, Stanford, CA, USA
- 2Department of Pathology, Stanford University, Stanford, CA, USA
Granuloma formation, bringing into close proximity highly activated macrophages and T cells, is a typical event in inflammatory blood vessel diseases, and is noted in the name of several of the vasculitides. It is not known whether specific properties of the microenvironment in the blood vessel wall or the immediate surroundings of blood vessels contribute to granuloma formation and, in some cases, generation of multinucleated giant cells. Granulomas provide a specialized niche to optimize macrophage–T cell interactions, strongly activating both cell types. This is mirrored by the intensity of the systemic inflammation encountered in patients with vasculitis, often presenting with malaise, weight loss, fever, and strongly upregulated acute phase responses. As a sophisticated and highly organized structure, granulomas can serve as an ideal site to induce differentiation and maturation of T cells. The granulomas possibly seed aberrant Th1 and Th17 cells into the circulation, which are known to be the main pathogenic cells in vasculitis. Through the induction of memory T cells, aberrant innate immune responses can imprint the host immune system for decades to come and promote chronicity of the disease process. Improved understanding of T cell–macrophage interactions will redefine pathogenic models in the vasculitides and provide new avenues for immunomodulatory therapy.
Introduction
Protecting the host from infection and maintaining tissue integrity relies on two highly complex and evolutionary distinguished systems, the innate and adaptive immune system. The two arms of the immune system have developed a sophisticated and efficient crosstalk to defend the host. Monocytes that come into contact with penetrating pathogens differentiate into specialized antigen-presenting cells (APC), such as macrophages and dendritic cells (DC) (1). After phagocytosis and degradation of the pathogen, proteins are presented to specialized cells of the adaptive immune system, specifically T cells. Interactions between macrophages and T cells are critical in the communication between innate and adaptive immunity. Errors in this interaction may result in immunodeficiency, failure to destroy invading pathogens, or damage to host–tissues in the form of autoimmunity. Although the principal function of macrophages was recognized a long time ago (2), the precise mechanisms of macrophage physiology are only now beginning to be unraveled.
Chronic (aberrant) macrophage–T cell interaction leads to the formation of organized lymphoid organ structures, such as granulomas. Granulomas are typically formed during infection, especially when the host has difficulties to eliminate the infectious organism. Classic examples are granulomas induced by Mycobacterium tuberculosis infection, often considered a mechanism to contain the infectious organism (3). Granuloma formation is equally important in non-infectious disease states, such as inflammatory blood vessel disease. In giant-cell arteritis (GCA; formerly known as temporal arteritis), granulomas are an almost obligatory part of the disease process. In granulomatosis with polyangiitis (GPA; formerly known as Wegener’s granulomatosis), granuloma formation is captured in the disease name. An important issue in granulomatous diseases is whether the highly activated macrophages building the granulomatous structures have primarily a protective function or whether they are key drivers of tissue damage and disease propagation (4).
In the current review, we compare and contrast the interaction of macrophages and/or DC with T cells in the context of granuloma formation and vasculitis and focus on GCA and GPA as quintessential model systems of how the interface between innate and adaptive immunity contributes to disease pathogenesis.
Macrophages and Dendritic Cells Influence T Cells
Monocytes relocate to inflammatory lesions upon sensing a chemokine gradient (5) and can differentiate into distinct types of APC on site. A discussion of the similarities and differences between DC and macrophages is beyond the scope of this review (6). Macrophage subtypes form two main groups: M1 or classically activated macrophages (CAM) and M2 or alternatively activated macrophages (AAM). M1 generally specialize in amplifying inflammatory reactions and produce high levels of TNFα, IL-6, and IL-1β. In contrast, M2 are primarily active in tissue repair and their product profile includes IL-10, TGF-β, and growth factors. An active TGF-β pathway results in suppression of inducible nitric oxide synthase (iNOS) expression and NO secretion in macrophages, deviating the cells away from M1 differentiation (7). M1 have been described as “fighting” or “soldier” cells and M2 as “fixing” or “repair” cells (8, 9). The M2 or AAM subtype is not as well defined and much debated (4). It is plausible that monocytes can differentiate into macrophage subtypes positioned somewhere on the M1–M2 or CAM–AAM continuum and are endowed with varying adaptability and plasticity (8, 10).
Antigen Recognition and Presentation
Macrophages recognize pathogens through so-called pathogen associated molecular patterns, which are detected through Toll-like receptors (TLR) (11, 12), thus distinguishing between self and non-self. As critical recognition structures, TLR enable the build-up of a defensive immune response, they also participate in shaping immune responses underlying autoimmunity (13, 14). To orchestrate tissue cleanup and repair, macrophages must be able to recognize and remove modified host proteins and lipids, e.g., oxidized proteins and lipids. Such products are often described as danger-associated molecular patterns and require competent TLR as recognition structures (15). Oxidation of host proteins, lipids, and nucleic acids results from the action of reactive oxygen species (ROS), often derived from activated macrophages themselves. The latter process has been implicated in the development and propagation of atherosclerosis (16). Importantly, T cells also express TLR, but it is currently unknown what the precise role of these receptors is in modulating T cell function (14, 17).
Macrophage-Induced Polarization of T Cell Differentiation
Macrophages are principal regulators of immunity by processing and presenting antigens to T cells (18), which are charged with distinguishing self from non-self (19). Antigen recognition by T cells involves the highly polymorphic major histocompatibility complex (MHC) molecules classes I and II (20, 21), which selectively bind antigen peptides and present them on the surface of APC. While T cell receptors bind to HLA–peptide complexes, costimulatory molecules such as CD28 are co-ligated, a mechanism that is mandatory for a more powerful induction of T cell activation (22–24). After entering the T cell activation cascade, T cells differentiate into distinct functional lineages. Some of them become effector cells; others specialize as memory T cells and position themselves in lymphoid storage sites (25). The fate of individual T cells is ultimately shaped by the microenvironment, composed of cytokines, chemokines, and tissue-specific signals (26). The exact mechanism by which macrophages induce activation, proliferation, and differentiation of T cells is incompletely understood (27). Antigen dose, the type of APC and the contact between APC and T cell are all important variables (28). It has been proposed that DC are more powerful partners of naïve T cells and preferentially interact with T cells in organized lymphoid tissue. Conversely, macrophages function as APC for naïve and memory T cells, encountering them in peripheral tissue lesions (29). DC are thought to skew CD4+ cells toward Th1 differentiation in an IL-12 dependent manner (30). Other studies have demonstrated a similar effect of macrophages on CD4+ cells (31). DC that have been activated by inflammatory mediators can stimulate Th1 proliferation in vitro, but these same DC could not do so in vivo, possibly due to lacking pathogen contact, resulting in diminished IL-12p40 production (32).
Importantly, one study found inflammatory DC to be more potent inducers of Th17 cells when compared to inflammatory macrophages. The authors concluded that this difference was reflective of differential IL-23 production, which was observed in inflammatory DC but not in macrophages (33). In contrast, other DC subsets have been implicated in inducing regulatory T cells by virtue of producing TGF-β or expressing PD-L1 (34). Suppression of T cell function and proliferation may also result from the local action of IL-10 and TGF-β, typically secreted by M2 macrophages. Gut-residing macrophages have been implicated in inducing regulatory T cells, whereas DC were found to induce Th17 cells by secreting IL-6 combined with TGF-β and possibly IL-23 (35).
Macrophage-Induced Inhibition of T Cells
Generally, macrophages inhibit the proliferation of T cells via cell–cell contact. Abundantly studied inhibitory mechanisms in T cells are the programed death (PD1) and cytotoxic T lymphocyte antigen (CTLA) pathways. PD1 and CTLA-4 are found on T cells and mediate inhibitory signals when engaged by their respective ligands expressed on the surface of interacting macrophages (36). Malfunctioning of these inhibitory signals, e.g., due to polymorphisms, increases susceptibility for autoimmunity (37–39). Both DC and macrophages express membrane-integrated ligands for PD-1 and CTLA-4 (24). Blockade of PD-L1 on DC is a powerful mechanism to enhance T cell proliferation and cytokine release (34, 40). Besides polymorphisms in PD-1 and CTLA-4, a series of gene polymorphisms, including genes relevant for cytoplasmic signaling pathways, have been associated with susceptibility for autoimmunity (41, 42). As minor variations in threshold settings of cytoplasmic signaling cascades have the potential to bias immune interactions profoundly, it is likely that they impact macrophage–T cell interactions both by accelerating as well as downregulating immune responses.
Another concept has been that resting macrophages preferentially dampen immune responses. Accordingly, resting macrophages have been reported to induce allogeneic T cell anergy, partly by enhancing regulatory T cells. In one study, T cells proliferated when co-cultured with immature DC, became anergic when in a second co-culture with macrophages and proliferated when co-cultured with mature DC in a third co-culture, finally producing IL-2 and IFN-γ. Immature and mature DC both expressed high levels of MHC class II (HLA-DR), but macrophages did not. The costimulatory molecules CD80 and CD86 were present at higher density on mature DC than on immature DC or on macrophages (43). These studies support the notion that APC functions of macrophages and DC are fundamentally distinct.
In some infectious settings, specifically in filarial and yeast infections, the pathogen undermines protective immune responses by inducing macrophages and DC that are able to suppress T cell activation (44, 45). This mechanism is dependent on the production of TGF-β and/or IL-10 in combination with a lack of IL-6. Also, co-culturing allogeneic naïve CD4+ T cells with immature DC has been reported to lead to the expansion of IL-10 producing T cells, whereas mature DC promote the proliferation of Th1 cells (46).
Under hypoxic conditions, macrophages were found to suppress the proliferation of T cells via hypoxia-inducible factor 1α (HIF-1α). More HIF-1α knockout macrophages were necessary to suppress T cells as compared to wild-type macrophages (47). HIF-1α may enhance the production of nitric oxide species, which directly suppress T cell proliferation. In contrast, under hypoxic conditions, which enrich the environment for macrophage-derived oxidative species, T cells preferentially differentiated into Th17 cells. HIF-1α-dependent proteosomal degradation of the transcription factor Foxp3 and enhancement of IL-17 expression via RORγt and Stat3 have been proposed as the underlying mechanism (48). The HIF-1α dependent processes have been shown to play an important role in rheumatoid arthritis and may be of importance in other autoimmune diseases (49).
T Cells Regulate the Maturation of Macrophages and Dendritic Cells
The differentiation of monocytes into macrophages is a rapid process controlled by cytokines in the environment and cell–cell interactions (10). Thus, neighboring cells can effectively regulate the induction, functional differentiation, and the survival of macrophages (1, 50). It is believed that the polarization of monocytes into M1 or M2 can occur in the absence of T cells (51). However, generally it is assumed that Th1 cells skew monocytes toward M1 whereas Th2 cells skew monocytes toward M2 (4). The lineage commitment of M1 and M2 cells has been associated with the induction of distinct arginine metabolical pathways (52). How T cells regulate this process, however, is not understood.
IL-17 was shown to induce macrophages to produce high levels of IL-6, IL-1β, and TNFα, as well as lower levels of IL-10, IL-12, and PGE2 (53), suggesting that Th17 cells bias monocytes toward an M1-like phenotype. Another study demonstrated that pretreatment of monocytes with IFN-γ (in addition to IL-10 and glucocorticoids) prevented the differentiation of monocytes into M2c cells. Instead, IFN-γ-treated monocytes had a higher expression of Fas and were apoptosis susceptible. Interestingly, monocytes treated with IL-17 (in addition to IL-10 and glucocorticoids) differentiated into M2c and had enhanced phagocytic capacity (54). In essence, Th17 cells may regulate phagocytic effector functions. In vivo, however, the source of IL-17 can be heterogeneous since neutrophils, DC, and macrophages are all capable to produce IL-17, although in low amounts (55, 56).
In mice, CD4+CD25+ regulatory T cells exert a potent suppressive effect on splenic APC, which cannot be overcome by preactivation (57). In humans, CD4+CD25+Foxp3+ regulatory T cells were found to direct monocytes toward M2; characterized by high surface expression of CD206 and CD163 but low levels of HLA-DR (58). Also, human CD4+CD25+ T cells decrease the production of TNFα and IL-6 and increase the production of IL-10 in co-cultured monocytes (59). Murine CD4+CD25−Foxp3−cells can temper the production of proinflammatory cytokines in macrophages via close cell–cell contact. Biologic relevance of this mechanism is suggested by the observation that in the absence of CD4+ T cells, innate immune responses are so vigorous that they cause a cytokine storm and death (60). A more recent murine study has demonstrated that both memory and effector CD4+ T cells decrease IL-1β production in bone marrow-derived macrophages without affecting TNFα or IL-6 production, possibly by selective inhibition of the inflammasomes NLRP3 and NLRP1 (61).
In summary, selected macrophages and DC can shape T cell differentiation through the secretion of IL-1β, IL-6, and TNFα, whereas other macrophages and DC can suppress T cells via cell–cell contact and the secretion of IL-10 and TGF-β (Table 1). Vice versa, Th1 and Th17 activate macrophages via IFN-γ and IL-17, respectively. Regulatory T cells can suppress the activity of macrophages by secreting IL-10, thus driving them toward the M2 phenotype (Table 2).
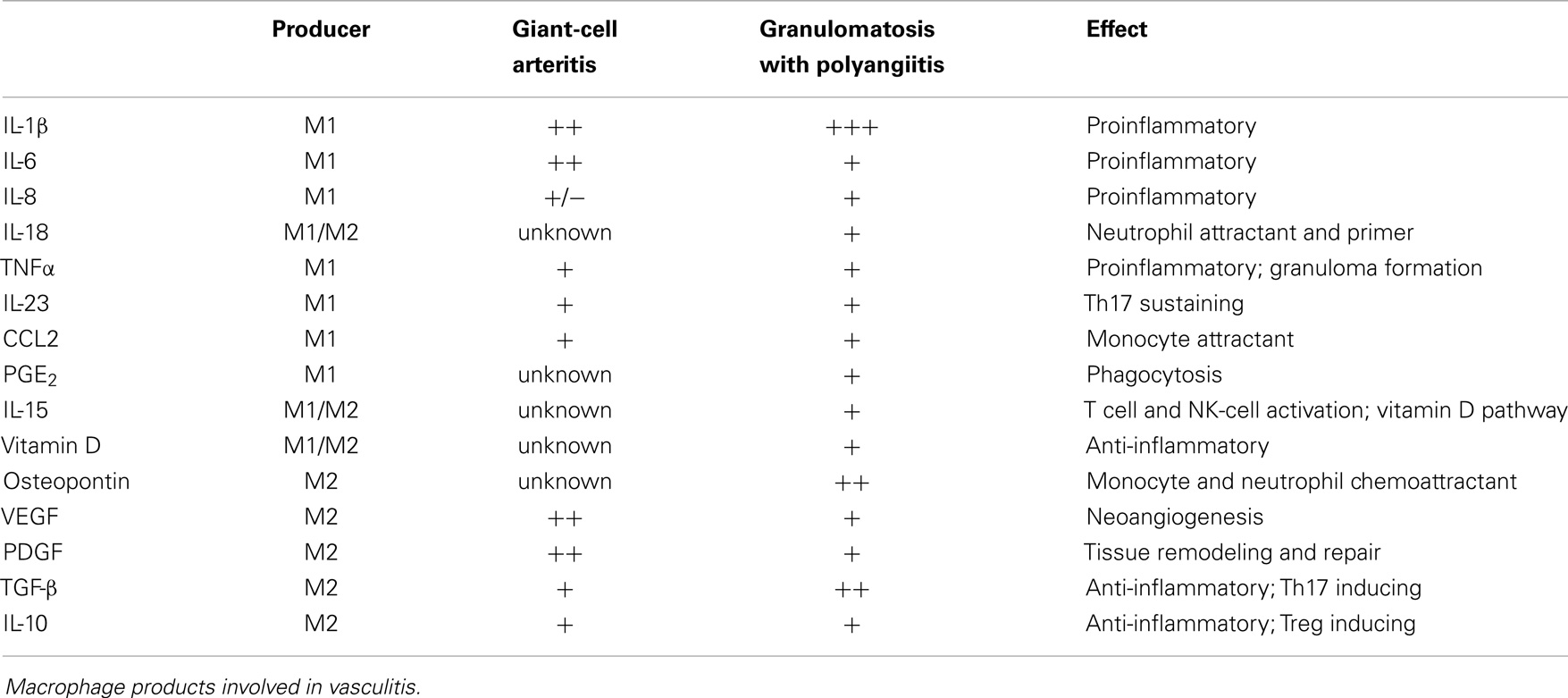
Table 1. Summary of macrophage products in relation to possible effects on T cells.
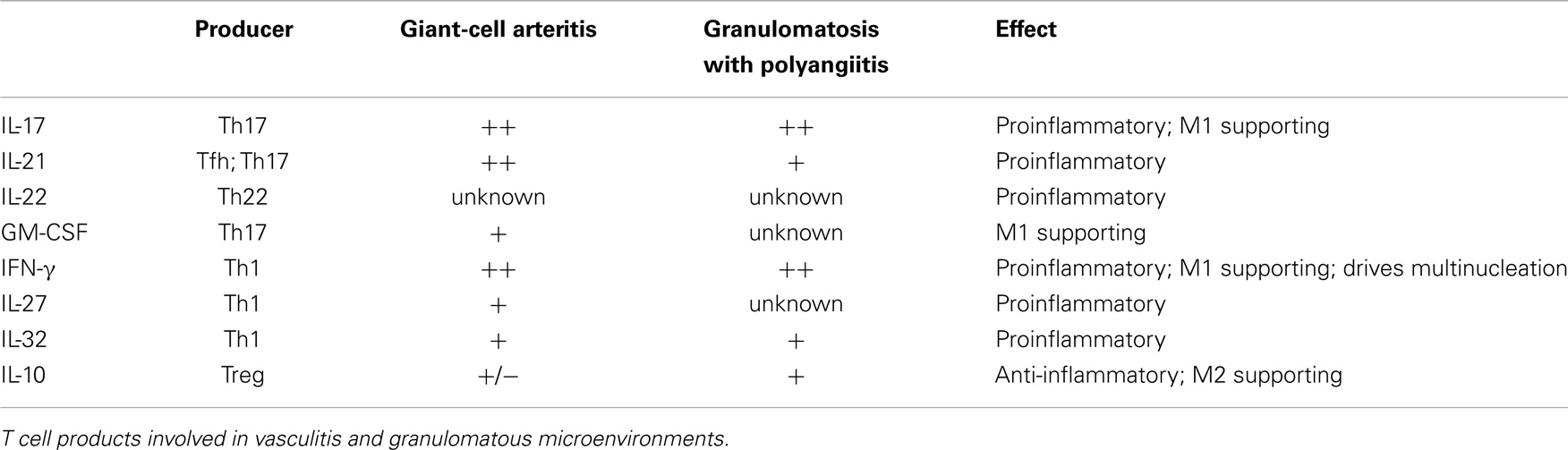
Table 2. Summary of T cell products in relation to possible effects on macrophages.
Granuloma Formation
Designed to protect the host from infection and cancer, the adaptive immune system displays complex microarchitectures to optimize immune responses. One of these lymphoid microstructures has been named granuloma and typically consists of a sphere of highly activated macrophages, surrounded by a shell, i.e., a peripheral layer of T lymphocytes (Figure 1) (62). The current paradigm holds that antigens that are difficult-to-eliminate are prone to elicit granuloma formation. Persistent particulate substances, such as silica, beryllium, or zirconium, but also suture material, are often considered as typical triggers of non-infectious granulomas (63). Difficult-to-eliminate antigens eventually induce the palisading of monocytes/macrophages, which depend on activating signals from other cell populations to form the sophisticated structure of a granuloma. Lymphocytes, especially CD4+ T cells, and DC consistently participate in granulomatous infiltrates, but neutrophils, eosinophils, and B cells have also been described (64). Cells are attracted to granulomas by chemokines, cytokines such as interleukins and complement breakdown products. Over time granulomas mature, resulting in the formation of multinucleated giant-cells and epithelioid cells. The precise composition of granulomas may differ according to the inciting agent or pathogen, but the overall architecture of granulomas is usually maintained.
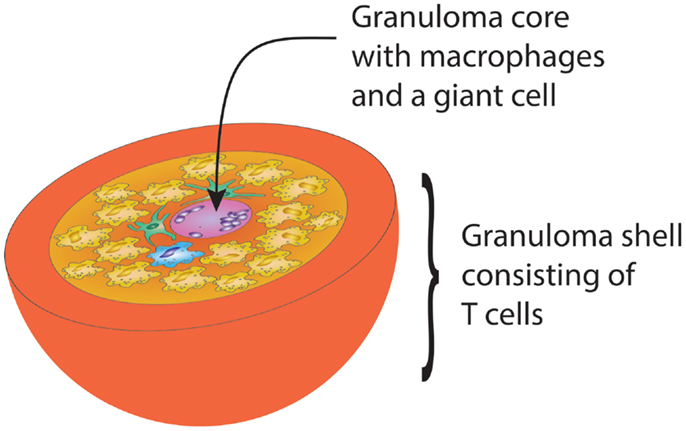
Figure 1. Schematic drawing of the gross architecture of a granuloma with macrophages, dendritic cells, and multinucleate giant-cells forming the core of the sphere, surrounded by a shell of lymphocytes. By clustering innate and adaptive immune cells, the granuloma has great potential to induce and perpetuate immune responses, but is equally powerful in causing damage to surrounding tissues.
Much of the knowledge on granulomas originates from studying the model system of M. tuberculosis infection (3), which remains one of the most prevalent and lethal infectious diseases on the planet (65). It is believed that tuberculous granuloma formation reflects a strategy of the immune system to encapsulate the infection and prevent spreading throughout the body. Tuberculous granulomas have been a rich source of information on the bi-directional interaction between macrophages and T cells (66, 67). One of the hallmark events on the side of macrophages and DC are cell–cell fusions, resulting in multinucleated giant cells. Why and under what circumstances these phagocytes fuse is incompletely understood. Culturing monocytes with IFN-γ in vitro reliably results in multinucleation, emphasizing the role of Th1 cells in the formation of giant cells (68). Th1 cells are critical drivers in the M1 differentiation of macrophages and M1 cells have been implicated in the formation and/or maintenance of granulomas (69). One study has suggested that DC fusion takes place under the influence of autocrine IL-17 and exogenous IFN-γ (56), proposing that the concerted action of several cytokines is necessary to promote the optimal granuloma function. In sarcoidosis, believed to be a Th1-dependent disease, granulomas are more inflammatory than suture granulomas or fungal granulomas, based on significantly higher production of IL-6, CCL2, IFN-γ, and Nox2 (70).
The role of TNFα in the formation of granulomas is debated. In tuberculosis, some studies have shown that TNFα induces the formation of granulomas (71) and that TNFα deficient mice have a more severe M. tuberculosis infection, possibly due to deficient granuloma formation and thus inability to contain the infection (72). Administering anti-TNFα medication in humans, however, does not appear to suppress granulomatous inflammation (73) or cause disassembly of existing granulomas more effectively than corticosteroids alone (74).
Increased numbers of IL-17 positive cells have been found in granulomas in patients with sarcoidosis (75), suggesting that Th17 cells may participate in the assembly of sterile granulomas. Alternatively, the cytokine milieu of granulomas may provide ideal conditions to polarize peripheral T cells toward the Th17 lineage.
Over time, granulomas can become necrotic or fibrotic. It is unknown which factors regulate the progression of the granulomatous reaction. Differences in the architecture, in the tissue distribution, and in the persistence of granulomas strongly support the notion that granuloma formation is distinct in different disease states. As a common rule, granulomatous infiltrates reflect an intense immune response, associated with marked inflammation and potential of tissue damage.
Efferocytosis
Besides being a site of concentrated cytokine production, granulomas have a critical function in debris removal, including the removal of apoptotic cells; a process named efferocytosis (76). It is conceivable that the inefficiency of efferocytosis could lead to the persistence of the granulomatous reaction and this could be particularly important in disease states characterized by sterile granulomas. The notion of ineffective phagocytosis in granulomatous disease was suggested in the past (77). Impaired efferocytosis has been described in chronic granulomatous disease, typically associated with a hyperinflammatory state (78). The deficiency of efferocytosis has been related to a defect in phosphatidylserine (PS) signaling and has been reversed by treating macrophages with IL-4, essentially skewing them toward M2.
Granuloma Formation and Hypervitaminosis D
Patients with granulomatous disease can present with hypercalcemia, considered a result of hypervitaminosis D. This excess vitamin D originates from the granulomas, most likely from the macrophages within. In sarcoidosis, macrophages can produce vitamin D and this production is increased upon stimulation with IFN-γ (79). In line with these findings, a TLR-mediated microbicidal pathway has been reported to upregulate the vitamin D receptor (VDR). The enzyme converting 25-hydroxyvitamin D into the active form of vitamin D, 1,25-hydroxyvitamin D, known as CYP27b1, is also upregulated upon activation of the above mentioned microbicidal pathway (80). In tuberculosis, IFN-γ can increase the production of autocrine IL-15, upregulating CYP27b1 and expression of the VDR. Upon inhibiting the VDR on monocytes, a reduction in autophagy as measured by LC3-positive vesicles was noted. The authors, therefore, suggest that enhanced production of vitamin D and the upregulation of the VDR in monocytes after IFN-γ stimulation result in maturation of the phagosome (81). It is unknown whether the vitamin D pathway is impaired or hyperactive in patients with sterile granulomatous disease.
Interestingly, in GPA and in sarcoidosis variations of disease prevalence patterns according to hours of sunlight per year (and thus dermal vitamin D production) have been discussed. Also, less exposure to sunlight and/or low levels of vitamin D increases the risk for developing GPA or a disease relapse (82, 83).
Giant-Cell Arteritis
Giant-cell arteritis is a medium- to large-vessel vasculitis almost exclusively diagnosed in patients older than 50 years (84). In brief, presenting symptoms are such of an intense acute phase response; e.g., fever, malaise, weight loss, and laboratory abnormalities of systemic inflammation combined with manifestations of tissue ischemia. It mainly affects extracranial and upper extremity branches of the aorta (e.g., temporal arteries, axillary arteries) and the aorta itself (85). The gold standard for diagnosis remains the biopsy of the temporal artery (86). Typical histopathological findings are granulomatous lesions and/or lymphomonocytic infiltrations in the vessel-wall layers, often containing multinucleated giant cells (87) (Figure 2). In contrast to tuberculous granulomas and granulomas found in Crohn’s disease [the latter assumed to result from a host–commensal bacteria homeostasis gone awry (88, 89)], granulomatous lesions in GCA have not been connected to an infectious agent (90). Reported associations between GCA and pre-existing infection with parvovirus B19, Chlamydia trachomatis and Mycoplasma pneumonia have raised the possibility of an infectious trigger, but subsequent studies could not always confirm these associations (91). Nevertheless, there is strong evidence for antigen-driven chronic T cell responses (92), particularly Th1 and Th17 responses (93). The induction of Th1 cells in GCA may originate from excessive IL-12 production, which has been found increased in GCA lesions during active disease and during remission (94). The source of IL-12 in GCA remains obscure, but DC have recently been described to produce high levels of IL-12 (95).
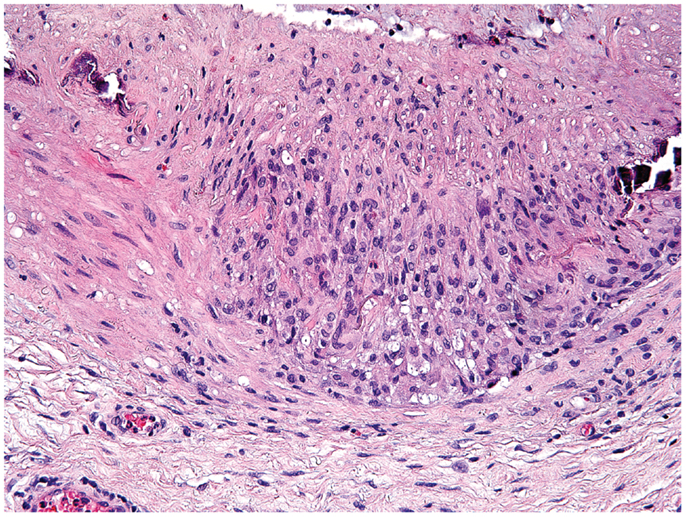
Figure 2. Histological section showing a granulomatous infiltrate in the temporal artery of a 77-year-old patient with giant-cell arteritis. The granulomatous reaction is localized at the media-intima border and includes fragments of the lamina elastica externa. Lymphocytes are surrounding highly activated macrophages and giant cells (hematoxylin and eosin staining).
Dendritic cells and macrophages are obligatory components of the granulomatous infiltrates in the wall of GCA-affected arteries (96). DC are thought to act as gate-keepers of the vasculitis by inducing T cell activation, and display a phenotype of strong immune-stimulatory APC (92). It has been demonstrated that vessel-wall residing DC are specifically activated via TLR4 or TLR5 (97), enabling them to activate p38 MAPK, and activate downstream TNFα and IL-1β (98).
Accordingly, serum IL-6 and IL-1β levels have been found strongly increased in patients with active GCA (74, 99) and associated with disease activity (100, 101). While the precise cellular source of the excess IL-6 and IL-1β has not been determined, highly activated macrophages and DC in the vasculitic lesions emerge as the likely producers (102). There is evidence that the granulomatous infiltrates in GCA contribute to IL-6 secretion (103), together with production of TGF-β, another well-known product of activated macrophages (104). IL-6 levels in GCA patients decrease upon treatment with corticosteroids, but remain higher in some patients when compared to healthy controls, indicating a more chronic course of disease (99).
Recently, IL-33 was reported to be increased in vessel-wall lesions of patients with GCA (105). IL-33 is secreted by endothelial cells when under stress or becoming apoptotic. This stress possibly results from high levels of circulating IL-6. Importantly, IL-33 has been found to favor M2 polarization of macrophages, implicating this macrophage subset in the pathogenesis of GCA (106).
Giant-cell arteritis is considered an antigen-dependent disease in which Th1 cytokines dominate and Th2 cytokines are generally absent. Moreover, during early and untreated disease, Th1 and Th17 cells co-exist in the vasculitic lesions (93) (Figure 2). The granulomatous inflammation may present the platform that allows differentiation of entering naïve T cells toward Th1 and Th17 under the influence of macrophage and DC products (Table 1). By seeding Th1 and Th17 cells into the periphery, granulomas could have a major impact on the composition of the overall immune system. Since differentiated memory cells are long-lived cells, even a temporary granulomatous reaction could permanently remodel the immune system and have long-term implications for the host. During corticosteroid treatment, Th17 cells decrease, but Th1 cells appear to persist (93). In line with these findings, increased serum levels of IL-12, IL-17, IL-21, IL-23, IL-27, IL-32, and IFN-γ have been observed in patients with active GCA (74, 107). Granulomas may represent an important source of IL-17, with elevated protein levels and mRNA observed within the granulomatous vessel-wall infiltrates (108, 109). It is important to consider that the overall frequencies of Th17 cells are low, outnumbered by Th1 cells by a factor of 10 or higher. This may be particularly relevant during the chronic phase of GCA, when persistent inflammation relies on Th1 cells and their major product, IFN-γ. Other Th1 products produced in the inflamed vessel wall include IL-27 and IL-32. Both have been reported to induce M1 cells (107).
Since the differentiation of T cells into distinct lineages results from the exposure of non-committed T cells to antigen plus polarizing cytokine environment, it is highly likely that macrophages and DC residing in the vessel wall ultimately shape vasculitogenic T cell responses (Figure 3). Differentiation of Th17 cells depends on the combined action of IL-6, TGF-β, and IL-23 (110, 111). Recent data have given rise to the concept that Th17 cells are plastic and, dependent on environmental signals, can be redirected to acquire regulatory T cell (Treg) functions (112). There is some evidence that Tregs may be underrepresented in GCA patients, perhaps explaining the inability of affected patients to clear granulomatous lesions (109).
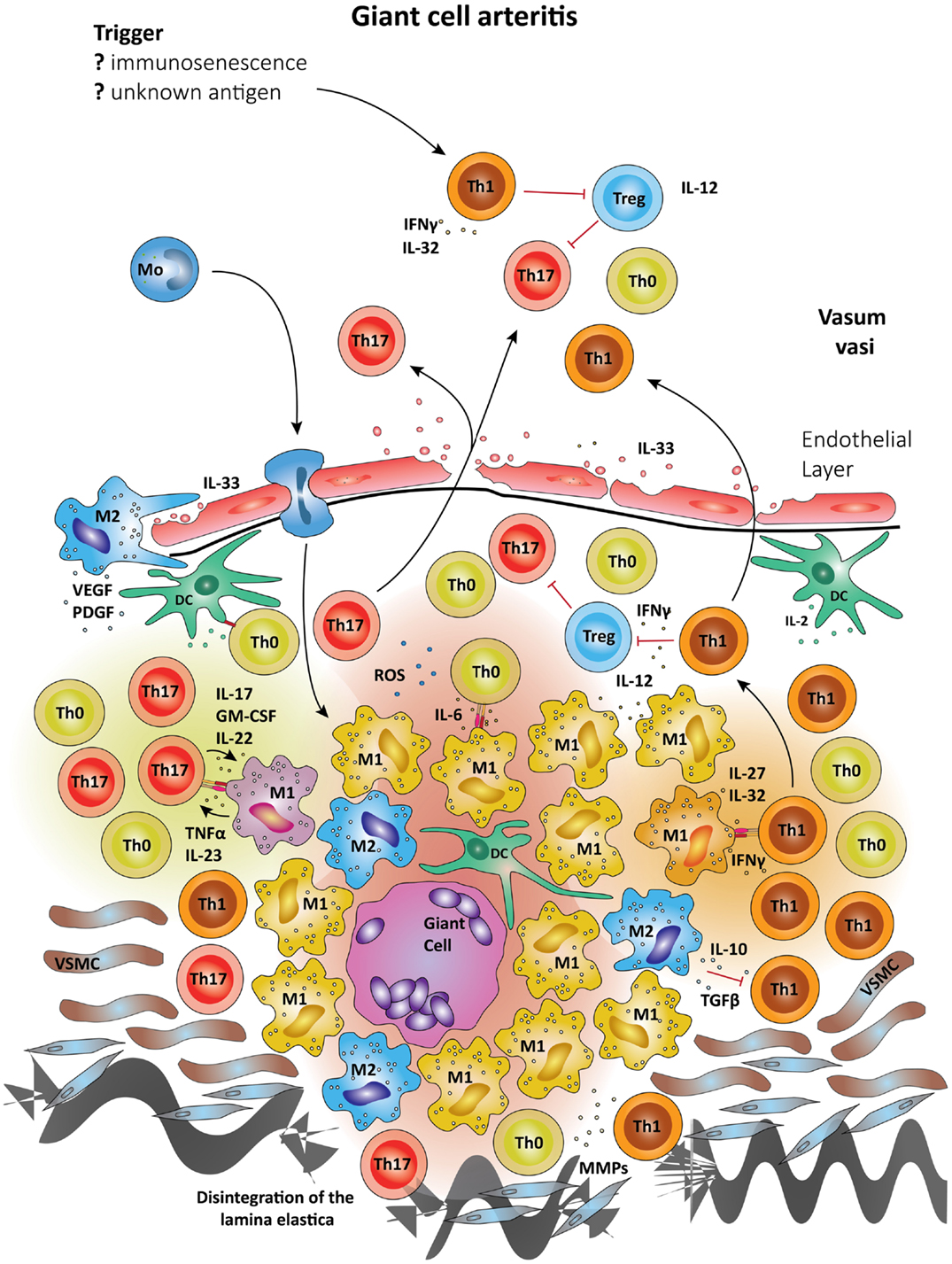
Figure 3. Schematic summary of the pathogenic pathways implicated in granulomatous lesions in giant-cell arteritis. The upper panel represents pathogenic events in GCA. All immune recognition events and tissue damage occur within the vessel wall, not the perivascular tissue. Wall-resident DC, so-called vasDC, coordinate the recruitment and the differentiation of macrophages and T cells. IL-12 is instrumental in biasing T cells toward the Th1 lineage; IL-6, TGF-β, and IL-23 provide signals for Th17 differentiation. Dependent on their positioning in the vessel-wall monocytes commit to distinct functional profiles; e.g., metalloproteinases production, release of ROS, secretion of cytokines. Treg are underrepresented, partially due to inhibitory effects from IFN-γ. The lower panel shows the typical localization of granulomatous infiltrates on the adventitia side of the vessel wall. Monocytes (Mo) enter these lesions via the vasa vasorum and mature in the lesions under the influence of specific microenvironments. Different M1 cells are presumed to influence Th0 cells into Th1 and Th17 cells via cell–cell contact and the secretion of cytokines. Activated macrophages and DC may fuse and form multinucleate giant cells, a hallmark of GCA. The granulomatous infiltrate is a highly inflammatory microenvironment, which promotes the differentiation of Th0 to Th1 and Th17, which are then seeded into the circulation. Due to their localization, granulomatous infiltrates in GCA influence vascular smooth muscle cells (VSMCs) and myofibroblasts. The latter expand in number, migrate and result in concentric media hypertrophy. Also, the external lamina elastica disintegrates as a result of damage from ROS and matrix metalloproteases (MMPs). Although M1 are assumed the most frequent in granulomatous infiltrates, M2 cells have been proposed as a counter-mechanism and implicated in supporting tissue repair. Vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) have been described in the vasculitic lesions of GCA and networks of neoangiogenic microvessels typically accompany the process of intimal hyperplasia (167, 168).
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is distinctly elevated in the serum of patients with GCA. Terrier et al. have reported that GM-CSF is essential for the production of IL-6 and IL-23 by DC; thus indirectly promoting the generation of Th17 cells (113). DC in GM-CSF−/− mice produce lower levels of IL-6, resulting in deficient proliferation of T cells in general and reduced Th17 differentiation. More recently, it has been reported that Th17 cells produce GM-CSF when stimulated with IL-23 and that this production is upregulated by IL-1β (114).
Given the central role of granulomatous infiltrates in GCA, disrupting granuloma formation should be a valuable therapeutic target. Surprisingly, anti-TNF therapy has failed to reduce steroid requirements or prevent disease flares in GCA patients when combined with corticosteroids (115).
With the knowledge of macrophages and DC forming the basis of granuloma formation, inhibiting these cells directly may prove beneficial in treating GCA; bearing in mind, however, the risk of infectious complications associated with a deficient innate immune system. As mentioned earlier, TLR play an important role in the activation of APC in infectious and in sterile inflammatory settings. TLR may, therefore, represent a therapeutic target to treat sterile granulomatous inflammation by inhibiting granuloma formation and reducing tissue damage (116). Other receptors and APC markers are being studied for their suitability in new therapeutic interventions. Targeting CD14 with anti-CD14 antibodies has been attempted in septic models, but not in autoimmune settings (117). In murine sepsis, blocking the innate immune system results in less inflammation, less intense cytokine storms, and a higher survival rate.
Granulomatosis with Polyangiitis
Small-vessel vasculitides associated with the production of autoantibodies reactive to proteinase-3 (PR3) or myeloperoxidase (MPO) are collectively called anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitides (AAV). The group of AAV encompasses GPA, microscopic polyangiitis, and eosinophilic GPA (118). The pathogenesis of these vasculitides is incompletely understood, but great progress has been made in diagnosis and therapy of these autoimmune diseases. Yearly incidence rates are estimated at 20 cases per million individuals (119). A high mortality and (co)morbidity unfortunately persists despite improvement of therapy and knowledge of the disease process (120–122). In GPA, granulomas are typically found in the ear, nose, and throat region, in the lungs, periglomerularly in the kidneys, and more rarely in other organs (123) (Figure 4). The autoantigens recognized by the autoantibodies in AAV are intracellular enzymes produced by neutrophils and monocytes. In both cell types, PR3 and MPO are expressed on the cell membrane in low levels in healthy controls and in aberrantly high levels in AAV patients, especially during active disease (124, 125). Over 75% of GPA patients are PR3-ANCA positive with the remainder being either MPO-ANCA positive or ANCA-negative, especially in patients of Caucasian descent (126). Interestingly, when monocytes differentiate into macrophages they lose expression of PR3 and MPO on their cell membrane. Thus, monocytes can be activated by ANCA (127), whereas mature macrophages cannot (128), placing antibody-dependent disease mechanisms early into the pathogenic immune response (Figure 5).
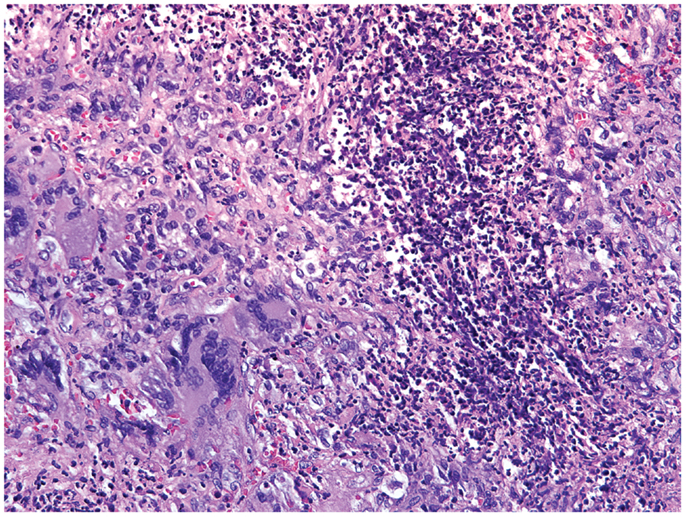
Figure 4. Histological section of a granulomatous lesion in the lung of a 56-year-old patient with granulomatosis with polyangiitis. The core of the granuloma consists of multinucleate giant cells, macrophages, and neutrophils and is encircled by a rim of lymphocytes (hematoxylin and eosin staining).
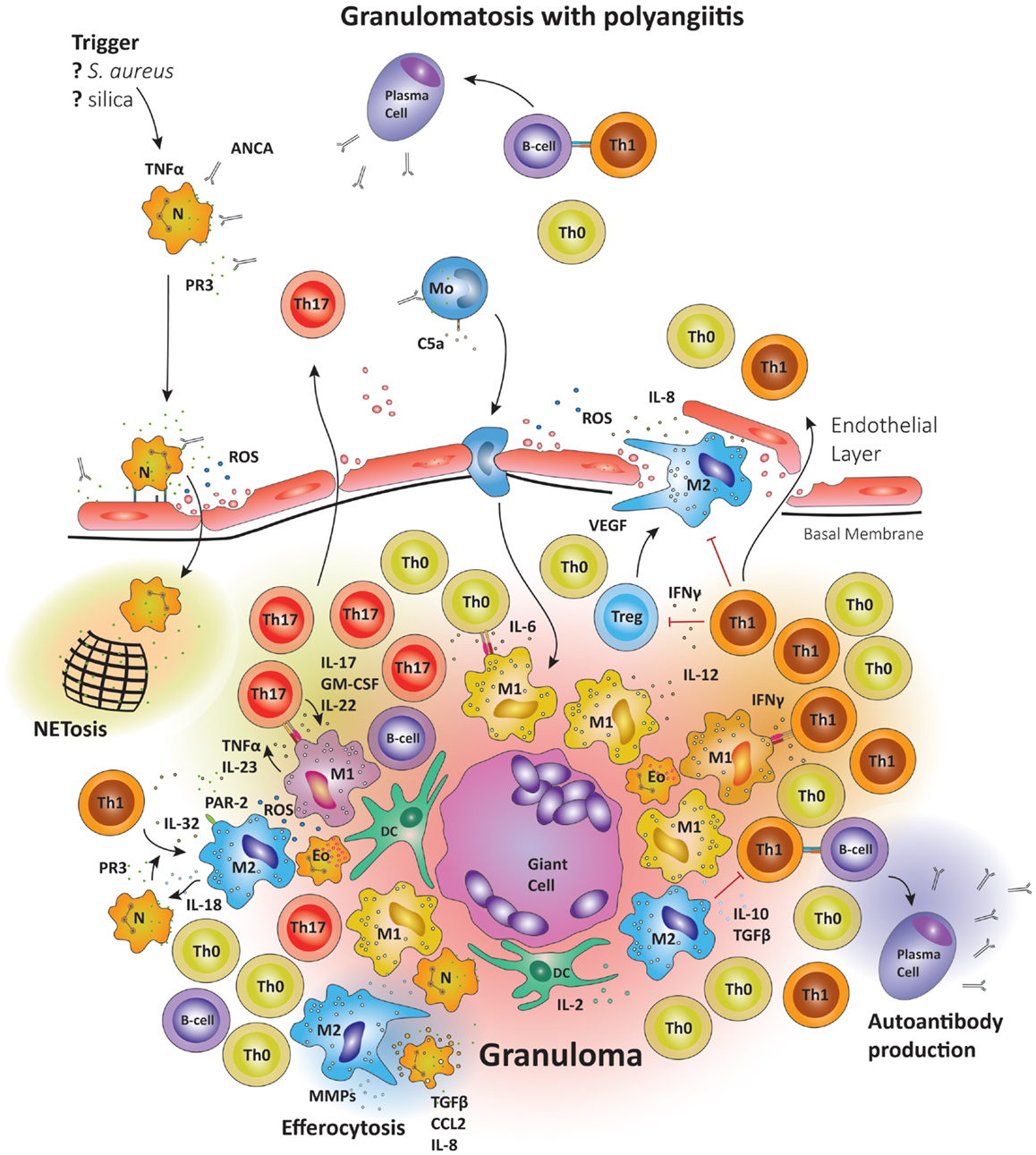
Figure 5. Schematic summary of the pathogenic pathways implicated in granuloma formation in granulomatosis with polyangiitis. The upper panel represents the pathogenesis of granulomatosis with polyangiitis (GPA). Upon priming neutrophils bring to their surface high levels of proteinase-3 (PR3) and myeloperoxidase (MPO), the autoantigens recognized by ANCA. The priming process is believed to be mediated by TNFα. TNFα production may be induced by a variety of triggers; e.g., S. aureus, silica, etc. Anti-PR3 or anti-MPO antibodies are then able to bind these enzymes on the cell surface, causing neutrophils to degranulate, bind to endothelial cells, enter the perivascular tissue, and release ROS; thereby damaging the vessel wall. In the tissue, neutrophils releases so-called neutrophil extracellular traps (NETs), which are networks of fibers and DNA to trap pathogens. Many of the highly activated neutrophils become apoptotic and are then phagocytosed by resident M2 macrophages, a process that has been named efferocytosis and that may be deficient in GPA. During efferocytosis, macrophages release TGF-β, IL-8, and CCL2. Monocytes may also be activated by circulating ANCA, enhancing their chemotactic responsiveness and enabling them to participate in granuloma formation. The lower panel represents the formation of a sterile granuloma in extravascular tissue. Monocytes and CD4+ cells enter the granuloma following a gradient of chemokines and cytokines; a process sufficient to transform monocytes into macrophages. Commitment to the M1 or M2 lineage is dependent on the specific cytokine environment. Neutrophils and eosinophils are commonly found in granulomas in GPA. Also, B cells have been reported in the surroundings of granulomas, where they may undergo further maturation. Multinucleate giant cells are present within the granulomas, resulting from the fusion of either macrophages or dendritic cells. The organized arrangement of the granuloma provides an ideal platform for macrophage–T cell interaction. CD4+ cells coming into contact with IL-6 and TGF-β producing M1 cells are skewed toward the Th17 lineage. M1 also secrete IL-23; sustaining the Th17 population. In turn, Th17 cells can secrete granulocyte and GM-CSF in addition to IL-17 and IL-22, thus stabilizing M1 differentiation. However, M2 cells are equally represented in granulomas and secrete IL-10 and TGF-β. M2 cells are a source of vascular endothelial growth factor (VEGF) and support the outgrowth of microvessels, critically important as the granuloma grows in size.
The neutrophil, playing a central role in the pathogenesis of GPA, degranulates once ANCA bind to surface PR3 or MPO (129). Neutrophils then become apoptotic and are cleared by macrophages. While dying, neutrophils excrete so-called neutrophil extracellular traps (NETs), physiologically meant to “trap” pathogens in a network of decondensed chromatin, MPO, PR3, and other enzymes, but causing damage in the case of GPA by releasing more autoantigen into the granulomatous lesion (130). The clearance of apoptotic neutrophils by macrophages has been termed efferocytosis (see above) and is associated with secretion of a variety of cytokines and chemokines, including TNFα and prostaglandin E2 (PGE2) (131). Strongly activated macrophages have been localized within and around glomeruli affected by pauci-immune necrotizing crescentic glomerulonephritis, the pathognomonic lesion of AAV (132). These macrophages have been found to produce IL-18, thus attracting more neutrophils to the granulomatous inflammation (133). Such, activated macrophages express high levels of HLA class II molecules, enabling them to act as highly efficient APC (132). Of the leukocytes infiltrating the glomeruli and the tubulointerstitium, macrophages are the most prevalent, followed by granulocytes (134), differentiating GPA from GCA, in which granulocytes essentially do not participate. In renal biopsies from patients with active GPA, DC, and macrophages are localized in glomeruli and in periglomerular infiltrates, whereas DC and macrophages are absent from normal renal tissue. DC and T cells appear to interact within the periglomerular infiltrates (135), consistent with antigen recognition events orchestrating the tissue-damaging immune responses.
Moderately elevated levels of serum IL-1β and IL-6 have been reported in GPA patients during active disease, in line with persistent activation of macrophages in the tissue lesions (136–138). IL-1β and IL-6 are produced by M1 (138), but also by damaged endothelial cells (139). Serum levels of IL-1β, TNFα, and sIL-2R have been correlated with the presence of corresponding mRNA in tissue lesions, suggesting that mononuclear cells in vasculitic lesions are the origin of these proinflammatory mediators (140). In a mouse model of small-vessel vasculitis, IL-1β produced by monocytes has been associated with glomerulonephritis and blocking the IL-1β receptor with anakinra has resulted in a decrease of cellular crescents and hematuria (141).
The enzyme PR3 may play a central role in granuloma formation since it was found to be capable of cleaving IL-32, thereby enhancing its activity. IL-32 is produced by Th1 cells and its active form results in macrophage activation, leading to TNFα and IL-8 production (142). In addition, PR3 can cleave the protease-activated receptor 2 located on the cell surface of macrophages, leading to downstream inflammation, particularly IL-18, CXCL2, and IL-8 (142, 143). IL-18 is a known neutrophil attractant (Figure 5).
In search for the mechanisms through which autoantibodies mediate pathology in GPA, PR3-ANCA have been shown to induce upregulation of TLR2, 3, 4, 7, and 9, as well as NOD-1 and NOD-2 (144). Nucleotide-binding oligomerization domain-containing protein 2 (NOD-2) is an intracellular pattern-recognition molecule enabling macrophages to recognize bacterial molecules that contain muramyl dipeptide (MDP). In contrast to Crohn’s disease, where mutations in NOD2 have been implicated as disease risk factors (145), no such mutations were found in GPA patients (146). Interestingly, however, another study showed an association between mutations in the TLR9 gene and PR3-ANCA positivity as opposed to MPO-ANCA positivity (147). In mice, ligands for TLR2 and TLR9 have both been implicated in kicking off autoimmunity. Notably, ligands for TLR2 have led to an expansion of Th17 cells, whereas ligands for TLR9 preferentially facilitate the expansion of Th1 cells (148). A possible role of TLR2 ligands in the pathogenesis of GPA is supportive of an involvement of Staphylococcus aureus in the development (83) as well as the risk for relapse in GPA (149), as S. aureus is a known ligand for TLR2 (150). In support of this notion, monocytes from patients with GPA have been found to express higher levels of TLR2 on the cell surface (151). Genetic factors are likely to be associated with the process of granuloma formation: these factors remain difficult to establish. One study found an association between the PTPN22 R620W polymorphism and the presence of granulomatous lesions (especially in the ENT region) specifically in patients with GPA, as opposed to patients with MPA or EGPA (152).
A macrophage product previously associated with granuloma formation in tuberculosis and silicosis is osteopontin (153). Interestingly, osteopontin is elevated in patients with active GPA (154) and has been detected in crescentic lesions in glomeruli (155). Osteopontin production has been associated with macrophage activation and it has been proposed that this monokine functions as a monocyte chemoattractant, securing the influx of fresh monocytes into the granulomatous lesions. Importantly, osteopontin synthesis is stimulated by active vitamin D, the latter, as mentioned above, a signifying product of active granulomas (156). There is evidence that osteopontin may play a role in TLR4-mediated IL-10 production in T cells, which suppress macrophages (157), delineating a negative feedback loop via which T cells can minimize granuloma-associated tissue damage.
Considering the destructive consequences of granuloma formation in GPA, it would be advantageous to treat GPA patients with therapies inhibiting this process. Studies on the use of anti-TNFα treatment in GPA patients have suggested that it may have a place in induction therapy (158), but patients remained at a higher risk for relapse (159). Depletion of B cells by anti-CD20 antibodies provides effective immunosuppression in patients with GPA (160). Whether this therapeutic approach functions by depressing autoantibody-dependent mechanisms or whether B cells provide other disease relevant functions, such as cytokine production and antigen presentation, remains speculative. It is conceivable that B cells are critically involved in toning the immune system and that their depletion impairs innate as well as adaptive immunity. Directly targeting macrophages may hold promise in immunosuppressing patients with GPA, although loss of macrophage function may further weaken their ability to mount protective immunity, especially against microbial pathogens.
Future Directions
It is currently unknown whether macrophages trapped in granulomas can be easily assigned to a functional lineage, e.g., M1 and M2, or whether residence in a granuloma directs macrophages into a separate differentiation program. Circulating cytokines in granulomatous disease favors the concept that the majority of macrophages may be M1. However, M2 have been localized in granulomatous lesions. This may simply be a negative feedback mechanism to temper inflammation in order to prevent excessive tissue damage. Indeed, one study found both M1 and M2 in the vessel lesions in GCA patients (105). A distinguishing feature of granulomas is the high cell turnover, giving rise to the need to clear apoptotic short-lived cells, such as neutrophils and macrophages. Effective removal of apoptotic bodies relies on the process of efferocytosis, again placing macrophages at a center stage. The current paradigm suggests that mainly M2 are responsible for effective efferocytosis. The intactness of these mechanisms in granulomatous vasculitis is unknown, but it could be hypothesized that a major defect lies in the inability of the patients to turn down M1 macrophage activation and bring to bear M2 macrophages.
Interestingly, GCA and GPA are distinct diseases affecting different sizes of blood vessels, but they are both characterized by granulomatous lesions. The granulomatous infiltrates in GCA are predominantly located in the vessel wall, where monocytes arrive via the vasa vasorum (87). In contrast, the granulomatous lesions in GPA are more often extravascular and, in the case of renal involvement, periglomerular (161). The classic granuloma with palisading as described by Godman and Churg in 1954 (162), can be found in GPA but not in GCA. The overall architecture of granulomas, characterized by an inner core of macrophages and DC surrounded by T cells, however, is present in both vasculitides. Importantly, the cellular composition of GCA- and GPA-associated granulomas seems to be different. Due to the intramural localization of the granulomatous lesion in GCA, vascular smooth muscle cells (VSMCs) and myofibroblasts are in intimate relationship to the granuloma-forming immune cells. Neutrophils are found exclusively in granulomas of GPA patients, as well as eosinophils. Also, B cells are absent from lesions in GCA (163) but they can be found in granulomas of GPA patients (164), where they may be able to mature and contribute to B cell dependent pathology. It has been proposed that the granuloma in GPA may participate in autoantibody production (165).
Granulomatous vasculitides reflect abnormalities in both, the innate and adaptive arm of the immune system. Activation products of innate cells, in particular cytokines, have attracted much attention as potential biomarkers of disease and effort has been invested to test whether they can help in quantifying disease burden. Similarities in the abnormal immune reactions of distinct vasculitides make it unlikely that a single cytokine will emerge as a disease-specific biomarker. However, cocktails of cytokines may have value in assessing how active the disease is in individual patients. Quantifying adaptive immunity beyond antibody formation has been challenging. In AAV, autoantibodies against PR3 and MPO have served an important diagnostic role, as they can help in rapidly reducing differential diagnosis in acutely sick patients. There is currently insufficient evidence that the titer of these autoantibodies is a good marker of disease activity. During the chronic course of AAV, autoantibody titers have limited use in helping make therapeutic decisions (166).
The possibility remains that granulomas will guide the search for the disease inducing antigens; as such antigens should be enriched in these tissue sites. Understanding the mechanisms of granuloma formation and the role of these lymphoid microstructures in perpetuating pathology could greatly enhance the spectrum of therapeutic targets. In all inflammatory vasculopathies, corticosteroids remain a cornerstone of therapy. Their therapeutic benefit may mainly result from their ability to suppress macrophage function. Temporary suppression of macrophage function, while effective in reducing acute phase responses, has little impact on the long-lived cells of the adaptive immune system, and thus fails to induce durable remission.
There remain considerable challenges in optimizing the management of patients with inflammatory blood vessel disease, despite enormous progress in deciphering processes of innate and adaptive immunity. The initial triggers derailing host protective immunity are undetermined. Given the importance of granuloma formation in protective and pathogenic immunity, speculations about infectious agents setting off vasculitis have held steady over decades. Hopes for the identification of such a disease inducer have not been met with success. Clustering of risk in geographic regions and populations have nurtured the belief that genetic risk factors are important, but could equally well support the role of environmental determinants. Most of the vasculitides are HLA associated diseases, providing further support for a critical contribution of antigen recognition and adaptive immunity in pathogenesis. Granulomas remain fascinating structures that bring together innate and adaptive immune cells and may ultimately hold the key to understanding why the power of immune protection is misused to harm the host.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Linda Arneson for excellent secretarial assistance. This work was supported by the National Institutes of Health (R01 HL117913, R01 AI108906, R01 AR042527, and P01 HL058000 to Cornelia M. Weyand). Marc Hilhorst and Tsuyoshi Shirai received fellowship support from the Govenar Discovery Fund. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the NIH under Award R01 AR042547. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
1. Krutzik SR, Tan B, Li H, Ochoa MT, Liu PT, Sharfstein SE, et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med (2005) 11:653–60. doi:10.1038/nm1246
3. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol (2012) 12:352–66. doi:10.1038/nri3211
4. Gordon S. Alternative activation of macrophages. Nat Rev Immunol (2003) 3:23–35. doi:10.1038/nri978
5. Shi C, Pamer E. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11:762–74. doi:10.1038/nri3070
6. Hume D. Macrophages as APC and the dendritic cell myth. J Immunol (2008) 181:5829–35. doi:10.4049/jimmunol.181.9.5829
7. Vodovotz Y, Bogdan C, Paik J, Xie QW, Nathan C. Mechanisms of suppression of macrophage nitric oxide release by transforming growth factor beta. J Exp Med (1993) 178:605–13. doi:10.1084/jem.178.2.605
8. Mills C. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol (2012) 32:463–88. doi:10.1615/CritRevImmunol.v32.i6.10
9. Stout R. Editorial: macrophage functional phenotypes: no alternatives in dermal wound healing? J Leukoc Biol (2010) 87:19–21. doi:10.1189/jlb.0509311
10. Mosser D, Edwards J. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi:10.1038/nri2448
11. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi:10.1016/j.immuni.2011.05.006
12. Beutler B. TLRs and innate immunity. Blood (2009) 113:1399–407. doi:10.1182/blood-2008-07-019307
13. Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science (2010) 327:291–5. doi:10.1126/science.1183021
14. Mills K. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol (2011) 11:807–22. doi:10.1038/nri3095
15. Miller Y, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res (2011) 108:235–48. doi:10.1161/CIRCRESAHA.110.223875
16. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol (2013) 13:709–21. doi:10.1038/nri3520
17. Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, et al. Toll-like receptor 2 signaling in CD4+ T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity (2010) 32:692–702. doi:10.1016/j.immuni.2010.04.010
18. Unanue E. Antigen-presenting function of the macrophage. Annu Rev Immunol (1984) 2:395–428. doi:10.1146/annurev.iy.02.040184.002143
19. Janeway C. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today (1992) 13:11–6. doi:10.1016/0167-5699(92)90198-G
20. Rosenthal A, Shevach E. Function of macrophages in antigen recognition by guinea pig T lymphocytes. I. Requirement for histocompatible macrophages and lymphocytes. J Exp Med (1973) 138:1194–212. doi:10.1084/jem.138.5.1194
21. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol (2013) 31:443–73. doi:10.1146/annurev-immunol-032712-095910
22. Liu Y, Janeway C. Cells that present both specific ligand and costimulatory activity are the most efficient inducers of clonal expansion of normal CD4 T cells. Proc Natl Acad Sci U S A (1992) 89:3845–9. doi:10.1073/pnas.89.9.3845
23. Miga A, Masters S, Gonzalez M, Noelle RJ. The role of CD40-CD154 interactions in the regulation of cell mediated immunity. Immunol Invest (2000) 29:111–4. doi:10.3109/08820130009062292
24. Chen L, Flies D. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13:227–42. doi:10.1038/nri3405
25. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol (2013) 43:2797–809. doi:10.1002/eji.201343751
26. Zhu J, Paul W. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev (2010) 238:247–62. doi:10.1111/j.1600-065X.2010.00951.x
27. Pozzi LA, Maciaszek JW, Rock KL. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J Immunol (2005) 175:2071–81. doi:10.4049/jimmunol.175.4.2071
28. Rothoeft T, Gonschorek A, Bartz H, Anhenn O, Schauer U. Antigen dose, type of antigen-presenting cell and time of differentiation contribute to the T helper 1/T helper 2 polarization of naive T cells. Immunology (2003) 110:430–9. doi:10.1111/j.1365-2567.2003.01758.x
29. Itano A, Jenkins M. Antigen presentation to naive CD4 T cells in the lymph node. Nat Immunol (2003) 4:733–9. doi:10.1038/ni957
30. Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol (1995) 154:5071–9.
31. Hsieh C, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (1993) 260:547–9. doi:10.1126/science.8097338
32. Spörri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol (2005) 6:163–70. doi:10.1038/ni1162
33. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity (2013) 38:336–48. doi:10.1016/j.immuni.2012.10.018
34. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi:10.1084/jem.20090847
35. Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol (2007) 8:1086–94. doi:10.1038/ni1511
36. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1:405–13. doi:10.1016/1074-7613(94)90071-X
37. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature (2003) 423:506–11. doi:10.1038/nature01621
38. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of Lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51. doi:10.1016/S1074-7613(00)80089-8
39. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541. doi:10.1016/1074-7613(95)90125-6
40. Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol (2003) 170:1257–66. doi:10.4049/jimmunol.170.3.1257
41. Castiblanco J, Arcos-Burgos M, Anaya JM. What is next after the genes for autoimmunity? BMC Med (2013) 11:197. doi:10.1186/1741-7015-11-197
42. Rai E, Wakeland E. Genetic predisposition to autoimmunity – what have we learned? Sem Immunol (2011) 23:67–83. doi:10.1016/j.smim.2011.01.015
43. Hoves S, Krause SW, Schütz C, Halbritter D, Schölmerich J, Herfarth H, et al. Monocyte-derived human macrophages mediate anergy in allogeneic T cells and induce regulatory T cells. J Immunol (2006) 177:2691–8. doi:10.4049/jimmunol.177.4.2691
44. Taylor MD, Harris A, Nair MG, Maizels RM, Allen JE. F4/80+ alternatively activated macrophages control CD4+ T cell hyporesponsiveness at sites peripheral to filarial infection. J Immunol (2006) 176:6918–27. doi:10.4049/jimmunol.176.11.6918
45. Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest (2006) 116:916–28. doi:10.1172/JCI27203
46. Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10–producing, nonproliferating CD4+ T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med (2000) 192:1213–22. doi:10.1084/jem.192.9.1213
47. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Res (2010) 70:7465–75. doi:10.1158/0008-5472.CAN-10-1439
48. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
49. Konisti S, Kiriakidis S, Paleolog EM. Hypoxia – a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nat Rev Rheumatol (2012) 8:153–62. doi:10.1038/nrrheum.2011.205
50. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol (2005) 175:342–9. doi:10.4049/jimmunol.175.1.342
51. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164:6166–73. doi:10.4049/jimmunol.164.12.6166
52. Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by Th1 and Th2 cytokines. Eur J Immunol (1995) 25:1101–4. doi:10.1002/eji.1830250436
53. Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 Stimulates the production and expression of proinflammatory cytokines, IL-1b and TNF-a, by human macrophages. J Immunol (1998) 160:3513–21.
54. Zizzo G, Cohen P. IL-17 Stimulates differentiation of human anti-Inflammatory macrophages and phagocytosis of apoptotic neutrophils in response to IL-10 and glucocorticoids. J Immunol (2013) 190:5237–46. doi:10.4049/jimmunol.1203017
55. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol (2011) 187:490–500. doi:10.4049/jimmunol.1100123
56. Coury F, Annels N, Rivollier A, Olsson S, Santoro A, Speziani C, et al. Langerhans cell histiocytosis reveals a new IL-17A–dependent pathway of dendritic cell fusion. Nat Med (2008) 14:81–7. doi:10.1038/nm1694
57. Thornton A, Shevach E. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol (2000) 164:183–90. doi:10.4049/jimmunol.164.1.183
58. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A (2007) 104:19446–51. doi:10.1073/pnas.0706832104
59. Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC, Akbar AN, et al. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol (2005) 66:222–30. doi:10.1016/j.humimm.2004.12.006
60. Kim KD, Zhao J, Auh S, Yang X, Du P, Tang H, et al. Adaptive immune cells temper initial innate responses. Nat Med (2007) 13:1248–52. doi:10.1038/nm1633
61. Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature (2009) 460:269–74. doi:10.1038/nature08100
62. Petersen H, Smith A. The role of the innate immune system in granulomatous disorders. Front Immunol (2013) 4:120. doi:10.3389/fimmu.2013.00120
64. Williams G, Williams W. Granulomatous inflammation – a review. J Clin Pathol (1983) 36:723–33. doi:10.1136/jcp.36.7.723
65. Zumla A, Raviglione M, Hafner R, von Reyn CF. Tuberculosis. N Engl J Med (2013) 368:745–55. doi:10.1056/NEJMra1200894
66. Ulrichs T, Kaufmann S. New insights into the function of granulomas in human tuberculosis. J Pathol (2006) 208:261–9. doi:10.1002/path.1906
67. Saunders B, Britton W. Life and death in the granuloma: immunopathology of tuberculosis. Immunol Cell Biol (2007) 85:103–11. doi:10.1038/sj.icb.7100027
68. Weinberg JB, Hobbs MM, Misukonis MA. Recombinant human gamma-interferon induces human monocyte polykaryon formation. Proc Natl Acad Sci U S A (1984) 81:4554–7. doi:10.1073/pnas.81.14.4554
69. Broos C, van Nimwegen M, Hoogsteden HC, Hendriks RW, Kool M, van den Blink B. Granuloma formation in pulmonary sarcoidosis. Front Immunol (2013) 4:437. doi:10.3389/fimmu.2013.00437
70. Christophi GP, Caza T, Curtiss C, Gumber D, Massa PT, Landas SK. Gene expression profiles in granuloma tissue reveal novel diagnostic markers in sarcoidosis. Exp Mol Pathol (2014) 96:393–9. doi:10.1016/j.yexmp.2014.04.006
71. Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell (1989) 56:731–40. doi:10.1016/0092-8674(89)90676-4
72. Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, et al. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol (1999) 162:3504–11.
73. Garcia Vidal C, Rodríguez Fernández S, Martínez Lacasa J, Salavert M, Vidal R, Rodríguez Carballeira M, et al. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis (2005) 40:756–9. doi:10.1086/427941
74. Visvanathan S, Rahman MU, Hoffman GS, Xu S, García-Martínez A, Segarra M, et al. Tissue and serum markers of inflammation during the follow-up of patients with giant-cell arteritis – a prospective longitudinal study. Rheumatology (2011) 50:2061–70. doi:10.1093/rheumatology/ker163
75. Ten Berge B, Paats MS, Bergen IM, van denBlink B, Hoogsteden HC, Lambrecht BN, et al. Increased IL-17A expression in granulomas and in circulating memory T cells in sarcoidosis. Rheumatology (2012) 51:37–46. doi:10.1093/rheumatology/ker316
76. Rieber N, Hector A, Kuijpers T, Roos D, Hartl D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin Dev Immunol (2012) 2012:252460. doi:10.1155/2012/252460
77. Baehner R, Nathan D. Leukocyte oxidase: defective activity in chronic granulomatous disease. Science (1967) 155:835–6. doi:10.1126/science.155.3764.835
78. Fernandez-Boyanapalli RF, Frasch SC, McPhillips K, Vandivier RW, Harry BL, Riches DW, et al. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood (2009) 113:2047–55. doi:10.1182/blood-2008-05-160564
79. Adams J, Gacad M. Characterization of 1 alpha-hydroxylation of vitamin D3 sterols by cultured alveolar macrophages from patients with sarcoidosis. J Exp Med (1985) 161:755–65. doi:10.1084/jem.161.4.755
80. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin d-mediated human antimicrobial response. Science (2006) 311:1770–3. doi:10.1126/science.1123933
81. Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, et al. Vitamin D is required for IFN-γ-mediated antimicrobial activity of human macrophages. Sci Transl Med (2011) 3:104ra2. doi:10.1126/scitranslmed.3003045
82. Gatenby PA, Lucas RM, Engelsen O, Ponsonby AL, Clements M. Antineutrophil cytoplasmic antibody-associated vasculitides: could geographic patterns be explained by ambient ultraviolet radiation? Arthritis Rheum (2009) 61:1417–24. doi:10.1002/art.24790
83. de LindvanWijngaarden RA, van Rijn L, Hagen EC, Watts RA, Gregorini G, Tervaert JW, et al. Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody-associated vasculitis: the cause is hidden, but the result is known. Clin J Am Soc Nephrol (2008) 3:237–52. doi:10.2215/CJN.03550807
84. Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. Lancet (2008) 372:234–45. doi:10.1016/S0140-6736(08)61077-6
85. Weyand CM, Liao YJ, Goronzy JJ. The immunopathology of giant cell arteritis: diagnostic and therapeutic implications. J Neuroophthalmol (2012) 32:259–65. doi:10.1097/WNO.0b013e318268aa9b
86. Weyand C, Goronzy J. Clinical practice. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med (2014) 371:50–7. doi:10.1056/NEJMcp1214825
87. Weyand C, Goronzy J. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol (2013) 9:731–40. doi:10.1038/nrrheum.2013.161
88. Sewell GW, Marks DJ, Segal AW. The immunopathogenesis of Crohn’s disease: a three-stage model. Curr Opin Immunol (2009) 21:506–13. doi:10.1016/j.coi.2009.06.003
89. Abraham C, Cho J. Inflammatory bowel disease. N Engl J Med (2009) 361:2066–78. doi:10.1056/NEJMra0804647
90. Bhatt AS, Manzo VE, Pedamallu CS, Duke F, Cai D, Bienfang DC, et al. Brief report: in search of a candidate pathogen for giant cell arteritis: sequencing-based characterization of the giant cell arteritis microbiome. Arthritis Rheumatol (2014) 66:1939–44. doi:10.1002/art.38631
91. Rodríguez-Pla A, Stone JH. Vasculitis and systemic infections. Curr Opin Rheumatol (2006) 18:39–47. doi:10.1097/01.bor.0000197999.58073.2e
92. Krupa WM, Dewan M, Jeon MS, Kurtin PJ, Younge BR, Goronzy JJ, et al. Trapping of misdirected dendritic cells in the granulomatous lesions of giant cell arteritis. Am J Pathol (2002) 161:1815–23. doi:10.1016/S0002-9440(10)64458-6
93. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation (2010) 121:906–15. doi:10.1161/CIRCULATIONAHA.109.872903
94. Weyand CM, Younge BR, Goronzy JJ. IFN-γ and IL-17: the two faces of T-cell pathology in giant cell arteritis. Curr Opin Rheumatol (2011) 23:43–9. doi:10.1097/BOR.0b013e32833ee946
95. Nizzoli G, Krietsch J, Weick A, Steinfelder S, Facciotti F, Gruarin P, et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood (2013) 122:932–42. doi:10.1182/blood-2013-04-495424
96. Ghosh P, Borg FA, Dasgupta B. Current understanding and management of giant cell arteritis and polymyalgia rheumatica. Expert Rev Clin Immunol (2010) 6:913–28. doi:10.1586/eci.10.59
97. Deng J, Ma-Krupa W, Gewirtz AT, Younge BR, Goronzy JJ, Weyand CM. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res (2009) 104:488–95. doi:10.1161/CIRCRESAHA.108.185777
98. Baldassare JJ, Bi Y, Bellone CJ. The role of p38 mitogen-activated protein kinase in IL-1 β transcription. J Immunol (1999) 162:5367–73.
99. Dasgupta B, Panayi G. Interleukin-6 in serum of patients with polymyalgia rheumatica and giant cell arteritis. Br J Rheumatol (1990) 29:456–8. doi:10.1093/rheumatology/29.6.456
100. Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum (1993) 36:1286–94. doi:10.1002/art.1780360913
101. Baldini M, Maugeri N, Ramirez GA, Giacomassi C, Castiglioni A, Prieto-González S, et al. Selective up-regulation of the soluble pattern-recognition receptor pentraxin 3 and of vascular endothelial growth factor in giant cell arteritis. Arthritis Rheum (2012) 64:854–65. doi:10.1002/art.33411
102. Wagner AD, Goronzy JJ, Weyand CM. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. Evidence for two components of the disease. J Clin Invest (1994) 94:1134–40. doi:10.1172/JCI117428
103. Emilie D, Liozon E, Crevon MC, Lavignac C, Portier A, Liozon F, et al. Production of interleukin 6 by granulomas of giant cell arteritis. Hum Immunol (1994) 39:17–24. doi:10.1016/0198-8859(94)90096-5
104. Weyand CM, Wagner AD, Björnsson J, Goronzy JJ. Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J Clin Invest (1996) 98:1642–9. doi:10.1172/JCI118959
105. Ciccia F, Alessandro R, Rizzo A, Raimondo S, Giardina A, Raiata F, et al. IL-33 is overexpressed in the inflamed arteries of patients with giant cell arteritis. Ann Rheum Dis (2013) 72:258–64. doi:10.1136/annrheumdis-2012-201309
106. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol (2009) 183:6469–77. doi:10.4049/jimmunol.0901575
107. Ciccia F, Alessandro R, Rizzo A, Principe S, Raiata F, Cavazza A, et al. Expression of interleukin-32 in the inflamed arteries of patients with giant cell arteritis. Arthritis Rheum (2011) 63:2097–104. doi:10.1002/art.30374
108. Espígol-Frigolé G, Corbera-Bellalta M, Planas-Rigol E, Lozano E, Segarra M, García-Martínez A, et al. Increased IL-17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant-cell arteritis. Ann Rheum Dis (2013) 72:1481–7. doi:10.1136/annrheumdis-2012-201836
109. Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum (2012) 64:3788–98. doi:10.1002/art.34647
110. Mangan P, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-b induces development of the TH17 lineage. Nature (2006) 441:231–4. doi:10.1038/nature04754
111. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev (2014) 13:668–77. doi:10.1016/j.autrev.2013.12.004
112. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi:10.1038/nature04753
113. Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6 – dependent Th17 cell development and survival. J Exp Med (2008) 205:2281–94. doi:10.1084/jem.20071119
114. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12:568–76. doi:10.1038/ni.2031
115. Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med (2007) 146:621–30. doi:10.7326/0003-4819-146-9-200705010-00004
116. Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med (2007) 13:552–9. doi:10.1038/nm1589
117. Ulevitch R. New therapeutic targets revealed through investigations of innate immunity. Crit Care Med (2001) 29:S8–12. doi:10.1097/00003246-200107001-00004
118. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum (2013) 65:1–11. doi:10.1002/art.37715
119. Scott D, Watts R. Epidemiology and clinical features of systemic vasculitis. Clin Exp Nephrol (2013) 17:607–10. doi:10.1007/s10157-013-0830-8
120. Hilhorst M, Wilde B, van Paassen P, Winkens B, van Breda Vriesman P, Cohen Tervaert JW, et al. Improved outcome in anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis: a 30-year follow-up study. Nephrol Dial Transplant (2013) 28:373–9. doi:10.1093/ndt/gfs428
121. Ntatsaki E, Watts RA, Scott DG. Epidemiology of ANCA-associated vasculitis. Rheum Dis Clin North Am (2010) 36:447–61. doi:10.1016/j.rdc.2010.04.002
122. Harper L. L42. Morbidity in patients with ANCA-associated vasculitis. Presse Med (2013) 42:612–6. doi:10.1016/j.lpm.2013.01.040
123. Holl-Ulrich K. L18. Granuloma formation in granulomatosis with polyangiitis. Presse Med (2013) 42:555–8. doi:10.1016/j.lpm.2013.01.017
124. Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol (1994) 95:244–50. doi:10.1111/j.1365-2249.1994.tb06518.x
125. Weidner S, Neupert W, Goppelt-Struebe M, Rupprecht HD. Antineutrophil cytoplasmic antibodies induce human monocytes to produce oxygen radicals in vitro. Arthritis Rheum (2001) 44:1698–706. doi:10.1002/1529-0131(200107)44:7<1698::AID-ART294>3.0.CO;2-J
126. Tervaert JW, van der Woude FJ, Fauci AS, Ambrus JL, Velosa J, Keane WF, et al. Association between active Wegener’s granulomatosis and anticytoplasmic antibodies. Arch Int Med (1989) 149:2461–5. doi:10.1001/archinte.149.11.2461
127. Hattar K, van Bürck S, Bickenbach A, Grandel U, Maus U, Lohmeyer J, et al. Anti-proteinase 3 antibodies (c-ANCA) prime CD14-dependent leukocyte activation. J Leukoc Biol (2005) 78:992–1000. doi:10.1189/jlb.0902442
128. Rasmussen N, Petersen J. Reactivity of antineutrophil cytoplasmic antibodies with mononuclear phagocytes. J Leukoc Biol (1992) 51:65–8.
129. Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci U S A (1990) 87:4115–9. doi:10.1073/pnas.87.11.4115
130. Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood (2012) 120:3007–18. doi:10.1182/blood-2012-03-416156
131. Moosig F, Csernok E, Kumanovics G, Gross WL. Opsonization of apoptotic neutrophils by anti-neutrophil cytoplasmic antibodies (ANCA) leads to enhanced uptake by macrophages and increased release of tumour necrosis factor-alpha (TNF-a). Clin Exp Immunol (2000) 122:499–503. doi:10.1046/j.1365-2249.2000.01410.x
132. Rastaldi MP, Ferrario F, Crippa A, Dell’Antonio G, Casartelli D, Grillo C, et al. Glomerular monocyte-macrophage features in ANCA-positive renal vasculitis and cryoglobulinemic nephritis. J Am Soc Nephrol (2000) 11:2036–43.
133. Hewins P, Morgan MD, Holden N, Neil D, Williams JM, Savage CO, et al. IL-18 is upregulated in the kidney and primes neutrophil responsiveness in ANCA-associated vasculitis. Kidney Int (2006) 69:605–15. doi:10.1038/sj.ki.5000167
134. Weidner S, Carl M, Riess R, Rupprecht HD. Histologic analysis of renal leukocyte infiltration in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum (2004) 50:3651–7. doi:10.1002/art.20607
135. Wilde B, van Paassen P, Damoiseaux J, Heerings-Rewinkel P, van Rie H, Witzke O, et al. Dendritic cells in renal biopsies of patients with ANCA-associated vasculitis. Nephrol Dial Transplant (2009) 24:2151–6. doi:10.1093/ndt/gfp019
136. Nogueira E, Hamour S, Sawant D, Henderson S, Mansfield N, Chavele KM, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant (2010) 25:2209–17. doi:10.1093/ndt/gfp783
137. Grönhagen-Riska C, Teppo AM, Honkanen E, Ikäheimo R. Alpha-1-antitrypsin, CRP and interleukin-6 in ANCA-positive vasculitis. Adv Exp Med Biol (1993) 336:337–40. doi:10.1007/978-1-4757-9182-2_55
138. Wikman A, Fagergren A, Gunnar O, Johansson S, Lundahl J, Jacobson SH. Monocyte activation and relationship to anti-proteinase 3 in acute vasculitis. Nephrol Dial Transplant (2003) 18:1792–9. doi:10.1093/ndt/gfg216
139. Muller Kobold AC, van Wijk RT, Franssen CF, Molema G, Kallenberg CG, Tervaert JW. In vitro up-regulation of E-selectin and induction of interleukin-6 in endothelial cells by autoantibodies in Wegener’s granulomatosis and microscopic polyangiitis. Clin Exp Rheumatol (1999) 17:433–40.
140. Noronha IL, Krüger C, Andrassy K, Ritz E, Waldherr R. In situ production of TNF-alpha, IL-1 beta and IL-2R in ANCA-positive glomerulonephritis. Kidney Int (1993) 43:682–92. doi:10.1038/ki.1993.98
141. Schreiber A, Pham CT, Hu Y, Schneider W, Luft FC, Kettritz R. Neutrophil serine proteases promote IL-1b generation and injury in necrotizing crescentic glomerulonephritis. J Am Soc Nephrol (2012) 23:470–82. doi:10.1681/ASN.2010080892
142. Novick D, Rubinstein M, Azam T, Rabinkov A, Dinarello CA, Kim SH. Proteinase 3 is an IL-32 binding protein. Proc Natl Acad Sci U S A (2006) 103:3316–21. doi:10.1073/pnas.0511206103
143. Csernok E, Holle JU, Gross WL. Proteinase 3, protease-activated receptor-2 and interleukin-32: linking innate and autoimmunity in Wegener’s granulomatosis. Clin Exp Rheumatol (2008) 26:S112–7.
144. Uehara A, Sato T, Iwashiro A, Yokota S. PR3-ANCA in Wegener’s granulomatosis prime human mononuclear cells for enhanced activation via TLRs and NOD1/2. Diagn Pathol (2009) 4:23. doi:10.1186/1746-1596-4-23
145. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
146. Newman B, Rubin LA, Siminovitch KA. NOD2/CARD15 gene mutation is not associated with susceptibility to Wegener’s granulomatosis. J Rheumatol (2003) 30:305–7.
147. Husmann CA, Holle JU, Moosig F, Mueller S, Wilde B, Cohen Tervaert JW, et al. Genetics of toll like receptor 9 in ANCA associated vasculitides. Ann Rheum Dis (2014) 73:890–6. doi:10.1136/annrheumdis-2012-202803
148. Summers SA, Steinmetz OM, Gan PY, Ooi JD, Odobasic D, Kitching AR, et al. Toll-like receptor 2 induces Th17 myeloperoxidase autoimmunity while Toll-like receptor 9 drives Th1 autoimmunity in murine vasculitis. Arthritis Rheum (2011) 63:1124–35. doi:10.1002/art.30208
149. Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med (1994) 120:12–7. doi:10.7326/0003-4819-120-1-199401010-00003
150. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol (2003) 21:335–76. doi:10.1146/annurev.immunol.21.120601.141126
151. Tadema H, Abdulahad WH, Stegeman CA, Kallenberg CG, Heeringa P. Increased expression of Toll-like receptors by monocytes and natural killer cells in ANCA-associated vasculitis. PLoS One (2011) 6:e24315. doi:10.1371/journal.pone.0024315
152. Martorana D, Maritati F, Malerba G, Bonatti F, Alberici F, Oliva E, et al. PTPN22 R620W polymorphism in the ANCA-associated vasculitides. Rheumatology (2012) 51:805–12. doi:10.1093/rheumatology/ker446
153. Nau GJ, Guilfoile P, Chupp GL, Berman JS, Kim SJ, Kornfeld H, et al. A chemoattractant cytokine associated with granulomas in tuberculosis and silicosis. Proc Natl Acad Sci U S A (1997) 94:6414–9. doi:10.1073/pnas.94.12.6414
154. Lorenzen J, Lovric S, Krämer R, Haller H, Haubitz M. Osteopontin in antineutrophil cytoplasmic autoantibody-associated vasculitis: relation to disease activity, organ manifestation and immunosuppressive therapy. Ann Rheum Dis (2009) 69:1169–71. doi:10.1136/ard.2009.113621
155. Hudkins K, Giachelli CM, Eitner F, Couser WG, Johnson RJ, Alpers CE. Osteopontin expression in human crescentic glomerulonephritis. Kidney Int (2000) 57:105–16. doi:10.1046/j.1523-1755.2000.00813.x
156. Christakos S, Dhawan P, Benn B, Porta A, Hediger M, Oh GT, et al. Vitamin D: molecular mechanism of action. Ann N Y Acad Sci (2007) 1116:340–8. doi:10.1196/annals.1402.070
157. Inoue M, Arikawa T, Chen YH, Moriwaki Y, Price M, Brown M, et al. T cells down-regulate macrophage TNF production by IRAK1-mediated IL-10 expression and control innate hyperinflammation. Proc Natl Acad Sci U S A (2014) 111:5295–300. doi:10.1073/pnas.1321427111
158. Laurino S, Chaudhry A, Booth A, Conte G, Jayne D. Prospective study of TNFα blockade with adalimumab in ANCA-associated systemic vasculitis with renal involvement. Nephrol Dial Transplant (2010) 25:3307–14. doi:10.1093/ndt/gfq187
159. Wegener’s Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med (2005) 352:351–61. doi:10.1056/NEJMoa041884
160. Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med (2010) 363:211–20. doi:10.1056/NEJMoa0909169
161. Mueller A, Holl-Ulrich K, Gross WL. Granuloma in ANCA-associated vasculitides: another reason to distinguish between syndromes? Curr Rheumatol Rep (2013) 15:376. doi:10.1007/s11926-013-0376-5
162. Godman G, Churg J. Wegener’s granulomatosis: pathology and review of the literature. AMA Arch Pathol (1954) 58:533–53.
163. Martinez-Taboada V, Brack A, Hunder GG, Goronzy JJ, Weyand CM. The inflammatory infiltrate in giant cell arteritis selects against B lymphocytes. J Rheumatol (1996) 23:1011–4.
164. Voswinkel J, Assmann G, Held G, Pitann S, Gross WL, Holl-Ulrich K, et al. Single cell analysis of B lymphocytes from Wegener’s granulomatosis: B cell receptors display affinity maturation within the granulomatous lesions. Clin Exp Immunol (2008) 154:339–45. doi:10.1111/j.1365-2249.2008.03775.x
165. Voswinkel J, Müller A, Lamprecht P. Is PR3-ANCA formation initiated in Wegener’s granulomatosis lesions? Granulomas as potential lymphoid tissue maintaining autoantibody production. Ann N Y Acad Sci (2005) 1051:12–9. doi:10.1196/annals.1361.042
166. Tomasson G, Grayson PC, Mahr AD, Lavalley M, Merkel PA. Value of ANCA measurements during remission to predict a relapse of ANCA-associated vasculitis – a meta-analysis. Rheumatology (2012) 51:100–9. doi:10.1093/rheumatology/ker280
167. Kaiser M, Weyand CM, Björnsson J, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum (1998) 41:623–33. doi:10.1002/1529-0131(199804)41:4<623::AID-ART9>3.3.CO;2-Y
Keywords: macrophage, dendritic cell, T cell, granuloma, vasculitis
Citation: Hilhorst M, Shirai T, Berry G, Goronzy JJ and Weyand CM (2014) T cell–macrophage interactions and granuloma formation in vasculitis. Front. Immunol. 5:432. doi: 10.3389/fimmu.2014.00432
Received: 22 July 2014; Paper pending published: 17 August 2014;
Accepted: 23 August 2014; Published online: 12 September 2014.
Edited by:
Augusto Vaglio, University Hospital of Parma, ItalyReviewed by:
Augusto Vaglio, University Hospital of Parma, ItalyF. David Carmona, Instituto de Parasitología y Biomedicina López Neyra CSIC, Spain
Copyright: © 2014 Hilhorst, Shirai, Berry, Goronzy and Weyand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cornelia M. Weyand, Division of Immunology and Rheumatology, School of Medicine Stanford University, CCSR Building, Room 2225, Mail Code 5166, 269 Campus Drive West, Stanford, CA 94305-5166, USA e-mail: cweyand@stanford.edu